
Abstract
3,4-Methylenedioxypyrovalerone (MDPV) is a psychoactive component of so-called bath salts products that has caused serious medical consequences in humans. In this chapter, we review the neuropharmacology of MDPV and related analogs, and supplement the discussion with new results from our preclinical experiments. MDPV acts as a potent uptake inhibitor at plasma membrane transporters for dopamine (DAT) and norepinephrine (NET) in nervous tissue. The MDPV formulation in bath salts is a racemic mixture, and the S isomer is much more potent than the R isomer at blocking DAT and producing abuse-related effects. Elevations in brain extracellular dopamine produced by MDPV are likely to underlie its locomotor stimulant and addictive properties. MDPV displays rapid pharmacokinetics when injected into rats (0.5–2.0 mg/kg), with peak plasma concentrations achieved by 10–20 min and declining quickly thereafter. MDPV is metabolized to 3,4-dihydroxypyrovalerone (3,4-catechol-PV) and 4-hydroxy-3-methoxypyrovalerone (4-OH-3-MeO-PV) in vivo, but motor activation produced by the drug is positively correlated with plasma concentrations of parent drug and not its metabolites. 3,4-Catechol-PV is a potent uptake blocker at DAT in vitro but has little activity after administration in vivo. 4-OH-3-MeO-PV is the main MDPV metabolite but is weak at DAT and NET. MDPV analogs, such as α-pyrrolidinovalerophenone (α-PVP), display similar ability to inhibit DAT and increase extracellular dopamine concentrations. Taken together, these findings demonstrate that MDPV and its analogs represent a unique class of transporter inhibitors with a high propensity for abuse and addiction.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Drug abuse and addiction are persistent public health concerns, and an alarming new trend is the increased non-medical use of so-called designer drugs, legal highs, or research chemicals [1] [2, this volume], [3]. These drugs, collectively known as “new psychoactive substances” (NPS), are synthetic alternatives to more traditional illegal drugs of abuse. At the present time, there are popular NPS which mimic the effects of most types of abused drugs, including stimulants (e.g., “bath salts”), cannabinoids (e.g., “spice”), and hallucinogens (e.g., “NBOMes”). Most are manufactured by Asian laboratories and sold to consumers via the Internet or shipped to locations in Europe, the United States America (US), and elsewhere to be packaged for retail sale [4, 5]. NPS are marketed as non-drug products, given innocuous names, and labeled “not for human consumption” as a means to avoid legal scrutiny. Compared to traditional drugs of abuse, NPS are cheap, easy to obtain, and often not detectable by standard toxicology screens. As governments pass laws to ban specific NPS, clandestine chemists respond by quickly creating novel “replacement” analogs to stay one step ahead of law enforcement [6, 7]. The abuse of NPS is a global phenomenon fueled by information freely available on the Internet. Recent data from the United Nations indicate that 540 different NPS have been identified worldwide as of 2014, and this number is expected to rise [8].
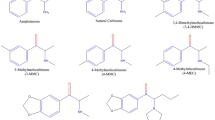
The first stimulant-like NPS to appear in the US were found in the so-called bath salts products which flooded the recreational drug marketplace beginning in late 2010 [9]. By early 2011, there was a dramatic spike in reports of bath salts intoxications to poison control centers, and an influx of patients admitted to emergency departments with toxic exposures [10–12]. Bath salts consist of powders or crystals that are administered intra-nasally or orally to produce their psychoactive effects. Low doses of bath salts induce typical psychomotor stimulant effects such as increased energy and mood elevation, but high doses or binge use can cause severe symptoms including hallucinations, psychosis, increased heart rate, high blood pressure and hyperthermia, often accompanied by combative or violent behaviors [9, 13]. The most serious syndrome induced by bath salts is known as “excited delirium,” a constellation of symptoms including elevated body temperature, delirium, agitation, breakdown of muscle tissue, and kidney failure, sometimes culminating in death [14, 15]. Forensic analysis of bath salts products in 2010 and 2011 identified three main synthetic compounds: 4-methyl-N-methylcathinone (mephedrone), 3,4-methylenedioxy-N-methylcathinone (methylone), and 3,4-methylenedioxypyrovalerone (MDPV) (Spiller et al. 2011; [7, 12]). These compounds are chemically similar to the naturally occurring substance cathinone, an amphetamine-like stimulant found in the khat plant, Catha edulis. Legislation passed in 2013 placed mephedrone, methylone, and MDPV into permanent Schedule I control, making the drugs illegal in the US [16]. Figure 1 depicts the chemical structures of bath salts cathinones compared to the related compounds amphetamine and cathinone.

Chemical structures of MDPV and related pyrrolidinophenones, and their relationship to amphetamine and cathinone
Although a number of different cathinones are found in bath salts products (e.g., [7, 12, 17]), MDPV appears more apt to cause life-threatening medical consequences (see [18]). For example, in the first study of patients reported to US poison control centers for “bath salts” overdose, the majority of subjects with blood and urine toxicology data were positive for MDPV but not mephedrone or methylone [12]. A more recent interrogation of a US clinical toxicology database found that all patients with confirmed synthetic cathinone exposure tested positive for MDPV [19]. Perhaps more importantly, MDPV was found in blood and urine from many fatal cases of drug overdose in the US and Europe [12, 14, 20–22]. Collectively, the clinical case data point to MDPV as the chief culprit in causing serious medical consequences. Given the widespread popularity of MDPV and the risks associated with its use, the purpose of the present chapter is to describe the neuropharmacology of MDPV, its metabolites, and related analogs. We review the literature on this topic and supplement the discussion with new data from preclinical experiments carried out at the Intramural Research Program (IRP) of the National Institute on Drug Abuse (NIDA).
2 Pharmacology of MDPV and Its Stereoisomers
2.1 Stimulant Drugs and Monoamine Transporters
As noted above, the psychoactive constituents of bath salts are chemically related to the parent compound cathinone, the β-keto analog of amphetamine (see Fig. 1 for structures). Mephedrone and methylone have functional groups attached to the phenyl ring and are considered ring-substituted cathinones, whereas MDPV is structurally more complex with a bulky nitrogen-containing pyrrolidine ring and a flexible alkyl chain extending from the α-carbon. MDPV and related compounds containing a pyrrolidine ring are collectively known as pyrrolidinophenones. Like other stimulant drugs, bath salts cathinones exert their effects by binding to transporter proteins on the surface of nerve cells that synthesize the monoamine neurotransmitters dopamine, norepinephrine, and serotonin (5-HT) [23] [24, this volume]. In order to understand the precise mechanism of action for cathinone analogs at the molecular level, it is essential to first consider the physiological role of monoamine transporters and the types of drugs targeting these proteins.
Under normal circumstances, the solute carrier 6 (SLC6) transporters for dopamine (DAT), norepinephrine (NET), and serotonin (SERT) are responsible for translocating previously released neurotransmitter molecules from the extracellular medium back into the neuronal cytoplasm, a process known as neurotransmitter “uptake” [25]. Transporter-mediated uptake is the principal mechanism for terminating the action of monoamine neurotransmitters, so drugs targeting these transporter proteins can have profound effects on cell-to-cell monoamine signaling. Accordingly, monoamine transporters are the principal sites of action for medications used to treat a range of psychiatric diseases such as depression, anxiety, and attention-deficit hyperactivity disorder [26, 27]. Drugs which preferentially interact at SERT are widely prescribed as efficacious treatments for major depression and anxiety disorders. By contrast, drugs which preferentially act at DAT and NET, like amphetamine and methamphetamine, have powerful psychomotor stimulant and addictive properties [28, 29].
Drugs that bind to monoamine transporters can be divided into two types based on their precise molecular mechanisms of action: (1) cocaine-like “inhibitors” – which bind to the neurotransmitter binding site on the transporter (i.e., orthosteric site), thereby blocking uptake of neurotransmitters from the extracellular medium, and (2) amphetamine-like “substrates” – which also bind to the orthosteric site, but are subsequently translocated through the transporter channel into the neuronal cytoplasm and trigger efflux of intracellular neurotransmitter molecules (i.e., transporter-mediated release) [30, 31]. Drugs that act as transporter substrates are often called “releasers” because they induce non-exocytotic transporter-mediated neurotransmitter release from neurons. Irrespective of molecular mechanism, all drugs which bind to transporters can dramatically increase extracellular concentrations of monoamines in vivo, amplifying cell-to-cell chemical signaling in various brain circuits. It is important to distinguish between transporter inhibitors versus substrates because substrates display a number of unique properties: they are translocated into cells along with sodium ions, they induce inward depolarizing sodium currents, and they reverse the normal direction of transporter flux to trigger non-exocytotic release of neurotransmitters (i.e., reverse transport) [30, 31]. Finally, because transporter substrate-type drugs are accumulated into the neuronal cytoplasm, they can produce intracellular deficits in monoamine neurons such as inhibition of neurotransmitter synthesis and disruption of vesicular storage, leading to long-term neurotransmitter depletions [32, this volume] [33] [34].
