
Abstract
Mitochondrial disorders are a heterogeneous group of often multisystemic and early fatal diseases caused by defects in the oxidative phosphorylation (OXPHOS) system. Given the complexity and intricacy of the OXPHOS system, it is not surprising that the underlying molecular defect remains unidentified in many patients with a mitochondrial disorder. Here, we report the clinical features and diagnostic workup leading to the elucidation of the genetic basis for a combined complex I and IV OXPHOS deficiency secondary to a mitochondrial translational defect in an infant who presented with rapidly progressive liver failure, encephalomyopathy, and severe refractory lactic acidemia. Sequencing of the GFM1 gene revealed two inherited novel, heterozygous mutations: a.539delG (p.Gly180AlafsX11) in exon 4 which resulted in a frameshift mutation, and a second c.688G > A (p.Gly230Ser) mutation in exon 5. This missense mutation is likely to be pathogenic since it affects an amino acid residue that is highly conserved across species and is absent from the dbSNP and 1,000 genomes databases. Review of literature and comparison were made with previously reported cases of this recently identified mitochondrial disorder encoded by a nuclear gene. Although limited in number, nuclear gene defects causing mitochondrial translation abnormalities represent a new, rapidly expanding field of mitochondrial medicine and should potentially be considered in the diagnostic investigation of infants with progressive hepatoencephalomyopathy and combined OXPHOS disorders.
Competing interests: None declared.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mitochondrial Protein Synthesis
- Mitochondrial Translation
- OXPHOS System
- Depletion Syndrome
- Multicystic Kidney
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Mitochondria are ubiquitous intracellular organelles present in virtually all eukaryotic cells. They are relics of an ancestral alpha-proteobacterial endosymbiont (Gray et al. 1999) that took permanent residence in our cells. Genetic disorders of mitochondrial respiratory chain are the most common group of inborn errors of metabolism, collectively affecting approximately 1 in 5,000 births (Skladal et al. 2003). Mitochondrial dysfunction encompasses an extraordinary assemblage of clinical phenotypes, commonly manifesting in tissues with high energy requirements, such as brain, retina, heart, muscle, liver, and endocrine systems (Cwerman-Thibault et al. 2011). It has also been implicated in a variety of diseases, including common multifactorial disorders such as diabetes (Gerbitz et al. 1996), Parkinson’s disease (Mizuno et al. 1995; Mandemakers et al. 2007), and cancer (Brandon et al. 2006).
The complex and intricate nature of the oxidative phosphorylation (OXPHOS) system, which consists of about 90 proteins encoded by both the nuclear and the mitochondrial genome, explains the clinical heterogeneity associated with genetic defects in OXPHOS (Munnich et al. 1992). In this report, we describe an infant presenting with early fatal hepatoencephalopathy resulting from combined deficiencies of complex I and IV due to mutations in a nuclear gene encoding mitochondrial translational factor EFG1.
