
Abstract
In glycogen storage disease type III (GSD III), deficiency of the debranching enzyme causes storage of an intermediate glycogen molecule (limit dextrin) in the affected tissues. In subtype IIIa hepatic tissue, skeletal- and cardiac muscle tissue is affected, while in subtype IIIb only hepatic tissue is affected. Cardiac storage of limit dextrin causes a form of cardiomyopathy, which resembles primary hypertrophic cardiomyopathy on cardiac ultrasound. We present a 32-year-old GSD IIIa patient with severe left ventricular hypertrophy (LVH) first diagnosed at the age of 8 years. LVH remained stable and symptomless until the patient presented at age 25 years with increasing dyspnea, fatigue, obesity, and NYHA (New York Heart Association) functional classification two out of four. Dyspnea, fatigue, and obesity progressed, and at age 28 years she was severely symptomatic with NYHA classification 3+ out of 4. On echocardiogram and electrocardiogram, the LVH had progressed as well. Initially, she was rejected for cardiac transplantation because of severe obesity. Therefore, a 900 cal, high protein diet providing 37% of total energy was prescribed during 4 months on which 10 kg weight loss was achieved. However, her symptoms as well as the electrocardiographic and echocardiographic LVH indices had improved dramatically – ultimately deferring cardiac transplantation. Thereafter, the caloric intake was increased to 1,370 cal per day, and the high protein intake was continued providing 43% of total energy. After 3 years of follow-up, the patient remains satisfied with reasonable exercise tolerance and minor symptoms in daily life.
Competing interests: None declared.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Left Ventricular Hypertrophy
- High Protein Diet
- Glycogen Storage Disease Type
- Concentric Left Ventricular Hypertrophy
- Debranching Enzyme
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Glycogen storage disease type III (GSD III) is an autosomal recessive disorder in which a mutation in the AGL gene causes deficiency of the debranching enzyme (DE). The DE consists of two active centers, which catalyze the last step in the conversion of glycogen to glucose (Smit et al. 2006). The absence of DE activity in GSD III patients causes storage of an intermediate form of glycogen, limit dextrin (LD) (Chen 2001). Eighty-five percent of the GSD III patients have subtype IIIa in which DE is deficient in muscle and liver tissue. Fifteen percent of the patients have subtype IIIb in which DE is deficient in the liver (Shen et al. 1996). In neonates and infants, the main features are hepatomegaly, keto-hypoglycemic episodes after short periods of fasting, and hyperlipidemia. Poorly treated neonates and children have developmental delay, growth retardation, and delayed puberty. Proximal and distal myopathy presents in adult GSD IIIa patients, which may be enforced by the development of peripheral neuropathy (Wolfsdorf and Weinstein 2003). Cardiomyopathy is a frequent complication in GSD IIIa since LD can also store in myocardial cells and between bundles of myofilaments (Moses et al. 1989; Smit et al. 1990; Labrune et al. 1991; Coleman et al. 1992; Carvalho et al. 1993; Talente et al. 1994). This causes a form of cardiomyopathy that echocardiographically resembles primary hypertrophic cardiomyopathy due to sarcomere gene mutations, but shows a different response to exercise testing, 24-h electrocardiographic monitoring and thallium-201 myocardial scintigraphy (Lee et al. 1997; Akazawa et al. 1997; Olson et al. 1984). The clinical significance and long-term consequences of GSD IIIa-related cardiomyopathy are unclear due to a lack of data and experience.
The aim of the dietary treatment of GSD III is to divide the carbohydrate intake throughout the day to maintain normoglycemia by taking frequent meals and regular cornstarch doses (Gremse et al. 1990). Protein supplementation is necessary as it serves as a substrate for gluconeogenesis during fasting conditions and improves myopathy and growth failure (Slonim et al. 1982, 1984; Kiechl et al. 1999). However, there is no consensus between centers on the usage of a high protein diet or the amount of cornstarch that should be provided.
