
Abstract
The catalytic and regulatory subunits of class I phosphoinositide 3-kinase (PI3K) have oncogenic potential. The catalytic subunit p110α and the regulatory subunit p85 undergo cancer-specific gain-of-function mutations that lead to enhanced enzymatic activity, ability to signal constitutively, and oncogenicity. The β, γ, and δ isoforms of p110 are cell-transforming as overexpressed wild-type proteins. Class I PI3Ks have the unique ability to generate phosphoinositide 3,4,5 trisphosphate (PIP3). Class II and class III PI3Ks lack this ability. Genetic and cell biological evidence suggests that PIP3 is essential for PI3K-mediated oncogenicity, explaining why class II and class III enzymes have not been linked to cancer. Mutational analysis reveals the existence of at least two distinct molecular mechanisms for the gain of function seen with cancer-specific mutations in p110α; one causing independence from upstream receptor tyrosine kinases, the other inducing independence from Ras. An essential component of the oncogenic signal that is initiated by PI3K is the TOR (target of rapamycin) kinase. TOR is an integrator of growth and of metabolic inputs. In complex with the raptor protein (TORC1), it controls cap-dependent translation, and this function is essential for PI3K-initiated oncogenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- PI3K Signaling
- Oncogenic Transformation
- Chicken Embryo Fibroblast
- Kinase Domain Mutant
- Catalytic Subunit P110
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Phosphatidylinositol 3-Kinases and Cancer
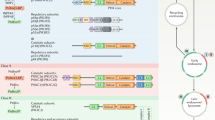
The phosphatidylinositol 3-kinases (PI3Ks) are grouped into three classes (I–III) which differ in structure and function (Fruman et al. 1998; Vanhaesebroeck et al. 1997; Vanhaesebroeck and Waterfield 1999). Class I enzymes have been intensely studied and have emerged as promising drug targets in cancer and in immune disorders (Brachmann et al. 2009; Ghigo and Hirsch 2008). They are heterodimeric enzymes consisting of a catalytic subunit p110 that associates with a regulatory subunit. The vertebrate genome codes for four isoforms of p110 (α–δ). Several regulatory subunits have been identified. For p110α, β, and δ, the regulatory subunit p85 is the most prevalent. p110γ associates with separate, specific regulatory subunits of which p101 is the most common (Stephens et al. 1997). Class I PI3Ks occur as obligatory dimers in the cell (Geering et al. 2007). Regulatory and catalytic subunits show distinct structure-function domains that are illustrated in Fig. 1 (Amzel et al. 2008; Huang et al. 2007; Walker et al. 1999). Class I PI3Ks act on three substrates, the nonphosphorylated phosphatidylinositol, PI, the inositol monophosphate (PI(4)P) and the bisphosphate (PI(4,5)P2), to add a phosphate group in the D-3 position of the inositol ring and generate PI(3)P, PI(3,4)P2 and PI(3,4,5)P3, respectively (Carpenter et al. 1990). The latter, also referred to as PIP3, functions as an important second messenger in the cell and is the predominant mediator of PI3K activity. The phosphatase PTEN (phosphatase and tensin homolog deleted on chromosome 10) removes the phosphate group from the D-3 position of phosphatidylinositol, acting as the direct catalytic antagonist of PI3K (Li et al. 1997; Maehama and Dixon 1998).

Domain structure of catalytic and regulatory subunits of PI3K.
ABD adaptor-binding domain; RBD RAS-binding domain; C2 C2 domain; HELICAL helical domain; KINASE kinase domain; C-SH2 C-terminal SH2 domain; iSH2 inter-SH2 domain; N-SH2 N-terminal SH2 domain; RhoGAP Rho GTPase-activating protein homology domain; SH3 SH3 domain
Class I PI3Ks have a long history of association with cancer (Yuan and Cantley 2008; Zhao and Roberts 2006; Zhao and Vogt 2008a). Extensive studies in the 1980s have documented a tight link of PI3K activity with tyrosine kinase oncoproteins and with the polyoma virus middle T oncoprotein (Sugimoto et al. 1984; Whitman et al. 1985). A representative interaction of this type is the binding of middle T to the Src oncoprotein, leading to an activation of the Src kinase which results in the phosphorylation of several tyrosine residues on middle T and the subsequent recruitment and activation of the p85-p110α dimer (Courtneidge and Smith 1984; Utermark et al. 2007). The cell-transforming activity of tyrosine kinase oncoproteins is correlated with their ability to associate with PI3K (Engelman et al. 2006; Kaplan et al. 1989; Schaffhausen and Roberts 2009). In 1997, the gene encoding the p110α catalytic subunit of PI3K was identified as the cell-derived oncogene in an avian retrovirus and shown to be constitutively activated by N-terminal fusion to viral sequences (Chang et al. 1997; Aoki et al. 2000). The isolation of this avian retrovirus documented the direct oncogenic potential of p110α.
2 Cancer-Specific Mutations in PI3K
In 2004, the discovery of cancer-specific mutations in PIK3CA, the gene encoding the catalytic subunit p110 of PI3K, put PI3Ks in the limelight as clinically relevant oncoproteins and as drug targets (Liu and Roberts 2006; Samuels and Velculescu 2004; Samuels et al. 2004; Stephens et al. 2005). Mutations have now been identified in the genes coding for both subunits of PI3K, in PIK3CA and in PIK3R1, the gene encoding p85 (Fig. 2) (Cancer Genome Atlas Research Network 2008; Samuels et al. 2004). These mutations occur at frequencies extending from 5 to 25% in several common cancers, including cancers of the breast, endometrium, and the large intestine (http://www.sanger.ac.uk). The PI3K antagonist PTEN functions as an important tumor suppressor and is frequently inactivated by mutation or deletion in cancer (Di Cristofano and Pandolfi 2000).

Cancer derived and engineered gain-of-function mutations in p110α and in p85. p110α: The hot-spot mutations are in red, rare mutations in blue; engineered gain-of-function mutations are marked by an asterisk. p85: The engineered gain-of-function mutation is marked by an asterisk
The PIK3CA mutations are concentrated in three hot spots in the coding sequence (Samuels et al. 2004). Two of these hot spots are located in the helical domain of p110α, and one is situated in the catalytic domain. These hot spot mutations are single nucleotide substitutions that lead to the amino acid substitutions: E542K, E545K and H1047R (Samuels et al. 2004). The preferential mapping of cancer-specific mutations to hot spots suggested immediately a strong positive selection for such mutations, possibly reflecting a powerful replicative advantage of mutant-carrying cells. Studies of the mutant proteins rapidly revealed a mutation-induced gain of function as compared to the wild-type enzyme. The PIP3-generating lipid kinase activity of the mutants is increased several fold (Carson et al. 2008; Chaussade et al. 2009; Ikenoue et al. 2005; Kang et al. 2005; Sugita et al. 2008; Zhao et al. 2005). Downstream signaling is no longer dependent on upstream stimulation by growth factors. It is constitutive and operates in serum-starved cells. This downstream signaling manifests itself in the phosphorylation of AKT (murine thymoma viral oncoprotein homolog) at T308 and S473, of the eukaryotic initiation factor 4E-binding protein (4E-BP) at T37 and T46 and of p70 S6 kinase (S6K) at T389. The three hot spot mutations activate the oncogenic potential of p110α. Expression of wild-type p110α does not detectably affect the growth behavior and the morphology of the cell. In contrast, expression of the hot spot mutants induces oncogenic transformation in avian and in mammalian cell culture (Ikenoue et al. 2005; Isakoff et al. 2005; Kang et al. 2005; Zhang et al. 2008; Zhao et al. 2005). The transformed cells are also tumorigenic in animal model systems (Bader et al. 2006; Zhao et al. 2005). In a mouse model, transgenic expression of the H1047R mutant p110α in the lung induces adenocarcinomas (Engelman et al. 2008). These data on enhanced enzymatic activity, constitutive downstream signaling and oncogenic potency strongly suggest that the hot spot mutations function as “drivers” in human cancer, responsible for at least part of the oncogenic phenotype of the cancer cell.