Case Report
The patient was the second child of a healthy, non-consanguineous Chinese couple, born at term via Cesarean section for intrauterine growth retardation and abnormal lie. The Apgar scores were good. Her weight of 2.03 kg and her head circumference of 30 cm were both below the third percentile, while her length was 49 cm, at the 25th percentile. On day 2 of life, she was noted to be lethargic, tachypnoeic, and hypoglycemic, with a venous blood sugar of 1.7 mmol/L. She had mild hyperammonemia, 167 μmol/L (normal <50), refractory raised anion gap metabolic acidosis (~22–30) with a pH of 6.8, greatly elevated serum lactate of 17–25 mmol/L (normal <2.4), and CSF lactate of 12 mmol/L (normal <2.1). She was ventilated and had single volume exchange transfusion followed by peritoneal dialysis performed in an effort to control the acidosis. Her urine organic acids showed excessive excretion of lactate, 3-OH butyric, 2-OH butyric acids, 4-OH phenyllactate, and ketonuria. Her acylcarnitines were normal and her serum amino acids revealed elevated alanine at 1,228 μmol/L (normal 122–546), glutamine 1,114 μmol/L (normal 59–561), methionine 128 μmol/L (normal 10–79), phenylalanine 165 μmol/L (normal 31–157), and tyrosine 569 μmol/L (normal 5–167). Liver function test showed hypoproteinemia (protein 42 and albumin 24 g/L), raised alkaline phosphatase 516 IU/L (age-related reference range 0–341), gamma-glutamyl transferase 523 IU/L (normal 11–50), mildly elevated alanine aminotransferase 66 U/L (normal 0–54), aspartate aminotransferase 86 U/L (normal 0–82), and total bilirubin of 108 μmol/L (normal 0–17). She responded to peritoneal dialysis with normalization of acidosis and a reduction of serum lactate to 6 mmol/L. Echocardiogram was normal and ultrasound brain showed dilated ventricles. She had notable generalized hypotonia with myopathic facies and subtle dysmorphism including flat nasal bridge, low-set ears, high, broad forehead, and smooth philtrum. She was suspected to have a possible mitochondrial DNA (mtDNA) depletion syndrome and commenced on oral Coenzyme Q 10, oral thiamine, riboflavin, biotin, and vitamin E. She was discharged home at 2 weeks of age on breast feeding with normal blood gas and lactate.
Oral sodium bicarbonate was supplemented at 4 weeks of life with the recurrence of systemic acidosis and lacticacidemia. She developed persistent vomiting and steatorrhoea. Clinically an enlarged liver was palpable 5 cm below the right costal margin. Progressive deterioration of liver function occurred with conjugated hyperbilirubinemia (direct bilirubin 70 μmol/L), total 134 (normal 0–17), worsening hypoalbuminemia, and transaminemia.
In the following months, she remained hypotonic with failure to thrive and globally delayed developmental milestones. She was readmitted at 10 weeks of age with escalating lethargy and inactivity precipitated by recurrent vomiting and loose stools. Clinically, she demonstrated features of circulatory compromise with cold, cyanosed peripheries. Biochemical parameters included markedly raised lactate at 15 mmol/L and compensated metabolic acidosis (pH 7.35, bicarbonate 12.7, base excess −13). Brain MRI showed global cystic changes in the subcortical white matter, T2 hyperintensities in the putamen, globus pallidi, and ventricular dilatation with septatation (Fig. 1a–c). She was discharged home after several days with anti-reflux infant formula, oral ranitidine, and domperidone.

(a, b) MRI brain T1 and T2 weighted axial images showing global, cystic changes and hyperintensities in putamen and glubus pallidi. (c) MRI brain showing subcortical cystic changes and dilated lateral ventricles with ventricular septae
Shortly thereafter, she was readmitted with a similar episode of recurrent vomiting and systemic acidosis, and was subsequently referred to a tertiary center for further management. Features noted at initial assessment here showed an emaciated, pale, and mildly jaundiced infant with a weight of only 2.9 kg at 4 months of age. She had reduced spontaneous movements, hypotonia with severe head lag, myopathic facies, and reduced deep tendon reflexes. Intermittent roving nystagmus and mild ptosis were also noted. A soft ejection systolic murmur was auscultated over her upper left sternal edge. Echocardiography showed a small, 0.21 cm, atrial septal defect, with good contractility and ejection fraction of 65.8%. Her liver was palpable 6 cm below the right costal margin. Her hemoglobin was 9.5 g/dL with normochromic normocytic cells on film and adequate reticulocyte response. The other cell lines were normal. Hepatic dysfunction was evident with mild coagulopathy; activated partial thromboplastin time 50 s (control 30.9–45.9 s), prothrombin time 19 s (control 11.9–14 s), serum albumin 27 g/L (normal 30–54), and mildly elevated transaminases; alanine aminotransferase 170 U/L (normal 0–33), aspartate aminotransferase 116 (normal 0–82). Total bilirubin was 115 μmol/L (normal 0–17) with conjugated bilirubin 90 μmol/L, raised alkaline phosphatase 954 U/L (age-related reference range 0–356), gamma-glutamyl transferase 289 U/L (normal 11–50), and ammonia 65 μmol/L (normal <50). Serum lactate was 17.7 mmol/L, pyruvate 445.6 μmol/L (normal 30–80), blood pH 7.151, pCO2 12.1 mmHg, pO2 168.8 mmHg, bicarbonate 4, and base excess −24.5. Serum creatine kinase, very long chain fatty acids, carnitine, and transferrin isoforms were normal. Plasma alanine and proline were raised at 1,116 μmol/L (normal 132–455) and 508 μmol/L (normal 77–329), respectively. Urine organic acids showed increased excretion of lactate, ketones, citramalic, and fumaric acid.
