
Abstract
Krabbe disease is a lysosomal storage disease caused by galactosylceramidase deficiency, resulting in neurodegeneration with a rapid clinical downhill course within the first months of life in the classic infantile form. This process may be triggered by the accumulation of galactosylceramide (GalCer) in nervous tissues. Both the enzyme galactosylceramidase and its in vivo activator molecule, saposin A, are essential during GalCer degradation. A clinical manifestation almost identical to Krabbe disease is observed when, instead of the galactosylceramidase protein, the saposin A molecule is defective. Saposin A results from posttranslational processing of the precursor molecule, prosaposin, encoded by the PSAP gene. Clinical and neuroimaging findings in a 7-month-old child strongly suggested Krabbe disease, but this condition was excluded by enzymatic and genetic testing. However, at whole exome sequencing, the previously undescribed homozygous, obviously pathogenic PSAP gene NM_002778.3:c.209T>G(p.Val70Gly) variant was determined in the saposin A domain of the PSAP gene. Fibroblast studies showed GalCer accumulation and the activation of autophagy for the first time in a case of human saposin A deficiency. Our patient represents the second known case in the literature and provides new information concerning the pathophysiology of saposin A deficiency and its intralysosomal effects.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Introduction
Lysosomal degradation of glycosphingolipids begins with the cleavage of monosaccharide units from the nonreducing ends of the oligosaccharide chain. This reaction is catalyzed by exohydrolases exhibiting activity in acidic pH optima. Several enzymes require glycoprotein molecules known as low molecular weight activator proteins for these reactions (Kolter and Sandhoff 2005; Sandhoff 2016)
Activator proteins are glycoproteins that provide the structural modifications necessary for binding the enzyme and substrate during degradation of lysosomal glycosphingolipids, which combine with the enzyme or the substrate or sometimes with both. These permit selective degradation of membrane lipids in the intraendosomal and intralysosomal membrane pool without compromise of lysosomal integrity (Spiegel et al. 2005).
These proteins are divided into two main groups, sphingolipid activator proteins (SAPs) and GM2 activator protein (Kolter and Sandhoff 2005). With posttranslational modification, the molecule prosaposin encoded by the PSAP gene is separated into four homologous mature proteins – saposins A–D. These are acidic, enzymatically inactive, thermostable, protease-resistant glycoproteins, 8–11 kDa in size. Despite all their structural similarities, they possess distinct, specific activator functions (Kishimoto et al. 1992).
Inherited activator protein function deficiency leads to the accumulation of undegraded membrane glycosphingolipids in the lysosomal compartments and to lysosomal storage disease (Kolter and Sandhoff 2005). Saposin A is responsible for the activation and stabilization of galactosylceramide beta-galactosidase (GalCer degradation) (Harzer et al. 1997; Morimoto et al. 1989; Pankiv et al. 2007) Deficiency of saposin A results in a Krabbe-like manifestation (Kolter and Sandhoff 2005; Matsuda et al. 2001; Spiegel et al. 2005).
Rather than being the last stage in degradation, the lysosome has been shown to have cellular functions. These include coordinating several intracellular signal pathways, including autophagy, a mechanism necessary for cellular survival (Matsuda et al. 2001; Seranova et al. 2017; Settembre et al. 2008). The impaired degradation mechanism in lysosomal storage diseases compromises autophagic flux, and this has been shown to be one of the mechanisms in the etiopathogenesis of lysosomal storage diseases (Lieberman et al. 2012). Mouse studies have been performed on this subject, but there has been very little examination of saposin A deficiency and its effect on autophagy (Sun and Grabowski 2013).
We report a case of a newly described homozygous mutation in the saposin A domain in the PSAP gene in an infant exhibiting clinical findings of Krabbe disease. To the best of our knowledge, this is the second case following Spiegel et al.’s case report published in 2005, and the first case to be reported from Turkey. This study also presents, for the first time, autophagic findings in human saposin A deficiency.
