
Abstract
Systemic inflammation rapidly impairs mood, motivation, and cognition inducing a stereotyped cluster of symptoms collectively known as “sickness behaviors.” When inflammation is severe or chronic, these behavioral changes can appear indistinguishable from major depressive disorder (MDD). Human and rodent neuroimaging combined with experimental inflammatory challenges has clarified the neural circuitry associated with many of the key features of inflammation-induced-sickness behavior, and in so doing revealed often-remarkable commonalities with circuit abnormalities observed in MDD. This review aims to provide the first synthesis of this work illustrating areas of convergence and divergence with the MDD literature as well as highlighting areas for future study.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Human and animal studies implicate systemic inflammation in the pathogenesis of depression [1]. In healthy mammals, systemic infection triggers profound behavioral changes, including cognitive and mood symptoms (e.g., memory impairment, social withdrawal, anxiety, and depression), change in motivation (anorexia, adipsia, and anhedonia), and neurovegetative symptoms (sleep disturbance, fatigue, and psychomotor slowing) [2–4] known as sickness behaviors. Clinical and pre-clinical studies suggest that inflammatory cytokines play a central role in mediating these sickness-related behaviors by communicating peripheral inflammation to the brain. These cytokine-induced sickness behaviors show striking similarity to symptoms of major depression [1] supporting a role for immune–brain interactions in the etiology of at least some patients with major depressive disorder (MDD).
In rodents, systemic administration of interleukin (IL)-1β or bacterial lipopolysaccharide (LPS), a potent stimulant of cytokine release, can rapidly elicit a depression-like syndrome characterized by a reduction in positively motivated approach behaviors such as exploration, social interaction, and operant behaviors for food reward [5–7]. Similarly, experimental induction of inflammation in healthy human participants using either LPS [8, 9] or typhoid vaccination [10–12] induces symptoms of fatigue, psychomotor slowing, mild cognitive confusion, memory impairment, social withdrawal, anxiety, and deterioration in mood that mirror features of depression. However, arguably, the most powerful empirical support for an etiological role for inflammation in depression comes from studies of patients with chronic Hepatitis-C infection treated with interferon-alpha (IFN-α)-based therapies, up to 50% of whom develop major depressive episodes [13]. Moreover, in patients with MDD, the presence of high levels of pro-inflammatory cytokines (in particular IL-6) [14] and acute phase proteins [15] suggest that inflammatory mediators might contribute to the pathophysiology of depression even in the absence of medical illness.
Far from being a unitary construct, depression is recognized to be a multi-componential disorder involving changes in motivation, cognition, attention, memory, and mood; as well as features such as disturbed appetite, sleep, and sexual dysfunction and physiological changes including cardiovascular and metabolic change. In studies of MDD, patients are typically recruited when symptoms are well established across each of these domains. However, following chronic IFN-α administration, individual features of clinical depression evolve with characteristic time-courses allowing a unique opportunity to investigate the temporal evolution of individual symptom domains. For example, changes in mood, motivation, and fatigue (and in some cases feelings of social connection and spatial memory) can be readily observed within hours of IFN-α administration [16] and/or other experimental inflammatory challenges such as typhoid vaccination [11, 12, 17, 18] and LPS injection [8, 9, 19]. Changes in physiology, including altered central autonomic regulation of heart rate variability (similar to that observed in MDD) also occur acutely [20]. However in contrast, subjective reports of depressed mood, anxiety, and irritability assessed with clinical depression scales typically develop later, between the first and third months of IFN-α therapy [21].
This differential evolution of individual features of inflammation-associated clinical depression also provides a unique opportunity to characterize the neural circuitry underpinning specific components of inflammation-induced depression. To date, rodent and human brain imaging studies have successfully identified a discrete set of cortical and sub-cortical structures that appear particularly sensitive to changes in peripheral inflammation. These include the amygdala, striatum (particularly ventral regions), substantia nigra, insula, sub-genual and dorsal anterior cingulate, orbitofrontal cortex, and hippocampus/parahippocampus. Some of these structures appear to play relatively specific roles in particular aspects of inflammation-associated behavioral change. For example, actions on the ventral striatum [8, 22, 23] are associated with impaired reward sensitivity and hippocampus/parahippocampus acute memory impairment [4, 20] whereas other regions such as the insula, anterior and sub-genual cingulate, and amygdala appear to play broader less circumscribed roles [11, 16]. Common to many of these regions is that they form part of the extended limbic circuitry critical to complex motivational behavior, emotion, learning, and memory and the integration of behavioral and physiological allostatic responses to infection [24, 25].
Together, these studies are beginning to clarify how changes in peripheral inflammation are communicated to the brain. As discussed in more detail in the following sections they are also beginning to identify how actions of inflammation on discrete neural circuits induce individual components of this coordinated behavioral reorientation, which when chronic may evolve to MDD.
2 Communicating Inflammation to the Brain
In rodents, both the early central communication of peripheral inflammatory signals [26] and the subsequent motivational reorientation appear dependent upon the integrity of interoceptive visceral afferents traveling in the vagus nerve, visceral terminals of which express cytokine binding sites [27]. Early in the inflammatory response, antigen-presenting cells cluster in the vicinity of vagus nerve afferents and act as immune chemosensory elements signaling to vagal neurons via cytokine-dependent [28] and -independent mechanisms [29].
Immunohistochemical studies using the immediate early gene c-Fos to index neural activation confirm that peripheral inflammation and specifically binding of pro-inflammatory cytokines to vagus nerve receptors activate a network of brain structures implicated in homeostasis and the representation of internal bodily state (interoception) [26]. This afferent signaling is rapid; in the rat peripheral inflammation induces c-Fos expression in the primary projection nucleus of the vagus nerve (nucleus tractus solitarius – NTS) and secondary projection regions (including parabrachial, paraventricular and supraoptic hypothalamic nuclei, central amygdala, and bed nucleus of the stria terminalis) within an hour of peripheral inflammatory challenge [26].
In addition to signaling via the vagus nerve, central signaling of peripheral inflammation may also occur via interoceptive information conveyed via the spinal cord. For example, information traveling through spinal lamina I is predominantly tuned to motivationally salient sensations, including pain [30, 31], temperature [32], itch [33], and sensual touch [34], and converges with afferents traveling in the vagus nerve within the brainstem and thalamus [35]. In humans, cortical projections of convergent vagus and spinal interoceptive pathways to the posterior then mid/anterior insula cortex have been proposed to support a consciously accessible representation of physical wellbeing [36] and provide a neural substrate for subjective emotional feelings [30, 36].
We have previously demonstrated that this human interoceptive pathway is also activated by mild systemic inflammation [12]. Specifically, we demonstrated increased activity on fMRI within bilateral thalamic (basal (VMb) and posterior (VMpo) ventromedial nuclei) and dorsal mid and anterior insula components of this interoceptive pathway within 3 h of typhoid vaccine induced inflammation. The location of these activations is noteworthy as both the VMb (which receives predominantly vagal fibers) and VMpo (predominantly sympathetic inputs) project to dorsal mid/posterior insula in a rostrocaudal topographic manner (with vagal projections extending more rostrally) [30]. Following inflammation activations occurred in more rostral regions of interoceptive insula cortex than those reported for thermal sensation [32], noxious pain, and itch (non-vagal) but close to the region previously reported to be activated by antigen-induced airways inflammation in asthmatic patients [37].
Insula (and cingulate cortices) also played a key role in mediating subjective responses to inflammation, particularly fatigue (Fig. 1). For example, inflammation but not placebo-associated fatigue was predicted by activity changes within bilateral mid/posterior insula and right anterior cingulate (pACC/aMCC). These findings highlight a degree of specificity of the neural mechanisms underlying inflammation-associated fatigue that is not seen in more general placebo-associated fatigue (which might result from more heterogeneous mechanisms).