Abdominal ultrasound showed a left multicystic kidney with a dilated distal left ureter and enlarged right kidney with bipolar length of 5.5 cm. The liver was enlarged with a smooth outline and absence of any focal lesions. Repeat brain ultrasound revealed multiple cystic changes predominantly observed in bifrontal lobes, and periventricular regions with ex-vacuo dilatation of the lateral ventricles. Her thyroid function studies were abnormal; free T4 12.1 pmol/L (14.1–19.2), TSH 7.55 mU/L (0.98–5.3). Ophthalmological assessment reported the presence of alternating exotropia with poor fixation, but normal fundus, possibly indicating cortical visual impairment. Treatment included attempts to correct electrolyte imbalances, metabolic derangements with oral sodium bicarbonate, and addressing feeding difficulties with nasogastric feeding of anti-reflux infant formula with supplemented medium chain triglycerides (MCT). Nevertheless her weight failed to pick up and at 8+ months of age, she only weighed 4.2 kg, well below the third percentile. She developed increasingly severe liver impairment, systemic metabolic and lactic acidosis. Her parents opted for a conservative management and she succumbed at home from respiratory failure at 8 months of age.
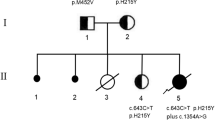
Meanwhile, further investigations were carried out to identify the precise cause of her illness. Biochemical examination cultured fibroblasts had demonstrated reduced respiratory chain enzyme activities (OXPHOS) of complex I and IV with 68% and 47% residual activities expressed as a percent of the lowest control value respectively. The other OXPHOS enzymes complex II, complex III, and complex V showed a normal activity. Assays to quantify OXPHOS enzyme activities were based on spectrophotometry (Janssen et al. 2007; Mourmans et al. 1997; Cooperstein and Lazarow 1951; Jonckheere et al. 2008; Srere 1969). Muscle and liver OXPHOS assays would perhaps have yielded more representative results of the tissue-specific involvement observed in mitochondrial respiratory chain disorders; however, her parents had declined these tests due to concerns over her fragile state. Molecular genetic testing of a total of 12 point mutations in mtDNA isolated from blood for five genes associated with Leigh disease and seven genes associated with MELAS failed to demonstrate any pathogenic mutations. Postmortem liver biopsy (Figs. 2 and 3) and histopathology displayed lobular disarray, portal fibrosis, micro and macrovesicular steatosis, and intrahepatic and intracanicular cholestasis. Due to financial constraints, we were unable to analyze OXPHOS assays on postmortem liver tissue or perform direct sequencing of mtDNA. mtDNA depletion studies were not performed as the finding of reduced enzyme deficiencies in fibroblasts make a depletion syndrome less likely. Her parents had not consented for postmortem muscle biopsy. The associated biochemical evidence of a combined enzyme deficiency in cultured fibroblasts, in addition to the early, severe, and rapid progression of a predominant hepatocerebral disease in our patient, suggested a possible candidate nuclear gene defect such as GFM1 mutation which had only recently been described in a handful of patients with a similar phenotype. Subsequent sequencing of GFM1 gene was performed (Smits et al. 2010a) and revealed the heterozygous changes c.539delG (p.Gly180AlafsX11) in exon 4 which was inherited from the father, and c.688G > A (p. Gly230Ser) in exon 5, inherited from the mother.