Material and Methods
Case Report
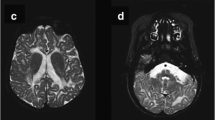
A 7-month-old girl, with first-degree consanguinity between her parents, presented to our clinic due to refractory convulsions. Head control was present at the age of 1 month, but this was lost at the age of 4 months, while feeding difficulty occurred at 5 months and generalized tonic convulsions started at 6 months. At examination on arrival, her head circumference was in the 3rd percentile, weight in the 3rd to 10th percentile, and height in the 25th percentile. No marked dysmorphic feature was observed, and no organomegaly was present. Deep tendon reflexes were increased, and hypertonicity was present. Ammonia, lactate, plasma amino acids, blood spot carnitine and acylcarnitine, and urine organic acid analyses were normal. Cerebrospinal fluid (CSF) proteins were 135 mg/dL (normal for age, 20–50 mg/dL). Bilateral ventricular enlargement, hyperintense lesions in cerebral white matter, and thickening in the optic nerves were determined at cerebral imaging. No increase in cerebral metabolites was observed at cerebral magnetic resonance spectroscopy (Fig. 1). Severe axonal polyneuropathy was determined by electromyography. Galactosylceramidase enzyme analysis was performed due to compatibility with Krabbe disease. Galactosylceramidase activity in dried blood was low (Table 1). No mutation was determined at GALC gene analysis performed due to clinical and cerebral imaging findings compatible with Krabbe disease. Intraleukocytic galactosylceramidase levels investigated for confirmation of enzyme levels were low (Table 2). However, the patient’s intraleukocytic galactosylceramidase enzyme activity was higher than the enzyme levels of patients with Krabbe disease. When comparison with other lysosomal enzymes was performed, glucocerebrosidase, beta-galactosidase, hexosaminidase A, hexosaminidase B, alpha-glucosidase, and sphingomyelinase enzyme levels were normal. Assessed in the light of the MR and clinical findings, the suspicion of Krabbe disease was very powerful, but since no mutation was determined at genetic analysis, whole exome sequencing was investigated with a preliminary diagnosis of saposin A deficiency, known to follow a similar clinical course to that of Krabbe disease. At whole exome sequencing, the pathogenic NM_002778.3:c.209T>G (p.Val70Gly) variant not previously described in the PSAP gene was identified and determined as homozygous. This pathogenic variant was confirmed at PSAP gene investigation using Sanger sequencing. Eight pathogenic predictions were demonstrated from DANN, GERP, dbNSFP.FATHMM, MetaLR, MetaSVM, MutationAssessor, MutationTaster, and PROVEAN (vs one benign prediction from LRT) by in silico assessment tools. Allele was not found in Broad gnomAD exomes. Screening of the patient’s mother, father, and sibling identified the same pathogenic variant as heterozygous. The patient’s and family members’ PSAP mutation Sanger sequencing patterns and the patient’s Integrative Genomics Viewer presentation are shown in Fig. 2.

Magnetic resonance imaging of the patient. (a) Axial T2 image; hyperintensity at the periventricular white matter and centrum semiovale. (b) Axial T1 IR (inversion recovery) image; hypointensity at the periventricular white matter and centrum semiovale. (c) Coronal FLAIR image; hyperintensity at deep white matter, subcortical u-fiber is spared. (d) Multivoxel proton MR spectroscopy; choline peak at the level of right centrum semiovale


Sanger sequencing images of proband (a), father (b), mother (c), brother (d), and Integrative Genomics Viewer presentation of PSAP gene of proband (e)
Biochemical Studies
Enzyme Assay
Galactosylceramidase activity was measured by UHPLC MS/MS (Waters Acquity™ UPLC I-Class system) method (Orsini et al. 2012). Substrate was kindly provided by CDC.
Cell Culture
Fibroblasts were obtained from forearm skin with punch biopsy. Fibroblasts were cultured and maintained in DMEM high glucose (Gibco) supplemented with 20% FBS (Gibco) and 1% (vol/vol) penicillin/streptomycin (Gibco). Passage-matched fibroblasts (passages 3–5) were used in all experiments.
Glycosphingolipid (GSL) Extraction and Thin Layer Chromatography
GSLs were extracted from proband and healthy control primary skin fibroblasts, separated and chemically stained as described previously (Sandhoff et al. 2002) with minor modifications. The cells were washed with 1 × PBS and resuspended in 1 mL distilled water. They were then subjected to three successive rounds of freeze-thawing by incubation in a −80°C freezer for 5 min and thawing in cold water. The sample was sonicated for 3 min, followed by lyophilization and extraction two times with 2 mL of acetone. The pellets were re-extracted with solvent mixtures of chloroform/methanol/water, twice in a ratio of 10:10:1 and twice in a ratio of 30:60:8, and all supernatants were pooled. Neutral GSLs were separated on DEAE A-25 as the flow-through and methanol wash. The solvent was evaporated, and the dried lipids were dissolved in the solvent mixture of chloroform/methanol/water in a ratio of 10:10:1. TLC plates, loaded with lipid extracts, were developed with chloroform/methanol/0.2% aqueous CaCl2 (60/35/8) and then dried and stained with orcinol to reveal sugar containing compounds.