Inflammation-induced insula activity predicts subjective fatigue. (a) Increase in bilateral insula activity on fMRI during performance of a color word Stroop task after inflammation compared to placebo. Lower panel shows that left insula cortex activity predicted experience of inflammation-induced fatigue (blue) but not fatigue associated with placebo (red). Data from Harrison et al. [12]. (b) Increase in bilateral resting glucose metabolism (FDG-PET) after typhoid vaccine induced inflammation compared to placebo. Lower panel shows correlation between change in left insula glucose uptake and subjective fatigue. Data from Harrison et al. [23]. (c) Left insula region showing an increase in magnetization transfer constant k f after typhoid vaccine induced inflammation compared to placebo (yellow), correlation with fatigue (blue), and overlap of these regions (green). Data from Harrison et al. [23]
Previous studies showing insula responses to subjective experience of graded cooling [32], itch [38], and intensity of dynamic exercise [39] support the hypothesis that the subjective experience of inflammation-associated fatigue results from an insula-based interoceptive mechanism. This hypothesis has also been reinforced by three more recent studies. The first, using typhoid vaccine induced inflammation and fluorodeoxyglucose PET (FDG-PET) imaging, which replicated associations between changes in mid insula activity (in this case glucose metabolism) and the subjective experience of inflammation-induced fatigue [23]. The two other studies used a more potent model of inflammation (0.8–0.6 ng/kg LPS) and either FDG-PET or resting state fMRI which is a powerful technique for identifying functionally connected brain networks [40]. The first demonstrated an increase in right anterior insula glucose metabolism that correlated with loss of social interest (but not fatigue) [41] while the latter demonstrated correlations between subjective feelings of both inflammation-induced malaise and discomfort and heightened functional connectivity between the left anterior insula and mid-cingulate cortex [42]. Together, these studies highlight the importance of this interoceptive pathway projecting to insula in the central communication of inflammation induced using experimental models of bacterial infection. They also emphasize the likely importance of the insula in translating these interoceptive signals into negative subjective experiences of inflammation-induced fatigue and malaise (and possibly social disconnect) that develop early after the onset of inflammation.
Interestingly, patients with left or right insular strokes describe significantly greater subjective anergia with under activity and tiredness than patients with strokes sparing the insula region [43]. Furthermore, increased insula metabolism and altered interoceptive processing have also emerged as key features of MDD [44, 45]. Increases in insula (particularly anterior insula) glucose metabolism occur following sadness induction with a converse reduction in insula glucose metabolism observed following successful depression remission [44]. Insula functional connectivity [46] and regional homogeneity (ReHo) (a measure of the temporal homogeneity of neural activity within this region) are also impaired in MDD with the later correlating with retardation components of depression [47]. These findings have been interpreted as consonant with neurovegetative features of MDD (such as fatigue) and associated changes in autonomic function [44].
In addition to this neurally mediated pathway, LPS-induced peripheral inflammation also results in a rapid (within 3 h) and diffuse increase in the central nervous system (CNS) expression of translocator protein (TSPO) [48]. In the CNS, TSPO is predominantly expressed in activated microglial cells suggesting that systemic inflammation may be translated into a central microglial inflammatory signal through diffuse actions at the cerebrovascular endothelium. However to date, relatively few participants have undergone TSPO imaging post-LPS and no association has been identified between regionally specific increases in TSPO uptake and subjective experiences of inflammation-induced fatigue.
Curiously, activation of interoceptive projections to insula appears less critical in mediating subjective responses to inflammation induced using mimics of viral infection such as IFN-α. Indeed, IFN-α does not appear to be associated with substantial changes in insula microstructure or glucose metabolism either acutely [16] or when chronically administered [49] suggesting that visceral afferents may not be the principle pathway mediating IFN-α-induced fatigue. Why such marked differences exist between models of bacterial and virally induced infection is currently unclear though may be usefully informed by the pre-clinical rodent and non-human primate literature. For example, though IFN-α injection results in a rapid increase in circulating and cerebrospinal fluid (CSF) concentrations of type-I interferon [50] other pro-inflammatory cytokines such as IL-6, TNF-alpha, and IL-1 are only modestly elevated [16]. Furthermore, in rodents profound CNS induction of IFN-inducible genes is observed within hours of intraperitoneal IFN injection [51], indicating that IFN-α likely gains rapid access to the CNS where its actions may be more directly transduced.
To summarize, the dorsal insula represents the ultimate projection of interoceptive pathways and is believed to provide a cortical representation of all aspects of bodily physiology including changes in peripheral inflammation [24]. Progressive posterior to anterior projections are proposed to integrate and translate this information into experiential feeling states such as feelings of warmth, malaise, or fatigue. Experimental models of bacterially induced inflammation result in rapid structural and functional changes in posterior, mid, and anterior insula cortices that correlate with concomitant increases in subjective fatigue and malaise. Similar changes in insula function are also well described in MDD where they correlate with neurovegetative symptoms of depression including fatigue illustrating striking commonalities between inflammation-induced fatigue/malaise and neurovegetative symptoms of depression.
3 Motivational Change
Impairment in reward-related behavior is a core feature of the motivational reorientation characteristic of both inflammation-induced sickness behavior [2] and idiopathic depression [52]. In the context of sickness, this motivational shift is proposed to efficiently prioritize whole organism responses to clearing the infecting agent. However when inflammation is severe or prolonged this persistent motivational reorientation may predispose to the development of MDD [1].
A wealth of human and rodent studies have identified the ventral striatum as a critical structure for mammalian reward-related processing and appetitive motivation [53]. Single cell recordings demonstrate that a subgroup of dopaminergic cells within the midbrain encode a reward prediction error, increasing (or decreasing) firing rate if a reward is higher (or lower) than predicted [54]. Dopaminergic projections from the midbrain to the ventral striatum serve to update estimates of the value of different available options and bias behavioral choice so that long-term future reward is maximized. Reinforcement learning algorithms such as temporal difference models [55] have provided a powerful framework for modeling this dopaminergic prediction error signal that is proposed to mediate learning of associations between stimuli, responses, and outcomes [56]. They also allow interrogation of brain imaging data to identify brain regions whose activity correlates with these reward prediction error signals [57]. Dopamine firing may also contribute to “incentive salience,” the process by which a stimulus grasps attention and motivates goal-directed behavior by its association with reinforcing events [58].
Interestingly, both MDD patients [59] and previously healthy participants given an inflammatory challenge show a reduction in ventral striatal responses to reward outcomes [22, 60]. Inflammation has also been linked to acute reductions in ventral striatal reactivity to cues predicting rewards [19], though this is less convincingly reported in MDD [60]. In the context of inflammation, reduced ventral striatal responses to both reward cues and reward outcomes also correlate with induced anhedonia [19, 22]. Patients with MDD have also been shown to exhibit reduced reward prediction error encoding in the striatum and midbrain [59]. This change also correlated with the severity of anhedonia symptoms suggesting that abnormal encoding of prediction errors in MDD could result in anhedonia by altering the learning and salience of rewarding events [59].
Using a similar probabilistic instrumental learning task we have recently demonstrated that mild inflammatory challenge (induced with typhoid vaccination) also results in a relative impairment in sensitivity to rewards compared to punishments [61]. Furthermore, this motivational reorientation was associated with opposing actions on ventral striatal reward (and right anterior insula punishment) prediction error encoding [61]. Similar to patients with MDD, inflammation was associated with reduced striatal reward prediction error encoding. Behaviorally, after inflammation participants in this study also showed a reduced propensity to choose rewarded options but enhanced avoidance of punished ones. This behavioral change was captured computationally as a significant condition (gain, loss) by inflammation (vaccine, placebo) interaction for the subjective value of rewards compared with punishments.
Though dopamine activity was not measured in this (or Eisenberger’s study demonstrating effects of inflammation on reward cues), a similar reduction in striatal reward prediction error magnitude (and propensity to choose the most rewarded action) has been reported on this task after haloperidol (a dopamine receptor-2 antagonist) [57]. This suggests that effects of inflammation on striatal prediction errors were likely mediated by actions on dopamine release. Supporting this, inflammation has been linked to altered nucleus accumbens dopamine efflux in rodents [62] and disrupted presynaptic dopamine synthesis/release in humans [22]. After LPS challenge monkeys also exhibit significantly lower cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid [50].
However, exactly how inflammation modulates dopamine function is currently unclear. Cytokines such as interferon-alpha have been shown to inhibit dopamine synthesis by reducing CNS tetrahydrobiopterin, an essential cofactor for tyrosine hydroxylase, the rate-limiting step in dopamine synthesis [63]. Inflammation can also decrease synaptic dopamine by increasing expression of the monoamine reuptake transporter [63–66]. Inflammation may further influence dopamine neurotransmission via activation of the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase and resultant formation of neurotoxic kynurenine metabolites [1].
To date most studies investigating the basis of the motivational reorientation associated with inflammation have focused on reward-related processing. However, in our recent study we also showed that inflammation significantly enhances sensitivity to punishment. This was captured in the data modeling as a significant increase in the subjective (negative) value of punishment; i.e., the magnitude of the potential punishment was experienced as being greater after inflammation [61]. This behavioral change was also associated with greater encoding of negative punishment prediction error in the right anterior insula.
Increasing punishment prediction error is one way to increase the subjective value of punishment and may serve as the computational mechanism by which the anterior insula drives this improvement in avoidance behavior. This interpretation is in keeping with theories proposing that brain areas like the insula involved with somatic affective representations (as discussed above) are causally involved in choice behaviors [24, 30, 57]; particularly in the context of potential losses [67]. This association between punishment sensitivity and insula activity also complements an earlier study showing impaired punishment sensitivity in patients with selective insula lesions [68] and suggests that relative sensitivity to reward versus punishment is a state rather than a trait-dependent attitude; flexibly enhancing loss minimization in the context of a serious threat (such as an infection) yet maximizing responses to gains when in good health.
