
Abstract
The present review briefly explores the neurotoxic properties of methcathinone, mephedrone, methylone, and methylenedioxypyrovalerone (MDPV), four synthetic cathinones most commonly found in “bath salts.” Cathinones are β-keto analogs of the commonly abused amphetamines and display pharmacological effects resembling cocaine and amphetamines, but despite their commonalities in chemical structures, synthetic cathinones possess distinct neuropharmacological profiles and produce unique effects. Among the similarities of synthetic cathinones with their non-keto analogs are their targeting of monoamine systems, the release of neurotransmitters, and their stimulant properties. Most of the literature on synthetic cathinones has focused on describing their properties as psychostimulants, their behavioral effects on locomotion, memory, and potential for abuse, whereas descriptions of their neurotoxic properties are not abundant. The biochemical gauges of neurotoxicity induced by non-keto analogs are well studied in humans and experimental animals and include their ability to induce neuroinflammation, oxidative stress, excitotoxicity, temperature alterations as well as dysregulation of neurotransmitter systems and induce changes in monoamine transporters and receptors. These neurotoxicity gauges will serve as parameters to discuss the effects of the four previously mentioned synthetic cathinones alone or in combination with either another cathinone or with some of their non-keto analogs. Bath salts are not a defined combination of drugs and may consist of one synthetic cathinone compound or combinations of more cathinones. Furthermore, this review also presents some of the mechanisms that are thought to underlie this toxicity. A better understanding of the cellular and molecular mechanisms involved in the synthetic cathinones-induced neurotoxicity should contribute to generate modern therapeutic approaches to prevent or attenuate the adverse consequences of use of these drugs in humans.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

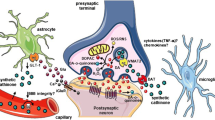

Keywords
1 Neuroinflammation
One of the most relevant issues related to non-ketoamphetamines-induced neurotoxicity is that they can trigger inflammatory processes in those brain areas that exhibit terminal degeneration [1]. Studies have demonstrated that glial activation participates in the events that induce neuronal damage, since chronic neuroinflammation elevates the levels of glia-derived cytokines that exert neurotoxic effects on vulnerable neurons [2]. Microglia and astrocytes are the primary modulators of inflammation in the CNS and have been associated with the toxicity induced by administration of methamphetamine [3], amphetamine and parachloroamphetamine [4], and 3,4-methylenedioxy-methamphetamine (MDMA) [5]. Studies of the effects of β-ketoamphetamines on neuroinflammation are summarized in Table 1.
To the best of our knowledge, no reports have been made on the neuroinflammatory effects of methcathinone.
With regard to mephedrone, in vivo studies reported that there were no signs of striatal [17] or cortical [25] astroglial activation after administration of mephedrone in a binge paradigm. Similarly, no signs of microglial activation were observed in the striatum at 2 or 7 days after administration of mephedrone [17]. Lopez-Arnau and colleagues measured [3H]PK11195-specific binding to investigate the microglial activation after neuronal injury in rats killed 24 h post-treatment with mephedrone. PK11195 is an isoquinoline carboxamide that purportedly binds to microglia in conditions of brain injury. In these animals, no increase in the density of [3H]PK11195 binding sites was detected, indicating a lack of microglial activation [20]. However, Martinez-Clemente reported an increase in reactive astrocytes in the dentate gyrus of the hippocampus of mice treated with mephedrone at 7 days after a binge of 3 doses of 25 mg/kg for 2 days [25].
There were no signs of striatal, hippocampal, or cortical microgliosis in methylone-treated rats using a regimen of 4 doses of 20 mg/kg every 3 h [39]. However, a significant increase of reactive astrocytes was reported in the frontal cortex of methylone-treated rats. By contrast, no significant differences were found in the expression of the astroglial marker GFAP in striatum or any subregion of the hippocampus (dentate gyrus, CA1 or CA3) [39]. MDPV was not found to elicit GFAP increases when administered in a binge regimen of 4 doses of 30 mg/kg every 2 h in mice [19].