In our laboratory, we developed in vitro functional assays to assess the ability of test drugs to act as inhibitors or substrates at DAT, NET, and SERT [35, 36]. We employ two types of assays: (1) uptake inhibition and (2) release. The assays are carried out in synaptosomes derived from rat brain tissue and are designed to rapidly assess potency and efficacy of drugs at all three transporters under similar conditions. Synaptosomes consist of sealed vesicle-filled nerve endings with their plasma membrane leaflets oriented in a manner akin to neurons in vivo. For the uptake inhibition assays, radiolabeled substrate (i.e., [3H]neurotransmitter) and test drug are co-incubated with synaptosomes for a brief period, and the reaction is stopped by vacuum filtration. If test drugs are transporter inhibitors, the accumulation of [3H]neurotransmitter into the synaptosomes (i.e., uptake) is blocked because the test drug and neurotransmitter compete for the same orthosteric binding site on the transporter protein. It is noteworthy that uptake inhibition assays cannot distinguish between inhibitors and substrates because both types of drugs will effectively reduce the accumulation of [3H]neurotransmitter into synaptosomes.
In order to definitively identify substrate-type drugs, we use release assays. For the release assays, synaptosomes are first incubated with radiolabeled substrate molecules in order to fill or “preload” the interior of the synaptosomes. [3H]1-Methyl-4-phenylpyridinium ([3H]MPP+) is used as the radiolabeled substrate for DAT and NET release assays, whereas [3H]5-HT is used for SERT release assays. Once synaptosomes are preloaded, test drug is added for a brief incubation period, and the reaction is stopped by vacuum filtration. If test drugs are transporter substrates, efflux of [3H]MPP+ or [3H]5-HT out of the synaptosomes is induced (i.e., release) by reversal of the normal direction of transporter flux. Drugs that act as pure transporter inhibitors will not evoke substantial release of [3H]MPP+ or [3H]5-HT from preloaded synaptosomes. Therefore, by testing drugs in the combined uptake inhibition and release assay procedures, the precise molecular mechanism of drug action can be ascertained.
2.2 Molecular Mechanisms of Action
Prior to the bath salts phenomenon in 2010–2011, few scientific investigations had examined the pharmacology of ring-substituted cathinones or pyrrolidinophenones. Studies from the 1980s demonstrated that cathinone and methcathinone release dopamine from rat brain tissue by an amphetamine-like mechanism [37, 38], and subsequent reports revealed methcathinone acts as a substrate for DAT, NET, and SERT [36, 39]. Cozzi et al. [39] first demonstrated that methylone acts as an uptake inhibitor at monoamine transporters [39], while other investigations showed the drug is a transporter substrate capable of releasing dopamine, norepinephrine, and 5-HT from rat brain tissue [40]. Studies from the 1990s revealed that pyrovalerone, a structural analog of MDPV (see Fig. 1), is a potent dopamine uptake blocker which produces psychomotor stimulant effects when administered to rodents [41, 42]. A comprehensive study by Meltzer et al. [43] examined the monoamine transporter activities for several pyrovalerone analogs and showed these agents are potent inhibitors of DAT and NET with minimal activity at SERT [43]. Importantly, the study of Meltzer and colleagues did not address the possibility of whether pyrovalerone analogs might act as transporter substrates, and no assessment of MDPV activity was included.
Hadlock et al. [44] carried out the first detailed investigation of mephedrone pharmacology, and found the drug inhibits dopamine uptake and stimulates dopamine release from rat brain synaptosomes [44]. López-Arnau et al. [45] reported that mephedrone and methylone both inhibit uptake at DAT and SERT, but no transporter release data were reported in their study [45]. Our laboratory extended the findings of Lopez-Arnau and colleagues by showing that mephedrone and methylone act as non-selective transporter substrates that evoke release of [3H]MPP+ from DAT and NET, and release of [3H]5-HT from SERT [46]. The non-selective substrate activity of mephedrone and methylone at monoamine transporters is similar to the molecular mechanism of action for the club drug 3,4-methylenedioxy-N-methylamphetamine (MDMA). In assay systems using human transporters expressed in human embryonic kidney (HEK) cells, mephedrone and methylone act as substrates for DAT, NET, and SERT [47, 48], consistent with the findings in synaptosomes. Taken together, results from studies using rat and human transporters agree that ring-substituted cathinones like mephedrone and methylone are non-selective transporter substrates capable of inducing transmitter release via DAT, NET, and SERT.
We examined the in vitro transporter activity of MDPV in rat brain synaptosomes and showed the drug displays potent uptake inhibition at DAT (IC50 = 4.1 nM) and NET (IC50 = 26 nM), with much weaker activity at SERT (IC50 = 3,349 nM) [49]. Table 1 summarizes the uptake inhibition potencies at DAT, NET, and SERT for MDPV and a number of other stimulant drugs discussed in this chapter. The in vitro results with MDPV agree with prior data of Meltzer et al. (2006) showing that pyrovalerone analogs are potent inhibitors of DAT and NET. When compared to the prototypical uptake inhibitor cocaine, MDPV is 50-fold more potent as an inhibitor at DAT, tenfold more potent at NET and tenfold less potent at SERT. We found that MDPV does not act as a substrate for monoamine transporters, probably because the drug molecule is sterically too bulky to fit through the transporter channel. In an informative structure-activity study, Kolanos et al. [52] “deconstructed” the MDPV molecule piece-by-piece to determine which structural features govern activity at DAT. They found that the bulky pyrrolidine ring and the flexible α-carbon chain are critical attributes for potent uptake inhibition at DAT, whereas the 3,4-methylenedioxy ring moiety is of little consequence in this regard.
In mouse striatal slices, MDPV is a potent and efficacious inhibitor of DAT-mediated dopamine clearance (i.e., dopamine uptake) as measured by fast-scan cyclic voltammetry [49]. In assays using HEK cells expressing human transporters, Eshleman et al. [47] and Simmler et al. [48] confirmed that MDPV is a potent inhibitor at DAT and NET, but not SERT, and the drug does not evoke transporter-mediated release. These same investigators examined the potency of MDPV at various G protein-coupled receptor subtypes and found no significant affinity of the drug for non-transporter sites of action [47, 48]. Cameron et al. [53] provided definitive evidence that MDPV is not a substrate at DAT by comparing the electrophysiological effects of mephedrone and MDPV in Xenopus oocytes expressing human DAT [53]. They found that mephedrone induces a DAT-mediated inward depolarizing current, consistent with the action of a transportable substrate, whereas MDPV does not produce this effect. In fact, MDPV induces a DAT-mediated outward hyperpolarizing current due to the inhibition of an inward “leak” current. Overall, the in vitro findings from a variety of different assay methods in native tissues and transporter-expressing cells indicate that MDPV is a potent inhibitor at DAT and NET, which lacks significant activity at SERT and non-transporter sites of action.
The formulation of MDPV available in the recreational drug marketplace is a racemic mixture of S and R isomers, which poses a logical question about whether these isomers have stereoselective biological effects. Meltzer et al. [43] showed that S-pyrovalerone is much more potent as an inhibitor at DAT and NET when compared to R-pyrovalerone, suggesting MDPV isomers might exhibit a similar degree of transporter selectivity. Kolanos et al. [50] reported the stereoselective synthesis of MDPV enantiomers using S- and R-norvaline as starting materials [50], whereas Suzuki et al. [54] resolved MDPV enantiomers from the racemic mixture [54]. In the study of Kolanos et al. [50], S-MDPV was 100-times more potent at inhibiting DAT when compared to R-MDPV (see Table 1). Therefore, similar to the findings reported for pyrovalerone, the biological activity of racemic MDPV resides primarily with the S isomer. In agreement with the in vitro transporter results, S-MDPV is much more potent than R-MDPV in eliciting locomotor stimulant and reinforcing effects in both rats and mice [50, 55].
2.3 In Vivo Pharmacological Effects
Drugs which act as inhibitors or substrates at DAT, NET, and SERT increase the extracellular concentrations of dopamine, norepinephrine, and 5-HT in the brain to enhance monoamine signaling [28, 29]. In our laboratory, we developed in vivo methods to simultaneously examine the neurochemical and behavioral effects of transporter ligands in rats [56, 57]. Specifically, we use in vivo microdialysis perfusion to collect samples of extracellular fluid (i.e., dialysate samples) from the brains of conscious freely behaving rats. The microdialysis probes are placed into the nucleus accumbens, a brain region implicated in the locomotor stimulant and reinforcing effects of abused drugs [58, 59], and dialysate samples are analyzed for concentrations of dopamine and 5-HT using high-performance liquid chromatography coupled to electrochemical detection (HPLC-ECD). Rats undergoing microdialysis are housed in chambers equipped with photo-beam arrays sensitive to locomotor activity in the horizontal plane (i.e., ambulation) and repetitive back-and-forth movements of the head, trunk, and limbs (i.e., stereotypy). Our methods allow for the assessment of relationships between extracellular monoamines and behavior. For example, in previous studies, we found a significant positive correlation between the amount of dialysate dopamine in the nucleus accumbens and the extent of locomotor activation produced by stimulant drugs [56, 60]. Furthermore, data reveal that elevations in dialysate 5-HT alone are not sufficient to produce locomotor activation [61], but elevations in extracellular 5-HT can dampen the motor stimulant effects mediated by concurrent elevations in extracellular dopamine [56, 57].