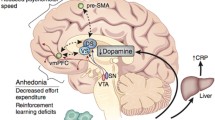
As predicted by models of learned helplessness, dysfunctional responses to negative feedback were some of the earliest cognitive changes described in depression [69]. More recently, meta-analysis of computationally modeled reinforcement learning tasks in patients with current or past history of MDD has reported a selective reduction in subjective reward value (rather than reward learning rate) [52] similar to what has been observed in the context of inflammation. Relatively selective actions on reward/punishment magnitude have also been reported following dopamine manipulation and insular damage [57, 68]. This rapid cognitive adaptation following inflammation serves to heighten relative sensitivity to punishment versus reward raising the intriguing possibility that while this may be beneficial in the context of an infective challenge when metabolic resources are diverted to fighting the infecting organism, when chronic, it may predispose to developing the maladaptive changes in motivation observed in depression. Evidence for common neural mechanisms mediating motivational change and anhedonia in MDD and inflammation have been further strengthened by a recent paper in unmedicated MDD patients showing that decreased connectivity between the ventral striatum and ventromedial prefrontal cortex (vmPFC) also mediates observed associations between raised CRP and anhedonia [70].
4 Psychomotor Retardation
Psychomotor retardation, defined as a slowing-down of thought and physical movements, is a core feature of depression [71] and readily induced following systemic administration of pro-inflammatory cytokines or other experimental models of inflammation [72].
In rodents, systemic administration of bacterial LPS or inflammatory cytokines (notably IL-1β) consistently suppresses locomotor and motivational behaviors, resulting in increased periods of immobility [73]. These depressant effects of peripheral IL-1β on behavior are potentiated by IL-6 [74] and motor-suppressing effects of inflammatory challenge are reduced by antibodies against IL-6 [75, 76] and attenuated in IL-6 knockout mice [77]. Psychomotor retardation expressed as prolonged motor reaction times is also a feature of human sickness behaviors [78, 79], and is observed even after relatively mild inflammatory challenges [10].
In the first study to directly investigate the neural mechanisms underlying the psychomotor consequences of peripheral inflammation Brydon et al. [10] recorded whole brain responses to performance of a simple motor task (button press). They demonstrated that low-level inflammation (induced using typhoid vaccine) selectively modulated substantia nigra reactivity to performance of a button press task during both a low-level visual stimulation (flashing checkerboard) task and a more demanding cognitive (color word Stroop) task. They also observed a striking correlation between peripheral IL-6 responses and motor response time on the Stroop task for both low level (congruent) and attentionally demanding (incongruent) trials suggesting an action on low-level pre-cognitive processes. Supporting this, both peripheral IL-6 responses and changes in left substantia nigra reactivity predicted inter-individual differences in sensitivity to the motor impairing effects of inflammation (Fig. 2).

Inflammation induces psychomotor slowing through actions on substantia nigra. (a) Effect of inflammation on substantia nigra reactivity during a low-grade visual stimulation task (flashing checkerboard). (b) Correlation between IL-6 response to typhoid vaccination and reaction time on a color word Stroop task. (c) Correlation between IL-6 response to typhoid vaccine and increase in left substantia nigra activity during Stroop task performance. (d) Correlation between mean Stroop reaction time and left substantia nigra reactivity following inflammation. Reproduced from Brydon et al. [10] with permission
Located in the midbrain, the substantia nigra is the major source of dopamine in the brain with striatal projections playing a pivotal role in the facilitation of movement [80]. Nigral dopaminergic projections within striatal target regions also modulate sensorimotor processing in response to stimulus salience [81] and have been linked to the reduction in novelty salience observed during inflammation [17]. Agonists that potentiate dopaminergic neurotransmission improve the speed of motor responses in animals, whereas striatal dopamine depletion and selective blockade of dopamine D1 or D2 receptors have been shown to significantly impair performance on reaction time tasks [82, 83]. Similar to effects of inflammation, impaired task performance following inhibition of dopamine is due to lengthened response latencies rather than deficits in the accuracy of responses [83]. Lower levels of striatal dopamine transporter have also been associated with slower motor reactions in healthy elderly humans [84]. Brydon’s results extend these observations and provide empirical evidence for involvement of the dopamine system in behavioral consequences of peripheral inflammation, highlighting a role for IL-6 and substantia nigra neural activity in infection-related psychomotor impairments.
As discussed in the previous section, brain dopamine levels are modulated by peripheral administration of IFN-α and other inflammatory cytokines in rodents [66]. Human patients receiving IFN-α-based immunotherapy also experience marked psychomotor slowing [72] which has been shown to correlate with abnormalities in left dorsal striatal glucose metabolism [49]. These patients also demonstrate bilateral reductions in striatal 18F-DOPA turnover on PET imaging suggesting decreased presynaptic dopamine synthesis or release, though associations with psychomotor responses were not reported in this study [22]. Interestingly, elevated circulating IL-6 and altered striatal dopaminergic neurotransmission are also associated with psychomotor slowing in people with MDD [85, 86]. However, these studies have tended to focus on changes within striatal projection regions (rather than the substantia nigra) that are typically easier to image in human functional imaging studies. Interestingly, a recent study has shown that older individuals appear particularly susceptible to the psychomotor effects of IFN-α treatment. In this study, increased choice movement time correlated with changes in both peripherally induced TNF and left basal ganglia glutamate (reflected by glutamate/creatine ratio (Glu/Cr)), the other major neurochemical input to the striatum [87].
Correspondingly, decreased left striatal presynaptic dopamine function has been described in depressed patients presenting with marked psychomotor retardation [86]. Left dorsal striatum (caudate) lesions are also associated with a higher frequency and severity of post-stroke depression [88]. In their study using 18F-DOPA PET Martinot et al. [86] demonstrated a reduction in left caudate tracer uptake in MDD patients with psychomotor retardation but not MDD patients with high impulsivity or comparison control participants, providing direct evidence of a link between striatal dopamine hypofunction and psychomotor retardation. The importance of the left dorsal striatum to psychomotor retardation associated with MDD and inflammation has been further strengthened by a recent paper demonstrating a link between plasma and CSF levels of C-reactive protein (CRP), left basal ganglia glutamate, and psychomotor slowing in untreated depressed patients [89].
Together, these data support a central role for bottom-up dopaminergic (substantia nigra) and top-down glutaminergic inputs into the dorsal striatum in both inflammation- and MDD-associated psychomotor retardation. Curiously, most of these studies also report strikingly left lateralized effects. Though robustly reported this finding is currently poorly understood and will require further investigation in future studies. During an infection, psychomotor slowing may serve to minimize energy expenditure and conserve heat, thereby enhancing immune function. Convergent findings in inflammation and MDD-associated psychomotor retardation suggest that chronic activation of these mechanisms may differentiate MDD patients presenting with predominant psychomotor retardation or impulsivity/anxiety symptoms. Whether this difference in presentation also relates to differences in peripheral inflammatory markers is the focus of ongoing studies.
5 Autonomic Responses
Physiological changes including hyperactivity of the hypothalamic-pituitary-adrenal axis [90] and disturbance in the autonomic control of the heart and vasculature [91] are another feature of MDD, with the later proposed to mediate the relationship between MDD and risk for cardiovascular disease. Inflammation is also increasingly implicated in cardiovascular disease and has been highlighted as a potentially modifiable risk factor by the American Heart Association (AHA) and Centers for Disease Control (CDC) [92]. In this context it is therefore noteworthy that, in addition to impairing mood, experimentally induced inflammation can also perturb both local cardiovascular reactivity [93] and the central autonomic control of the cardiovasculature [20].
One of the most consistent physiological changes reported in MDD is a change in heart rate variability (HRV) which provides an index of beat-to-beat changes in heart rate [91]. Briefly, high frequency (HF) variation in heart rate is mediated by parasympathetic tone and is believed to maintain cardiac stability and protect against myocardial infarction and heart failure. In contrast low frequency (LF) variation tends to reflect sympathetic tone which is associated with an increased risk of malignant arrhythmias and sudden cardiac death. The ratio of these measures (LF/HF) provides a composite measure of HRV with lower values reflecting healthy cardiac function [94]. Meta-analysis demonstrates a significantly higher LF/HF ratio in MDD patients, suggesting an increase in sympathetic and reciprocal decrease in parasympathetic activity [91] that may underlie the association with increased cardiovascular risk.
