
Abstract
Infantile Refsum disease (IRD) is one of the less severe of Zellweger spectrum disorders (ZSDs), a group of peroxisomal biogenesis disorders resulting from a generalized peroxisomal function impairment. Increased plasma levels of very long chain fatty acids (VLCFA) and phytanic acid are biomarkers used in IRD diagnosis. Furthermore, an increased plasma level of phytanic acid is known to be associated with neurologic damage. Treatment of IRD is symptomatic and multidisciplinary.
The authors report a 3-year-old child, born from consanguineous parents, who presented with developmental delay, retinitis pigmentosa, sensorineural deafness and craniofacial dysmorphisms. While the relative level of plasma C26:0 was slightly increased, other VLCFA were normal. Thus, a detailed characterization of the phenotype was essential to point to a ZSD. Repeatedly increased levels of plasma VLCFA, along with phytanic acid and pristanic acid, deficient dihydroxyacetone phosphate acyltransferase activity in fibroblasts and identification of the homozygous pathogenic mutation c.2528G>A (p.Gly843Asp) in the PEX1 gene, confirmed this diagnosis. Nutritional advice and follow-up was proposed aiming phytanic acid dietary intake reduction. During dietary treatment, plasma levels of phytanic acid decreased to normal, and the patient’s development evaluation showed slow progressive acquisition of new competences.
This case report highlights the relevance of considering a ZSD in any child with developmental delay who manifests hearing and visual impairment and of performing a systematic biochemical investigation, when plasma VLCFA are mildly increased. During dietary intervention, a biochemical improvement was observed, and the long-term clinical effect of this approach needs to be evaluated.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Dietary treatment
- Infantile Refsum disease
- Peroxisomal biogenesis disorder
- Phytanic acid
- Psychomotor development
- Very long chain fatty acids
Introduction
Infantile Refsum disease (IRD; OMIM#601539) is the less severe form of Zellweger spectrum disorders (ZSDs), which are a continuum of phenotypes with clinical, biochemical and genetic overlap – Zellweger syndrome, neonatal adrenoleukodystrophy and IRD. Presenting neonatally with hypotonia, large fontanels, failure to thrive and cholestatic jaundice, IRD is later recognizable by developmental delay, retinitis pigmentosa, sensorineural deafness, hepatomegaly, growth retardation and milder dysmorphic features (Baumgartner et al. 1998; Poll-The et al. 2004). The disease progresses slowly and some patients may survive into adulthood, depending on the level of clinical care (Poll-The et al. 2004; Crane et al. 2005).
The IRD diagnosis is made by a combination of clinical manifestations with biochemical and molecular testing, as well as macro- and microscopic examinations and immunocytochemistry analysis of peroxisomes (Aubourg and Wanders 2013). Diagnostic tools rely on the fact that IRD is an autosomal recessive peroxisomal biogenesis disorder (PBD) caused by pathogenic variants in the PEX genes, which encode peroxins that are essential to peroxisomal assembly and the protein import system, resulting in none or a few abnormally formed peroxisomes, whose functions are generally impaired (Aubourg and Wanders 2013). Since very long chain fatty acids (VLCFA) beta-oxidation is a unique metabolic function of peroxisomes, the identification of an increased level of VLCFA in plasma, fibroblasts and amniotic fluid cells is a biomarker of ZSD (Wanders 2014). Elevated plasma levels of phytanic and pristanic acids and bile acid precursors (BAP), as well as reduced plasmalogen levels in erythrocytes, are additional biochemical abnormalities that point to a ZSD (Wanders 2014). A reduced dihydroxyacetone phosphate acyltransferase (DHAP-AT) activity in fibroblasts and amniocytes confirms the postnatal and prenatal diagnosis of ZSD (Wanders et al. 1995). Finally, identification of pathogenic mutations in a PEX gene (http://www.dbpex.org) is useful for diagnosis, prognosis, and management of a ZSD, but it also enables carrier testing of at-risk relatives and prenatal or preimplantation diagnosis (Ebberink et al. 2011).
Management of IRD is multidisciplinary and treatment remains symptomatic (Braverman et al. 2013). Reports of treatment impact in disease progression are limited. However, sporadically reports have showed that changes in diet lead to specific biochemical effects (Robertson et al. 1988; Moser et al. 1999).