The hot spot mutations account for about 80% of the mutated PIK3CA genes in cancer. But there are also numerous cancer-specific mutations that occur at lower frequencies. Most of these are again single nucleotide substitutions, but recently two in-frame deletions have also been identified (Cancer Genome Atlas Research Network 2008). An investigation of seventeen rare point mutations led to the surprising finding that most of these (16 out of 17) also show a gain of function (Gymnopoulos et al. 2007). However, compared with the hot spot mutants, the rare mutations induce smaller gains of function. The mutant proteins show lower enzymatic activity, mediate lower levels of downstream phosphorylation and induce decreased oncogenic transformation in cell culture as measured by the number of transformed cell foci per ng of DNA. These lesser gains of function may explain the low frequencies at which such mutants are found in cancer. The broad distribution of rare, cancer-specific mutations over almost the entire coding sequence of p110α, with the notable and so far unexplained exception of the RAS-binding domain, raised the possibility that any random mutation may induce a gain of function, perhaps by triggering a conformational change. However, several random mutations introduced into PIK3CA had no phenotype, suggesting that cancer-specific mutations, no matter how rare, are still the result of positive selection (Gymnopoulos et al. 2007).
The mutations in PIK3R1 are also clustered (Cancer Genome Atlas Research Network 2008). Most occur within a stretch of six residues (560–565) located in the inter-SH2 domain of p85 (Cancer Genome Atlas Research Network 2008). This portion of p85 includes the contact points with residues in the C2 domain of p110α. The mutations in PIK3R1 interfere with the proper binding to p110α, relieving an inhibitory interaction. Several of these mutations in the inter-SH2 domain of p85 have recently been shown to induce a gain of function in PI3K including enhanced signaling to Akt, stimulation of cell replication and oncogenic transformation (Jaiswal et al. 2009; Wu et al. 2009; Sun et al. 2010, submitted). A cancer-derived PIK3R1 mutation in the N-terminal SH2 domain of p85 (G376R) may reduce the inhibitory interaction with the helical domain of p110α as is the case with the engineered p85 mutation K379E (Sun et al. 2010, submitted). Thus, the mutations in the inter-SH2 domain of p85 may be functionally equivalent to the mutations in the C2 domain of p110α, and the p85 mutations in the N-terminal SH2 domain may have the same effect as the helical domain mutations in p110α.
The map of the gain-of-function mutations on the structure of p110α (Amzel et al. 2008; Huang et al. 2007, 2008; Miled et al. 2007) reveals two properties that are shared by several mutants: location on the surface of the protein and a change from an acidic to a basic amino acid. This observation suggests that many of the gain-of-function mutations change the surface properties of the enzyme, probably affecting the interaction with other proteins or with membranes. In fact, of three engineered mutants inducing an acidic to basic change on the protein surface, two showed oncogenic activity (Gymnopoulos et al. 2007).
3 Several Molecular Mechanisms Can Induce a Gain of Function in p110
The occurrence of gain-of-function mutations distributed over several domains of p110α raises the question of the molecular mechanism responsible for increased activity. Do all these mutants operate by the same mechanism, or are there several distinct ways of enhancing p110α function? The available evidence strongly favors the existence of several molecular mechanisms leading to a gain of function. The combination of two hot spot mutations, one from the helical and the other from the kinase domain, in one protein has a strong synergistic effect on p110α activity. In contrast, introducing both helical domain mutations into the same molecule results in an only moderately additive effect (Zhao and Vogt 2008b). These results suggest that helical and kinase domain mutations work by different mechanisms that can cooperate. Several additional mutant combinations have been studied. Combinations of mutations located in different domains of p110α often show a synergistic effect, but combinations of mutations in the same domain are merely additive. There is also one pair of mutations, E545K/Y1021C, that shows loss of function when introduced in the same molecule, indicating incompatibility of the combined mutation-induced changes with p110α function (Gymnopoulos and Vogt 2009, to be submitted).
The distinction between helical and kinase domain mutations is further illuminated by investigations that explore the interactions of the mutant proteins with the p85 regulatory subunit and with RAS (Zhao and Vogt 2008b). An N-terminal deletion of p110α that still permits expression of the protein but eliminates binding to p85, has contrasting effects on helical and kinase domain mutants. The oncogenic activity of the kinase domain mutant that lacks p85 binding is completely inactivated, and its downstream signaling is greatly reduced. The two helical domain mutants are much less affected by a lack of p85 binding. Their oncogenic activity in cell culture is only moderately reduced, and the effect on signaling is also minor. For wild-type p110α, deletion of the p85-binding domain has an activating effect, resulting in constitutive signaling and oncogenicity (Zhao et al. 2005). This somewhat surprising observation is explained by the fact that in cells devoid of upstream signaling, the p85-p110α interaction is both inhibitory and stabilizing for p110α. Upon growth factor stimulation, the SH2 domains of p85 interact with phosphorylated tyrosine on upstream signaling molecules, relieving the inhibition on p110α. Deletion of the p85-binding domain has a similar disinhibitory effect on p110α and reveals its latent oncogenic activity and signaling potential.
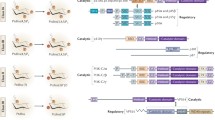
A mutational inactivation of the ability of p110α to interact with RAS has the opposite effect on the hot spot mutants. Interference with RAS binding decreases the oncogenicity of the helical domain mutants and drastically diminishes their downstream signaling. The kinase domain mutant is largely independent of RAS binding. Its oncogenicity is preserved in the absence of RAS binding, and signaling to AKT is only mildly affected. Complementary data have emerged from a study of mutant enzyme kinetics (Chaussade et al. 2009). The V max values of the hot spot p110α mutants are significantly above that of the wild-type. Wild-type p110α can be activated by a PDGFR (platelet-derived growth factor receptor)-derived diphosphoryl peptide (Cuevas et al. 2001; Shekar et al. 2005). The phosphorylated tyrosines of this peptide interact with the N-terminal SH2 domain of p85 and thereby alleviate p85-mediated inhibition of p110α. Significantly, this PDGFR-derived peptide has no effect on the activity of the helical domain mutants, but it inhibits the kinase domain mutant (Chaussade et al. 2009). These data on the functional differences between helical and kinase domain mutations suggest that the kinase domain mutant is independent of activation by RAS; the mutation appears to have induced the same or a similar activating change that in the wild-type enzyme and in the helical domain mutants is achieved by the interaction with RAS. However, the kinase domain mutant still remains critically dependent on an interaction with p85, and this dependence requires further clarification. One possibility is suggested the crystal structure of the p110α-p85 complex which reveals an unexpected interaction between the p85-binding domain and the kinase domain (Huang et al. 2007, 2008). This interaction might be important for the active conformation induced by H1047R and could explain the sensitivity of H1047R to a loss of p85 binding. Interactions between p85 and p110α that are relevant to the properties of the mutant proteins are summarized in Fig. 3.