A similar acute increase in LF/HF ratio has also been observed following induction of mild inflammation using typhoid vaccination [20] and shown to mediate associated changes in blood pressure. Within the brain, inflammation-induced shifts in LF/HF balance are associated with changes in glucose metabolism (FDG-PET) within three discrete regions: dorsal anterior cingulate, posterior cingulate, and pons. Each of these regions is implicated in regulating stressor-evoked blood pressure reactivity. Their recruitment following inflammation and role in mediating effects on blood pressure further illustrates how central effects of inflammation can also contribute to peripheral physiological changes. Together, these findings demonstrate that commonalities between MDD and inflammation extend beyond mood and motivational changes to include changes in physiological function and highlight the brain mechanisms that bind psychological and physiological wellbeing in these conditions.
6 Attention and Executive Function
Cognitive alterations are a prominent feature of sickness behaviors and manifest predominantly as disturbances in attention, memory, and higher-level executive function [4, 95]. Following experimentally induced influenza infection performance is impaired on oddball type tasks that demand sustained attention, though hand-eye coordination and logical reasoning are unaffected [95]. Similar impairments on attentionally demanding tasks are also observed after acute [96] and chronic administration of interferon-alpha [78]. Patients receiving chronic interferon-alpha also show impaired performance on tests of cognitive speed, verbal memory, and executive functions indicating likely impairment of frontal-sub-cortical brain function [97]. This interpretation is supported by EEG which shows focal changes in frontal lobe regions after acute IFN-alpha administration [98].
Cognitive impairments are an important feature of MDD with deficits particularly prominent in measures of sustained attention, memory, and executive function [99, 100]. Deficits in executive function appear wide-ranging and include impairments in inhibition, set shifting, updating, working memory, and planning indicating frontal-sub-cortical dysfunction [100]. Focusing on inhibition, which has been most closely investigated in inflammation, meta-analysis of color word Stroop studies (where target words are presented in congruent or incongruent font colors) shows a relatively selective impairment on incongruent trials that demand inhibition of pre-potent lexical responses [100]. Studies investigating this effect using neuroimaging suggest that MDD patients show greater interference because they fail to adequately recruit left dorsolateral prefrontal cortex (DLPFC) [101] or require greater left DLPFC activation to achieve performance levels observed in controls [102]. Interestingly, mid-DLPFC is implicated in selecting and biasing attention to the most task-relevant representation indicating a potential circumscribed cognitive deficit in MDD patients [100].
In the first neuroimaging study of inflammation and cognition Capuron et al. [103] used a variation of the CANTAB response time (RT) task to investigate effects of IFN-α on visuospatial attention. Interferon-alpha did not impair task performance or recruitment of the parieto–occipital attention network. However, IFN-α treated patients did recruit an additional dorsal anterior cingulate cortex (ACC) region that was not observed in controls. ACC is implicated in conflict monitoring and its activation is proposed to reflect the degree of intentional effort or willed control needed to perform a task [104]. ACC activation in IFN-treated patients therefore potentially reflects a need to exert greater cognitive control to maintain normal levels of task performance in the face of inflammation.
Interestingly, a similar pattern of behavioral and neuroimaging changes also emerged in a study using the color word Stroop task to investigate effects on mild inflammation (typhoid vaccination) on cognitive inhibition [12]. Again, inflammation was not associated with any significant change in task performance. However, it did have a striking impact on the network of brain regions recruited during attentionally demanding incongruent trials (that require inhibition of pre-potent responses) including DLPFC and mid-cingulate (aMCC/pMCC) cortices. Because participants showed no performance differences, these effects likely represent a need for additional neural resources to maintain task performance under inflammation. As mentioned above, activity within both DLPFC and dorsal ACC is typically enhanced with increasing cognitive demands. Both regions also show an increase in activity during performance of a cognitively demanding visual task in the face of cross-modal auditory distracters [105]. Their concurrent activation in states of inflammation therefore suggests that interdependent cognitive/attentional [106] and somatic (autonomic) [107] mechanisms may be invoked to maintain performance in the face of increased conflict from interoceptive processing.
In sum, these studies indicate the need to recruit additional neural resources, notably DLPFC and ACC to maintain cognitive performance in the face of systemic inflammation. Similar to findings observed in MDD, this is particularly marked in tasks with high attentional demand or requiring inhibition of pre-potent responses. Exactly how closely the cognitive deficits induced by inflammation relate to those observed in MDD is yet to be fully determined. Future research will need to further clarify the origin of impairments in executive function observed in MDD and dissect the neurobiological and cognitive mechanisms underlying the broad cognitive deficits reported in both MDD and inflammation. Neurobiological differences including changes in inflammatory state and its impact on neurotransmitters such as serotonin or kynurenine breakdown products are one possible cause for these changes and recent studies are now beginning to characterize this [108].
7 Memory
Work in rodents has demonstrated that inflammatory cytokines modulate a number of neuronal processes including long-term potentiation (LTP) [109, 110], synaptic plasticity [111], and neurogenesis [112] that are critical to learning and memory. In health, immune mechanisms play a role in each of these processes and contribute to the remodeling of neural circuits that promote learning and memory [4, 111]. However, during systemic infection this positive regulatory function is disrupted resulting in acute memory impairments [113]. When inflammation is severe, cognitive impairment may also become persistent [114] and when it is chronic typical age-related impairments in cognition are accelerated [115].
Medial temporal lobe (MTL) structures appear to be particularly sensitive to the effects of inflammation. This may reflect their relatively high receptor and messenger RNA expression for pro-inflammatory cytokines [116, 117] and their connectivity to regions such as the insula [118] that support cortical representations of peripheral inflammatory states [24]. Rodent studies have particularly emphasized the role of the hippocampus in inflammation-associated memory impairments. For example, IL-1 administration into the hippocampus selectively impairs spatial and contextual memory processes and contextual, but not (hippocampus independent) auditory-cued, fear conditioning [4, 119, 120]. Similarly, over-expression of IL-1 mRNA within the hippocampus has been associated with delayed acquisition of spatial memory [120]. LTP is arguably the key neuronal mechanism for synaptic plasticity that underlies memory encoding and recall. It is therefore noteworthy that IL-1 compromises both hippocampal and dentate gyrus LTP [109, 121, 122]. Peripheral inflammatory challenges also induce IL-1 expression within brain regions, including the MTL [123] and can replicate many of the direct actions of inflammatory cytokines on MTL-dependent memory [124, 125].
In humans, individuals suffering from acute flu-like symptoms have been shown to exhibit impaired memory on tests of immediate and delayed verbal (and delayed picture) recall [126]. However, retrieval of semantic information consolidated in long-term store is unimpaired [126]. Similarly, inflammation induced experimentally with LPS impairs both verbal and nonverbal, declarative memory [9]. Importantly, these memory impairments remain prominent even when mood normalizes suggesting an effect induced by inflammation rather than being secondary to associated changes in mood [9]. Impaired verbal memory is also reported following IFN-α [97]. These studies, which have focused on declarative memory, broadly support the MTL sensitivity to inflammation observed in rodents. However, marked differences in the memory testing paradigms used in rodents and humans limit finer-grained translational inferences.
This difficulty has been partially mitigated (in the context of spatial memory) by the recent translation of the Morris water maze, used extensively to assess rodent spatial memory, for human use [127]. This task requires participants to remember the identity and spatial location of objects in a virtual arena and has been recently used to demonstrate that low-level inflammation also selectively impairs human spatial memory [18]. In particular, inflammation was associated with a selective impairment in remembering object location but not object identity. Furthermore, inflammation did not impair motor skill learning (indexed by performance on a mirror tracing task), a form of procedural memory that relies on a separate dorsal striatum-based memory system independent of the MTL [127]. Interestingly, inflammation was also associated with bilateral reductions in MTL resting glucose metabolism (measured using FDG-PET) with changes in the right parahippocampus significantly mediating the inflammation-induced impairment in spatial memory [18] (Fig. 3). The location of this effect is noteworthy as studies in rodents associate similar spatial memory impairments with changes localized to the hippocampus not the parahippocampus [4].

Inflammation impairs spatial memory via actions on medial temporal lobe glucose metabolism. (a) Virtual reality object-location task. Inset shows that object-location accuracy (y axis) improves after placebo (red) but deteriorates after typhoid vaccine induced inflammation (blue). (b) Control mirror tracing task. Inset shows equivalent improvement in performance after both placebo (red) and vaccine (blue). (c) Decrease in medial temporal lobe (MTL) fluorodeoxyglucose (FDG) uptake in participants given typhoid vaccine after scan 1 (V1 to V2 red) compared to controls (blue). (d) Similar reduction in MTL FDG uptake in participants given typhoid vaccine after scan 2 (P2 to P3 blue). Note participants given typhoid vaccine after scan 1 (red) show a sustained reduction in FDG uptake at scans 2 and 3 (V2 to V3). (e) Reduction in MTL FDG uptake after typhoid vaccination (blue), correlation between object-location accuracy and FDG uptake (yellow), and area correlating with interaction between task performance and inflammation illustrated in a (red). Data from Harrison et al. [18]
Why these inter-species differences exist may be usefully informed by human lesion studies, that show that human performance on Morris water maze type tasks and direct tests of object-location memory can be more strongly dependent on right parahippocampal than hippocampal integrity [128, 129]. Right parahippocampal activity during object-location encoding has also been shown to predict subsequent retrieval success with a spatial cue [130]. Furthermore one-trial memory for object-place associations also appears to be critically dependent on posterior parahippocampus rather than the hippocampus in monkeys [131].