In this case report, we present a GSD IIIa patient with severely symptomatic hypertrophic cardiomyopathy, which was reversed after initiating a low calorie, high protein diet to achieve weight loss for a cardiac transplantation preparation program. Subsequently, her cardiomyopathy-related symptoms and signs improved dramatically and cardiac transplantation could be deferred.
Case Report
The patient reported is a 32-year-old Turkish female born to consanguineous parents after an uncomplicated pregnancy and birth. She was diagnosed with glycogen storage disease in Turkey at a young age, but subtyping for GSD III was only initiated at the age of 8 years after she moved to the Netherlands. Enzymatic measurements confirmed absent DE activity in muscle- and liver tissue, confirming GSD IIIa. Genetic mutation analysis revealed a pathologic homozygote four basepair deletion in exon 7 of the AGL gene (GeneBank genomic reference sequence NW_012865) c.753_756delCAGA causing a frameshift (Lucchiari et al. 2006). Upon clinical evaluation, the main findings were hepatomegaly with elevated aspartate transaminase (203 U/L), alanine transferase (253 U/L), triglycerides (1.7 mmol/L), and creatine kinase values (2,112 U/L). A grade III out of VI systolic cardiac murmur was heard in the 4th left intercostal space. Further cardiac evaluation revealed hypertrophic cardiomyopathy with concentric left ventricular hypertrophy (LVH). On the ECG, no rhythm- or conduction disturbances were seen. Following diagnosis, dietary treatment was initiated with protein-enriched frequent meals during the day and one late night meal. She responded well to the treatment and no hypoglycaemic episodes requiring hospitalization have occurred since. During puberty, the liver size normalized, but the transaminase values remained elevated. Echocardiographically the LVH remained stable and symptomless; therefore, no further cardiologic follow-up was deemed necessary.
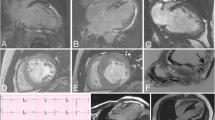
At age 25 years, the patient presented in the outpatient-ward with increasing dyspnea, fatigue, obesity, and functional classification according to the New York Heart Association (NYHA) two out of four (Table 1). On physical examination, her heart rate was 69 bpm, blood pressure 100/60 mmHg, with mild jugular venous pressure elevation. Her electrocardiogram showed increased QRS-voltage and -duration with negative T-waves comparable with severe LVH (Fig. 1). The Sokolow-Lyon-, Cornell Voltage- and Romhilt-Estes electrocardiographic indices for LVH were all positive (Fig. 1). The echocardiogram showed severe concentric LVH with intraventricular septum (IVS) thickness of 22 mm, and a posterior wall (PW) thickness of 18 mm with normal dimensions (Table 1). She was treated with low-dose furosemide, fluid restriction, and her diet was adjusted to provide extra protein during the day. Also, a late night feed combined with cornstarch was added to ensure normoglycemia during the night. In the following years, the symptoms of dyspnea and fatigue slowly progressed, and she gained weight. At age 28 years, she was severely symptomatic with a NYHA functional class of 3+ out of 4. The patient’s BMI increased to 32.2 because she avoided physical exercise as this provoked palpitations and chest pain. The electrocardiographic and echocardiographic LVH indices worsened accordingly (IVS thickness 32 mm, PW thickness 25 mm). At that time low-dose perindopril and carvedilol were added to the furosemide, and a prophylactic implantable cardioverter defibrillator was placed due to an increased risk of sudden cardiac death.