Liver H&E X40: Liver tissue displaying moderate microvesicular steatosis and patchy macrovesicular steatosis with intrahepatic and intracanicular cholestasis. The portal tracts are expanded with accompanying portal fibrosis and portal to portal bridging fibrosis

Liver MT X10: Lobular disarray of liver tissue is evident with the nodular pattern emphasized by Masson’s Trichrome stain
Discussion
OXPHOS disorders may occur as isolated enzyme deficiencies and have been reported in approximately 67% cases, while combined enzyme deficiencies account for 33% of all respiratory chain disorders (Smits et al. 2010b). Isolated OXPHOS deficiencies are generally caused by mutations in structural genes which encode subunits of the OXPHOS system, or in genes encoding proteins involved in the assembly of a specific OXPHOS enzyme complex (Zeviani and Di Donato 2004). Combined OXPHOS defects tend to involve genes required for mtDNA maintenance, mitochondrial transcription, or translation including posttranscriptional or posttranslational processes, import of nDNA-encoded proteins into the mitochondrion or mitochondrial membrane biogenesis (Smits et al. 2010b). Approximately 40% of all combined respiratory chain deficiencies occur as a result of mtDNA deletions and point mutations in mitochondrial transfer RNA genes, which more frequently affect adult patients (Kemp et al. 2011). Another 40% of combined OXPHOS deficiencies are related to mtDNA depletion, which predominantly affect young children and are caused by autosomal recessive mutations in nuclear genes (DGUOK, MPV17, POLG, TYMP, TK2, SUCLA2, SUCLG1, RRM2B, PEO1) influencing mtDNA replication and maintenance (Spinazzola et al. 2009). In the remaining 20% of combined respiratory chain deficiencies, after excluding mtDNA deletions, depletion, and point mutations, no clear diagnostic pathway is currently available to determine the cause of disease. The reduced enzyme activities in our patient’s cultured fibroblasts made the possibility of mtDNA depletion less likely as it has been reported that exponentially growing cells may not manifest any OXPHOS enzyme deficiencies, but instead have to be kept in a quiescent state in order to clearly demonstrate their phenotype (Pontarin et al. 2011; Gonzalez-Vioque et al. 2011). For several types of depletion syndromes, it has been shown that nucleotide metabolism plays an important role in this phenomenon (Gonzalez-Vioque et al. 2011). We used growing fibroblasts with a low passage number, and under these conditions fibroblasts from depletion syndrome patients usually do not show OXPHOS enzyme deficiencies.
A thorough clinical characterization of patients is imperative in identifying homogeneous patient groups and analyzing the complex molecular mechanisms behind combined respiratory chain deficiencies. When facilitated by linkage studies in consanguineous families and functional cell culture investigations conducted at various regulatory levels of mitochondrial function, transcription, translation, ribosome function, protein stability, sub-complex formation, it may provide a potentially promising approach in selecting novel candidates in combined respiratory chain deficiencies (Kemp et al. 2011).