Immunofluorescence
Proband and healthy control primary skin fibroblasts were grown on microscope slides. The cells were washed with 1 × PBS and fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature. They were then washed three times with PBS and permeabilized using PBS containing 0.3% TritonX100 at room temperature, followed by blocking for 1 h in PBS containing 10% goat serum and 0.3% TritonX100. Anti-LAMP1 (1:500, abcam – ab24170), Anti-SQSTM1/p62 (1:500, Thermo – PA5–20839), and Anti-hLC3/MAP 1LCA (1:20, R&D Systems – MAB8558) primary antibody incubation was performed overnight at 4°C. The cells were extensively washed with PBS containing 0.05% Tween 20. Alexa Fluor 488-conjugated goat anti-rabbit (1:500, Abcam – ab150077) and Alexa Fluor 568-conjugated goat anti-rat (1:250-Abcam- ab175476) secondary antibody incubation was performed for 1 h at room temperature. The cells were mounted with Fluoroshield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI, Abcam – ab104139). Images were acquired by fluorescence microscopy (OLYMPUS). Colocalization analysis between p62 and LC3A and between LC3A and LAMP1 was performed using Coloc 2 based on Pearson’s coefficients on ImageJ.
Statistical Analysis
GraphPad statistical software was used for statistical analysis. All values are expressed as mean ± S.E.M. Differences were tested using one-way ANOVA. A p-value of less than 0.05 was considered statistically significant.
Results
Altered Ganglioside Pattern in Saposin A-Deficient Fibroblasts
We showed the levels of neutral glycosphingolipids in fibroblasts from saposin A patient (proband) and healthy family members using thin layer chromatography (Fig. 3a). We detected significant increases in the levels of GalCer (Fig. 3b), LacCer (Fig. 3c), Cer (Fig. 3d), and GlcCer (Fig. 3e) in the proband fibroblast cell compared to the control group, 3.5-fold, 1.5-fold, 2-fold, and 1.4-fold, respectively.

Thin layer chromatographic analysis of neutral glycosphingolipid from fibroblasts of saposin A patient (proband) compared to healthy family members (father and brother) and control group (a). GalCer (b), LacCer (c), Cer (d), and GlcCer (e) ratio from proband and her healthy family controls with thin layer chromatography. The relative intensities of the bands on the TLC were analyzed using ImageJ. The data are represented as the mean ± S.E.M. One-way ANOVA was used for statistical analysis (*p < 0.05, **p < 0.025, ***p < 0.0025, and ****p < 0.001)
Increased Numbers of Lysosomes in Saposin A-Deficient Fibroblasts
Fluorescence microscopic imaging of lysosomal-associated membrane protein (LAMP1) revealed increased numbers of lysosomal vesicles in proband fibroblast (Fig. 4c), compared to healthy family members (Fig. 4a, b). The lysosomal vesicles in the fibroblast were measured using ImageJ. The lysosomal intensity measured per cell was 105 for the father, 96 for the son, and 207.5 for the proband. There are at least twofold increases in the proband fibroblast.

Increased number of lysosomes in saposin A patient fibroblasts (proband) compared to healthy controls (father and brother). Immunofluorescence signals of LAMP1 were enhanced in fibroblast of proband (c) compared with her healthy family members (a, b, respectively). Lysosomes (green) were stained with Anti-Lamp1 antibody. Nuclei (blue) were stained with DAPI. 20× microscopic images. An increased number of LAMP1 vesicles in proband fibroblasts were analyzed using ImageJ (d). The data are represented as the mean ± S.E.M. One-way ANOVA was used for statistical analysis (**p < 0.025)
Increased Autophagosome Numbers in Saposin A-Deficient Fibroblast
We determined twofold enhanced LC3 and p62 in the proband fibroblast (Fig. 5b, c, respectively) compared to healthy family members (father and son) (Fig. 5a). Autophagosome maturation was confirmed by colocalization of p62 and LC3 (Filimonenko et al. 2007; Pankiv et al. 2007; Aflaki et al. 2016) (Fig. 5a, d). Autophagosome-lysosome fusion is impaired in saposin A-deficient fibroblasts. We demonstrated that the extent of LAMP1/LC3A colocalization was reduced (ranging from 50 to 60%) in the proband fibroblast compared to those of the father and brother (Fig. 6a, b).

