
Abstract
Mitochondrial fatty acid oxidation is a vital biochemical process for energy metabolism. Among the known fatty-acid metabolism disorders, very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency and medium-chain acyl-CoA dehydrogenase (MCAD) deficiency count among the most frequent. Both are potentially very serious diseases as they carry a risk of severe neurological post-crisis sequelae, and even sudden death. Diagnosis relies on plasma acylcarnitine profile analysis and urine organic acid analysis, followed by genetic testing to confirm diagnosis. However, in some cases, it is crucial to run a specific diagnostic assay for enzyme activity, which is generally performed in leukocytes or fibroblasts. The aim of this study was to address this need, first by developing a MCAD and VLCAD enzyme activity-specific diagnostic assay in fibroblasts (by measuring the reaction products, i.e. enoyl-CoA) via a rapid LC–MS/MS-based technique, and then by testing MCAD-deficient patients (n = 6), VLCAD-deficient patients (n = 10), and control patients (n = 12). MCAD activity was significantly different in the MCAD-deficiency (MCADD) group (mean = 0.07 nmol C8:1 formed/min/mg protein) compared to the control group (mean = 0.36 nmol C8:1 formed/min/mg protein). All MCADD patients showed less than 35% residual MCAD activity. VLCAD activity was significantly decreased in the VLCADD group (mean = 0.06 nmol C16:1 formed/min/mg protein) compared to the control group (mean = 0.86 nmol C16:1 formed/min/mg protein, respectively). All VLCADD patients showed less than 35% residual VLCAD activity. This technique allowed also to confirm that a novel ACADVL gene mutation (c.1400T>C) is responsible for a defective VLCAD activity (residual activity at 10%).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Introduction
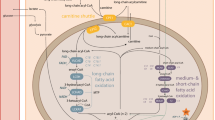
Mitochondrial fatty acid (FA) oxidation is essential for energy production (Houten and Wanders 2010), and a deficiency in the process leads to inability to utilize FA, often resulting in fasting hypoketotic hypoglycemia. Of the 24 known fatty-acid metabolism disorders, autosomal-recessive deficiencies in very-long-chain acyl-CoA dehydrogenase (VLCAD, EC1.3.8.9) (OMIM 609575) and medium-chain acyl-CoA dehydrogenase (MCAD, EC1.3.8.7) (OMIM 201450) count among the more frequent. Both enzymes catalyze the first step of the mitochondrial FA oxidation cycle leading to dehydrogenation of acyl-CoA to enoyl-CoA (Houten and Wanders 2010; Wanders et al. 1999). This step requires flavin adenine dinucleotide (FAD) bound to Electron Transfer Flavoprotein (ETF), which is then reduced into FADH2. VLCAD is bound to the inner mitochondrial membrane whereas MCAD is a matrix enzyme (Houten and Wanders 2010; Wanders et al. 1999). MCAD acts on medium-chain FA (4–14 carbon atoms, optimum activity with 6-carbon atoms). VLCAD acts on long-chain FA (12–22 carbon atoms, optimum activity with 16-carbon atoms) (Hashimoto 1992). Clinical presentation of VLCAD deficiency (VLCADD) may include cardiomyopathy, encephalopathy, hypoglycemia, and rhabdomyolysis (Arnold et al. 2009). MCAD deficiency (MCADD) is associated with hypoketotic hypoglycemia, hepatic symptoms that are predominantly related to intercurrent illnesses or prolonged fasting. Sudden death related to heartbeat disorders may also occur in adults (Feillet et al. 2012).
For VLCADD and MCADD, diagnosis is first suspected by plasma acylcarnitine and urine organic acid analyses, and has to be confirmed by molecular biology (Feillet et al. 2012; Vianey-Saban et al. 1998).
MCADD management specifies that enzyme activity must confirm diagnosis if molecular biology is not contributory. In addition, as a post mortem plasma acylcarnitine profile is very difficult to interpret, an enzyme activity assay can prove important in the exploration of sudden death (Feillet et al. 2012).
Strategy is similar for VLCADD where an enzyme activity assay may be even more valuable given the heterogeneity of ACADVL gene mutations (Vianey-Saban et al. 1998).
A method that measures the production of octenoyl-CoA (C8:1-CoA) (after addition of octanoyl-CoA as substrate) or hexadecenoyl-CoA (C16:1-CoA) (after addition of palmitoyl-CoA as substrate) by tandem mass spectrometry in lymphocytes was published (Tajima et al. 2008; ter Veld et al. 2009). The method uses ferrocenium hexafluorophosphate, an artificial electron acceptor for MCAD or VLCAD, to specifically determine MCAD and VLCAD activity within a day, but has never been further developed for analysis in fibroblasts.