p110α-p85 domain interactions of importance for gain-of-function mutations. Green arrows mark interactions with the kinase domain that could explain why the H1047R mutant is RAS-independent, but p85-dependent. The orange arrow signifies the inhibitory interaction between the N-terminal SH2 domain of p85 and the helical domain of p110α that is released by the helical domain mutation in p110α and the N-terminal SH2 domain mutation in p85. The orange and yellow arrows mark similar, probably inhibitory interactions between the inter-SH2 domain of p85 and the C2 or adaptor-binding domain of p110α, respectively. These interactions may be modulated by mutations in the respective domains of p110α
The kinase domain mutation H1047R maps to the hinge region of the activation loop. It could affect the position and the mobility of the activation loop. RAS also interacts with the kinase domain and could induce a change that is similar to the one caused by the H1047R mutation. The helical domain mutants remain dependent on binding to RAS, but are largely independent of p85-binding, probably because they mimic the activating event that follows the growth factor-induced relief of the helical domain-p85 interaction. The same effect of disrupting the helical domain-p85 binding probably also results from the cancer-specific mutation in the N-terminal SH2 domain of p85; it can also be achieved experimentally by mutating critical helical domain-interacting residues R340E and K379E in the p85 which are postulated to interact with E542 and E545 residues of p110α (Miled et al. 2007). Overexpression of such experimentally mutated p85 induces oncogenic transformation in cell culture (Sun et al. 2010, submitted).
The molecular mechanisms for the mutation-induced gain of function in helical and in kinase domain mutations are complementary and reciprocal. The helical domain mutations have gained activation through p85-independence, but still need the interaction with RAS. The kinase domain mutation is in a state of constitutive RAS activation, but still requires the interaction with p85. The helical-kinase domain double mutant is both independent of RAS and of p85.
Since the hot spot mutations account for about 80% of all cancer-specific mutations in p110α, inhibitors specific for these mutants could benefit the majority of the affected patients. Mutant-specific inhibitors would not induce side effects that can result from interfering with the life-sustaining functions of PI3K. The identification of small molecules that discriminate between mutant and wild-type is a challenging task for drug discovery. It would be greatly facilitated by structural information that is specific for the mutants.
4 Non-alpha Isoforms of Class I PI3K in Cancer
There are four isoforms of the catalytic subunit of class I PI3Ks: p110α, p110β, p110γ, and p110δ (Deane and Fruman 2004; Engelman et al. 2006; Fruman and Bismuth 2009; Hawkins et al. 2006; Stephens et al. 1996; Vanhaesebroeck et al. 2001). They are encoded by different genes, but share a basic domain structure. The α, β, and δ isoforms use the same regulatory subunits. The α and δ isoforms are linked primarily to upstream receptor tyrosine kinases, the upstream activation of p110β appears to be context-dependent, involving receptor tyrosine kinases and G-protein-coupled receptors (Ciraolo et al. 2008; Guillermet-Guibert et al. 2008). The p110γ isoform interacts with separate distinct regulatory subunits and is linked to G-protein-coupled receptors (Stoyanov et al. 1995; Yart et al. 2002). The α and β isoforms are expressed ubiquitously. Expression of γ and δ isoforms is restricted mainly to lymphocytes. Genetic inactivation of the α and β isoforms causes early embryonic lethality (Bi et al. 1999, 2002), whereas γ and δ knockouts are viable but suffer from immune deficiencies (Ali et al. 2004; Cantley 2002; Clayton et al. 2002; Hirsch et al. 2000; Ji et al. 2007; Jou et al. 2002; Laffargue et al. 2002; Okkenhaug et al. 2002; Rodriguez-Borlado et al. 2003; Sasaki et al. 2000). The functions of the p110 isoforms are overlapping, but they are clearly not redundant. Conditional and tissue-specific mutations have defined isoform-specific roles in cellular signaling. The principal roles of p110γ and p110δ are in the immune system (Alcazar et al. 2007; Ali et al. 2008; Ji et al. 2007; Okkenhaug et al. 2004; Patton et al. 2007), and p110α and p110β have distinct, complementary, context-dependent and cell type-dependent roles in the control of cell growth and metabolism (Graupera et al. 2008; Marone et al. 2008; Vanhaesebroeck et al. 2005). The functions of specific p110 isoforms are explored in greater detail in other chapters of this volume.
The oncogenic potential of p110α is well documented in experimental systems; the gain-of-function mutations in human cancer add significance to this activity (Samuels et al. 2004). For the non-alpha isoforms of class I PI3K, the connection to cancer is more tenuous. There are no cancer-specific mutations in these isoforms, but differential expression is observed in several cancers. The p110δ isoform is consistently overexpressed in acute myeloblastic leukemia, and inhibitors of p110δ specifically interfere with the growth of these leukemic cells, suggesting a role of p110δ in leukemogenesis (Samuels et al. 2004; Sujobert et al. 2005). Specific inhibitors of p110δ are in clinical trials for hematopoietic malignancies (http://clinicaltrials.gov/ct2/show/NCT00710528). Increased expression of p110γ is seen in chronic myeloid leukemia (Hickey and Cotter 2006; Knobbe et al. 2005). There are also data that suggest involvement of non-alpha isoforms of class I PI3K in solid tumors (Bénistant et al. 2000; Knobbe et al. 2005; Mizoguchi et al. 2004). The wild-type non-alpha isoforms have the ability to induce oncogenic transformation when overexpressed in cell culture, whereas wild-type p110α lacks this ability (Kang et al. 2006). The elevated expression of non-alpha isoforms in some cancers may therefore be a determinant of the oncogenic cellular phenotype. The oncogenic activity of wild-type non-alpha isoforms has so far been shown only in chicken embryo fibroblasts which are exquisitely sensitive to transformation by single oncoproteins. In this cell culture system, the various isoforms reveal distinctly different characteristics in their signaling through AKT, their interactions with RAS and sensitivity to inhibitors of the MAP kinase pathway (Denley et al. 2008). Overexpression of p110δ induces strong phosphorylation of AKT at T308 and S473 and of the downstream targets S6K, 4E-BP and GSK3β (glycogen and synthase kinase 3β). FOXO1 (forkhead box transcription factor O1) becomes undetectable in these cells. This signaling pattern closely resembles that of the p110α mutant H1047R. Expression of p110β and p110γ induces much lower levels of phosphorylation of AKT, S6K, 4E-BP, and GSK3β. The levels of FOXO1 are not significantly reduced in these cells. The contrasting properties of p110δ vs. p110β and p110γ are also seen in their responses to activation by RAS and to inhibition of the MAP kinase pathway. Introducing point mutations into the RAS binding domain that are designed to abolish RAS binding interferes with oncogenic transformation and signaling induced by p110β and p110γ, but does not affect p110δ. The activities of the mutated p110β and p110γ proteins can be restored by adding a myristylation signal to the N-terminus of the proteins, suggesting that the interaction with RAS mediates recruitment to the plasma membrane. The apparent independence of p110δ from RAS requires further examination and confirmation. Although the mutated residue in the RAS-binding domain of p110δ is conserved among the isoforms and is known to control RAS binding in p110α and in p110γ (Pirola et al. 2001; Rodriguez-Viciana et al. 1996), the effect of this mutation in p110δ on RAS binding has not been verified. Sensitivity to inhibitors also sets p110β and p110γ apart from p110δ. The MEK1/2 inhibitor U0126 effectively interferes with cellular transformation induced by p110β and p110γ, but does not affect the oncogenic activity of p110δ and of the H1047R mutant of p110α. The inhibitor of RAF, BAY 43-9006, shows a similar preferential effect on p110β and p110γ, but does not interfere with cellular transformation caused by p110δ. These observations single out p110δ as an exceptionally potent signaling protein, resistant to MAP kinase inhibition and capable of functioning preferentially through AKT. The similarities between p110β and p110γ are unexpected because these isoforms respond to different sets of upstream signaling: p110β has been linked to receptor tyrosine kinases and G-protein coupled receptors and p110γ exclusively to G-protein coupled receptors (Roche et al. 1998; Guillermet-Guibert et al. 2008). The similarities between p110β and p110γ revealed by the studies on oncogenicity and signaling in chicken embryo fibroblasts may extend to mammalian cells. An important role of non-alpha isoforms of PI3K in cancer is also emerging from new animal models and from studies in cell culture (Ciraolo et al. 2008; Jia et al. 2008; Torbett et al. 2008; Wee et al. 2008). These reveal isoform-specific enzymatic and nonenzymatic functions of p110β and are discussed in other chapters of this volume.