Herein, we describe an IRD patient with mild biochemical phenotype who had a reduction of plasma phytanic acid levels after onset of dietary management. Slight developmental progress was observed and retinopathy did not evolve significantly during follow-up.
Methods
The following peroxisomal biochemical analyses were performed, as previously reported, with minor modifications: plasma and fibroblast VLCFA quantification (Moser et al. 1999), plasma phytanic and pristanic acid level quantification (Dacremont et al. 1995), fibroblast DHAP-AT enzymatic activity measurement (Wanders et al. 1995), erythrocyte plasmalogen level quantification (Bjorkhem et al. 1986), plasma BAP measurement (Shimada et al. 2001), plasma polyunsaturated fatty acid level measurement (Bailey-Hall et al. 2008) and antibody anti-catalase immunofluorescence labelling (Wanders et al. 1989). Genomic DNA was extracted from peripheral blood, using Qiagen BioRobot EZ1 apparatus with EZ1 DNA blood 350 μl kit (according to the manufacturer’s instructions). PCR products of all PEX1 exons and flanking regions (reference number, NM_000466.2) were analysed by Sanger sequencing using BigDye Terminator v1.1 Cycle Sequencing Kit and 3130xl Genetic Analyzer (Applied Biosystems) (PCR primers and conditions are available upon request).
The nutritional management to reduce phytanic acid intake from the diet consisted in decreasing global fat intake, mainly from ruminant meats, dairy products and high fat content fish. Griffiths Mental Development Scales were used to evaluate psychomotor development.
Results
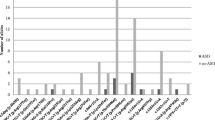
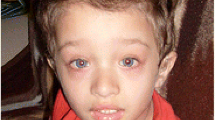
A 3-year- and 6-month-old girl was referred to the Medical Genetics outpatient clinic due to retinitis pigmentosa, sensorineural hearing loss (SNHL), developmental delay and dysmorphic craniofacial features (Fig. 1a). She was born from consanguineous healthy parents, who were first cousins, and she had two older twin brothers, who manifested hypotonia and psychomotor developmental delay associated with extreme prematurity. During gestation, nuchal translucency above the 95th percentile for gestational age prompted a fetal karyotype and a fetal echocardiography, which were unremarkable. She was born at 38 weeks, and, in the neonatal period, hypotonia and jaundice were diagnosed. Low-amplitude and high-frequency bilateral vertical nystagmus was first noticed at 3 months of age. At 6 months, bilateral pigment deposits became apparent in the retinal mid-periphery of both ocular fundi. The following months, these pigment deposits assumed round circular shapes instead of the more typical bone spicule form and approached the posterior pole, while the maculas acquired a salt and pepper appearance and eventually pigment scattering (Fig. 1b). Electroretinography was performed according to ISCEV standards at 4 years of age and demonstrated an extinguished rod response with a diminished maximum combined rod-cone response, compatible with retinitis pigmentosa. Bilateral and symmetric hyperopic astigmatism was also diagnosed and is currently corrected with glasses. Due to intellectual disability, visual acuity could not be reliably tested. Despite the severe and generalized photoreceptor dysfunction, both parents and other caretakers report good functional vision for indoor and outdoor activities, without tripping or collisions that suggested a markedly reduced visual field, and do not report significant limitations in scotopic conditions (nyctalopia), typical of severe retinitis pigmentosa. Both fundoscopic lesions and hyperopia remained stable during the last 3 years of follow-up. Cerebral MRI did not show brain abnormalities at 10 months old. Seizures were not reported. Profound bilateral SNHL at frequencies 2,000–4,000 Hz was confirmed at 2 years and 11 months old, by auditory evoked potentials. However, conditioned play audiometry showed a 40 dB loss at 250–2,000 Hz bilaterally with the use of hearing aids, placed at approximately 3 years old, and an increased social interaction was concomitantly observed. She seated alone at 8 months and started walking at 24 months. At 4 years, developmental evaluation showed a severe developmental delay across all areas (GQ = 31) (Fig. 1c). Height and weight progressed along the 5th percentile curve, while head circumference was growing along the 25th percentile curve. At 3 years old, electrocardiography was normal and echocardiography showed patent ductus arteriosus with small left-right shunt but good biventricular function.