Kehr et al. [62] first reported that subcutaneous (s.c.) administration of mephedrone to rats evokes concurrent elevations in extracellular dopamine and 5-HT in the nucleus accumbens [62], and other research groups confirmed these findings in rats receiving s.c. or intraperitoneal (i.p.) mephedrone injections [63, 64]. Intravenous (i.v.) administration of mephedrone or methylone produces dose-related increases in extracellular dopamine and 5-HT in rat nucleus accumbens, with mephedrone slightly more potent than methylone [46]. Interestingly, all microdialysis studies with mephedrone and methylone have found that the magnitude of increase in dialysate 5-HT exceeds the accompanying increase in dialysate dopamine. The profile of in vivo neurochemical effects produced by mephedrone and methylone is consistent with the substrate activity of these drugs at DAT and SERT, and mimics the known neurochemical effects of MDMA [46, 62, 65]. We reported that i.v. MDPV administration to rats produces dose-related increases in extracellular dopamine but not 5-HT, and MDPV is tenfold more potent than cocaine in its ability to increase dialysate dopamine [49, 66]. The selective rise in extracellular dopamine produced by MDPV is consistent with the potent inhibition of dopamine uptake produced by the drug in vitro. Figure 2 depicts unpublished data showing the effects of MDPV administration on extracellular dopamine and 5-HT, along with concurrent measures of ambulation. In these experiments, rats undergoing in vivo microdialysis in the nucleus accumbens were housed in chambers equipped with photobeams to allow for measurement of locomotor behaviors. After three baseline dialysate samples were obtained, rats received i.v. injection of 0.1 mg/kg MDPV at time zero, followed by 0.3 mg/kg 60 min later. Dialysate samples were collected at 20 min intervals before, during, and after drug injections. Data were analyzed by two-way (drug × time) ANOVA followed by Bonferroni post-hoc tests. The results show that MDPV produces significant dose-related increases in extracellular dopamine (F1,8 = 157.3, p < 0.0001), but not 5-HT (F1,8 = 1.6, NS), along with a parallel increases in ambulation (F1,8 = 198.7, p < 0.0001).

Neurochemical and behavioral effects of MDPV in male Sprague–Dawley rats undergoing microdialysis in nucleus accumbens. Rats received i.v. injection of 0.1 mg/kg at time zero, followed by 0.3 mg/kg 60 min later. Extracellular concentrations of monoamine transmitters (dopamine, 5-HT) and forward locomotion (ambulation) are expressed as % basal, determined from three time points prior to injection. Data are mean ± SEM, for N = 6–7 rats/group. *p < 0.05, **p < 0.01 compared to saline control at a given time point
The behavioral effects produced by MDPV have been recently reviewed [67], enabling brief consideration here, focusing on locomotor activity and drug self-administration studies. All of the synthetic cathinones examined thus far are known to stimulate locomotor activity when administered to rats [46, 68, 69] or mice [70–72]. In a representative study, Marusich et al. [72] showed that mephedrone, methylone, and MDPV produce dose-dependent increases in ambulation in mice, but MDPV is much more potent in this regard. We found that MDPV is about tenfold more potent than cocaine as a locomotor stimulant in rats, and MDPV is also more efficacious than cocaine, stimulating an overall greater magnitude of motor activation [49]. When MDPV is administered across a broad range of doses, the dose–response relationship for ambulation is an inverted U-shaped curve [68, 71]; the reduction in forward locomotion at higher MDPV doses is due to the emergence of focused stereotypies, such as in-place perseverative sniffing and head bobbing, as dose increases. In mice, the locomotor stimulation produced by MDPV is reduced by pretreatment with the dopamine D1 receptor antagonist SCH23390 [51]. Taken together with the microdialysis data, the available evidence indicates that MDPV elevates extracellular dopamine in critical brain circuits via DAT inhibition, and subsequent activation of D1 dopamine receptors by endogenous dopamine is responsible for locomotor stimulant effects of the drug.
The role of extracellular 5-HT in modulating the dopaminergic effects of synthetic cathinones is a topic of great interest. To this end, a recent investigation compared the neurochemical and locomotor effects of MDPV and methylone in rats [66]. It was found that i.v. doses of 0.3 mg/kg MDPV and 3.0 mg/kg methylone produce nearly identical threefold elevations in extracellular dopamine, whereas only methylone produces a dramatic tenfold elevation in extracellular 5-HT. At these same doses, MDPV elicits a much greater stimulation of ambulation and stereotypy when compared to methylone. One interpretation of these findings is that elevations in extracellular 5-HT tend to reduce locomotor stimulant effects mediated by extracellular dopamine. Indeed, substantial evidence indicates that high-affinity 5-HT2C receptor sites in the brain provide a strong inhibitory influence over dopamine-mediated behavioral effects [73]. Thus, MDPV’s powerful locomotor effects could be related to its potent DAT inhibition, coupled with its lack of activity at SERT and failure to increase extracellular 5-HT.
Drug self-administration is considered the “gold standard” behavioral test for determining the addictive potential of drugs, as most drugs self-administered by laboratory animals are abused by humans [74, this volume] [75]. In the rat drug self-administration paradigm, animals with surgically implanted i.v. catheters are trained to lever-press or nose-poke to obtain i.v. drug injections which are delivered via a computer-controlled infusion pump. A number of studies have shown that rats will self-administer mephedrone [44, 76, 77] and methylone [78–80], indicating these drugs have abuse liability. With regard to MDPV, Aarde et al. [68] reported that MDPV is readily self-administered by rats at i.v. training doses ranging from 0.01 to 0.5 mg/kg, and the drug is more potent and efficacious than methamphetamine, a known stimulant drug of abuse. Watterson et al. [82] found similar results with rats self-administering MDPV, and also showed the amount of drug administered displays robust escalation if rats are allowed prolonged access to the drug. Schindler et al. [66] directly compared the acquisition of i.v. self-administration behavior for MDPV (0.05 mg/kg) and methylone (0.5 mg/kg) in rats. It was found that MDPV self-administration is rapidly acquired within the first few days of training, whereas methylone self-administration takes much longer to develop. Additionally, the number of infusions per session is significantly greater for MDPV when compared to methylone. Based on the neurochemical effects of MDPV and methylone already mentioned, it is tempting to speculate that serotonergic effects of methylone function to counteract the positive reinforcing effects of this drug when compared to MDPV. In agreement with this idea, Bonano et al. [83] showed that MDPV is much more potent than methylone at facilitating intracranial self-stimulation (ICSS) in rats, an index of reinforcing effects of drugs. Furthermore, MDPV produces only abuse-related effects while methylone produces a mixture of abuse-related and abuse-limiting actions. Overall, the self-administration and ICSS data demonstrate that MDPV is a potent and efficacious reinforcer in rats, indicating the drug has a high potential for abuse and addiction in humans.
3 MDPV Pharmacokinetics and Metabolism
Pharmacokinetics (PK) describes the time course of drug concentrations in blood and tissues. Investigating the PK of synthetic cathinones and other NPS is important for the forensic detection of these substances and for evaluating their pharmacological/toxicological effects. When NPS first appear in the recreational drug marketplace, they must be identified and quantified in confiscated drug products and in biological specimens from subjects exposed to the drugs. As mentioned in the Introduction, most NPS are not detected by traditional toxicology screening methods, which rely on antibody-based technology (i.e., immunoassays) and recognize specific drugs and metabolites. Given the rapid increase in number and variety of NPS, the slow and cumbersome process of developing new immunoassays cannot keep pace with the appearance of new substances [84, 85]. Consequently, alternative analytical methods, particularly liquid chromatography (LC) coupled to mass spectrometry (MS) or high-resolution mass spectrometry (HRMS), are now being implemented to detect and quantify newly emerging drugs of abuse [86–88]. In vitro strategies using liver microsomes or hepatocytes are being exploited to quickly identify metabolites of NPS, since certain metabolites may be bioactive or have a much longer half-life than the parent compound, thereby serving as more persistent markers of drug exposure [89, 90]. Finally, because there are few controlled clinical studies examining the effects of NPS in humans, experiments in animal models must be employed to characterize in vivo PK and metabolism [85].