Eukaryotes contain two translational systems, one in the cytosol and the other in the mitochondria. The mitochondrial translation machinery comprises mtDNA-encoded rRNAs and tRNAs, in addition to various proteins encoded by the nuclear genome including two initiation factors, IF2 (MTIF) (Ma and Spremulli 1995), IF3 (MTIF3) (Koc and Spremulli 2002); three elongation factors, EFTu (TUFM) (Ling et al. 1997), EFTs (TSFM) (Xin et al. 1995), EFG1 (GFM1) (Gao et al. 2001); four release factors, RF1 (MTRF1) (Zhang and Spremulli 1998), RF1a (HMRF1L) (Zhang and Spremulli 1998), C12ORF65 (Antonicka et al. 2010), ICT1 (Richter et al. 2010); and two recycling factors, RRF (MRRF) (Soleimanpour-Lichaei et al. 2007) and EFG2, which has been renamed RRF2 (GFM2) (Zhang and Spremulli 1998; Rorbach et al. 2008; Tsuboi et al. 2009); mitochondrial ribosomal proteins (MRPs); mitochondrial aminoacyl-tRNA synthetases, and methionyl-tRNA transformylase (Smits et al. 2010b). Although most components of the mitochondrial translation system are nuclear encoded, the majority of patients harboring mutations in nuclear genes are very limited. Most mutations associated with mitochondrial protein synthesis defects to date have been reported in mtDNA.
Recently, mutations in GFM1, previously known as EFG1, a nuclear factor of the mitochondrial translation machinery, have been described (Smits et al. 2010a; Coenen et al. 2004; Antonicka et al. 2006; Valente et al. 2007). Mitochondrial GFM1 is a five-domain GTPase that catalyzes the translocation step of mitochondrial protein synthesis, during which peptidyl-tRNA moves from the ribosomal acceptor (amino acyl or A) site to the ribosomal peptidyl (P) site following removal of the deacylated tRNA from the ribosomal (P) site to the exit (E) site. This results in the concomitant advancement of the mRNA by one codon and exposure of the next codon in the A site preparing for a new elongation cycle (Wintermeyer et al. 2004). The compilation of biochemical evidence of a combined enzyme deficiency in cultured fibroblasts of complex I and IV, which is a clear and well-known functional consequence of GFM1 mutations (Smits et al. 2010a; Coenen et al. 2004; Antonicka et al. 2006; Valente et al. 2007), in addition to the severe and early onset presentation with rapidly progressive hepatocerebral disease in our patient suggested a possible candidate nuclear gene defect such as GFM1 mutation which had only recently been described in a handful of patients with a predominant hepatic and/or neurological phenotype.
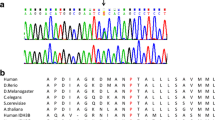
Sequence analysis of the complete coding region of the GFM1 gene in our patient revealed the heterozygous changes c.539delG (p.Gly180AlafsX11) in exon 4 which was inherited from the father had resulted in a frameshift mutation, while a second c.688G > A (p.Gly230Ser) mutation in exon 5 was inherited from the mother. The pathogenicity of the second novel missense mutation was suggested by its highly conserved nature in all nine organisms of which a GFM1 sequence could be found, and its absence in the dbSNP database. In addition, both these mutations were not present in the 1,000 genomes database which includes 60 Chinese genomes and 194 South-East Asian genomes.
Previously, four other GFM1 defects have been described. A review of their clinical features and investigations has been tabulated in Table 1. The first report of two siblings of consanguineous Lebanese parents harbored homozygous mutations, c.521A > G in the GTP-binding domain I (Coenen et al. 2004). The index patient had presented with intrauterine growth retardation, mild microcephaly, hypertonicity with reduced spontaneous movements, and she soon developed profound metabolic acidosis and lactic academia with raised lactate: pyruvate ratio of 38 (normal 12–18) by day 10 of life. Rapidly progressing liver failure occurred leading to death on day 27 of life.
Antonicka and colleagues described similar clinical phenotypes in two siblings with significant growth retardation, lactic acidosis, fatal hepatopathy, small or undeveloped corpus callosum, and severe respiratory deficiency in skeletal muscle and liver, without any evidence of clinical cardiac dysfunction (Antonicka et al. 2006). The molecular basis for the novel mutations in GFM1 demonstrated that the translation defect resulting from these mutations was associated with unique, tissue-specific patterns of OXPHOS deficiency. The severity of the OXPHOS defect correlates with the residual levels of the mutant EFG1 protein in different tissues. In addition, there appears to be an adaptive response that involves transcriptional upregulation of EFTu, another translation elongation factor, with significant quantitative differences in the ratios of the translation elongation factors. This may reasonably reflect the different tissue-specific demands for the mitochondrially encoded polypeptides and explain how the heart uniquely possesses mechanisms to circumvent the disruption of mitochondrial translation caused by mutations in the nuclear-encoded components of the translation machinery.