Episodic memory is also one of the cognitive functions most susceptible to depression suggesting a relatively selective impairment in MTL function [132, 133]. Supporting this, meta-analyses of structural MRI studies have shown an 8–10% reduction in hippocampal volume in MDD [134, 135]. Similar to participants receiving LPS or experiencing flu-like symptoms, studies conducted on large populations of MDD patients (>8,000) report impaired performance on the delayed paragraph recall test of verbal declarative memory [136]. MDD patients have also been shown to perform significantly worse on a virtual reality measure of spatial memory [137]. Though to date most studies investigating memory function in MDD have focused on the hippocampus, evidence suggests that MDD may also be associated with abnormalities in broader MTL structures [138]. Future studies characterizing the specific pattern of memory deficits associated with MDD and inflammation and their underlying neural substrates will be essential to furthering our understanding of how inflammatory processes contribute to the memory deficits observed in MDD.
8 Social Responses
Another feature of sickness behaviors is social withdrawal and social disconnection [139]. Feelings of social disconnection (experienced as loneliness) contribute to the development and maintenance of depression [140]. The observation that inflammation-induced social withdrawal can be reversed by antidepressant treatment [141] has motivated a number of recent human studies seeking to understanding how inflammation leads to social disconnection as a way of potentially understanding the mechanistic relationship between inflammation and depression.
In an early study, Eisenberger et al. [8] showed that inflammation increases feelings of social disconnection and furthermore that this change mediated the relationship between inflammatory activity and depressed mood. The questionnaire used to assess feelings of social disconnection in this study included items reflecting both a desire to withdraw socially and items that reflected a feeling of being socially isolated or disconnected from others. Both types of items were altered following LPS suggesting potentially dissociable effects on motivational processes (“I want to be alone”) as well as processes involved in social cognition and social perception. Addressing this, Moieni et al. have recently demonstrated that LPS induced inflammation impairs performance on the “Reading the Mind in the Eyes” test of theory of mind [142]. This task evaluates how accurately participants can identify another’s emotional state by looking only at their eyes [143] and suggests that in addition to effects on motivation, inflammation can additionally alter social processes central to our ability to correctly infer others mental and emotional states.
Another important feature of MDD is that it is twice as common in women as men. One factor proposed to mediate womens’ increased vulnerability is their greater exposure and reactivity to interpersonal stressors [144]. It is therefore noteworthy that though women do not show convincingly greater pro-inflammatory cytokine responses to LPS they do report greater increases in feelings of social disconnection and depressed mood suggesting that inflammation may play a role in mediating sex differences in rates of MDD [145]. This is supported by another study that combined LPS and the Cyberball task during fMRI to investigate effects of inflammation on social exclusion [146]. In this study, LPS-induced increases in IL-6 correlated with increased activity within a matrix of brain regions including dorsomedial prefrontal cortex (PFC), posterior superior temporal sulcus (STS), dorsal anterior cingulate (dACC), and insula that are implicated in social processing. However, though this relationship was observed across all participants only in women did it significantly mediate associations between inflammation and depressed mood.
Brain regions such as the posterior superior temporal sulcus (pSTS) and medial prefrontal cortex are strongly implicated in tasks of social cognition that involve extracting socially meaningful information and inferring another’s mental state [147]. It is therefore noteworthy that each of these regions showed heightened activity in proportion to induced inflammation [146]. This interpretation is also in keeping with reported impairments in performance on the “Mind in the Eyes” test on theory of mind [142]. Supporting these findings, inflammation has also been shown to disrupt the functional connectivity of both the pSTS and medial prefrontal cortex to sub-genual cingulate (sACC) (a region central to integrating social, emotional, and physiological responses) during an implicit emotional face processing task [11]. Together, these studies demonstrate effects of inflammation on social processing including increased feelings of social disconnection and impaired theory of mind. Accumulating evidence suggests that in some participants, particularly women, these changes may mediate associated impairments in mood and illustrate a potential mechanism linking inflammation to depression.
9 Network Connectivity
The preceding sections have focused on dissecting effects of inflammation and MDD on discrete emotional, cognitive, behavioral, and physiological changes and relating this to regional changes in brain function. However, a small number of recent studies have begun to look at the effects of inflammation on network connectivity, driven by the recognition that even the simplest cognitive functions involve highly distributed processing [148]. The first such study used a simple psychophysiological interaction (PPI)-based approach to investigate the effects of systemic inflammation on connectivity of the sub-genual cingulate (sACC) [11]. In so doing, this study showed that inflammation-associated changes in total mood modulated not just sACC activity but also its functional connectivity to the nucleus accumbens, amygdala and superior temporal sulcus, regions central to the processing of reward, and emotionally and socially salient information, respectively. Furthermore, inflammation-induced reductions in the effective connectivity of the sACC to each of these regions predicted the associated deterioration in total mood.
This study is noteworthy as the sACC is recognized as a key node in functional and anatomical models of mood regulation [149] and the coordination of emotional processing. It is also strongly implicated in the pathophysiology of MDD [44]. Increased sACC activity seen in depression has also been shown to reverse with successful depression treatment with a selective serotonin reuptake inhibitor [44, 150], deep brain stimulation [151] of adjacent white matter tracts, and even placebo [150]. Importantly, the sACC is a region that is strongly implicated in integrating multiple components of mood homeostasis. Its recruitment in inflammation-induced mood change suggests that inflammation-associated changes in mood recruits a network of brain regions similar to that implicated in primary depression.
More recently three further studies have investigated the effects of inflammation on brain functional connectivity networks observed at rest. In the first using low dose LPS (0.4 ng/kg), seed-based analysis revealed a rapid and widespread reduction in the functional coupling of the amygdala, insula, and cingulate cortices to multiple brain networks involved in affective-emotional, motivational, and cognitive-modulatory processes [152]. Similar to the PPI analysis of task-related fMRI [11], LPS was associated with reduced connectivity between the amygdala and prefrontal structures, though these authors were unable to show any significant relationship to changes in mood. The second study used a slightly higher dose (0.6 ng/kg) of LPS and specifically investigated effects on connectivity between anterior and posterior insula seeds and orbitofrontal and cingulate (anterior and middle). This study identified a specific increase in left anterior insula to left mid-cingulate cortex that additionally predicted LPS associated back pain and global sickness [42]. These regions form key components of the pain matrix, and it is noteworthy that they have also been previously linked to LPS induced increases in visceral pain sensitivity [153].
The third study adopted a different approach and investigated effects of IFN-α on measures of network function derived from graph theory [154]. Briefly, graph theory provides a powerful mathematical approach for analyzing the structure of complex networks, and application to the human brain has revealed insights unavailable from conventional approaches. For example, it has shown that similar to other complex networks, the brain utilizes an efficient “small-world” connectivity architecture that serves to minimize wiring cost while maintaining robustness to random damage to individual regions (nodes) or connections (edges) [155]. Within 4 h of administration, IFN-α was associated with a striking reduction in global network connectivity and network efficiency indicating a global reduction in information transfer among nodes forming the whole brain network. Furthermore, these changes in global network connectivity and efficiency of information exchange correlated strongly with IFN-α induced changes in mood, confusion, fatigue, and tension/anxiety.
How peripherally administered IFN-α or LPS can so rapidly impair the functional connectivity of such large-scale brain networks is currently uncertain. However, the observation that these actions are effected on a global scale points towards a likely role for neuromodulators such as dopamine, norepinephrine, or serotonin that can rapidly alter diverse and widespread neuronal populations rather than a more regionally targeted effect. In support of this, inflammation has been linked to altered nucleus accumbens dopamine efflux in rodents [62], decreased striatal dopamine release in rhesus monkeys [156, 157], and reduced presynaptic dopamine synthesis or release in humans [22]. Further, monkeys showing behavioral impairment after inflammatory challenge with lipopolysaccharide exhibit significantly lower cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid [50].