5 Class II and III PI3Ks
The family of PI3Ks encompasses three distinct classes that differ in structure and function (Vanhaesebroeck et al. 1997; Vanhaesebroeck and Waterfield 1999). One of the defining criteria for each class of PI3Ks is substrate recognition and hence the spectrum of products. Class I PI3Ks can utilize the nonphosphorylated phosphatidylinositol (PI), the monophosphate (PI(4)P), and the bisphosphate (PI(4,5)P2) phosphatidylinositols, giving rise to PI(3)P, PI(3,4)P2 and PI(3,4,5)P3, also referred to as PIP, PIP2 and PIP3, respectively. Class II PI3Ks recognize PI and PI(4)P, but not PI(4,5)P2 as substrates to produce PIP and PIP2. Class III PI3Ks can use only PI and convert it to PIP (Pirola et al. 2001). In addition, class I PI3Ks can function as serine protein kinases (Dhand et al. 1994). One of the protein substrates of class I PI3Ks is the regulatory subunit p85. This phosphorylation represents an autoregulatory mechanism (Foukas et al. 2004). The protein kinase activity of p110 is, however, not sufficient for oncogenic transformation induced by class I p110 mutants and isoforms (Denley et al. 2009; Kang et al. 2006). In the canonical PI3K signaling pathway, PIP2 and PIP3 are recognized by the pleckstrin homology domains of PDK-1 (phosphoinositide-dependent kinase) and of AKT (Alessi et al. 1996a, b; Klippel et al. 1997; Nicholson and Anderson 2002). These interactions recruit PDK-1 and AKT to the plasma membrane, resulting in the phosphorylation of AKT by PDK-1, catalytic activation of AKT, and phosphorylation of downstream targets (Alessi et al. 1997; Currie et al. 1999; Filippa et al. 2000; McManus et al. 2004; Milburn et al. 2003; Vanhaesebroeck and Alessi 2000). The structure of the AKT pleckstrin homology domain bound to IP4, the headgroup of PIP3, shows critical ionic interactions between basic pleckstrin homology domain residues and the phosphates at the D-3 and D-4 positions of IP4 (Milburn et al. 2003). The phosphate at the D-5 position does not participate in the interaction with the AKT pleckstrin homology domain. Hence, AKT has a lower affinity for PIP2 (PI(4,5)P2) than for PIP3.
So far, only class I PI3Ks have been firmly involved in cancer, although there are some observations that suggest class II may also play a role (Low et al. 2008). The defining characteristic of class I PI3Ks is the generation of PIP3 (Vanhaesebroeck et al. 1997; Vanhaesebroeck and Waterfield 1999). This ability may therefore constitute a prerequisite for the oncogenicity of lipid kinases. Emerging evidence supports this suggestion. A short sequence in the activation loop of PI3Ks determines substrate recognition and product specificity (Bondeva et al. 1998; Pirola et al. 2001). Substitution of this sequence in class I PI3K with the corresponding sequence of class II or of class III generates enzymes that produce PIP and PI(3,4)P2 or PIP, respectively, but fail to make PIP3. These constructs do not induce oncogenic transformation in cell culture and show greatly reduced signaling through AKT. Expression of the wild-type or the myristylated form of hVps34 class III PI3K fails to induce oncogenic transformation in cultures of chicken embryo fibroblasts and does not increase the phosphorylation status of Akt, p70 S6 kinase, 4E-BP, and glycogen synthase kinase-3β or cause a change in the level of FoxO1 (Denley et al. 2009). The production of PIP3 and PI3K signaling are reduced in the presence of the PIPP phosphatase which removes the phosphate from the D-5 position of phosphatidylinositol. Expression of PIPP also interferes with oncogenic transformation induced by the four isoforms of class I PI3K, reduces the levels of AKT phosphorylation and attenuates the degradation of FOXO1 (Denley et al. 2009). These observations support the conclusion that the ability to produce PIP3 is essential for the oncogenic activity of PI3K.
6 PI3K-Driven Oncogenic Transformation: Mechanistic Considerations
PI3K signaling affects numerous downstream targets, not all will be essential for oncogenic transformation. The canonical signaling cascade proceeds through AKT, the TSC (tuberous sclerosis complex), RHEB (Ras homolog enriched in brain) to TOR and from there to additional targets. In this pathway, two components stand out as particularly significant for oncogenicity: AKT and TOR. AKT is an important signal branching point that can direct PI3K signals into many different directions; TOR is of importance because it functions as integrator, receiving input from multiple sources. Thus, AKT and TOR link the canonical PI3K pathway to other regulatory activities in the cell.
In the canonical pathway, AKT phosphorylates and thereby inhibits TSC2 (Dan et al. 2002; Inoki et al. 2002; Manning et al. 2002). The TSC complex functions as GTPase-activating protein for RHEB (Castro et al. 2003; Garami et al. 2003; Inoki et al. 2003; Tee et al. 2003; Zhang et al. 2003b); reduction of GTPase activation by AKT activates RHEB. The GTP-bound RHEB then directly interacts with TOR and activates this target. AKT and RHEB are oncogenic when constitutively activated (Ahmed et al. 1993; Aoki et al. 1998; Jiang and Vogt 2008). TOR is a PI 3-kinase related protein kinase (PIKK) (Abraham 2004) that fulfills numerous tasks in cell growth and metabolism.
TOR functions in two distinct multiprotein complexes, TORC1 and TORC2 (Jacinto et al. 2004). TORC1 contains the proteins RAPTOR, LST8 and PRAS40 (Kim et al. 2002). TORC2 consists of LST8, RICTOR and SIN1 (Sarbassov et al. 2004). TORC1 and TORC2 are differentially regulated and have distinct functions. TORC1 can be activated by AKT-dependent and AKT-independent signals. AKT stimulates TORC1 by inducing an inhibition of the GTPase-activating protein activity of the TSC complex that targets RHEB and by phosphorylating and thereby inactivating PRAS40, a negative regulator of TORC1. AKT-independent regulation of TOR can also be mediated by the TSC complex. TSC is a sensor and integrator of signals that originate from energy deprivation, hypoxia or stimulation of growth. For instance, AMPK (AMP kinase), activated by an increase of cellular AMP, activates TSC2 and thereby inhibits TOR. Rag (Ras-related small GTP-binding proteins) activate TORC1 in response to the availability of amino acids (Sancak et al. 2008). Another AKT-independent pathway to TORC1 has been identified in glioblastoma and is mediated by PKCα (protein kinase Cα) (Fan et al. 2009). The principal downstream targets of TORC1 are 4E-BP1 (eukaryotic initiation factor 4E-binding protein) and S6K1 (p70 ribosomal protein S6 kinase). They will be considered below.
In contrast to the various AKT-dependent and AKT-independent ways that have been identified for the regulation of TORC1 (Cheng et al. 2009; Memmott and Dennis 2009; Vasudevan et al. 2009), the regulation of TORC2 is not fully understood. A distinguishing mark of TORC2 activation is the requirement for an active TSC complex, one that is not phosphorylated by AKT and opposite to the requirement for TORC1 activation (Huang and Manning 2009). Several targets have been identified for TORC2 (Huang et al. 2009). Among these, the phosphorylation of AKT at S473 appears potentially relevant to PI3K signaling (Sarbassov et al. 2005). This phosphorylation event achieves maximal activation of AKT, and it also expands the spectrum of AKT targets to include PRAS40 and FOXO (Guertin et al. 2006). However, it is doubtful whether the additional targets that can be addressed by the S473-phosphorylated AKT play an important role in transformation (see below).