(a) Facial dysmorphic features observed in the proband, at 4 years old, included high forehead, absent orbital ridges, and micrognathia. (b) Bilateral retinographies, obtained at 4 years old, showing diffuse pigment epithelium changes in the retinal mid-periphery, as well as mottled appearance in the macular area, compatible with retinitis pigmentosa. (c) Performance level (age of development in months) in five subscales of the Griffiths Mental Development Scales at 4, 5 and 6 years of age
Diagnosis of ZSD was considered, due to hearing and visual impairment associated with developmental delay and mild dysmorphic features. At 16 months, quantification of plasma levels of VLCFA did not clearly support this diagnosis, since C26:0 and C24:0/C22:0 ratios were within the normal range and the C26:0/C22:0 ratio was slightly increased (Table 1). However, at 33 years and 11 months old, increased plasma levels of VLCFA in combination with increased phytanic acid and pristanic acid levels supported the hypothesis of a peroxisomal disorder, either a ZSD or a D-bifunctional protein deficiency. The decreased activity of DHAP-AT in fibroblasts was in accordance with the diagnosis of a ZSD. A pathogenic homozygous missense mutation c.2528G>A, p.(Gly843Asp), was detected in the PEX1 gene, establishing a ZSD diagnosis for this patient. Parents are carriers of the same pathogenic variant. Catalase immunofluorescence analysis showed a punctuate fluorescence pattern in control cells, due to the catalase presence within the peroxisome compartment. In this patient, however, the catalase is predominately scattered in the cytosol, and this leads to a diffuse rather than a punctuate fluorescence pattern (Fig. 2). From this spectrum, considering the clinical phenotype, this patient can be classified as an IRD.

Distribution of peroxisomes in cultured skin fibroblasts from a healthy control (a) and from the patient (b), visualized by immunofluorescence microscopy using an anti-catalase antibody
After diagnosis, nutritional advice and follow-up was proposed. In the first nutritional appointment, at four years and two months old, the fat intake recorded was 29% of the total energy intake (1800 kcal/day). The nutritional prescription reduced the total of fat into 15% of the total energy intake. Diet was also supplemented with maltodextrins, in order to prevent catabolism, resulting in a higher total energy intake (1900 kcal/day). At 7, 12, 18 and 25 months after starting the dietary intervention, plasma levels of phytanic acid lowered to normal range. The subsequent nutritional appointments confirmed the excellent compliance with the proposed diet. The successive developmental evaluations revealed slight progress in all evaluated areas (at 6 years, GQ = 34), most significantly in locomotor and autonomy/sociability (Fig. 1c). However, her performance was significantly conditioned by a short attention span in more structured tasks.
Discussion
We report a 3-year-old child with IRD, in whom a thorough characterization of the clinical phenotype was critical to support the hypothesis of a peroxisomal disorder, in face of first-tier mild biochemical abnormalities. IRD was considered due to developmental delay associated with hearing loss and progressive retinopathy, since facial dysmorphic features were misleading. Although neurological dysfunction observed in the neonatal period resembled a neuromuscular disorder, later in life, our patient’s phenotype also resembled Usher type II, congenital defects of glycosylation or mitochondrial respiratory chain defects. Diagnosing IRD may be complicated due to its phenotypic as well as genotypic heterogeneity, leading eventually to an underestimation of its true prevalence. Since the onset and severity of manifestations is variable and numerous clinical differential diagnoses exist, a clinical suspicion of IRD – based on neurologic, developmental and sensory deficits – should prompt a systematic biochemical investigation of PBD (Baumgartner et al. 1998), including measurements in plasma, erythrocytes and skin fibroblasts, to show defects in the α-oxidation, β-oxidation and synthesis of ether phospholipids.