Autophagosomes produced both in proband fibroblasts and in healthy control group (father and brother) (a). Double immunocytochemical staining of fibroblast cells showed colocalization of LC3 with p62. LC3 protein was visualized in red, p62 in green, nuclei in blue, and autophagosomes in yellow. A yellow signal signifies colocalization of LC3A and p62. In order to better visualize the fluorescent signals present within the large merged images, zoom images were shown. In proband fibroblast increased the number of autophagy markers; LC3A (b) and p62 (c) were analyzed using ImageJ. Production of autophagosomes was shown healthy fibroblast cells and proband fibroblast cells. Colocalization analysis of LC3 with p62-labeled lysosomes was performed on ImageJ based on Coloc 2 analysis and Pearson value (d). The data are represented as the mean ± S.E.M. One-way ANOVA was used for statistical analysis (*p < 0.05, **p < 0.025)

Autophagosomes and lysosomes failed to fuse in saposin A patient fibroblasts compared to healthy controls (father and brother). (a) Double immunocytochemical staining of fibroblast cells showed the colocalization of LC3A with LAMP-1 antibodies by fluorescence microscopy. LC3A protein was visualized in red, LAMP1 in green, and nucleus in blue. Yellow signal signifies colocalization of LC3A and LAMP-1. In order to better visualize the fluorescent signals present within the large merged images, zoom images were shown. Fusion between autophagosomes and lysosomes (yellow) was evident in healthy fibroblast cells, but very little in proband fibroblast cells. Colocalization analysis of LC3 with LAMP-1-labeled lysosomes was performed on ImageJ based on Coloc 2 analysis and Pearson value (b). The data are represented as the mean ± S.E.M. One-way ANOVA was used for statistical analysis (**p < 0.025)
Discussion
This report describes a case of clinical Krabbe disease, uncommon for the molecular defect sparing the GALC gene but affecting the PSAP gene by a homozygous pathogenic variant in the gene’s saposin A domain (saposin A deficiency, OMIM #611722) highly compatible with infantile Krabbe disease. Krabbe disease (globoid cell leukodystrophy, OMIM #245200) was first described by Krabbe in 1916 and is an autosomal recessive lysosomal storage disease leading to globoid cell accumulation in brain tissue. Deficiency of the saposin A molecule leads to a Krabbe-like manifestation (Spiegel et al. 2005). Following the first detection of saposin B by Mehl and Jatzkewitz in 1964, diseases caused by deficiency of these small molecules with a very important role in glycosphingolipid degeneration also began being described (Kishimoto et al. 1992). Since saposin A activates galactosylceramidase together with saposin C in vitro, a deficiency of saposin A alone was not thought to lead to a manifestation of disease (Harzer et al. 1997). However, in their 2001 mouse study, Matsuda et al. identified saposin A as a major activator of galactosylceramidase in the brain and showed that saposin A deficiency results in a Krabbe disease-like clinical manifestation (Matsuda et al. 2001). Spiegel et al. reported the first human case of saposin A deficiency in 2005 (Spiegel et al. 2005). Interestingly, Matsuda et al. observed a late-onset clinical phenotype in the sap A−/− mice, while the Spiegel et al. case had an early infantile phenotype. Matsuda et al. attributed this difference to intact saposin C, which could, in addition to its other functions, have compensated for saposin A to some extent but was unable to perform its entire function (Matsuda et al. 2001). In contrast, classic infantile Krabbe findings were observed in Spiegel et al.’s case (2005). Classic infantile Krabbe findings were also present in our patient. Neuromotor development was normal until the 4th month, after which a severe neurodegenerative process commenced. The reason for this pronounced difference between human and mouse models is still unclear, although in a study from 2013, Sun et al. reported a shorter life span of sap A−/− mice and rapid neurodegeneration based on their own experiences (Sun and Grabowski 2013; Sun et al. 2003).