A subsequent report by Valente and colleagues on an infant affected by neonatal lactic acidosis, rapidly progressive encephalopathy, severely decreased mitochondrial protein synthesis, and combined OXPHOS deficiency was a compound heterozygote for two novel GFM1 mutations (Valente et al. 2007). No data are available concerning the expression of mitochondrial GFM1 in human brain, but the predominant neurological involvement with neuroradiological hallmarks of early onset Leigh syndrome including bilateral necrotizing lesions in the basal ganglia and brain stem overwhelmed, that of other organs notably, skeletal muscle, liver, and heart in this patient. It is postulated that the clinical variability may be attributed to various mechanisms, including the different sites of mutations in the EFG1 protein which may subsequently have tissue-specific effects on mitochondrial translation, adaptive processes (e.g., compensatory changes in other translational factor) which may act differently in different patients, and finally partial compensation by the second isoform of mitochondrial EFG, namely EFG2 which is highly expressed in energy-consuming tissues, such as skeletal muscle, heart, and fetal liver. The functional role of EFG2, however, is uncertain (Smits et al. 2010a).
Strikingly, in the recent report by Smits et al., a mutation in GFM1 was observed in a patient affected by severe, rapidly progressive mitochondrial encephalopathy; the decrease in enzyme activities of complex I, III, and IV detected in fibroblasts was not found in muscle tissue (Smits et al. 2010a). Reduced respiratory chain activities in fibroblasts with normal values in muscle tissue are an uncommon finding in mitochondrial disorders, whereas the opposite is often observed. The capacity of the mitochondrial energy-generating system, however, was clearly reduced in muscle tissue, indicated by impairments in pyruvate oxidation and ATP production rates. This evinces the importance of a thorough diagnostic biochemical analysis of muscle tissue and fibroblasts in patients as OXPHOS defects may be selectively expressed.
The clinical symptomatology in our patient is reminiscent of the severe phenotype of the first two reports (Coenen et al. 2004; Antonicka et al. 2006), characterized by intrauterine growth retardation, profound lactic acidosis, and progressive liver dysfunction, resulting in liver failure and death within the first weeks or months. In addition, our patient had a multicystic kidney and an atrial septal defect. Neuroradiological evidence of cystic changes in temporal lobes with white matter abnormalities and biochemical findings of high blood lactate levels of 25 mmol/L and lactate: pyruvate ratios of 38 suggest a more global and deleterious disturbance of mitochondrial functioning.
In summary, unique patterns of OXPHOS deficiency result when one of the nuclear-encoded components of the translation system fail. The clinical heterogeneity associated with mutations in the mitochondrial translation apparatus may reflect different tissue-specific demands for the mitochondrially encoded polypeptides with significant quantitative differences in the ratios of the translation elongation factors. Despite rapid advancements in our understanding of the mechanisms implicated in mitochondrial disease, it is far from complete. The intricacy and complexities of the OXPHOS system renders the identification of the genetic defect an arduous task. Rapid advances in technologies involving high-throughput sequencing which are much cheaper and faster than the conventional approach of polymerase chain reaction followed by capillary sequencing (Tucker et al. 2009) or exome sequencing (Ng et al. 2009) which is the targeted sequencing of all protein-coding regions when applied with appropriate bioinformatics tools potentially offer a promising approach in elucidating the genetic etiology of OXPHOS deficiencies. Clearly, much work remains to be done to fully comprehend the processes involved in mitochondrial translation and the biogenesis of the OXPHOS system and their roles in the pathogenesis of combined OXPHOS deficiencies.