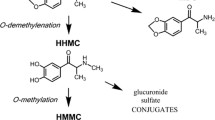
The chemical structure of MDPV displays a 3,4-methylenedioxy group on the phenyl ring, similar to the structure of methylone and MDMA. It is well established that the methylenedioxy moiety of MDMA is a primary target for metabolism by hepatic cytochrome P450 (CYP) enzymes, particularly CYP 2D6 [91–93]. Strano-Rossi et al. [94] reported the first description of MDPV metabolism in human liver microsomes in vitro. These investigators employed gas chromatography with MS for metabolite identification and LC-HRMS for definitive structural elucidation. It was found that MDPV is metabolized in a manner analogous to MDMA by O-demethylenation of the 3,4-methylenedioxy ring to form 3,4-dihydroxypyrovalerone (3,4-catechol-PV), followed by O-methylation to yield 4-hydroxy-3-methoxypyrovalerone (4-OH-3-MeO-PV) (see Fig. 1 for structures). Both of the phase I metabolites are conjugated to form phase II sulfates or glucuronides, which are subsequently excreted in urine. Meyer et al. [95] found that MDPV is metabolized in vitro by a number of mechanisms including demethylenation, aromatic and side-chain hydroxylation, and oxidation of the pyrrolidine ring, but 4-OH-3-MeO-PV is the major metabolite found in urine samples from rats and humans exposed to MDPV administration. Importantly, multiple hepatic enzymes including CYP 2C19, CYP 2D6, and CYP 1A2 were found to catalyze the primary O-demethylenation reaction forming 3,4-catechol-PV [95].
In our laboratory, we are interested in examining the in vivo PK and metabolism of MDPV in rats because data from controlled drug administration studies in humans are lacking. We have previously evaluated pharmacodynamic and PK parameters for MDMA in rats [96, 97], and used similar methods for examining the effects of MDPV [98]. As a first step, Anizan et al. [99] developed a fully validated analytical procedure to simultaneously detect and quantify MDPV, 3,4-catechol-PV and 4-OH-3-MeO-PV using LC-HRMS. The method involves specimen hydrolysis to cleave conjugated 3,4-catechol-PV and 4-OH-3-MeO-PV to their free forms, followed by protein precipitation prior to analysis. Limits of detection are 0.1 μg/L and the linear range is 0.25–1,000 μg/L. The high sensitivity for the assay is essential in order to quantify low analyte concentrations in the small volume of plasma obtained from catheterized rats. To examine PK of MDPV and its metabolites, Anizan et al. [98] administered s.c. doses of MDPV (0.5, 1, 2 mg/kg) to rats bearing surgically implanted i.v. catheters. Rats were placed into chambers equipped with photobeams to measure locomotor parameters, and connected to a tethering system which allowed free movement within the chamber. The i.v. catheters were attached to extension tubing that was threaded through the tether to facilitate stress-free blood withdrawal without any disturbance to the animal. Repeated blood samples (300 μL) were withdrawn via the catheter at various time points before and after injection. Blood samples were centrifuged and plasma specimens assayed for MDPV, 3,4-catechol-PV and 4-OH-3-MeO-PV using the LC-HRMS methods described above. Utilizing this strategy, we were able to simultaneously obtain pharmacodynamic measures (i.e., ambulation and stereotypy) and circulating concentrations of MDPV and its metabolites.
Results from the study of Anizan et al. [98] demonstrated that s.c. MDPV engenders rapid PK in rats, with maximal concentrations (Cmax) in plasma occurring within 15–20 min of injection and decreasing quickly thereafter. Upon injection of 2 mg/kg s.c. MDPV, the plasma Cmax for the drug is 271 μg/L (~1 μM) and the half-life (t1/2) is about 80 min. Plasma concentrations of the metabolites 3,4-catechol-PV and 4-OH-3-MeO-PV increase at a much slower rate, reaching Cmax between 3 and 4 h post-injection. Based on area-under-the-curve (AUC) values, 4-OH-3-MeO-PV is the major metabolite in rat plasma, in agreement with the findings of Meyer et al. [95], who found this to be the predominant metabolite in rat urine. As expected, s.c. MDPV produces dose-related stimulation of ambulation and stereotypy in catheterized rats, and plasma concentrations of MDPV are positively correlated with the extent of motor activation. Two additional findings from the study of Anizan et al. [98] are worth noting: (1) plasma MDPV concentrations display linear dose-proportional kinetics and (2) plasma MDPV metabolite concentrations are negatively correlated with locomotor activation produced by the drug. We found it surprising that MDPV displays linear PK in rats because other drugs exhibiting the 3,4-methylenedioxy moiety (e.g., MDMA) are known to cause sustained inhibition of CYP 2D6 in humans and CYP 2D1 in rats [100, 101], thereby leading to nonlinear accumulation of the parent drug in both species [97, 102, 103]. Indeed, recent evidence shows that MDPV inhibits CYP 2D6 in vitro with an IC50 of 1.3 μM [104]. The fact that MDPV metabolites are negatively correlated with locomotor stimulation suggests the compounds might be bioactive and counteract effects of the parent compound.
As a means to further explore the in vivo PK and metabolism of MDPV, we carried out a follow-up set of experiments to examine effects of i.p. MDPV administration in rats. The i.p. route of administration is expected to induce greater MDPV metabolism, leading to lower concentrations of the parent compound but higher concentrations of its metabolites. In these experiments, rats received i.p. MDPV (0.5, 1, 2 mg/kg), repeated blood samples were withdrawn at various time points, and all other aspects of the experiments were identical to those described above by Anizan and coworkers [98]. Figure 3 depicts new data showing the concentration-time profiles for MDPV and its metabolites after i.p. MDPV administration, while Table 2 summarizes the relevant PK parameters. Similar to the results with s.c. administration, i.p. MDPV engenders rapid PK, with Cmax being achieved within 10 min of injection. After 2 mg/kg i.p. MDPV, the Cmax for the drug is 135 μg/L (~0.5 μM) and plasma t1/2 is about 90 min. Our data demonstrate that i.p. MDPV yields circulating drug concentrations in rats which are about half that observed after s.c. administration of equivalent doses. It is noteworthy that MDPV Cmax values reported here for rats are in the same range as MDPV blood concentrations reported in human cases of non-fatal bath salts intoxication [12], but below those associated with fatal overdose [14, 22]. In contrast to the data with s.c. MDPV administration, i.p. administration appears to induce nonlinear PK. The results in Table 2 demonstrate that a fourfold increase in MDPV dose from 0.5 to 2.0 mg/kg is associated with an eightfold increase in MDPV AUC from 1,114 to 8,726 min μg/L, much greater than dose-proportional. The i.p. route of administration facilitates greater interaction of MDPV with hepatic enzymes when compared to the s.c. route. Thus, high i.p. doses of MDPV may produce nonlinear PK because in vivo drug concentrations in hepatic portal blood are close to the IC50 for inhibition of CYP 2D1. Future preclinical studies should explore PK parameters after the administration of higher doses of MDPV to rats.

Concentration-time profiles for MDPV and its metabolites, 3,4-catechol-PV and 4-OH-3-MeO-PV, after i.p. injection of MDPV in male Sprague–Dawley rats. Rats received i.p. injection of MDPV (0.5, 1 or 2 mg/kg) at time zero, and repeated blood samples (300 μL) were withdrawn immediately before and at 10, 20, 30, 60, 120, 240, and 480 min post-injection. Plasma specimens were assayed for concentrations of MDPV and its metabolites by LC-HRMS. Data are mean ± SEM for N = 6–7 rats/group
The data in Table 2 show that plasma concentrations of the metabolites 3,4-catechol-PV and 4-OH-3-MeO-PV display slow PK after i.p. MDPV administration, achieving Cmax between 3 and 4 h post-injection. Based on AUC values shown in Table 2, 4-OH-3-MeO-PV is the major metabolite in rat plasma. Intraperitoneal MDPV produces dose-related stimulation of ambulation and stereotypy, and the data in Fig. 4 show that both locomotor parameters are significantly correlated with circulating MDPV concentrations but not its metabolites. To generate the correlation plots depicted in Fig. 4, the pharmacodynamic data from the 20, 60, 120, and 240 min time points were plotted against simultaneously measured plasma MDPV or metabolite concentrations. The data matrix was subjected to Pearson correlation analysis. It was found that circulating MDPV concentrations positively correlate with the magnitude of ambulation (r = 0.747, p < 0.001) and stereotypy (r = 0.067, p < 0.001), whereas metabolites show no significant relationships with motor endpoints. The findings with i.p. MDPV indicate that the parent compound is the major factor contributing to the locomotor stimulant effects of the drug. In agreement with this idea, Novellas et al. [105] recently reported that MDPV concentrations in rat striatum are positively correlated with the extent of locomotor activation produced after MDPV administration [105]. These authors further speculated that MDPV-induced elevations in extracellular dopamine in striatal regions underlie behavioral effects observed in rats.