Since our patient’s disease was highly compatible with a clinical manifestation of Krabbe disease but incompatible with the results of enzymatic and GALC gene analysis, all enzymatic and genetic investigations were repeated twice due to a strong suspicion of that disease. Saposin A defect was primarily considered, and the whole exome sequencing method was employed to exclude any other Krabbe-like leukodystrophy. No other relevant change associated with the clinical manifestation was determined in addition to the pathogenic variant identified in the PSAP gene. The considerable similarities between the clinical manifestation in our case and that published by Spiegel et al. initially directed us to the hypothesis of saposin A deficiency (Spiegel et al. 2005). Another problematic point is that our patient’s enzyme level at dried blood and intraleukocytic investigation was higher than that expected in classic disease, but lower than that in normal controls. This is an unexpected finding considering the nature of the activator protein. For example, normal arylsulfatase B levels have been shown in cases of saposin B deficiency (Deconinck et al. 2008). However, in Spiegel et al.’s study, galactosylceramidase activity at intraleukocytic investigation was lower than that normally expected and was normal in fibroblast measurements. It has therefore been postulated that saposin A is required not only for activation but also for stabilization and that the enzyme requires an activator during intraleukocytic measurements. However, due to the use of in vitro detergents during fibroblast measurement, the activator does not affect the level of lack of protein (Spiegel et al. 2005). Additionally, Bradova et al. described low GALC activity in combined saposin A, B, C, and D deficiency (Bradova et al. 1993). Saposin A is very likely to have also in in vitro assays (despite the presence of detergents or liposomes with the GalCer substrate), not only in vivo, some activating influence on GALC activity. Thus, in saposin A deficiency, the missing influence (missing saposin A interaction with GALC and/or GalCer substrate) explains lowered GalCer degradation also in vitro, despite genetically intact GALC enzyme.
The identification of more new cases of saposin A and other activator protein deficiency will permit a better understanding of these proteins’ tissue levels and lysosomal functions and will help clarify the existing uncertainties.
Galactosylsphingosine (psychosine) accumulation is particularly responsible for neurotoxicity and central nervous system involvement in Krabbe disease. Psychosine accumulates, not alone, but together with GalCer. However, accumulation of psychosine has particularly been observed in brain tissue (Harzer et al. 2002). Psychosine accumulation could not be investigated in our study since the specimen taken from our patient was fibroblast tissue, and brain tissue specimens would be needed. Galactosylceramide levels are a significant and specific marker of insufficient galactosylceramidase activity. In vivo studies have shown that saposin A also plays a role in the degradation of GalCer to Cer and also of LacCer to GlcCer in saposin A-deficient mice (Sun et al. 2013). We detected significant increases in the levels of GalCer (Fig. 3b), LacCer (Fig. 3c), Cer (Fig. 3d), and GlcCer (Fig. 3e) in the proband fibroblast cell compared to the control group, 3.5-fold, 1.5-fold, 2-fold, and 1.4-fold, respectively. The absence of any mutation in the GALC gene that codes the protein galactosylceramidase is further evidence that GalCer accumulation is due to saposin A deficiency. Since the specimen taken from our patient was obtained from fibroblast tissue, we think that the accumulation might have been greater had brain tissue or peripheral nerve tissue specimens been investigated.
Galactosylceramidase-deficient (Krabbe) skin fibroblasts have been reported to contain increased LAMP1- and LAMP2-positive vesicles in their fibroblasts (Lim et al. 2016). We observed increased numbers of lysosomal vesicles in proband fibroblast (Fig. 4c), compared to healthy family members (Fig. 4a, b). Increases of at least twofold occur in the proband fibroblast for lysosomal intensity. These data clearly indicate increased expression of LAMP1 and lysosomal vesicles in our saposin A-deficient fibroblast patient, similar to other lysosomal storage diseases.
LC3 and p62 are widely used autophagy markers. LC3 binds specifically to autophagic membranes and remains bound throughout the pathway. p62 functions to deliver ubiquitinated substrates to the autophagosome. p62 interacts with LC3 and keeps it in the inner and outer autophagosome membranes. Autophagosomes then fuse with lysosomes to form autophagolysosomes, which act to degrade their contents. p62 is also degraded in the autophagolysosomes (Aflaki et al. 2016). Several studies have shown accumulation of autophagosome via increased levels of LC3 and p62 in various lysosomal storage disorders such as Fabry disease, Gaucher’s disease, and Pompe disease (Ward et al. 2016). GSL accumulations have also been shown to affect autophagy in lysosomal storage disorder, and saposin A deficiency is also the primary factor for autophagy. Enhancement of two autophagy markers, p62 and LC3, has been observed in the brains of saposin A-deficient mice (Sun and Grabowski 2013; Sun et al. 2013). Ours is the first report of impaired autophagy in the saposin A-deficient patient fibroblast. We determined twofold enhanced LC3 and p62 in the proband fibroblast (Fig. 5b, c, respectively) compared to healthy family members (father and son) (Fig. 5a). These features were attributed to a block in autophagic flux, since elevated levels of the autophagosomal marker LC3A and autophagic substrate p62 were detected in the proband fibroblast. Autophagosome maturation was confirmed by colocalization of p62 and LC3 (Aflaki et al. 2016; Filimonenko et al. 2007; Pankiv et al. 2007; Spiegel et al. 2005) (Fig. 5a, d). These results show that no problem occurred in the formation of autophagosomes in both healthy and patient fibroblasts.