References
Antonicka H, Sasarman F, Kennaway NG et al (2006) The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet 15:1835–1846
Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Anne Pedersen AM, Rodenburg RJ, van der Knaap MS, Smeitink JA, Chrzanowska-Lightowlers AM, Shoubridge EA (2010) Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 87:115–122
Brandon M, Baldi P, Wallace DC (2006) Mitochondrial mutations in cancer. Oncogene 25:4647–4662
Coenen MJ, Antonicka H, Ugalde C et al (2004) Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 351:2080–2086
Cooperstein SJ, Lazarow A (1951) A microspectrophotometric method for the determination of cytochrome oxidase. J Biol Chem 189(2):665–670
Cwerman-Thibault H, Sahel JA, Corral-Debrinski M (2011) Mitochondrial medicine: to a new era of gene therapy for mitochondrial DNA mutations. J Inherit Metab Dis 34(2):327–344
Gao J, Yu L, Zhang P et al (2001) Cloning and characterization of human and mouse mitochondrial elongation factor G, GFM and Gfm, and mapping of GFM to human chromosome 3q25.1-q26.2. Genomics 74:109–114
Gerbitz KD, Gempel K, Brdiczka D (1996) Mitochondria and diabetes: genetic, biochemical, and clinical implications of the cellular energy circuit. Diabetes 45(2):113–126
Gonzalez-Vioque E, Torres-Torronteras J, Andreu AL, Martı R (2011) Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS Genet 7(3):e1002035
Gray MW, Burger G, Lang BF (1999) Mitochondrial evolution. Science 283(5407):1476–1481
Janssen AJ, Trijbels FJ, Sengers RC et al (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734
Jonckheere AI, Hogeveen M, Nijtmans LG, van den Brand MA, Janssen AJ, Diepstra JH, van den Brandt FC, van den Heuvel LP, Hol FA, Hofste TG, Kapusta L, Dillmann U, Shamdeen MG, Smeitink JA, Rodenburg RJ (2008) A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet 45(3):129–133
Kemp J, Smith P, Pyle A et al (2011) Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain 134:183–195
Koc EC, Spremulli LL (2002) Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J Biol Chem 277:35541–35549
Ling M, Merante F, Chen HS et al (1997) The human mitochondrial elongation factor tu (EF-Tu) gene: cDNA sequence, genomic localization, genomic structure, and identification of a pseudogene. Gene 197:325–336
Ma L, Spremulli LL (1995) Cloning and sequence analysis of the human mitochondrial translational initiation factor 2 cDNA. J Biol Chem 270:1859–1865
Mandemakers W, Morais VA, De Strooper B (2007) A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci 120(10):1707–1716
Mizuno Y, Ikebe S, Hattori N et al (1995) Role of mitochondria in the etiology and pathogenesis of Parkinson’s disease. Biochim Biophys Acta 1271:265–274
Mourmans J, Wendel U, Bentlage HA, Trijbels JM, Smeitink JA, de Coo IF, Gabreëls FJ, Sengers RC, Ruitenbeek WJ (1997) Clinical heterogeneity in respiratory chain complex III deficiency in childhood. J Neurol Sci 149(1):111–117
Munnich A, Rustin P, Rötig A et al (1992) Clinical aspects of mitochondrial disorders. J Inherit Metab Dis 15(4):448–455
Ng SB, Turner EH, Robertson PD et al (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461(7261):272–276
Pontarin G, Ferraro P, Rampazzo C et al (2011) Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J Biol Chem 286(13):11132–11140
Richter R, Rorbach J, Pajak A, Smith PM, Wessels HJ, Huynen M, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM (2010) A functional peptidyltRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J 29:1116–1125
Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, Lightowlers R, Chrzanowska-Lightowler ZM (2008) The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res 36(18):5787–5799
Skladal D, Halliday J, Thorburn DR (2003) Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126:1905–1912
Smits P, Antonicka H, van Hasselt PM et al (2010a) Mutation in subdomain G′ of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum Genet. doi:10.1038/ejhg.2010.208