TOR is essential for the oncogenicity of PI3K and AKT (Jiang et al. 2000; Neshat et al. 2001; Podsypanina et al. 2001). The TOR inhibitor rapamycin strongly and specifically interferes with PI3K- and AKT-induced cellular transformation; yet it does not reduce transformation caused by 14 other oncogenes (Aoki et al. 2001). Exposure to resistance-inducing concentrations of rapamycin does not significantly affect cell replication. Whereas short-term treatment with rapamycin selectively inhibits TORC1, cells treated over prolonged periods of time (24 h or more) also show inhibition of TORC2 (Sarbassov et al. 2006). Since interference with oncogenic transformation by rapamycin results from long-term treatment with the drug, both TORC1 and TORC2 would be affected and could play an essential role in oncogenicity. However, at least one TORC2 activity, the phosphorylation of AKT on S473, appears to be dispensable for oncogenic transformation (Aoki et al. 1998).
The available data are compatible with the idea that TORC1, but not TORC2, plays the predominant role in oncogenesis (Guertin and Sabatini 2007). TORC1 functions as a positive regulator of protein synthesis. It phosphorylates and thereby activates S6K. It also phosphorylates 4E-BP1, releasing eIF4E (eukaryotic initiation factor 4E, the cap-binding protein) to become available for the assembly of the translation initiation complex. The TOR-dependent stimulation of protein synthesis preferentially affects mRNAs that have complex secondary structures in their 5′ untranslated regions (Culjkovic et al. 2005). These secondary structures require unwinding performed by the eIF4A (eukaryotic initiation factor 4A) helicase that together with the eukaryotic initiation factors eIF4E and eIF4G forms the eIF4F initiation complex. Numerous mRNAs that encode growth-promoting proteins are characterized by 5′ untranslated sequences with complex secondary structures, and their efficient translation is highly dependent on an abundance of eIF4E and the eIF4A helicase (Culjkovic et al. 2006). The enhancement of this activity by TORC1 could be a critical factor in the transformation process.
The importance of high efficiency translational initiation in PI3K- and AKT-induced oncogenicity is also documented by the effects of the YB-1 (Y Box binding protein) on the transformation process. The YB-1 protein is highly conserved in evolution with close relatives throughout prokaryotic and eukaryotic forms of life. It is abundantly and ubiquitously expressed, and with its cold shock domain, it binds both DNA and RNA, affecting transcription and translation (Evdokimova et al. 2006a, 2009; Izumi et al. 2001; MacDonald et al. 1995; Mertens et al. 1997; Zasedateleva et al. 2002). By binding to mRNA, YB-1 has the capacity to inhibit translation (Evdokimova et al. 2006a). In cells transformed by PI3K or AKT, YB-1 is downregulated transcriptionally and posttranscriptionally (Bader et al. 2003; Bader and Vogt 2005, 2008; Evdokimova et al. 2006b; Sutherland et al. 2005). This downregulation appears to be a necessary facilitator of transformation, because re-expression of YB-1 causes a strong and specific cellular resistance to PI3K- and AKT-induced oncogenic transformation, yet does not interfere with transformation induced by other oncogenes (Bader et al. 2003). These YB-1-expressing cells do not show a detectable reduction in the rate of replication. Phenotypically, YB-1 therefore acts like a rapamycin mimic, but it intervenes in transformation downstream of TORC1, at a level of mRNA (Bader and Vogt 2008). Interference with transformation depends on cytoplasmic localization of YB-1 and on the ability of YB-1 to bind to RNA (Bader and Vogt 2005). The interaction of YB-1 with RNA is not sequence-specific, and YB-1 can bind to multiple sites on the mRNA. However, for the inhibition of protein synthesis, binding at or near the cap structure of mRNA is essential. According to a recent model, YB-1 then interferes with binding of eIF4G (eukaryotic initiation factor 4G) to mRNA and thus competes with the assembly of the eIF4F initiation complex on the mRNA (Svitkin et al. 2009).
The specific sensitivity of PI3K-induced transformation to rapamycin and to expressed YB-1 supports the conclusion that the oncogenic effects of PI3K are mediated by TORC1 and that they involve stimulation of protein synthesis. This activity of TOR appears necessary for transformation, but it is probably not sufficient. A gain of function in TOR alone has so far not been found to transform cells. Cells lacking TSC1 or TSC2 show constitutive activation of TOR, but are not transformed (Kwiatkowski et al. 2002; Zhang et al. 2003a). Patients with heritable loss of function in the TSC complex develop hamartomas, but aggressive cancers are rare (Al-Saleem et al. 1998; Kwiatkowski and Manning 2005; Marcotte and Crino 2006). However, in rodent model systems, inactivating mutations of either TSC1 or TSC2 increases cancer incidence, possibly due to secondary mutations (Everitt et al. 1992; Kobayashi et al. 1999; Kwiatkowski et al. 2002).
If gain of function in TOR is necessary but not sufficient for transformation, what then are the other necessary, complementing oncogenic activities that originate with PI3K signaling? There likely will be several. A possible candidate is one of the multiple targets of AKT: NFκB. The transcriptional activity of NFκB is upregulated in AKT-transformed cells. This increased function is dependent on AKT and is abolished by small molecule inhibitors of AKT and by a dominant negative mutant of AKT (Bai et al. 2009). In AKT-transformed cells, the total amount of IκB inhibitor protein is dramatically decreased. Blocking NFκB activity with the super-repressor of NFκB (IκBSR) induces a cellular resistance that is selective for PI3K- and AKT-induced transformation. Thus, NFκB activity is essential for the oncogenicity of PI3K and AKT. Although there is no general agreement on how AKT communicates with NFκB, the balance of the evidence supports the idea of a phosphorylation cascade that connects the two proteins. AKT can phosphorylate IKKα (IκB kinase) in vivo (Ozes et al. 1999), and the activated IKK complex then phosphorylates the p65 subunit of NFκB (Sakurai et al. 1999), enhancing its transcriptional activity. It is, however, possible that the essential requirement for NFκB in PI3K-induced transformation can be satisfied with a basal level of activity and that the AKT-mediated gain of function represents a secondary consequence of transformation.
The identification of essential components in the oncogenic pathway is important for therapeutic considerations and helps define suitable drug targets. In this regard, the catalytic subunit p110 of PI3K remains a strong candidate, but more understanding of isoform-specific functions in various cancers and cell types and on different contributing genetic backgrounds (e.g., gain of function in receptor tyrosine kinases, loss of function in PTEN) is needed (Garcia-Echeverria and Sellers 2008; Kong and Yamori 2008; Maira et al. 2008; Wymann and Schneiter 2008; Yap et al. 2008). TOR emerges as another promising drug target (Guertin and Sabatini 2009). ATP-competitive inhibitors of TOR have recently been identified and are being characterized. They affect rapamycin-resistant functions of TOR (Feldman et al. 2009; GarcÚa-MartÚnez et al. 2009; Guertin and Sabatini 2009; Malagu et al. 2009; Nowak et al. 2009; Thoreen et al. 2009; Yu et al. 2009; Zask et al. 2009). The situation with AKT is more complex, because not all PI3K-driven tumors show AKT dependence (Vasudevan et al. 2009). Our understanding of the oncogenic signals emanating from PI3K is still evolving. New pathways and feedbacks are being characterized and novel interacting proteins discovered. Tissue- and cell-type specific differences in PI3K signaling are being defined. All these advances will eventually result in the recognition of new drug targets.