Correlations between motor parameters and plasma concentrations of MDPV and its metabolites after i.p. MDPV administration. To construct correlation plots, ambulation (cm) and stereotypy (episodes) measures obtained at 20, 60, 120, and 240 min post-injection were plotted against plasma concentrations of MDPV, 3,4-catechol-PV or 4-OH-3-MeO-PV (μg/L) at the same time points. Data were subjected to Pearson correlation analysis. Ambulation (r = 0.747, p < 0.001) and stereotypy (r = 0.607, p < 0.001) were significantly correlated with plasma MDPV but not its metabolites
The data considered thus far indicate that hydroxylated MDPV metabolites are probably not contributing to in vivo effects of systemically administered MDPV, especially since these metabolites exist as conjugated forms and are not “free” in the circulation. Nonetheless, we examined the possible biological activity of these metabolites because our previous work showed the 3,4-dihydroxy metabolite of MDMA is bioactive [106]. The effects of 3,4-catechol-PV and 4-OH-3-MeO-PV were first examined in uptake inhibition assays for DAT, NET, and SERT. Data in Table 1 demonstrate that 3,4-catechol-PV is a potent uptake blocker at DAT (IC50 = 11 nM) and NET (IC50 = 11 nM), whereas 4-OH-3-MeO-PV is much weaker in this regard. Neither of the metabolites displays measurable activity at inhibiting SERT, even at doses up to 10 μM. Data shown in Table 1 for 3,4-catechol-PV agree with previous findings of Meltzer et al. [43], who found that this compound is an uptake inhibitor at DAT and NET, with potency similar to pyrovalerone [43]. We next tested the metabolites of MDPV in the microdialysis paradigm to examine possible in vivo actions. Neither of the metabolites affected dialysate dopamine or behavior when administered at i.v. doses of 0.1 and 0.3 mg/kg; the same doses of MDPV elicit robust effects on both parameters (see Fig. 2). Given the in vitro potency of 3,4-catechol-PV at DAT, we examined the effects of higher doses of this metabolite in vivo. Figure 5 depicts new data that show i.v. administration of 3 mg/kg 3,4-catechol-PV produces small, albeit significant, elevations in extracellular dopamine but no change in ambulation. Taken together, the in vitro and in vivo findings with 3,4-catechol-PV indicate this compound may be too polar to readily penetrate the blood–brain barrier and achieve robust neurochemical effects. In support of this hypothesis, the total polar surface area of 3,4-catechol-PV is 60.77 compared to 38.78 for MDPV. The findings with 3,4-catechol-PV shown here serve as a cautionary reminder that inferring the mechanism of drug action should not rely on results from in vitro transporter/receptor profiling alone.

Neurochemical and behavioral effects of 3,4-catechol-PV in male Sprague–Dawley rats undergoing microdialysis in nucleus accumbens. Rats received i.v. injection of 1.0 mg/kg at time zero, followed by 3.0 mg/kg 60 min later. Extracellular concentrations of monoamine transmitters (dopamine, 5-HT) and forward locomotion (ambulation) are expressed as % basal, determined from three time points prior to injection. Data are mean ± SEM, for N = 6–7 rats/group. *p < 0.05, **p < 0.01 compared to saline control at a given time point
4 Pharmacology of “Replacement” Analogs of MDPV
As mentioned in the Introduction, legislation enacted in the US placed mephedrone, methylone, and MDPV into Schedule I control, rendering these drugs illegal. In response to this legislation, a number of “replacement” analogs appeared in the recreational drug marketplace, including several pyrrolidinophenone compounds. Perhaps the most notorious replacement analog of MDPV is α-pyrrolidinovalerophenone (α-PVP) (see Fig. 1). With regard to chemical structure, α-PVP is distinguished from MDPV by the absence of the 3,4-methylenedioxy moiety on the phenyl ring. α-PVP first appeared in the street drug marketplace in 2012 and quickly became a problematic drug of abuse in the US [6], especially in south Florida where the drug is known as “flakka” [107]. Many clinical cases of serious intoxication and death were attributed to overdose from α-PVP in the US and elsewhere [17, 40, 63, 108]. Meltzer et al. [43] first demonstrated that α-PVP is an inhibitor of dopamine and norepinephrine uptake, with potencies at DAT and NET in the same range as pyrovalerone. More recently, Marusich et al. [51] showed that α-PVP inhibits uptake at DAT and NET with IC50 values of 12 and 14 nM, respectively (Table 1). In studies carried out in HEK cells transfected with human transporters, α-PVP and a number of ring-substituted pyrrolidinophenones act as potent inhibitors of DAT and NET, but do not evoke release of preloaded [3H]substrates [109]. Thus, data from synaptosomes and cell systems agree that cathinone-related compounds which possess a pyrrolidine ring act as transporter inhibitors and not substrates.
The data in Table 1 show that removing the 3,4-methylenedioxy moiety from the phenyl ring of MDPV has little effect on drug potency at catecholamine transporters, consistent with the earlier findings of Kolanos and coworkers [52]. However, decreasing alkyl chain length at the α-carbon of α-PVP from propyl to ethyl for α-pyrrolidinobutiophenone (α-PBP), or methyl for α-pyrrolidinopropiophenone (α-PPP), produces a corresponding decrease in potencies at DAT and NET, but no change in transporter selectivity [51]. In a study which examined the structure–activity relationships for a series of α-PVP analogs, Kolanos et al. [110] found that increasing alkyl chain length at the α-carbon to four carbons, or even adding a hexane ring to this position, results in potent DAT inhibitors. Overall, the volume and lipophilicity of the α-carbon substituent of pyrrolidinophenone analogs are positively correlated with potency at DAT, indicating structural modifications at this position have a profound impact on biological activity of the compounds.
In one of the first investigations to examine in vivo α-PVP actions, Kaizaki et al. [111] reported that oral administration of 25 mg/kg α-PVP to male mice produces elevations in striatal extracellular dopamine, along with stimulation of ambulation. It was also found that motor stimulant effects of α-PVP are significantly reduced by pretreatment with antagonists for D1 or D2 dopamine receptor subtypes, implicating dopaminergic mechanisms in mediating behavioral activation. Subsequent reports confirmed α-PVP produces dose-related stimulation of ambulation in mice and rats [51, 112, 113]. In our laboratory, we recently examined the neurochemical effects of α-PVP in male rats undergoing microdialysis in the nucleus accumbens. For our experiments, rats received i.v. injection of 0.1 mg/kg α-PVP at time zero, followed by i.v. injection of 0.3 mg/kg 60 min later. Control rats received i.v. injections of saline vehicle on the same schedule. Microdialysis samples were collected at 20 min intervals before, during, and after drug injections, and dialysate concentrations of dopamine and 5-HT were assayed by HPLC-ECD. Data were analyzed by two-way ANOVA (drug × time) followed by Bonferroni post-hoc tests. The new data depicted in Fig. 6 illustrate that α-PVP causes dose-related increases in extracellular dopamine (F1,8 = 126.6, p < 0.0001) and concurrent stimulation of ambulation (F1,8 = 213.8, p < 0.0001) in rats. Interestingly, α-PVP also produces small, albeit significant, decreases in extracellular 5-HT in the same subjects (F1,8 = 3.5, p < 0.01). The increases in extracellular dopamine and motor activity produced by α-PVP are similar to the effects of MDPV, and are fully consistent with potent DAT blockade. While the decreases in 5-HT produced by α-PVP are more difficult to interpret, the drug is clearly not increasing serotonergic tone. Marusich et al. [51] showed that α-PVP, α-PBP, and α-PPP produce dose-related stimulation of locomotor activity in mice, and the rank order of in vivo potency (i.e., α-PVP > α-PBP > α-PPP) correlates with potency of the drugs at inhibiting DAT.

Neurochemical and behavioral effects of α-PVP in male Sprague–Dawley rats undergoing microdialysis in nucleus accumbens. Rats received i.v. injection of 0.1 mg/kg at time zero, followed by 0.3 mg/kg 60 min later. Extracellular concentrations of monoamine transmitters (dopamine, 5-HT) and forward locomotion (ambulation) are expressed as % basal, determined from three time points prior to injection. Data are mean ± SEM, for N = 6–7 rats/group. *p < 0.05, **p < 0.01 compared to saline control at a given time point
Recent studies examined the reinforcing effects of α-PVP using self-administration and ICSS assays in rats. Aarde et al. [112] directly compared effects of α-PVP and MDPV using i.v. self-administration in rats, and found a 0.05 mg/kg training dose produces similar patterns of acquisition for both drugs under a fixed-ratio schedule of reinforcement. In a progressive-ratio paradigm, it was shown that α-PVP and MDPV display nearly identical potency and efficacy, indicating similar abuse liability for the drugs. Watterson et al. [81] compared the effects of α-PVP and methamphetamine using ICSS, and noted both drugs produce dose-related reductions in self-stimulation thresholds, a measure of positive rewarding effects. Importantly, the potency of α-PVP in the ICSS model was identical to methamphetamine potency.