The repeated measurement of plasma VLCFA, along with phytanic acid and pristanic acid, proved valuable in the identification of a ZSD in this specific patient, which was subsequently confirmed by analysis of the DHAP-AT activity in fibroblasts. Biochemical diagnosis of IRD may be challenging in patients with normal or mildly increased plasma VLCFA levels (Gootjes et al. 2004; Zeharia et al. 2007). Indeed, older individuals were shown to have lower plasma ratios of C24:0/C22:0 and C26:0/C22:0 fatty acids when compared with children under one year (Hall et al. 1988). Since normal VLCFA plasma levels do not exclude the diagnosis of IRD, this clinical case highlights the need of repeated plasma VLCFA measurements or analysis of additional biochemical parameters, including plasma phytanic acid or erythrocyte plasmalogens, to increase the diagnostic rate of IRD patients (Krause et al. 2009).
The mild clinical and biochemical phenotype in this patient is likely to be explained by the type of mutation detected in the PEX1 gene, as well as by peroxisomal mosaicism, which was observed in the patient’s fibroblasts. This child is homozygous for the pathogenic missense variant c.2528G>A (p.Gly843Asp) in PEX1, affecting the second ATP-binding domain of the protein and which may enable transcription into mRNA and translation into protein to a certain extent (Crane et al. 2005). As a consequence, the binding between PEX1 and PEX6 is reduced but not abolished. This interaction is essential for the peroxisomal protein import system (Tamura et al. 2001). In addition, this pathogenic variant influences the PEX1 activity in a temperature-sensitive manner, i.e. while at 37°C import of matrix proteins into “ghost” peroxisomes is observed in some cells (peroxisomal mosaicism), at 30°C, peroxisomal import is almost completely recovered, as well as peroxisomal metabolic functions, and at 40°C, no peroxisomal import is observed (Imamura et al. 1998). Overall, this pathogenic variant retains a residual peroxisomal function which, along with peroxisomal mosaicism, results in less severe biochemical deficiencies, a milder clinical phenotype and prolonged survival (Osumi et al. 2000; Poll-The et al. 2004; Crane et al. 2005).
This IRD patient was treated with a phytanic acid-restricted diet, since there is increasing evidence on its toxicity, namely, disturbing normal lipid homeostasis (van den Brink and Wanders 2006). Additionally, carbohydrate supplementation was crucial to maintain an adequate energy intake. This led to persistent plasma phytanic acid level normalization after the onset of the dietary regimen. Similar biochemical results were previously reported in few patients with IRD treated with a low phytanic acid diet (Robertson et al. 1988; Pakzad-Vaezi and Maberley 2014). However, the long-term clinical benefit of this approach remains to be elucidated. Slow developmental progression was concomitantly observed in this patient. Nonetheless, since the prognosis of IRD is variable, the slow disease progression or halting of the disease progression may be due to the natural history of the disease. Since phytanic acid and pristanic acid originate exclusively from exogenous sources, its accumulation in ZSD patients is dependent on diet, as well as age (Wanders et al. 2011). Theoretically, the measurement of plasma phytanic acid levels may be regarded as a response marker to the low phytanic acid intake diet. Furthermore, accumulation of phytanic acid over time, through dietary intake, is a diagnostic marker of PBD (Aubourg and Wanders 2013). Nonetheless, since intermittent normalization of plasma phytanic acid levels may also be observed in IRD patients, possibly depending on their diet (Poll-The et al. 2004), its value as a biochemical diagnostic parameter may be less reliable, prompting the need to combine it with other biochemical diagnostic parameters.
The missense variant c.2528G>A (p.Gly843Asp) in PEX1 is the most commonly found pathogenic mutation in different patient cohorts (25–40% of the ZSD patients) (Ebberink et al. 2011) and is associated with a mild phenotype, meaning that IRD patients may live several decades (Crane et al. 2005). Accordingly, an earlier diagnosis will enable a more effective intervention in these patients. Systematic evaluation of long-term biochemical, clinical and developmental effects of a low phytanic acid intake diet in IRD patients may prove whether this could be a useful supplement to the recommended management of this disorder.