Network-based analyses have also been pursued in MDD and, similar to the findings of post-LPS, have shown reduced functional connectivity within broad prefrontal-limbic-thalamic areas, particular regions sub-served by the left amygdala-ACC and the right insula–precuneus Labrenz et al. [158]. Graph theoretic analyses of MDD have also demonstrated changes in network topology including abnormal small-world organization and network efficiency [159]. Though application of advanced connectivity analyses to both MDD and inflammation remains in its relatively infancy, similar marked changes in network connectivity, particularly within prefrontal and limbic regions across conditions, support the utility of this approach as a way of characterizing the relationship between inflammation and MDD. Demonstration of often-marked correlations between global measures of network function and mood/cognition suggests a likely role for neuromodulators such as dopamine and serotonin, that are implicated in the pathophysiology of both MDD and sickness behaviors. Further characterization of these associations will require combination of network-based analyses with PET imaging of specific neuromodulators and/or metabolomic approaches.
10 Summary
Rodent and human brain imaging studies have been successful in characterizing a discrete set of cortical and sub-cortical structures sensitive to changes in peripheral inflammation and highlighted their role in many of the key components of sickness behavior. These regions show a striking similarity to the network of areas implicated in the mood, motivational, and cognitive deficits characteristic of MDD. In the context of models of bacterial infection, peripheral inflammation rapidly recruits an interoceptive pathway projecting to insula. This pathway and its terminal projection to the insula provides a central representation of all aspects of bodily physiological state and its translation into consciously accessible feeling states. Following inflammation, recruitment of the insula cortex is implicated in feelings of fatigue and malaise (and possibly social disconnection) as well as a heightened sensitivity to punishment. Bidirectional connections between the insula and the anterior and mid-cingulate cortex provide a substrate for the heightened sensitivity to visceral and pressure pain. Similar to MDD, actions of inflammation on ventral striatal reward processing and reward prediction error encoding appear to underlie shifts in reward sensitivity and motivation. Cognitive deficits (particularly impaired MTL dependent memory and performance on attentionally demanding tasks) are characteristic of both MDD and inflammation and relate to disrupted hippocampal/parahippocampal processing and DLPFC, respectively. More recent functional connectivity studies highlight the distributed nature of cognitive processes and hint at the likely importance of broadly acting neuromodulators like dopamine and serotonin in the mood, motivational, and cognitive impairments characteristic of both MDD and inflammation.
Based on these foundations, future studies combining multiple functional neuroimaging techniques with metabolomic and proteomic approaches should help us move closer to the goal of a mechanistic understanding of the relationships between peripheral inflammation, regional brain structure/function, and discrete cognitive phenotypes observed in MDD.
References
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Hart BL (1987) Behavior of sick animals. Vet Clin North Am Food Anim Pract 3:383–391
Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, Broussard SR (2003) Cytokine-induced sickness behavior. Brain Behav Immun 17(Suppl 1):S112–S118
Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25:181–213
Merali Z, Brennan K, Brau P, Anisman H (2003) Dissociating anorexia and anhedonia elicited by interleukin-1beta: antidepressant and gender effects on responding for “free chow” and “earned” sucrose intake. Psychopharmacology (Berl) 165:413–418
Stone EA, Lehmann ML, Lin Y, Quartermain D (2006) Depressive behavior in mice due to immune stimulation is accompanied by reduced neural activity in brain regions involved in positively motivated behavior. Biol Psychiatry 60:803–811
Vichaya EG, Hunt SC, Dantzer R (2014) Lipopolysaccharide reduces incentive motivation while boosting preference for high reward in mice. Neuropsychopharmacology 39:2884–2890
Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR (2010) Inflammation and social experience: an inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav Immun 24:558–563
Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, Pollmacher T (2001) Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry 58:445–452
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD (2008) Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry 63:1022–1029
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009) Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66:407–414
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, Critchley HD (2009) Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry 66:415–422
Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS, Miller AH (2001) Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med 344:961–966
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67:446–457
Valkanova V, Ebmeier KP, Allan CL (2013) CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord 150:736–744
Dowell NG, Cooper EA, Tibble J, Voon V, Critchley HD, Cercignani M, Harrison NA (2016) Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol Psychiatry 79:320–328
Harrison NA, Cercignani M, Voon V, Critchley HD (2015) Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology 40:831–838
Harrison NA, Doeller CF, Voon V, Burgess N, Critchley HD (2014) Peripheral inflammation acutely impairs human spatial memory via actions on medial temporal lobe glucose metabolism. Biol Psychiatry 76:585–593
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010) Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68:748–754
Harrison NA, Cooper E, Voon V, Miles K, Critchley HD (2013) Central autonomic network mediates cardiovascular responses to acute inflammation: relevance to increased cardiovascular risk in depression? Brain Behav Immun 31:189–196
Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH (2002) Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26:643–652
Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, Miller AH (2012) Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 69:1044–1053
Harrison NA, Cooper E, Dowell NG, Keramida G, Voon V, Critchley HD, Cercignani M (2015) Quantitative magnetization transfer imaging as a biomarker for effects of systemic inflammation on the brain. Biol Psychiatry 78:49–57
Critchley HD, Harrison NA (2013) Visceral influences on brain and behavior. Neuron 77:624–638
McEwen BS, Gianaros PJ (2010) Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann N Y Acad Sci 1186:190–222
Wan W, Wetmore L, Sorensen CM, Greenberg AH, Nance DM (1994) Neural and biochemical mediators of endotoxin and stress-induced c-fos expression in the rat brain. Brain Res Bull 34:7–14
Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, Watkins LR (1997) Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull 43:357–364
Ek M, Kurosawa M, Lundeberg T, Ericsson A (1998) Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci 18:9471–9479
Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR (2000) Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci 85:49–59
Craig AD (2002) How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci 3:655–666
Craig AD, Krout K, Andrew D (2001) Quantitative response characteristics of thermoreceptive and nociceptive lamina I spinothalamic neurons in the cat. J Neurophysiol 86:1459–1480
Craig AD, Chen K, Bandy D, Reiman EM (2000) Thermosensory activation of insular cortex. Nat Neurosci 3:184–190
Andrew D, Craig AD (2001) Spinothalamic lamina I neurons selectively sensitive to histamine: a central neural pathway for itch. Nat Neurosci 4:72–77
Olausson H, Lamarre Y, Backlund H, Morin C, Wallin BG, Starck G, Bushnell MC (2002) Unmyelinated tactile afferents signal touch and project to insular cortex. Nat Neurosci 5:900–904
Craig AD, Blomqvist A (2002) Is there a specific lamina I spinothalamocortical pathway for pain and temperature sensations in primates? J Pain 3:95–101, discussion 113–104
Critchley HD, Wiens S, Rotshtein P, Ohman A, Dolan RJ (2004) Neural systems supporting interoceptive awareness. Nat Neurosci 7:189–195
Rosenkranz MA, Busse WW, Johnstone T, Swenson CA, Crisafi GM, Jackson MM, Davidson RJ (2005) Neural circuitry underlying the interaction between emotion and asthma symptom exacerbation. Proc Natl Acad Sci U S A 102:13319–13324
Drzezga A, Darsow U, Treede RD, Siebner H, Frisch M, Munz F, Bartenstein P (2001) Central activation by histamine-induced itch: analogies to pain processing: a correlational analysis of O-15 H2O positron emission tomography studies. Pain 92:295–305
Williamson JW, McColl R, Mathews D, Ginsburg M, Mitchell JH (1999) Activation of the insular cortex is affected by the intensity of exercise. J Appl Physiol (1985) 87:1213–1219
van den Heuvel MP, Pol HEH (2010) Exploring the brain network: a review on resting-state fMRI functional connectivity. Eur Neuropsychopharm 20:519–534
Hannestad J, Subramanyam K, Dellagioia N, Planeta-Wilson B, Weinzimmer D, Pittman B, Carson RE (2012) Glucose metabolism in the insula and cingulate is affected by systemic inflammation in humans. J Nucl Med 53:601–607