5 Summary
The findings reviewed in this chapter reveal that the pharmacology of MDPV differs substantially from the pharmacology of ring-substituted cathinones like mephedrone and methylone. MDPV is a potent inhibitor at DAT and NET, and the drug does not act as a transporter substrate like mephedrone and methylone. MDPV is highly selective for catecholamine transporters, whereas mephedrone and methylone are non-selective in this regard. The presence of a bulky pyrrolidine ring and a flexible α-carbon alkyl chain are the most critical structural elements governing potency of uptake inhibition at DAT and NET. S-MDPV is much more potent at inhibiting DAT and NET than R-MDPV, so the S isomer is responsible for pharmacological effects of the racemate. MDPV-induced increases in extracellular dopamine in mesolimbic reward circuits are likely responsible for the powerful stimulant and reinforcing actions of the drug. Upon systemic administration of MDPV, the circulating concentrations of the parent compound are positively correlated with the extent of locomotor activation, while concentrations of its metabolites are not. MDPV appears to induce nonlinear PK in rats after i.p. doses above 1 mg/kg, perhaps due to inhibition of CYP 2D1, and the phenomenon of nonlinear PK deserves further inquiry. Replacement analogs of MDPV like α-PVP, α-PBP, and α-PPP maintain potent and selective inhibition at DAT and NET, indicating these drugs have high abuse liability. Despite substantial knowledge about the pharmacology of MDPV and its analogs, a number of fundamental questions remain: What is the role of NET inhibition in the behavioral and cardiovascular effects of MDPV? Are there non-transporter targets of action for MDPV and its analogs? What are the molecular and cellular changes in the brain induced by chronic administration of MDPV, α-PVP, and related drugs? Finally, could certain pyrrolidinophenone analogs exhibit utility in treating dopamine deficit syndromes such as Parkinson’s disease? These and other questions warrant further consideration.
References
Baumann MH, Solis E Jr, Watterson LR, Marusich JA, Fantegrossi WE, Wiley JL (2014) Baths salts, spice, and related designer drugs: the science behind the headlines. J Neurosci 34:15150–151158
Madras BK (2016) The growing problem of new psychoactive substances (NPS). Curr Top Behav Neurosci (in press)
Zawilska JB, Andrzejczak D (2015) Next generation of novel psychoactive substances on the horizon – a complex problem to face. Drug Alcohol Depend 157:1–17
Baumann MH, Volkow ND (2016) Abuse of new psychoactive substances: threats and solutions. Neuropsychopharmacology 41:663–665
Brandt SD, King LA, Evans-Brown M (2014) The new drug phenomenon. Drug Test Anal 6:587–597
Drug Enforcement Administration, Office of Diversion Control (2014) Special report: synthetic cannabinoids and cathinones reported in NFLIS, 2010–2013. http://www.deadiversion.usdoj.gov/nflis/spec_rpt_CathCan_2013.pdf
Shanks KG, Dahn T, Behonick G, Terrell A (2012) Analysis of first and second generation legal highs for synthetic cannabinoids and synthetic stimulants by ultra-performance liquid chromatography and time of flight mass spectrometry. J Anal Toxicol 36:360–371
United Nations Office of Drugs and Crime. World Drug Report (2015). http://www.unodc.org/documents/wdr2015/World_Drug_Report_2015.pdf
Prosser JM, Nelson LS (2012) The toxicology of bath salts: a review of synthetic cathinones. J Med Toxicol 8:33–34
Centers for Disease Control and Prevention (CDC) (2011) Emergency department visits after use of a drug sold as “bath salts”--Michigan, November 13, 2010-March 31, 2011. MMWR Morb Mortal Wkly Rep 60:624–627
Murphy CM, Dulaney AR, Beuhler MC, Kacinko S (2013) “Bath salts” and “plant food” products: the experience of one regional US poison center. J Med Toxicol 9:42–48
Spiller HA, Ryan ML, Weston RG, Jansen J (2011) Clinical experience with and analytical confirmation of “bath salts” and “legal highs” (synthetic cathinones) in United States. Clin Toxicol (Phila) 49:499–505
Ross EA, Reisfield GM, Watson MC, Chronister CW, Goldberger BA (2012) Psychoactive “bath salts” intoxication with methylenedioxypyrovalerone. Am J Med 125:854–858
Kesha K, Boggs CL, Ripple MG, Allan CH, Levine B, Jufer-Phipps R, Doyon S, Chi P, Fowler DR (2013) Methylenedioxypyrovalerone (“bath salts”), related death: case report and review of the literature. J Forensic Sci 58:1654–1659
Penders TM, Gestring RE, Vilensky DA (2012) Intoxication delirium following use of synthetic cathinone derivatives. Am J Drug Alcohol Abuse 38:616–617
Drug Enforcement Administration (2013) Establishment of drug codes for 26 substances. Final rule. Fed Regist 78:664–666
Marinetti LJ, Antonides HM (2013) Analysis of synthetic cathinones commonly found in bath salts in human performance and postmortem toxicology: method development, drug distribution and interpretation of results. J Anal Toxicol 37:135–146
Karch SB (2015) Cathinone neurotoxicity (The “3Ms”). Curr Neuropharmacol 13:21–25
Froberg BA, Levine M, Beuhler MC, Judge BS, Moore PW, Engebretsen KM, Mckeown NJ, Rosenbaum CD, Young AC, Rusyniak DE; ACMT Toxicology Investigators Consortium (ToxIC) (2015) Acute methylenedioxypyrovalerone toxicity. J Med Toxicol 11:185–194
European Monitoring Centre for Drugs and Drug Addiction (2014) EMCDDA–Europol Joint Report on a new psychoactive substance: MDPV (3,4-methylenedioxypyrovalerone). http://www.emcdda.europa.eu/system/files/publications/819/TDAS14001ENN_466653.pdf
Murray BL, Murphy CM, Beuhler MC (2012) Death following recreational use of designer drug “bath salts” containing 3,4-Methylenedioxypyrovalerone (MDPV). J Med Toxicol 8:69–75
Wyman JF, Lavins ES, Engelhart D, Armstrong EJ, Snell KD, Boggs PD, Taylor SM, Norris RN, Miller FP (2013) Postmortem tissue distribution of MDPV following lethal intoxication by “bath salts”. J Anal Toxicol 37:182–185
Baumann MH (2014) Awash in a sea of ‘bath salts’: implications for biomedical research and public health. Addiction 109:1577–1579
Simmler LD, Liechti ME (2016) Interactions of cathinone NPS with human transporters and receptors in transfected cells. Curr Top Behav Neurosci (in press)
Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 63:585–640
Gorman JM, Kent JM (1999) SSRIs and SMRIs: broad spectrum of efficacy beyond major depression. J Clin Psychiatry 60(Suppl 4):33–38
Iversen L (2006) Neurotransmitter transporters and their impact on the development of psychopharmacology. Br J Pharmacol 147(Suppl1):S82–S88
Howell LL, Kimmel HL (2008) Monoamine transporters and psychostimulant addiction. Biochem Pharmacol 75:196–217
Rothman RB, Baumann MH (2003) Monoamine transporters and psychostimulant drugs. Eur J Pharmacol 479:23–40
Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL (2015) Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend 147:1–19
Sitte HH, Freissmuth M (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci 36:41–50
Angoa-Perez M, Anneken JH, Kuhn DM (2016) Neurotoxicology of synthetic cathinone analogs. Curr Top Behav Neurosci (in press)
Baumann MH, Wang X, Rothman RB (2007) 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacology (Berl) 189:407–424
Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR (2007) New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol 47:681–698
Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS (2001) Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39:32–41
Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA, Birkes J, Young R, Glennon RA (2003) In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther 307:138–145
Glennon RA, Yousif M, Naiman N, Kalix P (1987) Methcathinone: a new and potent amphetamine-like agent. Pharmacol Biochem Behav 26:547–551
Kalix P, Glennon RA (1986) Further evidence for an amphetamine-like mechanism of action of the alkaloid cathinone. Biochem Pharmacol 35:3015–3019
Cozzi NV, Sievert MK, Shulgin AT, Jacob P 3rd, Ruoho AE (1999) Inhibition of plasma membrane monoamine transporters by beta-ketoamphetamines. Eur J Pharmacol 381:63–69