Methods
Patients
Skin fibroblasts (75 cm2 flask per strain, i.e. around 6.106 cells) grown in Ham’s culture medium added with 12% fetal calf serum are harvested at confluence (Centre de Biotechnologie Cellulaire, Hospices Civils de Lyon, Bron). After trypsinization, washing in phosphate-buffered saline (PBS) and aspirating the supernatant, dry-harvested fibroblast pellets (12 controls, 10 known VLCAD-deficient patients, 6 known MCAD-deficient patients) were frozen at −80°C for storage.
The pellets were then resuspended with 300 μL of ammonium acetate (10 mM, pH = 8) and sonicated, in ice, for three × 15 sec bursts at 30 sec intervals.
Tests were also carried out in control-sample fibroblasts to evaluate analytical performance.
Reagents
Ammonium acetate and 2-(N-morpholino)ethanesulfonic acid (MES) were purchased from Merck (Darmstadt, Germany). Acetonitrile, octanoyl-CoA (C8:0), hexadecenoyl-CoA (C16:0), and ferrocenium hexafluorophosphate were obtained from Sigma-Aldrich (Deisenhofen, Germany).
Protein Assay
The total protein assay in sonicated fibroblast homogenates was performed in a 1:20 dilution on an ABX Pentra400® benchtop analyzer (Horiba Medical) using a colorimetric technique based on the bicinchoninic acid method (Thermo Scientific, Rockford, USA).
Procedures
Sonicated fibroblast homogenates, adjusted to a protein concentration of 0.2 g/L with ammonium acetate 10 mM, pH = 8, were diluted (1:1 V/V) in ferrocenium hexafluorophosphate solution (400 μM, pH = 8) to a final volume of 100 μL. The enzymatic reaction was started at 37°C by adding the substrate in MES buffer (20 mM, pH = 6): 4 μL of C8:0 (5 mM) to assay MCAD activity; 4 μL of C16:0 (5 mM) to assay VLCAD activity. After 15 min incubation, the reaction was stopped by adding 100 μL of ice-cold acetonitrile. A T0 timepoint measure was taken for each condition by simultaneously adding substrate and acetonitrile. After centrifugation at 14,000 g for 8 min at 4°C, the supernatant was transferred to autosampler vials with inserts for HPLC–mass spectrometry.
The analyte samples were injected (20 μL for the MCAD assay, 5 μL for the VLCAD assay) and chromatographic separation was carried out using a C18 Uptisphere column (50×2.1 mm, particle size: 3 μm) (Interchim, Montluçon, France). Flowrate was 200 μL/min. Eluent was a 70:30 (V/V) mixture (for MCAD assay) or 45:55 (V/V) mixture (for VLCAD assay) of ammonium acetate (10 mM, pH = 8) and acetonitrile. After chromatographic separation, the samples were analyzed by electrospray ionization–tandem mass spectrometry (ESI-MS/MS, Api 3200, Sciex, Les Ulis, France) in positive ion mode. The products derived directly from the MCAD-catalyzed reaction, i.e. octenoyl-CoA (C8:1), and the VLCAD-catalyzed reaction, i.e. hexadecenoyl-CoA (C16:1), were quantified. The products of the second step in the FA oxidation process were also assayed, i.e. 3-hydroxyoctanoyl-CoA (C8OH) and 3-hydroxypalmitoyl-CoA (C16OH), respectively. The product ions were studied in multiple reaction monitoring (MRM) mode, with each transition corresponding to substrate (C8:0 m/z 894.2>387.3, and C16:0 m/z 1006.4>499.4) or to products of the enzymatic reaction (C8:1 m/z 892.2>385.3, and C8OH m/z 910.2>403.3 or C16:1 m/z 1004.4>497.4 and C16OH m/z 1022.4>515.4). Working up from the peak areas obtained, the quantities of reaction products were calculated from substrate area at timepoint T0. As C8:1, C16:1, C8OH, and C16OH are not commercially available, response of the substrate and response of the enzymatic reaction products were considered identical.