Lekander M, Karshikoff B, Johansson E, Soop A, Fransson P, Lundstrom JN, Nilsonne G (2015) Intrinsic functional connectivity of insular cortex and symptoms of sickness during acute experimental inflammation. Brain Behav Immun. doi:10.1016/j.bbi.2015.12.018
Manes F, Paradiso S, Robinson RG (1999) Neuropsychiatric effects of insular stroke. J Nerv Ment Dis 187:707–712
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Fox PT (1999) Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 156:675–682
Paulus MP, Stein MB (2010) Interoception in anxiety and depression. Brain Struct Funct 214:451–463
Veer IM, Beckmann CF, van Tol MJ, Ferrarini L, Milles J, Veltman DJ, Rombouts SA (2010) Whole brain resting-state analysis reveals decreased functional connectivity in major depression. Front Syst Neurosci 4
Yao Z, Wang L, Lu Q, Liu H, Teng G (2009) Regional homogeneity in depression and its relationship with separate depressive symptom clusters: a resting-state fMRI study. J Affect Disord 115:430–438
Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, Lin SF, Cosgrove KP (2015) Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A 112:12468–12473
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, Miller AH (2007) Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 32:2384–2392
Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM, Miller AH (2007) Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry 62:1324–1333
Wang J, Campbell IL, Zhang H (2008) Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry 13:293–301
Huys QJ, Pizzagalli DA, Bogdan R, Dayan P (2013) Mapping anhedonia onto reinforcement learning: a behavioural meta-analysis. Biol Mood Anxiety Disord 3:12
Schultz W, Apicella P, Scarnati E, Ljungberg T (1992) Neuronal activity in monkey ventral striatum related to the expectation of reward. J Neurosci 12:4595–4610
Schultz W (1998) Predictive reward signal of dopamine neurons. J Neurophysiol 80:1–27
Dayan P, Abbott L (2001) Theoretical neuroscience: computational and mathematical modelling of neural systems. The MIT Press, Cambridge
Montague PR, Dayan P, Sejnowski TJ (1996) A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J Neurosci 16:1936–1947
Pessiglione M, Seymour B, Flandin G, Dolan RJ, Frith CD (2006) Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature 442:1042–1045
Berridge KC, Robinson TE (1998) What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 28:309–369
Gradin VB, Kumar P, Waiter G, Ahearn T, Stickle C, Milders M, Steele JD (2011) Expected value and prediction error abnormalities in depression and schizophrenia. Brain 134:1751–1764
Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R, Fava M (2009) Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry 166:702–710
Harrison NA, Voon V, Cercignani M, Cooper EA, Pessiglione M, Critchley HD (2015) A neurocomputational account of how inflammation enhances sensitivity to punishments versus rewards. Biol Psychiatry. doi:10.1016/j.bbi.2015.12.018
Borowski T, Kokkinidis L, Merali Z, Anisman H (1998) Lipopolysaccharide, central in vivo biogenic amine variations, and anhedonia. Neuroreport 9:3797–3802
Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T (2003) Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res 978:104–114
Felger JC, Miller AH (2012) Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol 33:315–327
Kamata M, Higuchi H, Yoshimoto M, Yoshida K, Shimizu T (2000) Effect of single intracerebroventricular injection of alpha-interferon on monoamine concentrations in the rat brain. Eur Neuropsychopharmacol 10:129–132
Shuto H, Kataoka Y, Horikawa T, Fujihara N, Oishi R (1997) Repeated interferon-alpha administration inhibits dopaminergic neural activity in the mouse brain. Brain Res 747:348–351
Paulus MP, Rogalsky C, Simmons A, Feinstein JS, Stein MB (2003) Increased activation in the right insula during risk-taking decision making is related to harm avoidance and neuroticism. Neuroimage 19:1439–1448
Palminteri S, Justo D, Jauffret C, Pavlicek B, Dauta A, Delmaire C, Pessiglione M (2012) Critical roles for anterior insula and dorsal striatum in punishment-based avoidance learning. Neuron 76:998–1009
Seligman ME (1972) Learned helplessness. Annu Rev Med 23:407–412
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, Miller AH (2015) Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. doi:10.1016/j.bbi.2015.12.018
Zung WW, Richards CB, Short MJ (1965) Self-rating depression scale in an outpatient clinic. Further validation of the SDS. Arch Gen Psychiatry 13:508–515
Capuron L, Miller AH (2004) Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry 56:819–824
Dantzer R, Bluthe R, Castanon N, Kelly K, Konsman J, Laye S (2007) Cytokines, sickness behavior, and depression. In: Ader R (ed) Psychoneuroimmunology, 4th edn. Elsevier, San Diego
Lenczowski MJ, Bluthe RM, Roth J, Rees GS, Rushforth DA, van Dam AM, Luheshi GN (1999) Central administration of rat IL-6 induces HPA activation and fever but not sickness behavior in rats. Am J Physiol 276:R652–R658
Harden LM, du Plessis I, Poole S, Laburn HP (2006) Interleukin-6 and leptin mediate lipopolysaccharide-induced fever and sickness behavior. Physiol Behav 89:146–155
Pang Y, Fan LW, Zheng B, Cai Z, Rhodes PG (2006) Role of interleukin-6 in lipopolysaccharide-induced brain injury and behavioral dysfunction in neonatal rats. Neuroscience 141:745–755
Bluthe RM, Michaud B, Poli V, Dantzer R (2000) Role of IL-6 in cytokine-induced sickness behavior: a study with IL-6 deficient mice. Physiol Behav 70:367–373
Capuron L, Ravaud A, Dantzer R (2001) Timing and specificity of the cognitive changes induced by interleukin-2 and interferon-alpha treatments in cancer patients. Psychosom Med 63:376–386
Smith AP, Tyrrell DA, Al-Nakib W, Coyle KB, Donovan CB, Higgins PG, Willman JS (1987) Effects of experimentally induced respiratory virus infections and illness on psychomotor performance. Neuropsychobiology 18:144–148
Graybiel AM, Aosaki T, Flaherty AW, Kimura M (1994) The basal ganglia and adaptive motor control. Science 265:1826–1831
Bunzeck N, Duzel E (2006) Absolute coding of stimulus novelty in the human substantia nigra/VTA. Neuron 51:369–379
Baunez C, Robbins TW (1999) Effects of dopamine depletion of the dorsal striatum and further interaction with subthalamic nucleus lesions in an attentional task in the rat. Neuroscience 92:1343–1356
Weed MR, Gold LH (1998) The effects of dopaminergic agents on reaction time in rhesus monkeys. Psychopharmacology (Berl) 137:33–42
van den Biggelaar AH, Gussekloo J, de Craen AJ, Frolich M, Stek ML, van der Mast RC, Westendorp RG (2007) Inflammation and interleukin-1 signaling network contribute to depressive symptoms but not cognitive decline in old age. Exp Gerontol 42:693–701
Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H (1997) Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 9:853–858
Martinot M, Bragulat V, Artiges E, Dolle F, Hinnen F, Jouvent R, Martinot J (2001) Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am J Psychiatry 158:314–316
Haroon E, Felger JC, Woolwine BJ, Chen X, Parekh S, Spivey JR, Miller AH (2015) Age-related increases in basal ganglia glutamate are associated with TNF, reduced motivation and decreased psychomotor speed during IFN-alpha treatment: preliminary findings. Brain Behav Immun 46:17–22
Starkstein SE, Robinson RG, Berthier ML, Parikh RM, Price TR (1988) Differential mood changes following basal ganglia vs thalamic lesions. Arch Neurol 45:725–730
Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, Miller AH (2016) Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. doi:10.1038/mp.2015.206
Pariante CM, Lightman SL (2008) The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31:464–468
Kemp AH, Quintana DS, Gray MA, Felmingham KL, Brown K, Gatt JM (2010) Impact of depression and antidepressant treatment on heart rate variability: a review and meta-analysis. Biol Psychiatry 67:1067–1074
Pearson TA, Bazzarre TL, Daniels SR, Fair JM, Fortmann SP, Franklin BA, Prevention S (2003) American Heart Association guide for improving cardiovascular health at the community level: a statement for public health practitioners, healthcare providers, and health policy makers from the American Heart Association Expert Panel on Population and Prevention Science. Circulation 107:645–651
Hingorani AD, Cross J, Kharbanda RK, Mullen MJ, Bhagat K, Taylor M, Vallance P (2000) Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 102:994–999
Dekker JM, Crow RS, Folsom AR, Hannan PJ, Liao D, Swenne CA, Schouten EG (2000) Low heart rate variability in a 2-minute rhythm strip predicts risk of coronary heart disease and mortality from several causes: the ARIC Study. Atherosclerosis Risk In Communities. Circulation 102:1239–1244
Smith AP, Tyrrell DA, Al-Nakib W, Coyle KB, Donovan CB, Higgins PG, Willman JS (1988) The effects of experimentally induced respiratory virus infections on performance. Psychol Med 18:65–71
Smith A, Tyrrell D, Coyle K, Higgins P (1988) Effects of interferon alpha on performance in man: a preliminary report. Psychopharmacology (Berl) 96:414–416
Pavol MA, Meyers CA, Rexer JL, Valentine AD, Mattis PJ, Talpaz M (1995) Pattern of neurobehavioral deficits associated with interferon alfa therapy for leukemia. Neurology 45:947–950
Adams F, Quesada JR, Gutterman JU (1984) Neuropsychiatric manifestations of human leukocyte interferon therapy in patients with cancer. JAMA 252:938–941
Paelecke-Habermann Y, Pohl J, Leplow B (2005) Attention and executive functions in remitted major depression patients. J Affect Disord 89:125–135