References
Aubourg P, Wanders R (2013) Peroxisomal disorders. In: Dulac O, Lassonde M, Sarnat HB (eds) Handbook of clinical neurology. Elsevier B.V, Amsterdam, pp 1593–1609
Bailey-Hall E, Nelson EB, Ryan AS (2008) Validation of a rapid measure of blood PUFA levels in humans. Lipids 43:181–186
Baumgartner MR, Poll-The BT, Verhoeven NM et al (1998) Clinical approach to inherited peroxisomal disorders: a series of 27 patients. Ann Neurol 44:720–730
Bjorkhem I, Sisfontes L, Bostrom B, Kase BF, Blomstrand R (1986) Simple diagnosis of the Zellweger syndrome by gas-liquid chromatography of dimethylacetals. J Lipid Res 27:786–791
Braverman NE, D'Agostino MD, Maclean GE (2013) Peroxisome biogenesis disorders: biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev 17:187–196
Crane DI, Maxwell MA, Paton BC (2005) PEX1 mutations in the Zellweger spectrum of the peroxisome biogenesis disorders. Hum Mutat 26:167–175
Dacremont G, Cocquyt G, Vincent G (1995) Measurement of very long-chain fatty acids, phytanic and pristanic acid in plasma and cultured fibroblasts by gas chromatography. J Inherit Metab Dis 18(Suppl 1):76–83
Ebberink MS, Mooijer PA, Gootjes J, Koster J, Wanders RJ, Waterham HR (2011) Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat 32:59–69
Gootjes J, Skovby F, Christensen E, Wanders RJ, Ferdinandusse S (2004) Reinvestigation of trihydroxycholestanoic acidemia reveals a peroxisome biogenesis disorder. Neurology 62:2077–2081
Hall NA, Lynes GW, Hjelm NM (1988) Ratios for very-long-chain fatty acids in plasma of subjects with peroxisomal disorders, as determined by HPLC and validated by gas chromatography-mass spectrometry. Clin Chem 34:1041–1045
Imamura A, Tamura S, Shimozawa N et al (1998) Temperature-sensitive mutation in PEX1 moderates the phenotypes of peroxisome deficiency disorders. Hum Mol Genet 7:2089–2094
Krause C, Rosewich H, Gartner J (2009) Rational diagnostic strategy for Zellweger syndrome spectrum patients. Eur J Hum Genet 17:741–748
Moser AB, Jones DS, Raymond GV, Moser HW (1999) Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res 24:187–197
Osumi T, Imamura A, Tsukamoto T et al (2000) Temperature sensitivity in peroxisome assembly processes characterizes milder forms of peroxisome biogenesis disorders. Cell Biochem Biophys 32:165–170
Pakzad-Vaezi KL, Maberley DA (2014) Infantile Refsum disease in a young adult: case presentation and brief review. Retin Cases Brief Rep 8:56–59
Poll-The BT, Gootjes J, Duran M et al (2004) Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients. Am J Med Genet A 126A(4):333–338
Robertson EF, Poulos A, Sharp P et al (1988) Treatment of infantile phytanic acid storage disease: clinical, biochemical and ultrastructural findings in two children treated for 2 years. Eur J Pediatr 147:133–142
Shimada K, Mitamura K, Higashi T (2001) Gas chromatography and high-performance liquid chromatography of natural steroids. J Chromatogr A 935:141–172
Tamura S, Matsumoto N, Imamura A et al (2001) Phenotype-genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p-Pex6p interaction. Biochem J 357:417–426
van den Brink DM, Wanders RJ (2006) Phytanic acid: production from phytol, its breakdown and role in human disease. Cell Mol Life Sci 63:1752–1765
Wanders RJ (2014) Metabolic functions of peroxisomes in health and disease. Biochimie 98:36–44
Wanders RJ, Wiemer EA, Brul S, Schutgens RB, van den Bosch H, Tager JM (1989) Prenatal diagnosis of Zellweger syndrome by direct visualization of peroxisomes in chorionic villus fibroblasts by immunofluorescence microscopy. J Inherit Metab Dis 12(Suppl 2):301–304
Wanders RJ, Ofman R, Romeijn GJ et al (1995) Measurement of dihydroxyacetone-phosphate acyltransferase (DHAPAT) in chorionic villous samples, blood cells and cultured cells. J Inherit Metab Dis 18(Suppl 1):90–100
Wanders RJ, Komen J, Ferdinandusse S (2011) Phytanic acid metabolism in health and disease. Biochim Biophys Acta 1811:498–507
Zeharia A, Ebberink MS, Wanders RJ et al (2007) A novel PEX12 mutation identified as the cause of a peroxisomal biogenesis disorder with mild clinical phenotype, mild biochemical abnormalities in fibroblasts and a mosaic catalase immunofluorescence pattern, even at 40 degrees C. J Hum Genet 52:599–606
Acknowledgements