Expression of Results
Enzymatic activities are expressed in nmol C8:1 (MCAD) or C16:1 (VLCAD) formed per minute per mg of proteins. For patients with MCADD or VLCADD, residual activity was evaluated as a ratio of the activity level of the control enzyme, i.e. VLCAD for MCAD-deficient patients and MCAD for VLCAD-deficient patients. The MCAD-to-VLCAD or VLCAD-to-MCAD ratios are then normalized by the mean of the ratios of the control group tested in the same experiment, thus giving (MCAD-to-VLCAD)-to-control and (VLCAD-to-MCAD)-to-control ratios.
Statistical Analysis
Group-wise results (control, MCADD, VLCADD) for MCAD or VLCAD enzymatic activity are reported as means (SD; CI95%). Between-group comparisons (control, MCADD, VLCADD) of MCAD (or VLCAD) activity were performed by one-way ANOVA, followed by a Tukey’s post hoc test for pairwise comparisons between significantly different groups. All statistical analyses were performed using GraphPad® Prism 5 software. Statistical significance was set at p <0.05.
Results
Optimization of the Method
The final selected analytical parameters were set as follows: protein concentration = 0.1 g/L, incubation time = 15 min, ferrocenium hexafluorophosphate concentration = 200 μM, final substrate (C8 or C16) concentration = 0.2 mmol/L.
Intra-assay coefficient of variation (CV) assessed via 10 repeated measures on the same control cell line was 4.3% (mean 0.47 nmol/min/mg protein) for MCAD activity and 10.8% (mean 0.74 nmol/min/mg protein) for VLCAD activity. Inter-assay CV assessed via 5 measurements carried out over 5 days on the same control cell line was 19% (mean 0.41 nmol/min/mg protein) for MCAD activity and 31% (mean 0.92 nmol/min/mg protein) for VLCAD activity.
Figure 1 illustrates chromatograms obtained in MRM mode for a patient with MCADD (Fig. 1a) or VLCADD (Fig. 1b), compared to a control patient with a decrease in signal intensity for the products C8:1 or C16:1 and C8OH or C16OH without concomitant change in signal intensity for substrate C8 or C16 (in surplus).

MRM-mode chromatograms obtained after MCAD (a) and VLCAD (b) enzyme reactions. For each molecule, peaks are shown at identical scale between control patient and known MCAD-deficient (a) or known VLCAD-deficient (b) patient. (a) Substrate octanoyl-CoA (C8) and products octenoyl-CoA (C8:1) and 3-hydroxyoctanoyl-CoA (C8OH) were assayed. (b) Substrate palmitoyl-CoA (C16) and products hexadecenoyl-CoA (C16:1) and 3-hydroxypalmitoyl-CoA (C16OH) were assayed
Population Studied
Three patient groups were studied: a group of 12 control patients, a group of 10 previously diagnosed VLCAD-deficient patients (P1–P10), and a group of six previously diagnosed MCAD-deficient patients (P11–P16). For each patient, MCAD and VLCAD activity assays were performed in duplicate within a same series and over two independent experimental runs.
Key clinical data on patients P1–P16 are reported in Table 1. All enzyme deficiencies were confirmed by molecular biology except for patient P11 who was only heterozygous for the frequent c.985A>G mutation. Diagnosis was confirmed for this patient by an enzymatic activity assay using ETF as electron acceptor (Bertrand et al. 1992). All the identified mutations (Table 1) have been described as deleterious in the literature and/or by predictions software (Alamut®, Interactive biosoftware).
Enzymatic Activities in the Study Population
Mean MCAD activity in the control group was 0.36 nmol C8:1 formed/min/mg protein (SD: 0.07; CI95%: 0.31–0.40). Mean MCAD activity in known VLCAD-deficient patients was 0.30 nmol C8:1 formed/min/mg protein (SD: 0.09; CI95%: 0.25–0.35). Mean MCAD activity in known MCAD-deficient patients was 0.07 nmol C8:1 formed/min/mg protein (SD: 0.08; CI95%: 0.00–0.13), which is significantly different to the control and “VLCAD-deficiency” groups (p <0.0001) (Fig. 2a).

Mean MCAD (a) and VLCAD (b) enzyme activities per patient group: control patients (n = 12), known VLCAD-deficient patients (n = 10) (VLCADD) and known MCAD-deficient patients (n = 6) (MCADD). For each patient, enzymatic reaction was performed in duplicate over two independent experiments. *p < 0.0001
Mean VLCAD activity in the control group was 0.86 nmol C16:1 formed/min/mg protein (SD: 0.028; CI95%: 0.68–1.04). Mean VLCAD activity in known MCAD-deficient patients was 0.73 nmol C16:1 formed/min/mg protein (SD: 0.49; CI95%: 0.35–1.11). Mean VLCAD activity in known VLCAD-deficient patients was 0.06 nmol C16:1 formed/min/mg protein (SD: 0.07; CI95%: 0.02–0.10), which is significantly different to the control and “MCAD-deficiency” groups (p <0.0001) (Fig. 2b).