Smits P, Smeitink J, van den Heuvel L (2010) Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol (Article ID 737385, 24 pages, Epub 2010 Apr 13). doi:10.1155/2010/737385
Soleimanpour-Lichaei HR, Kuhl I, Gaisne M, Passos JF, Wydro M, Rorbach J, Temperley R, Bonnefoy N, Tate W, Lightowlers R, Chrzanowska-Lightowler ZMA (2007) mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol Cell 27:745–757
Spinazzola A, Invernizzi F, Carrara F et al (2009) Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis 32:143–158
Srere PA (1969) Citrate synthase, EC 4.1.3.7 citrate oxaloacetate lyase (CoA-acetylating). Methods Enzymol 13:3–11
Tsuboi M, Morita H, Nozaki Y, Nozaki Y, Akama K, Ueda T, Ito K, Nierhaus KH, Takeuchi N (2009) EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol Cell 35(4):502–510
Tucker T, Marra M, Friedman JM (2009) Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet 85(2):142–154
Valente L, Tiranti V, Marsano RM et al (2007) Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet 80:44–58
Wintermeyer W, Peske F, Beringer M et al (2004) Mechanisms of elongation on the ribosome: dynamics of a macromolecular machine. Biochem Soc Trans 32:733–737
Xin H, Woriax V, Burkhart W et al (1995) Cloning and expression of mitochondrial translational elongation factor Ts from bovine and human liver. J Biol Chem 270:17243–17249
Zeviani M, Di Donato S (2004) Mitochondrial disorders. Brain 127(10):2153–2172
Zhang Y, Spremulli LL (1998) Identification and cloning of human mitochondrial translational release factor 1 and the ribosome recycling factor. Biochim Biophys Acta 1443:245–250
Acknowledgments
The authors would like to express their sincere gratitude to the Director General of Health, Ministry of Health, Malaysia for allowing the publication of this case report.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Shamima Rahman.
Appendices
Contributions of Authors
1. Balasubramaniam S – Clinical management of the patient, draft of manuscript, and completed version.
2. YS Choy – Clinical management of patient, review of the draft, and contribution to the completed article.
3. Talib A – Histopathological analysis of liver biopsy, review of the draft, and contribution to the completed article.
4. Norsiah MD – Molecular testing of parental DNA for GFM1 mutation, review of the draft, and contribution to the completed article.
5. van den Heuvel LP – Biochemical analyses of OXPHOS assay and molecular testing in the proband, review of the draft, and contribution to the completed article.
6. Rodenburg RJ – E Biochemical analyses of OXPHOS assay and molecular testing in the proband, review of the draft, and contribution to the completed article.
One Sentence Take Home Message
This report demonstrates a combined OXPHOS deficiency detected in patient fibroblasts, occurring as a result of a nuclear-encoded mitochondrial translational defect secondary to GFM1 mutation, ultimately leading to mitochondrial hepatoencephalomyopathy and death at 8 months of age.
All authors declare that the answers to all questions on the JIMD competing interest form are “no” and therefore have nothing to declare.
Rights and permissions
Copyright information
© 2011 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Balasubramaniam, S., Choy, Y.S., Talib, A., Norsiah, M.D., van den Heuvel, L.P., Rodenburg, R.J. (2011). Infantile Progressive Hepatoencephalomyopathy with Combined OXPHOS Deficiency due to Mutations in the Mitochondrial Translation Elongation Factor Gene GFM1 . In: JIMD Reports - Case and Research Reports, 2012/2. JIMD Reports, vol 5. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2011_107
Download citation
DOI: https://doi.org/10.1007/8904_2011_107
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-28095-5
Online ISBN: 978-3-642-28096-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)