The authors thank the patient and her family for their invaluable contribution to this study. The authors are very thankful to Arjan P.M. de Brouwer for his critical revision of this manuscript. The authors are grateful to Amplifon, in particular Wendy Lopes, for providing the results of the tonal audiometry of the patient. This work is part of the Clinical Genetics fellowship of Maria João Nabais Sá in the Department of Medical Genetics, Centro de Genética Médica Dr. Jacinto de Magalhães/Centro Hospitalar do Porto, Porto, Portugal. Parts of these data were presented at the European Human Genetics Conference 2013 (Paris, France) as a poster (P12.007), as well as at the Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium 2014, both as poster (P-316; Abstract ID: LATTAK-301994-795734-SSIEM2014), and at the SSIEM-Dietetics Group Meeting 2014, as oral communication.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Jutta Gaertner
Appendices
Synopsis
In spite of being a peroxisomal biogenesis disorder, infantile Refsum disease may have residual peroxisomal activity once we obtained a decrease of phytanic acid plasma levels, along with a low phytanic acid diet, in a 3-year-old child with the common pathogenic p.(Gly843Asp) mutation in the PEX1 gene.
Compliance with Ethics Guidelines
Conflict of Interest
Maria João Nabais Sá, Júlio C. Rocha, Manuela F. Almeida, Carla Carmona, Esmeralda Martins, Vasco Miranda, Miguel Coutinho, Rita Ferreira, Sara Pacheco, Francisco Laranjeira, Isaura Ribeiro, Ana Maria Fortuna and Lúcia Lacerda declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the patient’s parents for being included in the study. Proof that informed consent was obtained is available upon request.
Details of the Contributions of Individual Authors
Maria João Nabais Sá, Júlio C. Rocha and Lúcia Lacerda contributed pertinent aspects of the planning, conducting and reporting of the work described in the article. Maria João Nabais Sá wrote the first draft of this manuscript. All the authors critically revised the original draft manuscript for intellectual content and approved the version submitted for publication.
Maria João Nabais Sá made the clinical diagnosis, requested and interpreted the necessary diagnostic procedures, conducted the biochemical investigation, is in charge of the follow-up of the patient, performed the family screening and counselling, collected the protocol baseline and historical and follow-up clinical data of the patient and performed and selected the corresponding photographs, included in Fig. 1a of this manuscript.
Lúcia Lacerda supervised the diagnostic biochemical and molecular studies of the peroxisomal biogenesis disorder and was responsible for their interpretation and reporting.
Esmeralda Martins, Vasco Miranda and Miguel Coutinho did the necessary diagnostic procedures, namely, the biochemical investigation, ophthalmologic evaluation and audiological evaluation, respectively, are in charge of the follow-up and treatment of the patients and collected the protocol baseline and historical and follow-up clinical data of the patient.
Vasco Miranda performed and selected the photographs included in Fig. 1b of this manuscript.
Júlio C. Rocha and Manuela F. Almeida are in charge of the nutritional follow-up and treatment of the patient.
Carla Carmona conducted the psychomotor development assessment of the patient and designed the bar chart included in Fig. 1c.
Rita Ferreira, Sara Pacheco and Isaura Ribeiro performed the biochemical assays and were responsible for their interpretation and reporting. Isaura Ribeiro performed and selected the photograph included in Fig. 2 of this manuscript.
Francisco Laranjeira performed the molecular analysis of the PEX1 gene and was responsible for its interpretation and reporting.
Ana Maria Fortuna supervised the clinical and laboratory content of this manuscript, as Head of the Department of Medical Genetics, Centro de Genética Médica Dr. Jacinto de Magalhães/Centro Hospitalar do Porto, Porto, Portugal.
Rights and permissions
Copyright information
© 2015 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sá, M.J.N. et al. (2015). Infantile Refsum Disease: Influence of Dietary Treatment on Plasma Phytanic Acid Levels. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 26. JIMD Reports, vol 26. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2015_487
Download citation
DOI: https://doi.org/10.1007/8904_2015_487
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-49832-3
Online ISBN: 978-3-662-49833-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)