All the known VLCADD and MCADD patients showed less than 35% residual VLCAD or MCAD activity (Table 1).
Discussion
MCADD and VLCADD are autosomal-recessive inborn errors of metabolism. Diagnosis is first suggested by plasma acylcarnitine profile analysis and urine organic acid analysis, and generally has to be confirmed by molecular biology. But sometimes a specific MCAD and VLCAD enzyme activity assay may prove necessary in cases involving non-contributive genetics or cases where patients refuse consent for genetic testing. In this context we developed a tandem mass spectrometry assay of MCAD/VLCAD activity in fibroblasts.
We first optimized the technique originally developed by Tajima et al. (2008) and ter Veld et al. (2009) in lymphocytes. Wanders et al. (2010) also used the ferrocenium hexafluorophosphate in a HPLC coupled to UV-detection assay of MCAD/VLCAD activity in fibroblasts. Although we ultimately retained the same ferrocenium hexafluorophosphate concentration (200 μM) and the same amount of substrate (20 nmol) as the original method, we opted for a 15 min incubation time at 37°C (instead of 5 min originally) and a 0.1 g/L protein concentration (instead of 0.03 g/L originally). Analytical performances for VLCAD activity are weaker than for MCAD activity, while the CVs remain acceptable for a tandem mass spectrometry technique. This can be explained by the fact that VLCAD is a membrane bound enzyme and therefore offers more random substrate availability than MCAD which is a matrix-soluble enzyme, despite the sonication step.
We applied this new method to analyze fibroblasts from MCAD-deficient or VLCAD-deficient patients. The activity of the deficient enzyme was significantly lower in the corresponding patient groups. Each individual patient with MCADD or VLCADD showed a significant decrease in the corresponding enzyme activity.
However, three particular cases draw our attention.
In patient P1, who died at day 2 of life with only post mortem organic acid profile evocative of long-chain FAO defect, two mutations were identified in ACADVL gene. Mutation c.1837C>T (p.Arg613Trp) has previously been described in the literature (Souri et al. 1996). The second substitution, c.1400T>C (p.Ile467Thr), has never been described (zero Pubmed, Ensembl, Clinvar, or ExAc Browser hits) and was classified as intermediate-severity by the prediction software. Both c.1837C>T and c.1400T>C mutations were properly segregated in the parents. The enzyme activity assay demonstrates its full utility in this case and enables diagnosis to be confirmed with a mean 10% residual VLCAD activity.
For patient P11, presenting with MCADD of good prognosis, only one heterozygous mutation was identified in ACADM gene. No other mutation was found in exons and flanking intronic regions of the second allele. The MCAD residual activity of 8% confirms plasma acylcarnitine and urine organic acid analyses. This method is easier and shorter to achieve than the previous method using ETF (Bertrand et al. 1992).
For patient P13, presenting with MCADD of good prognosis, two mutations were identified in the ACADM gene, both of which have been described in the literature (McKinney et al. 2004, Waddell et al. 2006). Mutation c.127G>A (p.Glu43Lys) has been described in homozygous state in a patient who also presented a mutation in heterozygous state on the gene coding for SCAD (McKinney et al. 2004). Mutation c.617G>A, which leads to substitution of a positively charged side-chain amino acid (arginine) at position 206 by a same-family amino acid (histidine), has been described in a compound heterozygous c.617G>A/c.985A>G patient (Waddell et al. 2006). Both mutations are classified as intermediate-severity by prediction software, which is an understandable conclusion given how c.617G>A is a conservative mutation. The enzyme activity assay demonstrates its utility and enables diagnosis to be confirmed with a mean 33% residual MCAD activity compared to controls. French consensus guidelines on diagnostic screening have set the confirmatory cut-off as less than 25% residual activity (Feillet et al. 2012). However, the guidelines were put together based on results obtained with the reference technique using ETF in fluorescence analysis, and it is now time to redefine a new threshold (possibly 35%) with this technique. Furthermore, the clinical picture and type of mutations point towards a moderate form of MCAD deficiency and explain why residual activity was found to be around 33%. Analysis of other patients is needed to define this new threshold.