Nagai F, Nonaka R, Satoh Hisashi Kamimura K (2007) The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur J Pharmacol 559:132–137
Héron C, Costentin J, Bonnet JJ (1994) Evidence that pure uptake inhibitors including cocaine interact slowly with the dopamine neuronal carrier. Eur J Pharmacol 264:391–398
Vaugeois JM, Bonnet JJ, Duterte-Boucher D, Costentin J (1993) In vivo occupancy of the striatal dopamine uptake complex by various inhibitors does not predict their effects on locomotion. Eur J Pharmacol 230:195–201
Meltzer PC, Butler D, Deschamps JR, Madras BK (2006) 1(4-Methylphenyl)-2-pyrrolidin-1-yl-petan-1-one (pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem 49:1420–1432
Hadlock GC, Webb KM, McFadden LM, Chu PW, Ellis JD, Allen SC, Andrenyak DM, Vieira-Brock PL, German CL, Conrad KM, Hoonakker AJ, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE (2011) 4-Methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J Pharmacol Exp Ther 339:530–536
López-Arnau R, Martínez-Clemente J, Pubill D, Escubedo E, Camarasa J (2012) Comparative neuropharmacology of three psychostimulant cathinone derivatives: butylone, mephedrone and methylone. Br J Pharmacol 167:407–420
Baumann MH, Ayestas MA Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203
Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A (2013) Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85:1803–1815
Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME (2013) Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168:458–470
Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW (2013) Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology 38:552–562
Kolanos R, Partilla JS, Baumann MH, Hutsell BA, Banks ML, Negus SS, Glennon RA (2015) Stereoselective actions of methylenedioxypyrovalerone (MDPV) to inhibit dopamine and norepinephrine transporters and facilitate intracranial self-stimulation in rats. ACS Chem Neurosci 6:771–777
Marusich JA, Antonazzo KR, Wiley JL, Blough BE, Partilla JS, Baumann MH (2014) Pharmacology of novel synthetic stimulants structurally related to the “bath salts” constituent 3,4-methylenedioxypyrovalerone (MDPV). Neuropharmacology 87:206–213
Kolanos R, Solis E Jr, Sakloth F, De Felice LJ, Glennon RA (2013) “Deconstruction” of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS Chem Neurosci 4:1524–1529
Cameron KN, Kolanos R, Solis E Jr, Glennon RA, De Felice LJ (2013) Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. Br J Pharmacol 168:1750–1757
Suzuki M, Deschamps JR, Jacobson AE, Rice KC (2015) Chiral resolution and absolute configuration of the enantiomers of the psychoactive “designer drug” 3,4-methylenedioxypyrovalerone. Chirality 27:287–293
Gannon BM, Williamson A, Suzuki M, Rice KC, Fantegrossi WE (2016) Stereoselective effects of abused “bath salt” constituent 3,4-methylenedioxypyrovalerone in mice: drug discrimination, locomotor activity, and thermoregulation. J Pharmacol Exp Ther 356:615–623
Baumann MH, Clark RD, Woolverton WL, Wee S, Blough BE, Rothman RB (2011) In vivo effects of amphetamine analogs reveal evidence for serotonergic inhibition of mesolimbic dopamine transmission in the rat. J Pharmacol Exp Ther 337:218–2125
Rothman RB, Blough BE, Woolverton WL, Anderson KG, Negus SS, Mello NK, Roth BL, Baumann MH (2005) Development of a rationally designed, low abuse potential, biogenic amine releaser that suppresses cocaine self-administration. J Pharmacol Exp Ther 313:1361–1369
Ikemoto S (2007) Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev 56:27–78
Willuhn I, Wanat MJ, Clark JJ, Phillips PE (2010) Dopamine signaling in the nucleus accumbens of animals self-administering drugs of abuse. Curr Top Behav Neurosci 3:29–71
Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V, Prisinzano TE, Baumann MH (2009) Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil. J Pharmacol Exp Ther 329:738–746
Cozzi NV, Brandt SD, Daley PF, Partilla JS, Rothman RB, Tulzer A, Sitte HH, Baumann MH (2013) Pharmacological examination of trifluoromethyl ring-substituted methcathinone analogs. Eur J Pharmacol 699:180–187
Kehr J, Ichinose F, Yoshitake S, Goiny M, Sievertsson T, Nyberg F, Yoshitake T (2011) Mephedrone, compared with MDMA (ecstasy) and amphetamine, rapidly increases both dopamine and 5-HT levels in nucleus accumbens of awake rats. Br J Pharmacol 164:1949–1958
Wright MJ Jr, Angrish D, Aarde SM, Barlow DJ, Buczynski MW, Creehan KM, Vandewater SA, Parsons LH, Houseknecht KL, Dickerson TJ, Taffe MA (2012) Effect of ambient temperature on the thermoregulatory and locomotor stimulant effects of 4-methylmethcathinone in Wistar and Sprague-Dawley rats. PLoS One 7, e44652
Suyama JA, Sakloth F, Kolanos R, Glennon RA, Lazenka MF, Negus SS, Banks ML (2016) Abuse-related neurochemical effects of para-substituted methcathinone analogs in rats: microdialysis studies of nucleus accumbens dopamine and serotine. J Pharmacol Exp Ther 356:182–190
Baumann MH, Clark RD, Franken FH, Rutter JJ, Rothman RB (2008) Tolerance to 3,4-methylenedioxymethamphetamine in rats exposed to single high-dose binges. Neuroscience 152:773–784
Schindler CW, Thorndike EB, Goldberg SR, Lehner KR, Cozzi NV, Brandt SD, Baumann MH (2016) Reinforcing and neurochemical effects of the “bath salts” constituents 3,4-methylenedioxypyrovalerone (MDPV) and 3,4-methylenedioxy-N-methylcathinone (methylone) in male rats. Psychopharmacology (Berl) 233:1981–1990
Glennon RA, Young R (2016) Neurobiology of 3,4-methylenedioxypyrovalerone (MDPV) and α-pyrrolidinovalerophenone (α-PVP). Brain Res Bull 126:111–126
Aarde SM, Huang PK, Creehan KM, Dickerson TJ, Taffe MA (2013) The novel recreational drug 3,4-methylenedioxypyrovalerone (MDPV) is a potent psychomotor stimulant: self-administration and locomotor activity in rats. Neuropharmacology 71:130–140
Huang PK, Aarde SM, Angrish D, Houseknecht KL, Dickerson TJ, Taffe MA (2012) Contrasting effects of d-methamphetamine, 3,4-methylenedioxymethamphetamine, 3,4-methylenedioxypyrovalerone, and 4-methylmethcathinone on wheel activity in rats. Drug Alcohol Depend 126:168–175
Fantegrossi WE, Gannon BM, Zimmerman SM, Rice KC (2013) In vivo effects of abused ‘bath salt’ constituent 3,4-methylenedioxypyrovalerone (MDPV) in mice: drug discrimination, thermoregulation, and locomotor activity. Neuropsychopharmacology 38:563–573
Gatch MB, Taylor CM, Forster MJ (2013) Locomotor stimulant and discriminative stimulus effects of ‘bath salt’ cathinones. Behav Pharmacol 24:437–447
Marusich JA, Grant KR, Blough BE, Wiley JL (2012) Effects of synthetic cathinones contained in “bath salts” on motor behavior and a functional observational battery in mice. Neurotoxicology 33:1305–1313
Howell LL, Cunningham KA (2015) Serotonin 5-HT2 receptor interactions with dopamine function: implications for therapeutics in cocaine use disorder. Pharmacol Rev 67:176–1797
Watterson LR, Olive MF (2016) Reinforcing effects of cathinone NPS in the i.v. drug self-administration paradigm. Curr Top Behav Neurosci (in press)
Watterson LR, Watterson E, Olive MF (2013) Abuse liability of novel ‘legal high’ designer stimulants: evidence from animal models. Behav Pharmacol 24:341–355
Aarde SM, Angrish D, Barlow DJ, Wright MJ Jr, Vandewater SA, Creehan KM, Houseknecht KL, Dickerson TJ, Taffe MA (2013) Mephedrone (4-methylmethcathinone) supports i.v. self-administration in Sprague-Dawley and Wistar rats. Addict Biol 18:786–799
Motbey CP, Clemens KJ, Apetz N, Winstock AR, Ramsey J, Li KM, Wyatt N, Callaghan PD, Bowen MT, Cornish JL, McGregor IS (2013) High levels of i.v. mephedrone (4-methylmethcathinone) self-administration in rats: neural consequences and comparison with methamphetamine. J Psychopharmacol 27:823–836
Creehan KM, Vandewater SA, Taffe MA (2015) I.v. self-administration of mephedrone, methylone and MDMA in female rats. Neuropharmacology 92:90–97
Vandewater SA, Creehan KM, Taffe MA (2015) I.v. self-administration of entactogen-class stimulants in male rats. Neuropharmacology 99:538–545