Twelve-lead electrocardiograpy showing severe left ventricular hypertrophy with secondary repolarization abnormalities at the age of 25, 29, 30, and 32 years old
Due to the worsening situation, the patient was evaluated for heart transplantation (HTX) at the age of 30 years. Therefore, a pre-HTX program was set up consisting of: (1) evaluation of the status of liver and skeletal muscle, (2) a new dietary regimen to reduce weight by 10 kg, (3) peri-operative advice regarding the management of GSD III during the HTX. The new dietary regimen consisted of 24-h protein-enriched naso-gastric drip feeding containing 900 cal per day, with protein providing 37%, carbohydrates 61%, and lipids 2% providing of total energy. After following the new dietary regimen for 4 months, her BMI decreased to 27.7, along with a significant improvement of her complaints of constant fatigue and exercise intolerance. The NYHA classification decreased accordingly to two out of four. On cardiac ultrasound, concentric LVH was still present but the PW and IVS thickness had decreased to 25 mm and 24 mm, respectively, along with the electrocardiographic LVH indices. Consequently, the patient was taken off the pre-HTX program, and cardiac transplantation was deferred. The caloric intake was increased to 1,370 cal per day, and the continuous naso-gastric drip feeding was stopped. The dietary regimen was switched to seven meals at daytime with 2-h intervals. Five of these meals were drinks, and two were normal meals, which were comparable in composition of energy and protein to the drinks. In the nocturnal period, two gifts of cornstarch were added to reach a 4-h interval between meals. The high protein nature of the diet was maintained, and even increased to provide 43% of total energy per day compared to 37% protein per day during the pre-HTX program. After 3 years of follow-up, she remains satisfied with reasonable exercise tolerance and minor symptoms in daily life.
Discussion
We report a severely symptomatic GSD IIIa patient with cardiomyopathy who improves clinically and objectively on a high protein diet with a limited supply of carbohydrates. To our knowledge, a similar case has been reported recently in a pediatric patient (Valayannopoulos et al. 2011), and once in an adult patient by Dagli et al. in 2009 (Dagli et al. 2009). The latter describes a 22-year-old male with severe GSD III-related cardiomyopathy treated with a high protein diet in which overtreatment with cornstarch was avoided. Their patient improved dramatically with reversal of symptoms and echocardiographic signs of hypertrophic cardiomyopathy.
The physiology of the reversal of extreme LVH in GSD IIIa is not clear. A direct effect, where the limited supplementation of carbohydrate and increased usage of protein in gluconeogenesis reduces the cardiac storage of LD, is feasible. An indirect effect is also feasible where the weight reduction reduces fat disposition in skeletal muscles, improving the condition and possible workload of the skeletal muscles and reducing the workload on the heart (Kelley et al. 1991; Goodpaster et al. 1999). A combination of these effects is also possible.
The majority of the patients seem to remain asymptomatic and the cardiomyopathy seems to be nonprogressive (Lee et al. 1997). Therefore, severe cases such as these emphasize the need for regular cardiac follow-up in patients with GSD IIIa-related cardiomyopathy, along with the benefits of a high protein diet. In our population of GSD III patients, a high protein diet is implemented at a young age and continued through into adulthood. Through these dietary regimens, GSD III patients usually derive 20–30% of total energy from protein, which varies from 4 g of protein per kilogram body weight in children and adolescents to 2 g of protein per kilogram body weight in adult patients. Therefore, renal function is also closely monitored during follow-up of these patients.
This report presents the first case of a GSD IIIa patient in whom cardiac transplantation could be deferred after initiating a low calorie, high protein diet. In younger unaffected GSD IIIa patients, this approach may prevent the development of symptomatic cardiomyopathy.