In most lysosomal storage disorders, the autophagic pathway is impaired by lysosomal dysfunction. Blocking of the autophagic pathway occurs as a consequence of a decreased ability of lysosomes to fuse with autophagosomes (Filimonenko et al. 2007; Settembre et al. 2008; Ward et al. 2016). Clearance of autophagosomes occurs via fusion with lysosomes. Here, we investigated the subcellular localization of the lysosomal marker (LAMP1) and the autophagosomal marker LC3A using fluorescence microscopy and demonstrated that the extent of LAMP1/LC3A colocalization was significantly reduced (ranging from 50 to 60%) in the proband fibroblast compared to those of the father and brother (Fig. 6a, b). Our data suggest that accumulation of autophagosomes (Fig. 5b, c) in saposin A-deficient fibroblasts is due to defective clearance caused by impaired autophagosome-lysosome fusion (Fig. 6a, b).
Autophagy has been identified as an important cause of neurodegeneration in neurodegenerative lysosomal storage diseases, including Krabbe disease (Ribbens et al. 2014). Activated autophagy is a significant indicator that the intracellular coordinated lysosomal expression and regulation (CLEAR) system is also compromised. The neuropathology is thought to occur, not solely in association with GalCer or glycosphingolipid accumulation but also with impairment of the CLEAR system (Palmieri et al. 2011). Ours is also the first study to show the presence of autophagy in a case of human saposin A deficiency.
De Duve, who first described the lysosome, said that very little mystery remained regarding its place in genetic diseases. However, many uncertainties in fact remain. The role of all activator proteins, including saposin A, inside the cell, in all likelihood exceeds what we currently know. Much still remains to be explained concerning the roles of activator proteins in the lysosomal and degradation pathways, their interactions with other proteins, and whether or not these will have a role to play in the treatment of lysosomal storage diseases in the future. Further studies and new cases are now required for these questions to be answered. Saposin A deficiency may be accurately described as one of the rarest forms of inborn errors of metabolism. Our case is the second known report in the literature. However, there are thought to be a large number of leukodystrophies still awaiting identification. The most important step in the diagnosis of saposin A and other activator protein deficiencies is clinical suspicion, and it is not possible to identify these diseases in the absence of such suspicion. Investigation directed toward saposin A deficiency must be considered when clinical and radiological diagnosis of Krabbe disease is not supported by enzymatic and genetic methods.