Conclusion
The tandem mass spectrometry assay of MCAD/VLCAD activity in fibroblasts developed here offers powerful selective diagnostic screening of MCAD-deficient and VLCAD-deficient patients.
Abbreviations
- ETF:
-
Electron Transfer Flavoprotein
- FA:
-
Fatty acids
- FAD:
-
Flavin adenine dinucleotide
- HPLC:
-
High-performance liquid chromatography
- MCAD:
-
Medium-chain acyl-CoA dehydrogenase
- MCADD:
-
MCAD deficiency
- MES:
-
2-(N-morpholino)ethanesulfonic acid
- OMIM:
-
Online Mendelian Inheritance in Man
- VLCAD:
-
Very-long-chain acyl-CoA dehydrogenase
- VLCADD:
-
VLCAD deficiency
References
Arnold GL, Van Hove J, Freedenberg D et al (2009) A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 96(3):85–90
Bertrand C, Dumoulin R, Divry P, Mathieu M, Vianey-Saban C (1992) Purification of electron transfer flavoprotein from pig liver mitochondria and its application to the diagnosis of deficiencies of acyl-CoA dehydrogenases in human fibroblasts. Clin Chim Acta 210(1-2):75–91
Feillet F, Ogier H, Cheillan D et al (2012) Medium-chain acyl-CoA-dehydrogenase (MCAD) deficiency: french consensus for neonatal screening, diagnosis, and management. Arch Pediatr 19(2):184–193
Hashimoto T (1992) Peroxisomal and mitochondrial enzymes. Prog Clin Biol Res 375:19–32
Houten SM, Wanders RJA (2010) A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis 33:469–477
McKinney JT, Longo N, Hahn SH et al (2004) Rapid, comprehensive screening of the human medium chain acyl-CoA dehydrogenase gene. Mol Genet Metab 82(2):112–120
Souri M, Aoyama T, Orii K, Yamaguchi S, Hashimoto T (1996) Mutation analysis of very-long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency: identification and characterization of mutant VLCAD cDNAs from four patients. Am J Hum Genet 58(1):97–106
Tajima G, Sakura N, Shirao K et al (2008) Development of a new enzymatic diagnosis method for very-long-chain Acyl-CoA dehydrogenase deficiency by detecting 2-hexadecenoyl-CoA production and its application in tandem mass spectrometry-based selective screening and newborn screening in Japan. Pediatr Res 64(6):667–672
ter Veld F, Mueller M, Kramer S et al (2009) A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns. PLoS One 4:e6449
Vianey-Saban C, Divry P, Brivet M et al (1998) Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta 269(1):43–62
Waddell L, Wiley V, Carpenter K et al (2006) Medium-chain acyl-CoA dehydrogenase deficiency: genotype-biochemical phenotype correlations. Mol Genet Metab 87(1):32–39
Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, IJlst L (1999) Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis 22(4):442–487
Wanders RJ, Ruiter JP, IJLst L, Waterham HR, Houten SM (2010) The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis 33(5):479–494
Acknowledgments
The authors thank A.T.T. (Auvergne Traduction Technique®) for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Cornelis Jakobs
Appendices
Take-Home Message
The reader will learn about the usefulness of MCAD and VLCAD activity measurement and the performance of the method developed in fibroblasts.
Conflict of Interest
Damien Bouvier, Christine Vianey-Saban, Séverine Ruet, and Cécile Acquaviva declare that they have no conflict of interest.
Compliance with ethics guidelines: Informed consent was obtained from all patients for being included in the study.
Authors Contribution
Damien Bouvier and Severine Ruet have performed technical part of the study.
Damien Bouvier, Christine Vianey-Saban, Severine Ruet, and Cécile Acquaviva have performed analysis and interpretation of the data.
Damien Bouvier drafted the article.
Christine Vianey-Saban and Cécile Acquaviva revised the article.
Rights and permissions
Copyright information
© 2016 Society for the Study of Inborn Errors of Metabolism (SSIEM)
About this chapter
Cite this chapter
Bouvier, D., Vianey-Saban, C., Ruet, S., Acquaviva, C. (2016). Development of a Tandem Mass Spectrometry Method for Rapid Measurement of Medium- and Very-Long-Chain Acyl-CoA Dehydrogenase Activity in Fibroblasts. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 35. JIMD Reports, vol 35. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2016_22
Download citation
DOI: https://doi.org/10.1007/8904_2016_22
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-55832-4
Online ISBN: 978-3-662-55833-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)