7 Conclusion
PI3Ks have oncogenic potential. The requisite gain of function can be achieved by mutation or by differential expression. Oncogenicity is mainly associated with class I PI3Ks and correlated with the ability to produce PIP3. PIP3 is the critical PI3K product that links lipid kinase activity to a network of downstream signals originating in AKT. In sensitive experimental systems, gain of function in a single PI3K isoform is sufficient to induce oncogenic transformation. These systems can be used for quantitative determination of oncogenicity and serve as models for investigations of transformation-associated changes in the cellular phenotype. Transformed focus assays in cell culture remain the gold standard for measuring and comparing oncogenic activity; such assays include all four isoforms of class I PI3K and can yield valuable data on the antioncogenic potency of drug candidates. The experimental systems of PI3K-induced oncogenic transformation also allow determination of specific changes in signaling pathways, cell behavior and metabolism.
The PI3K pathway is deregulated in the majority of human cancers. In sporadic tumors and in cancer cell lines, there are numerous other genetic and epigenetic changes that have been extensively documented by the human cancer genome project (Cancer Genome Atlas Research Network 2008; He et al. 2008; Jones et al. 2008; Parsons et al. 2008; Wood et al. 2007). In these situations, PI3K signaling can be expected to make a contribution to the oncogenic phenotype of the cell, but rarely will it function as the sole or dominant transforming event. The oncogenic phenotype of human cancer is the composite of all genetic and epigenetic changes. Experience with inhibitors of PI3K reflects this complexity. The growth of cells experimentally transformed by PI3K is generally highly sensitive to PI3K inhibitors. Few human cancer cell lines show such sensitivity. However, combinations of inhibitors that target critical nodes in cellular signaling or single inhibitors that target a critical combination of oncoproteins can be very effective (Cheng et al. 2009; Fan et al. 2006, 2007, 2009; Fan and Weiss 2006; Jaiswal et al. 2009b; Nelander et al. 2008). These observations are detailed in other chapters of this book.
The greatest challenge in the area of PI3K oncogenicity remains the identification of mutant-specific inhibitors that have drug-like properties. The highly targeted therapeutic potential of such inhibitors justifies intense efforts by industry and in academic laboratories.
References
Abraham RT (2004) PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 3:883–887
Ahmed NN, Franke TF, Bellacosa A, Datta K, Gonzalez-Portal ME, Taguchi T, Testa JR, Tsichlis PN (1993) The proteins encoded by c-akt and v-akt differ in post-translational modification, subcellular localization and oncogenic potential. Oncogene 8:1957–1963
Alcazar I, Marques M, Kumar A, Hirsch E, Wymann M, Carrera AC, Barber DF (2007) Phosphoinositide 3-kinase gamma participates in T cell receptor-induced T cell activation. J Exp Med 204:2977–2987
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996a) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551
Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P (1996b) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7:261–269
Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, Kuehn N, Gray A, Giddings J, Peskett E, Fox R, Bruce I, Walker C, Sawyer C, Okkenhaug K, Finan P, Vanhaesebroeck B (2004) Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature 431:1007–1011
Ali K, Camps M, Pearce WP, Ji H, Rückle T, Kuehn N, Pasquali C, Chabert C, Rommel C, Vanhaesebroeck B (2008) Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J Immunol 180:2538–2544
Al-Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ, Fujikawa K, Bjornsson J, Bernstein J, Henske EP (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83:2208–2216
Amzel LM, Huang CH, Mandelker D, Lengauer C, Gabelli SB, Vogelstein B (2008) Structural comparisons of class I phosphoinositide 3-kinases. Nat Rev Cancer 8:665–669
Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK (1998) The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci USA 95:14950–14955
Aoki M, Schetter C, Himly M, Batista O, Chang HW, Vogt PK (2000) The catalytic subunit of phosphoinositide 3-kinase: requirements for oncogenicity. J Biol Chem 275:6267–6275
Aoki M, Blazek E, Vogt P (2001) A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA 98:136–141
Bader AG, Vogt PK (2005) Inhibition of protein synthesis by Y box-binding protein 1 blocks oncogenic cell transformation. Mol Cell Biol 25:2095–2106
Bader AG, Vogt P (2008) Phosphorylation by Akt disables the anti-oncogenic activity of YB-1. Oncogene 27:1179–1182
Bader AG, Felts KA, Jiang N, Chang HW, Vogt PK (2003) Y box-binding protein 1 induces resistance to oncogenic transformation by the phosphatidylinositol 3-kinase pathway. Proc Natl Acad Sci USA 100:12384–12389
Bader AG, Kang S, Vogt PK (2006) Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci USA 103:1475–1479
Bai D, Ueno L, Vogt PK (2009) Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer 125:2863–2870
Bénistant C, Chapuis H, Roche S (2000) A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 19:5083–5090
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem 274:10963–10968
Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome 13:169–172
Bondeva T, Pirola L, Bulgarelli-Leva G, Rubio I, Wetzker R, Wymann MP (1998) Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science 282:293–296
Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C (2009) PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol 21:194–198
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657
Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC (1990) Purification and characterization of phosphoinositide 3-kinase from rat liver. J Biol Chem 265:19704–19711
Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, Luo L (2008) Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J 409:519–524
Castro AF, Rebhun JF, Clark GJ, Quilliam LA (2003) Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 278:32493–32496
Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM, Vogt PK (1997) Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science 276:1848–1850
Chaussade C, Cho K, Mawson C, Rewcastle GW, Shepherd PR (2009) Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun 381:577–581
Cheng CK, Fan QW, Weiss WA (2009) PI3K signaling in glioma–animal models and therapeutic challenges. Brain Pathol 19:112–120
Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastrù W, Martin EL, Silengo L, Altruda F, Turco E, Lanzetti L, Musiani P, Rückle T, Rommel C, Backer JM, Forni G, Wymann MP, Hirsch E (2008) Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1:ra3
Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, Rawlings D, Reynolds H, Vigorito E, Turner M (2002) A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med 196:753–763
Courtneidge SA, Smith AE (1984) The complex of polyoma virus middle-T antigen and pp 60c-src. EMBO J 3:585–591
Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K, Mills GB (2001) Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem 276:27455–27461
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL (2005) eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3’UTR. J Cell Biol 169:245–256
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL (2006) eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol 175:415–426
Currie R, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J (1999) Role of phosphatidylinositol 3, 4, 5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 337(pt 3):575–583
Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, Nellist M, Yeung RS, Halley DJ, Nicosia SV, Pledger WJ, Cheng JQ (2002) Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem 277:35364–35370
Deane JA, Fruman DA (2004) Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol 22:563–598
Denley A, Kang S, Karst U, Vogt P (2008) Oncogenic signaling of class I PI3K isoforms. Oncogene 27:2561–2574
Denley A, Gymnopoulos M, Kang S, Mitchell C, Vogt PK (2009) Requirement of phosphatidylinositol(3, 4, 5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol Cancer Res 7:1132–1138
Dhand R, Hiles I, Panayotou G, Roche S, Fry MJ, Gout I, Totty NF, Truong O, Vicendo P, Yonezawa K et al (1994) PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. EMBO J 13:522–533
Di Cristofano A, Pandolfi PP (2000) The multiple roles of PTEN in tumor suppression. Cell 100:387–390
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, García-Echeverría C, Weissleder R, Mahmood U, Cantley L, Wong KK (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14:1351–1356
Evdokimova V, Ovchinnikov LP, Sorensen PH (2006a) Y-box binding protein 1: providing a new angle on translational regulation. Cell Cycle 5:1143–1147
Evdokimova V, Ruzanov P, Anglesio MS, Sorokin A, Ovchinnikov L, Buckley J, Triche TJ, Sonenberg N, Sorensen P (2006b) Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol Cell Biol 26:277–292
Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, Sorensen PH (2009) Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15:402–415
Everitt JI, Goldsworthy TL, Wolf DC, Walker CL (1992) Hereditary renal cell carcinoma in the Eker rat: a rodent familial cancer syndrome. J Urol 148:1932–1936
Fan QW, Weiss WA (2006) Isoform specific inhibitors of PI3 kinase in glioma. Cell Cycle 5:2301–2305
Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9:341–349
Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA (2007) A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res 67:7960–7965
Fan QW, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD, McCormick F, Shokat KM, Weiss WA (2009) EGFR Signals to mTOR Through PKC and Independently of Akt in Glioma. Science Signal 2:ra4
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7:e38
Filippa N, Sable CL, Hemmings BA, Van Obberghen E (2000) Effect of phosphoinositide-dependent kinase 1 on protein kinase B translocation and its subsequent activation. Mol Cell Biol 20:5712–5721
Foukas LC, Beeton CA, Jensen J, Phillips WA, Shepherd PR (2004) Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol Cell Biol 24:966–975
Fruman DA, Bismuth G (2009) Fine tuning the immune response with PI3K. Immunol Rev 228:253–272
Fruman DA, Meyers RE, Cantley L (1998) Phosphoinositide kinases. Annu Rev Biochem 67:481–507
Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11:1457–1466
Garcia-Echeverria C, Sellers WR (2008) Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 27:5511–5526
GarcÚa-MartÚnez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR (2009) Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J 421:29–42
Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B (2007) Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA 104:7809–7814
Ghigo A, Hirsch E (2008) Isoform selective phosphoinositide 3-kinase gamma and delta inhibitors and their therapeutic potential. Recent Pat Inflamm Allergy Drug Discov 2:1–10
Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B (2008) Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453:662–666
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12:9–22
Guertin DA, Sabatini DM (2009) The pharmacology of mTOR inhibition. Science Signal 2:pe24
Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11:859–871
Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K, Vanhaesebroeck B (2008) The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci USA 105:8292–8297
Gymnopoulos M, Elsliger MA, Vogt PK (2007) Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA 104:5569–5574
Hawkins PT, Anderson KE, Davidson K, Stephens LR (2006) Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 34:647–662
He X, Zhu Z, Johnson C, Stoops J, Eaker A, Bowen W, Defrances M (2008) PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res 68:5591–5598
Hickey FB, Cotter TG (2006) BCR-ABL regulates phosphatidylinositol 3-kinase-p110gamma transcription and activation and is required for proliferation and drug resistance. J Biol Chem 281:2441–2450
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287:1049–1053
Huang J, Manning BD (2009) A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 37:217–222
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM (2007) The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318:1744–1748
Huang CH, Mandelker D, Gabelli SB, Amzel LM (2008) Insights into the oncogenic effects of PIK3CA mutations from the structure of p110alpha/p85alpha. Cell Cycle 7:1151–1156
Huang J, Wu S, Wu CL, Manning BD (2009) Signaling events downstream of mammalian target of rapamycin complex 2 are attenuated in cells and tumors deficient for the tuberous sclerosis complex tumor suppressors. Cancer Res 69:6107–6114
Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65:4562–4567
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657
Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834
Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS (2005) Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 65:10992–11000
Izumi H, Imamura T, Nagatani G, Ise T, Murakami T, Uramoto H, Torigoe T, Ishiguchi H, Yoshida Y, Nomoto M, Okamoto T, Uchiumi T, Kuwano M, Funa K, Kohno K (2001) Y box-binding protein-1 binds preferentially to single-stranded nucleic acids and exhibits 3′– > 5′ exonuclease activity. Nucleic Acids Res 29:1200–1207
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6:1122–1128
Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J, Modrusan Z, Stinson J, Eby M, Yue P, Kaminker JS, de Sauvage FJ, Backer JM, Seshagiri S (2009a) Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 16:463–474
Jaiswal BS, Janakiraman V, Kljavin NM, Eastham-Anderson J, Cupp JE, Liang Y, Davis DP, Hoeflich KP, Seshagiri S (2009b) Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS ONE 4:e5717
Ji H, Rintelen F, Waltzinger C, Bertschy Meier D, Bilancio A, Pearce W, Hirsch E, Wymann MP, Rückle T, Camps M, Vanhaesebroeck B, Okkenhaug K, Rommel C (2007) Inactivation of PI3Kgamma and PI3Kdelta distorts T-cell development and causes multiple organ inflammation. Blood 110:2940–2947
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ (2008) Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454:776–779
Jiang H, Vogt PK (2008) Constitutively active Rheb induces oncogenic transformation. Oncogene 27:5729–5740
Jiang B, Zheng JZ, Aoki M, Vogt PK (2000) Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci USA 97:1749–1753
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321:1801–1806
Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Carpino N, Wang D, Ihle JN (2002) Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol 22:8580–8591
Kang S, Bader AG, Vogt PK (2005) Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 102:802–807
Kang S, Denley A, Vanhaesebroeck B, Vogt PK (2006) Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA 103:1289–1294
Kaplan DR, Pallas DC, Morgan W, Schaffhausen B, Roberts TM (1989) Mechanisms of transformation by polyoma virus middle T antigen. Biochim Biophys Acta 948:345–364
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175
Klippel A, Kavanaugh WM, Pot D, Williams LT (1997) A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol 17:338–344
Knobbe CB, Trampe-Kieslich A, Reifenberger G (2005) Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol 31:486–490
Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T (1999) Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res 59:1206–1211
Kong D, Yamori T (2008) Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci 99:1734–1740
Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14 Spec No. 2:R251–R258
Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H (2002) A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet 11:525–534
Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann MP (2002) Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 16:441–451
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947
Liu Z, Roberts TM (2006) Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle 5:675–677
Low S, Vougioukas VI, Hielscher T, Schmidt U, Unterberg A, Halatsch ME (2008) Pathogenetic pathways leading to glioblastoma multiforme: association between gene expressions and resistance to erlotinib. Anticancer Res 28:3729–3732
MacDonald GH, Itoh-Lindstrom Y, Ting JP (1995) The transcriptional regulatory protein, YB-1, promotes single-stranded regions in the DRA promoter. J Biol Chem 270:3527–3533
Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J Biol Chem 273:13375–13378
Maira S, Voliva C, García-Echeverría C (2008) Class IA phosphatidylinositol 3-kinase: from their biologic implication in human cancers to drug discovery. Expert Opin Ther Targets 12:223–238
Malagu K, Duggan H, Menear K, Hummersone M, Gomez S, Bailey C, Edwards P, Drzewiecki J, Leroux F, Quesada MJ, Hermann G, Maine S, Molyneaux CA, Le Gall A, Pullen J, Hickson I, Smith L, Maguire S, Martin N, Smith G, Pass M (2009) The discovery and optimisation of pyrido[2,3-d]pyrimidine-2,4-diamines as potent and selective inhibitors of mTOR kinase. Bioorg Med Chem Lett 19:5950–5953
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10:151–162
Marcotte L, Crino PB (2006) The neurobiology of the tuberous sclerosis complex. Neuromolecular Med 8:531–546
Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784:159–185
McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J 23:2071–2082
Memmott RM, Dennis PA (2009) Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 21:656–664
Mertens PR, Harendza S, Pollock AS, Lovett DH (1997) Glomerular mesangial cell-specific transactivation of matrix metalloproteinase 2 transcription is mediated by YB-1. J Biol Chem 272:22905–22912
Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, van Aalten DM (2003) Binding of phosphatidylinositol 3, 4, 5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 375:531–538
Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL (2007) Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317:239–242
Mizoguchi M, Nutt CL, Mohapatra G, Louis DN (2004) Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 14:372–377