Watterson LR, Hood L, Sewalia K, Tomek SE, Yahn S, Johnson CT, Wegner S, Blough BE, Marusich JA, Olive MF (2012) The reinforcing and rewarding effects of methylone, a synthetic cathinone commonly found in “bath salts”. J Addict Res Ther Suppl 9:002
Watterson LR, Burrows BT, Hernandez RD, Moore KN, Grabenauer M, Marusich JA, Olive MF (2014) Effects of α-pyrrolidinopentiophenone and 4-methyl-N-ethylcathinone, two synthetic cathinones commonly found in second-generation “bath salts,” on intracranial self-stimulation thresholds in rats. Int J Neuropsychopharmacol 18 pii:pyu014
Watterson LR, Kufahl PR, Nemirovsky NE, Sewalia K, Grabenauer M, Thomas BF, Marusich JA, Wegner S, Olive MF (2014) Potent rewarding and reinforcing effects of the synthetic cathinone 3,4-methylenedioxypyrovalerone (MDPV). Addict Biol 19:165–174
Bonano JS, Glennon RA, De Felice LJ, Banks ML, Negus SS (2014) Abuse-related and abuse-limiting effects of methcathinone and the synthetic “bath salts” cathinone analogs methylenedioxypyrovalerone (MDPV), methylone and mephedrone on intracranial self-stimulation in rats. Psychopharmacology (Berl) 231:199–207
Tettey J, Crean C (2015) New psychoactive substances: catalysing a shift in forensic science practice? Philos Trans R Soc Lond B Biol Sci 370:20140265
Ellefsen KN, Concherio M, Huestis MA (2016) Synthetic cathinone pharmacokinetics, analytical methods, and toxicological findings from human performance and postmortem cases. Drug Metab Rev 48:237–265
Concheiro M, Castaneto M, Kronstrand R, Huestis MA (2015) Simultaneous determination of 40 novel psychoactive stimulants in urine by liquid chromatography-high resolution mass spectrometry and library matching. J Chromatogr A 1397:32–42
Favretto D, Pascali JP, Tagliaro F (2013) New challenges and innovation in forensic toxicology: focus on the “New Psychoactive Substances”. J Chromatogr A 1287:84–95
Smith JP, Sutcliffe OB, Banks CE (2015) An overview of recent developments in the analytical detection of new psychoactive substances (NPSs). Analyst 140:4932–4948
Castaneto MS, Wohlfarth A, Desrosiers NA, Hartman RL, Gorelick DA, Huestis MA (2015) Synthetic cannabinoids pharmacokinetics and detection methods in biological matrices. Drug Metab Rev 47:124–174
Meyer MR, Maurer HH (2012) Current applications of high-resolution mass spectrometry in drug metabolism studies. Anal Bioanal Chem 403:1221–1231
Kreth K, Kovar K, Schwab M, Zanger UM (2000) Identification of the human cytochromes P450 involved in the oxidative metabolism of “Ecstasy”-related designer drugs. Biochem Pharmacol 59:1563–1571
Meyer MR, Peters FT, Maurer HH (2008) The role of human hepatic cytochrome P450 isozymes in the metabolism of racemic 3,4-methylenedioxy-methamphetamine and its enantiomers. Drug Metab Dispos 36:2345–2354
Tucker GT, Lennard MS, Ellis SW, Woods HF, Cho AK, Lin LY, Hiratsuka A, Schmitz DA, Chu TY (1994) The demethylenation of methylenedioxymethamphetamine (“ecstasy”) by debrisoquine hydroxylase (CYP2D6). Biochem Pharmacol 47:1151–1156
Strano-Rossi S, Cadwallader AB, de la Torre X, Botrè F (2010) Toxicological determination and in vitro metabolism of the designer drug methylenedioxypyrovalerone (MDPV) by gas chromatography/mass spectrometry and liquid chromatography/quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 24:2706–2714
Meyer MR, Du P, Schuster F, Maurer HH (2010) Studies on the metabolism of the α-pyrrolidinophenone designer drug methylenedioxy-pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC-MS and LC-high-resolution MS and its detectability in urine by GC-MS. J Mass Spectrom 45:1426–1442
Baumann MH, Zolkowska D, Kim I, Scheidweiler KB, Rothman RB, Huestis MA (2009) Effects of dose and route of administration on pharmacokinetics of 3,4-methylenedioxymethamphetamine in the rat. Drug Metab Dispos 37:2163–2170
Concheiro M, Baumann MH, Scheidweiler KB, Rothman RB, Marrone GF, Huestis MA (2014) Nonlinear pharmacokinetics of (+/-)3,4-methylenedioxymethamphetamine (MDMA) and its pharmacodynamic consequences in the rat. Drug Metab Dispos 42:119–125
Anizan S, Concheiro M, Lehner KR, Bukhari MO, Suzuki M, Rice KC, Baumann MH, Huestis MA (2016) Linear pharmacokinetics of 3,4-methylenedioxypyrovalerone (MDPV) and its metabolites in the rat: relationship to pharmacodynamic effects. Addict Biol 21:339–347
Anizan S, Ellefsen K, Concheiro M, Suzuki M, Rice KC, Baumann MH, Huestis MA (2014) 3,4-Methylenedioxypyrovalerone (MDPV) and metabolites quantification in human and rat plasma by liquid chromatography-high resolution mass spectrometry. Anal Chim Acta 827:54–63
Delaforge M, Jaouen M, Bouille G (1999) Inhibitory metabolite complex formation of methylenedioxymethamphetamine with rat and human cytochrome P450. Particular involvement of CYP 2D. Environ Toxicol Pharmacol 7:153–158
Heydari A, Yeo KR, Lennard MS, Ellis SW, Tucker GT, Rostami-Hodjegan A (2004) Mechanism-based inactivation of CYP2D6 by methylenedioxymethamphetamine. Drug Metab Dispos 32:1213–1217
Chu T, Kumagai Y, DiStefano EW, Cho AK (1996) Disposition of methylenedioxymethamphetamine and three metabolites in the brains of different rat strains and their possible roles in acute serotonin depletion. Biochem Pharmacol 51:789–796
de la Torre R, Farré M, Ortuño J, Mas M, Brenneisen R, Roset PN, Segura J, Camí J (2000) Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans. Br J Clin Pharmacol 49:104–109
Dinger J, Meyer MR, Maurer HH (2016) In vitro cytochrome P450 inhibition potential of methylenedioxy-derived designer drugs studied with a two-cocktail approach. Arch Toxicol 90:305–318
Novellas J, López-Arnau R, Carbó ML, Pubill D, Camarasa J, Escubedo E (2015) Concentrations of MDPV in rat striatum correlate with the psychostimulant effect. J Psychopharmacol 29:1209–1218
Schindler CW, Thorndike EB, Blough BE, Tella SR, Goldberg SR, Baumann MH (2014) Effects of 3,4-methylenedioxymethamphetamine (MDMA) and its main metabolites on cardiovascular function in conscious rats. Br J Pharmacol 171:83–91
Crespi C (2016) Flakka-induced prolonged psychosis. Case Rep Psychiatry 2016:3460849
Sykutera M, Cychowska M, Bloch-Boguslawska E (2015) A fatal case of pentedrone and α-pyrrolidinovalerophenone poisoning. J Anal Toxicol 39:324–329
Rickli A, Hoener MC, Liechti ME (2015) Monoamine transporter and receptor interaction profiles of novel psychoactive substances: para-halogenated amphetamines and pyrovalerone cathinones. Eur Neuropsychopharmacol 25:365–376
Kolanos R, Sakloth F, Jain AD, Partilla JS, Baumann MH, Glennon RA (2015) Structural modification of the designer stimulant α-pyrrolidinovalerophenone (α-PVP) influences potency at dopamine transporters. ACS Chem Neurosci 6:1726–1731
Kaizaki A, Tanaka S, Numazawa S (2014) New recreational drug 1-phenyl-2-(1-pyrrolidinyl)-1-pentanone (alpha-PVP) activates central nervous system via dopaminergic neuron. J Toxicol Sci 39:1–6
Aarde SM, Creehan KM, Vandewater SA, Dickerson TJ, Taffe MA (2015) In vivo potency and efficacy of the cathinone α-pyrrolidinopentiophenone and 3,4-methylenedioxypyrovalerone: self-administration and locomotor stimulation in male rats. Psychopharmacology (Berl) 232:3045–3055
Gatch MB, Dolan SB, Forster MJ (2015) Comparative behavioral pharmacology of three pyrrolidine-containing synthetic cathinone derivatives. J Pharmacol Exp Ther 354:103–110
Funding and Disclosures
This research was generously supported by the Intramural Research Program (IRP) of the National Institute on Drug Abuse (NIDA) grant 1ZIADA000523-08. The authors have nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Baumann, M.H. et al. (2016). Neuropharmacology of 3,4-Methylenedioxypyrovalerone (MDPV), Its Metabolites, and Related Analogs. In: Baumann, M.H., Glennon, R.A., Wiley, J.L. (eds) Neuropharmacology of New Psychoactive Substances (NPS). Current Topics in Behavioral Neurosciences, vol 32. Springer, Cham. https://doi.org/10.1007/7854_2016_53
Download citation
DOI: https://doi.org/10.1007/7854_2016_53
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52442-9
Online ISBN: 978-3-319-52444-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)