Snyder HR (2013) Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: a meta-analysis and review. Psychol Bull 139:81–132
Holmes AJ, Pizzagalli DA (2007) Task feedback effects on conflict monitoring and executive control: relationship to subclinical measures of depression. Emotion 7:68–76
Wagner G, Sinsel E, Sobanski T, Kohler S, Marinou V, Mentzel HJ, Schlosser RG (2006) Cortical inefficiency in patients with unipolar depression: an event-related FMRI study with the Stroop task. Biol Psychiatry 59:958–965
Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS, Miller AH (2005) Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry 58:190–196
Duncan J, Owen AM (2000) Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci 23:475–483
Weissman DH, Warner LM, Woldorff MG (2004) The neural mechanisms for minimizing cross-modal distraction. J Neurosci 24:10941–10949
MacDonald AW 3rd, Cohen JD, Stenger VA, Carter CS (2000) Dissociating the role of the dorsolateral prefrontal and anterior cingulate cortex in cognitive control. Science 288:1835–1838
Critchley HD, Tang J, Glaser D, Butterworth B, Dolan RJ (2005) Anterior cingulate activity during error and autonomic response. Neuroimage 27:885–895
Meier TB, Drevets WC, Wurfel BE, Ford BN, Morris HM, Victor TA, Savitz J (2016) Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain Behav Immun 53:39–48
Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M (1990) Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol 181:323–326
Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO (1998) A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A 95:7778–7783
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100:13632–13637
Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Walker FR (2010) Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 24:1058–1068
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304:1787–1794
Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE (2002) Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 59:371–378
Ericsson A, Liu C, Hart RP, Sawchenko PE (1995) Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol 361:681–698
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, Jones AR (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489:391–399
Suzuki WA, Amaral DG (1994) Perirhinal and parahippocampal cortices of the macaque monkey: cortical afferents. J Comp Neurol 350:497–533
Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF (2002) Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res 134:291–298
Oitzl MS, van Oers H, Schobitz B, de Kloet ER (1993) Interleukin-1 beta, but not interleukin-6, impairs spatial navigation learning. Brain Res 613:160–163
Bellinger FP, Madamba S, Siggins GR (1993) Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res 628:227–234
Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ (1996) Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20
Ban E, Haour F, Lenstra R (1992) Brain interleukin 1 gene expression induced by peripheral lipopolysaccharide administration. Cytokine 4:48–54
Gibertini M (1996) IL1 beta impairs relational but not procedural rodent learning in a water maze task. Adv Exp Med Biol 402:207–217
Pugh CR, Kumagawa K, Fleshner M, Watkins LR, Maier SF, Rudy JW (1998) Selective effects of peripheral lipopolysaccharide administration on contextual and auditory-cue fear conditioning. Brain Behav Immun 12:212–229
Capuron L, Lamarque D, Dantzer R, Goodall G (1999) Attentional and mnemonic deficits associated with infectious disease in humans. Psychol Med 29:291–297
Doeller CF, King JA, Burgess N (2008) Parallel striatal and hippocampal systems for landmarks and boundaries in spatial memory. Proc Natl Acad Sci U S A 105:5915–5920
Bohbot VD, Corkin S (2007) Posterior parahippocampal place learning in H.M. Hippocampus 17:863–872
Ploner CJ, Gaymard BM, Rivaud-Pechoux S, Baulac M, Clemenceau S, Samson S, Pierrot-Deseilligny C (2000) Lesions affecting the parahippocampal cortex yield spatial memory deficits in humans. Cereb Cortex 10:1211–1216
Sommer T, Rose M, Glascher J, Wolbers T, Buchel C (2005) Dissociable contributions within the medial temporal lobe to encoding of object-location associations. Learn Mem 12:343–351
Malkova L, Mishkin M (2003) One-trial memory for object-place associations after separate lesions of hippocampus and posterior parahippocampal region in the monkey. J Neurosci 23:1956–1965
Airaksinen E, Larsson M, Lundberg I, Forsell Y (2004) Cognitive functions in depressive disorders: evidence from a population-based study. Psychol Med 34:83–91
Sweeney JA, Kmiec JA, Kupfer DJ (2000) Neuropsychologic impairments in bipolar and unipolar mood disorders on the CANTAB neurocognitive battery. Biol Psychiatry 48:674–684
Campbell S, Marriott M, Nahmias C, MacQueen GM (2004) Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry 161:598–607
Videbech P, Ravnkilde B (2004) Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 161:1957–1966
Gorwood P, Corruble E, Falissard B, Goodwin GM (2008) Toxic effects of depression on brain function: impairment of delayed recall and the cumulative length of depressive disorder in a large sample of depressed outpatients. Am J Psychiatry 165:731–739
Gould NF, Holmes MK, Fantie BD, Luckenbaugh DA, Pine DS, Gould TD, Zarate CA Jr (2007) Performance on a virtual reality spatial memory navigation task in depressed patients. Am J Psychiatry 164:516–519
Montag C, Weber B, Fliessbach K, Elger C, Reuter M (2009) The BDNF Val66Met polymorphism impacts parahippocampal and amygdala volume in healthy humans: incremental support for a genetic risk factor for depression. Psychol Med 39:1831–1839
Hart BL (1988) Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 12:123–137
Heinrich LM, Gullone E (2006) The clinical significance of loneliness: a literature review. Clin Psychol Rev 26(6):695–718
Yirmiya R (1996) Endotoxin produces a depressive-like episode in rats. Brain Res 7:163–174
Moieni M, Irwin MR, Jevtic I, Breen EC, Eisenberger NI (2015) Inflammation impairs social cognitive processing: a randomized controlled trial of endotoxin. Brain Behav Immun 48:132–138
Baron-Cohen S, Wheelwright S, Hill J, Raste Y, Plumb I (2001) The “Reading the Mind in the Eyes” Test revised version: a study with normal adults, and adults with Asperger syndrome or high-functioning autism. J Child Psychol Psychiatry 42:241–251
Cyranowski JM, Frank E, Young E, Shear MK (2000) Adolescent onset of the gender difference in lifetime rates of major depression: a theoretical model. Arch Gen Psychiatry 57:21–27
Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI (2015) Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40:1709–1716
Eisenberger NI, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2009) An fMRI study of cytokine-induced depressed mood and social pain: the role of sex differences. Neuroimage 47:881–890
Frith CD, Frith U (2012) Mechanisms of social cognition. Annu Rev Psychol 63:287–313
Johansen-Berg H (2013) Human connectomics – what will the future demand? Neuroimage 80:541–544
Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, Rafi-Tari S (2004) Limbic-frontal circuitry in major depression: a path modeling metaanalysis. Neuroimage 22:409–418
Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S, Jerabek PA (2000) Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry 48:830–843
Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Kennedy SH (2005) Deep brain stimulation for treatment-resistant depression. Neuron 45:651–660
Labrenz F, Wrede K, Forsting M, Engler H, Schedlowski M, Elsenbruch S, Benson S (2016) Alterations in functional connectivity of resting state networks during experimental endotoxemia – an exploratory study in healthy men. Brain Behav Immun 54:17–26
Benson S, Rebernik L, Wegner A, Kleine-Borgmann J, Engler H, Schlamann M, Elsenbruch S (2015) Neural circuitry mediating inflammation-induced central pain amplification in human experimental endotoxemia. Brain Behav Immun 48:222–231
Dipasquale O, Cooper EA, Tibble J, Voon V, Baglio F, Baselli G, Harrison NA (2015) Interferon-alpha acutely impairs whole-brain functional connectivity network architecture – a preliminary study. Brain Behav Immun. doi:10.1016/j.bbi.2015.12.018
Achard S, Salvador R, Whitcher B, Suckling J, Bullmore E (2006) A resilient, low-frequency, small-world human brain functional network with highly connected association cortical hubs. J Neurosci 26(1):63–72
Felger JC, Hernandez CR, Miller AH (2015) Levodopa reverses cytokine-induced reductions in striatal dopamine release. Int J Neuropsychopharmacol 18
Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, Miller AH (2013) Chronic interferon-alpha decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 38:2179–2187
Lui S, Wu Q, Qiu L, Yang X, Kuang W, Chan RC, Gong Q (2011) Resting-state functional connectivity in treatment-resistant depression. Am J Psychiatry 168:642–648
Zhang J, Wang J, Wu Q, Kuang W, Huang X, He Y, Gong Q (2011) Disrupted brain connectivity networks in drug-naive, first-episode major depressive disorder. Biol Psychiatry 70:334–342
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Harrison, N.A. (2016). Brain Structures Implicated in Inflammation-Associated Depression. In: Dantzer, R., Capuron, L. (eds) Inflammation-Associated Depression: Evidence, Mechanisms and Implications. Current Topics in Behavioral Neurosciences, vol 31. Springer, Cham. https://doi.org/10.1007/7854_2016_30
Download citation
DOI: https://doi.org/10.1007/7854_2016_30
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-51151-1
Online ISBN: 978-3-319-51152-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)