References
Akazawa H, Kuroda T, Kim S, Mito H, Kojo T, Shimada K (1997) Specific heart muscle disease associated with glycogen storage disease type III: clinical similarity to the dilated phase of hypertrophic cardiomyopathy. Eur Heart J 18:532–533
Carvalho JS, Matthews EE, Leonard JV, Deanfield J (1993) Cardiomyopathy of glycogen storage disease type III. Heart Vessels 8:155–159
Chen YT (2001) Glycogen storage diseases. In: Scriver CR et al (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill Professional, New York, pp 1521–1551
Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT (1992) Glycogen storage disease type III (glycogen debranching enzyme deficiency): correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med 116:896–900
Dagli AI, Zori RT, McCune H, Ivsic T, Maisenbacher MK, Weinstein DA (2009) Reversal of glycogen storage disease type IIIa-related cardiomyopathy with modification of diet. J Inherit Metab Dis Epub Mar 30
Goodpaster BH, Kelley DE, Wing RR, Meier A, Theate FL (1999) Effects of weight loss on insulin sensitivity in obesity – influence of regional adiposity. Diabetes 48:839–847
Gremse DA, Bucuvalas JC, Balisteri WF (1990) Efficacy of cornstarch therapy in type III glycogen-storage disease. Am J Clin Nutr 52:671–674
Kelley DE, Slasky S, Janosky J (1991) Effects of obesity and non-insulin dependent diabetes mellitus. Am J Clin Nutr 54:509–515
Kiechl S, Willeit J, Vogel W, Kohlendorfer U, Poewe W (1999) Reversible severe myopathy of respiratory muscles due to adult-onset type III glycogenosis. Neuromuscul Disord 9:408–410
Labrune P, Huguet P, Odievre M (1991) Cardiomyopathy in glycogen-storage disease type III: clinical and echographic study of 18 patients. Pediatr Cardiol 12:161–163
Lee PJ, Deanfield JE, Burch M, Baig K, McKenna WJ, Leonard JV (1997) Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am J Cardiol 79:834–838
Lucchiari S, Pagliarani S, Salani S et al (2006) Hepatic and neuromuscular forms of glycogenosis type III: nine mutations in AGL. Hum Mutat 27:600–601
Moses SW, Wanderman KL, Myroz A, Frydman M (1989) Cardiac involvement in glycogen storage disease type III. Eur J Pediatr 148:764–766
Olson LJ, Reeder GS, Noller KL, Edwards WD, Howell RR, Michels VV (1984) Cardiac involvement in glycogen storage disease III: morphologic and biochemical characterization with endomyocardial biopsy. Am J Cardiol 53:980–981
Shen J, Bao Y, Liu HM, Lee PJ, Leonard JV, Chen YT (1996) Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest 98:352–357
Slonim AE, Weisberg C, Benke P, Evans OB, Burr IM (1982) Reversal of debrancher deficiency myopathy by the use of high-protein nutrition. Ann Neurol 11:420–422
Slonim AE, Coleman RA, Moses SW (1984) Myopathy and growth failure in debrancher enzyme deficiency: improvement with high protein noctural enteral therapy. J Pediatr 105:906–911
Smit GP, Fernandes J, Leonard JV et al (1990) The long-term outcome of patients with glycogen storage diseases. J Inherit Metab Dis 13:411–418
Smit GPA, Rake JP, Akman HO, DiMauro S (2006) The glycogen storage diseases and related disorders. In: Fernandes J, Saudubray J-M, van den Berghe G, Walter JH (eds) Inborn metabolic diseases: diagnosis and treatment. Springer Medizin, Heidelberg, pp 103–116
Talente GM, Coleman RA, Alter C et al (1994) Glycogen storage disease in adults. Ann Intern Med 120:218–226
Valayannopoulos V, Bajolle F, Arnoux JB et al (2011) Successful treatment of severe cardiomyopathy in glycogen storage disease type III with D,L-3-hydroxybutyrate, ketogenic and high protein diet. Pediatr Res 70(6):638–641
Wolfsdorf JI, Weinstein DA (2003) Glycogen storage diseases. Rev Endocr Metab Disord 4:95–102
Acknowledgments
We thank Margreet van Rijn, Ph.D. and Els B. Haagsma, M.D., Ph.D. of the University Medical Centre Groningen for providing information on the natural course and dietary regimen.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Alberto B Burlina.
Synopsis
Synopsis
Hypertrophic cardiomyopathy in glycogen storage disease type IIIa may be reversible by a low calorie, high protein diet, even in severely symptomatic patients for whom cardiac transplantation is being considered.
Rights and permissions
Copyright information
© 2011 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sentner, C.P., Caliskan, K., Vletter, W.B., Smit, G.P.A. (2011). Heart Failure Due to Severe Hypertrophic Cardiomyopathy Reversed by Low Calorie, High Protein Dietary Adjustments in a Glycogen Storage Disease Type IIIa Patient. In: JIMD Reports - Case and Research Reports, 2012/2. JIMD Reports, vol 5. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2011_111
Download citation
DOI: https://doi.org/10.1007/8904_2011_111
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-28095-5
Online ISBN: 978-3-642-28096-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)