References
Aflaki E, Moaven N, Borger DK, Lopez G, Westbroek W, Chae JJ et al (2016) Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15(1):77–88. https://doi.org/10.1111/acel.12409
Bradova V, Smid F, Ulrich-Bott B, Roggendorf W, Paton BC, Harzer K (1993) Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Hum Genet 92(2):143–152
Deconinck N, Messaaoui A, Ziereisen F, Kadhim H, Sznajer Y, Pelc K et al (2008) Metachromatic leukodystrophy without arylsulfatase A deficiency: a new case of saposin-B deficiency. Eur J Paediatr Neurol 12(1):46–50. https://doi.org/10.1016/j.ejpn.2007.05.004
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179(3):485–500. https://doi.org/10.1083/jcb.200702115
Harzer K, Paton BC, Christomanou H, Chatelut M, Levade T, Hiraiwa M, O'Brien JS (1997) Saposins (sap) A and C activate the degradation of galactosylceramide in living cells. FEBS Lett 417(3):270–274
Harzer K, Knoblich R, Rolfs A, Bauer P, Eggers J (2002) Residual galactosylsphingosine (psychosine) beta-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta 317(1–2):77–84
Kishimoto Y, Hiraiwa M, O'Brien JS (1992) Saposins: structure, function, distribution, and molecular genetics. J Lipid Res 33(9):1255–1267
Kolter T, Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol 21:81–103. https://doi.org/10.1146/annurev.cellbio.21.122303.120013
Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A (2012) Autophagy in lysosomal storage disorders. Autophagy 8(5):719–730. https://doi.org/10.4161/auto.19469
Lim SM, Choi BO, Oh SI, Choi WJ, Oh KW, Nahm M et al (2016) Patient fibroblasts-derived induced neurons demonstrate autonomous neuronal defects in adult-onset Krabbe disease. Oncotarget 7(46):74496–74509. https://doi.org/10.18632/oncotarget.12812
Matsuda J, Vanier MT, Saito Y, Tohyama J, Suzuki K, Suzuki K (2001) A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum Mol Genet 10(11):1191–1199
Morimoto S, Martin BM, Yamamoto Y, Kretz KA, O'Brien JS, Kishimoto Y (1989) Saposin A: second cerebrosidase activator protein. Proc Natl Acad Sci U S A 86(9):3389–3393
Orsini JJ, Martin MM, Showers AL, Bodamer OA, Zhang XK, Gelb MH, Caggana M (2012) Lysosomal storage disorder 4+1 multiplex assay for newborn screening using tandem mass spectrometry: application to a small-scale population study for five lysosomal storage disorders. Clin Chim Acta 413(15–16):1270–1273. https://doi.org/10.1016/j.cca.2012.04.012
Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20(19):3852–3866. https://doi.org/10.1093/hmg/ddr306
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33):24131–24145. https://doi.org/10.1074/jbc.M702824200
Ribbens JJ, Moser AB, Hubbard WC, Bongarzone ER, Maegawa GH (2014) Characterization and application of a disease-cell model for a neurodegenerative lysosomal disease. Mol Genet Metab 111(2):172–183. https://doi.org/10.1016/j.ymgme.2013.09.011
Sandhoff K (2016) Neuronal sphingolipidoses: membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie 130:146–151. https://doi.org/10.1016/j.biochi.2016.05.004
Sandhoff R, Hepbildikler ST, Jennemann R, Geyer R, Gieselmann V, Proia RL et al (2002) Kidney sulfatides in mouse models of inherited glycosphingolipid disorders: determination by nano-electrospray ionization tandem mass spectrometry. J Biol Chem 277(23):20386–20398. https://doi.org/10.1074/jbc.M110641200
Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI, Sarkar S (2017) Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem 61(6):733–749. https://doi.org/10.1042/ebc20170055
Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D et al (2008) A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17(1):119–129. https://doi.org/10.1093/hmg/ddm289
Spiegel R, Bach G, Sury V, Mengistu G, Meidan B, Shalev S et al (2005) A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol Genet Metab 84(2):160–166
Sun Y, Grabowski GA (2013) Altered autophagy in the mice with a deficiency of saposin A and saposin B. Autophagy 9(7):1115–1116. https://doi.org/10.4161/auto.24919
Sun Y, Qi X, Grabowski GA (2003) Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J Biol Chem 278(34):31918–31923. https://doi.org/10.1074/jbc.M302752200
Sun Y, Zamzow M, Ran H, Zhang W, Quinn B, Barnes S et al (2013) Tissue-specific effects of saposin A and saposin B on glycosphingolipid degradation in mutant mice. Hum Mol Genet 22(12):2435–2450. https://doi.org/10.1093/hmg/ddt096
Ward C, Martinez-Lopez N, Otten EG, Carroll B, Maetzel D, Singh R et al (2016) Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim Biophys Acta 1861(4):269–284. https://doi.org/10.1016/j.bbalip.2016.01.006
Acknowledgments
We thank Centogene AG for whole exome sequencing analyses.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: John Christodoulou, MB BS PhD FRACP FRCPA
Appendices
Conflict of Interest
No conflict of interest was declared by the authors.
Financial Disclosure
The authors declared that this study has received no financial support.
Ethics
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Rights and permissions
Copyright information
© 2018 Society for the Study of Inborn Errors of Metabolism (SSIEM)
About this chapter

Cite this chapter
Kose, M. et al. (2018). The Second Case of Saposin A Deficiency and Altered Autophagy. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 44. JIMD Reports, vol 44. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2018_114
Download citation
DOI: https://doi.org/10.1007/8904_2018_114
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-58616-7
Online ISBN: 978-3-662-58617-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)