Nelander S, Wang W, Nilsson B, She QB, Pratilas C, Rosen N, Gennemark P, Sander C (2008) Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol 4:216
Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL (2001) Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA 98:10314–10319
Nicholson KM, Anderson NG (2002) The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 14:381–395
Nowak P, Cole DC, Brooijmans N, Bursavich MG, Curran KJ, Ellingboe JW, Gibbons JJ, Hollander I, Hu Y, Kaplan J, Malwitz DJ, Toral-Barza L, Verheijen JC, Zask A, Zhang WG, Yu K (2009) Discovery of potent and selective inhibitors of the mammalian target of rapamycin (mTOR) kinase. J Med Chem 52:7081–7089
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B (2002) Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297:1031–1034
Okkenhaug K, Bilancio A, Emery JL, Vanhaesebroeck B (2004) Phosphoinositide 3-kinase in T cell activation and survival. Biochem Soc Trans 32:332–335
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB (1999) NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401:82–85
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Patton DT, Garçon F, Okkenhaug K (2007) The PI3K p110delta controls T-cell development, differentiation and regulation. Biochem Soc Trans 35:167–171
Pirola L, Zvelebil MJ, Bulgarelli-Leva G, Van Obberghen E, Waterfield MD, Wymann MP (2001) Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha). Functions of lipid kinase-deficient PI3Kalpha in signaling. J Biol Chem 276:21544–21554
Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, Frost P, Dreisbach V, Blenis J, Gaciong Z, Fisher P, Sawyers C, Hedrick-Ellenson L, Parsons R (2001) An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc Natl Acad Sci USA 98:10320–10325
Roche S, Downward J, Raynal P, Courtneidge SA (1998) A function for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol 18:7119–7129
Rodriguez-Borlado L, Barber DF, Hernandez C, Rodriguez-Marcos MA, Sanchez A, Hirsch E, Wymann M, Martinez AC, Carrera AC (2003) Phosphatidylinositol 3-kinase regulates the CD4/CD8 T cell differentiation ratio. J Immunol 170:4475–4482
Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J (1996) Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J 15:2442–2451
Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W (1999) IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274:30353–30356
Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3:1221–1224
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14:1296–1302
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287:1040–1046
Schaffhausen BS, Roberts TM (2009) Lessons from polyoma middle T antigen on signaling and transformation: a DNA tumor virus contribution to the war on cancer. Virology 384:304–316
Shekar SC, Wu H, Fu Z, Yip SC, Nagajyothi CSM, Girvin ME, Backer JM (2005) Mechanism of constitutive phosphoinositide 3-kinase activation by oncogenic mutants of the p85 regulatory subunit. J Biol Chem 280:27850–27855
Stephens L, Hawkins PT, Eguinoa A, Cooke F (1996) A heterotrimeric GTPase-regulated isoform of PI3K and the regulation of its potential effectors. Philos Trans R Soc Lond B Biol Sci 351:211–215
Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89:105–114
Stephens L, Williams R, Hawkins P (2005) Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol 5:357–365
Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nürnberg B (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269:690–693
Sugimoto Y, Whitman M, Cantley L, Erikson RL (1984) Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proc Natl Acad Sci USA 81:2117–2121
Sugita H, Dan S, Kong D, Tomida A, Yamori T (2008) A new evaluation method for quantifying PI3K activity by HTRF assay. Biochem Biophys Res Commun 377:941–945
Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, Hunault-Berger M, Berthou C, Villemagne B, Jourdan E, Audhuy B, Solary E, Witz B, Harousseau JL, Himberlin C, Lamy T, Lioure B, Cahn JY, Dreyfus F, Mayeux P, Lacombe C, Bouscary D (2005) Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106:1063–1066
Sutherland BW, Kucab J, Wu J, Lee C, Cheang MC, Yorida E, Turbin D, Dedhar S, Nelson C, Pollak M, Leighton Grimes H, Miller K, Badve S, Huntsman D, Blake-Gilks C, Chen M, Pallen CJ, Dunn SE (2005) Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 24:4281–4292
Svitkin YV, Evdokimova VM, Brasey A, Pestova TV, Fantus D, Yanagiya A, Imataka H, Skabkin MA, Ovchinnikov LP, Merrick WC, Sonenberg N (2009) General RNA-binding proteins have a function in poly(A)-binding protein-dependent translation. EMBO J 28:58–68
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13:1259–1268
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032
Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, Shokat KM, Stokoe D (2008) A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J 415:97–110
Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ (2007) The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol 81:7069–7076
Vanhaesebroeck B, Alessi DR (2000) The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346(pt 3):561–576
Vanhaesebroeck B, Waterfield MD (1999) Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res 253:239–254
Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD (1997) Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci 22:267–272
Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70:535–602
Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC (2005) Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 30:194–204
Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA (2009) AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16:21–32
Walker EH, Perisic O, Ried C, Stephens L, Williams RL (1999) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402:313–320
Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C (2008) PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA 105:13057–13062
Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM (1985) Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B (2007) The genomic landscapes of human breast and colorectal cancers. Science 318:1108–1113
Wu H, Shekar SC, Flinn RJ, El-Sibai M, Jaiswal BS, Sen KI, Janakiraman V, Seshagiri S, Gerfen GJ, Girvin ME, Backer JM (2009) Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci USA. 106:20258–20263
Wymann MP, Schneiter R (2008) Lipid signalling in disease. Nat Rev Mol Cell Biol 9:162–176
Yap T, Garrett M, Walton M, Raynaud F, Debono J, Workman P (2008) Targeting the PI3K–AKT–mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol 8:393–412
Yart A, Chap H, Raynal P (2002) Phosphoinositide 3-kinases in lysophosphatidic acid signaling: regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim Biophys Acta 1582:107–111
Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, Nowak P, Zask A (2009) Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res 69:6232–6240
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510
Zasedateleva OA, Krylov AS, Prokopenko DV, Skabkin MA, Ovchinnikov LP, Kolchinsky A, Mirzabekov AD (2002) Specificity of mammalian Y-box binding protein p50 in interaction with ss and ds DNA analyzed with generic oligonucleotide microchip. J Mol Biol 324:73–87
Zask A, Verheijen JC, Curran K, Kaplan J, Richard DJ, Nowak P, Malwitz DJ, Brooijmans N, Bard J, Svenson K, Lucas J, Toral-Barza L, Zhang WG, Hollander I, Gibbons JJ, Abraham RT, Ayral-Kaloustian S, Mansour TS, Yu K (2009) ATP-Competitive Inhibitors of the Mammalian Target of Rapamycin: Design and Synthesis of Highly Potent and Selective Pyrazolopyrimidines. J Med Chem 52:5013–5016
Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ (2003a) Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 112:1223–1233
Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D (2003b) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5:578–581
Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G (2008) Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat 112:217–227
Zhao JJ, Roberts TM (2006) PI3 kinases in cancer: from oncogene artifact to leading cancer target. Sci STKE 2006:pe52
Zhao L, Vogt P (2008a) Class I PI3K in oncogenic cellular transformation. Oncogene 27:5486–5496
Zhao L, Vogt PK (2008b) Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA 105:2652–2657
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM (2005) The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci USA 102:18443–18448
Acknowledgment
Work of the authors is supported by grants from the National Cancer Institute. This is manuscript number 20111 of The Scripps Research Institute.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Vogt, P.K. et al. (2010). Phosphatidylinositol 3-Kinase: The Oncoprotein. In: Rommel, C., Vanhaesebroeck, B., Vogt, P. (eds) Phosphoinositide 3-kinase in Health and Disease. Current Topics in Microbiology and Immunology, vol 347. Springer, Berlin, Heidelberg. https://doi.org/10.1007/82_2010_80
Download citation
DOI: https://doi.org/10.1007/82_2010_80
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-14815-6
Online ISBN: 978-3-642-14816-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)