None of the β-ketoamphetamines mephedrone, methylone, or MDPV in combination with each other resulted in changes in the levels of GFAP in the striatum of mice [19]. However, methylone was able to enhance by approximately 50% the expression of GFAP induced by administration of methamphetamine [19]. MDPV, on the contrary, prevented the striatal increases in GFAP observed after administration of methamphetamine and MDMA [19].
2 Thermoregulation
A common adverse effect of non-ketoamphetamines such as methamphetamine [52], amphetamine, and MDMA [53] is an increase in body temperature. This hyperthermia is dependent on the frequency of exposure, dosing, age, ambient temperature, and route of exposure [52]. MDMA and methamphetamine produce hyperthermia following acute and repeated exposure at ambient temperature and elevated ambient temperature [14, 54–58]. Hyperthermia is an important factor known to exacerbate the deleterious effects of amphetamine-type drugs. Studies of the effects of β-ketoamphetamines on thermoregulation are summarized in Table 1.
A single dose of 10 mg/kg of methcathinone caused a sustained increase in rectal temperature that was not accompanied by any significant concomitant change in tail temperature in individually housed rats [7]. Similarly, an acute intoxication with a methcathinone infusion (5 mg/kg/min; 100 mg/mL) caused hyperthermia in rats [6]. By contrast, acute exposure to mephedrone produces hypothermia in rats [7, 59]. This hypothermic response at ambient and elevated temperatures is rat-strain specific, with the reduction in body temperature detected in Wistar but not Sprague-Dawley rats [27]. Alpha-1 adrenoreceptor and dopamine D1 receptor blockade seem to enhance the hypothermic response induced by mephedrone [7]. On the other hand, when mephedrone is administered repeatedly in a binge paradigm, it produces hyperthermia in both mice [17, 19, 25] and rats [24]. These studies indicate that mephedrone differs from MDMA and methamphetamine in its thermoregulatory effects despite their neuropharmacological similarity. Lopez-Arnau and colleagues showed that the effect of mephedrone changes with the dose using a 2-day binge paradigm of 3 doses of 25 mg/kg a day every 2 h [20]. On day 1, after receiving the first dose of mephedrone, the treated animals showed a significant transient reduction in body temperature; after the second dose, temperature increased over the saline values but this hyperthermic response was significant only after the third dose. On day 2, no significant hyperthermic response was evidenced. Consistent with that report, other studies have suggested that the mephedrone-induced hypothermia is attenuated with repeated dosing and that this response can be attenuated by 6-hydroxydopamine or abolished by 5,7-dihydroxytryptamine [26]. Taken together, mephedrone produces hypothermia following acute exposure, while producing hyperthermia following binge models of dosing [60].
Studies of self-administration of mephedrone in rats evidenced that its thermoregulatory effects also differed between rat strains, with the Sprague-Dawley rats being most sensitive. While in Sprague-Dawley rats the administration of mephedrone produced a dose-dependently decreased body temperature, in Wistar rats, the response was biphasic, starting with a decrease during the first 15 min, followed by an increase during the next 25–30 min [61]. Another study with these two strains of rats monitored the effects of the subcutaneous administration of mephedrone under conditions of low (23°C) and high (27°C) ambient temperature. A reliable reduction of body temperature was produced by mephedrone in Wistar rats at low and high temperatures with only minimal effect in Sprague-Dawley rats. Furthermore, hypothermia produced by serotonin (5-HT) 1A/7 receptor agonists was similar in each strain [27].
Similar to mephedrone, methylone produces hyperthermia following binge dosing [60]. In mice, 4 injections of methylone (20 mg/kg) every 3 h produced a robust hyperthermic response that reached a peak between 25 and 35 min after each drug administration. This effect of methylone increased significantly with the dose, so that the last dose induced a greater increase in body temperature than the first one [38]. In rats, methylone treatment (3 injections at 3 and 10 mg/kg every 2 h) caused significant hyperthermia from 2 h through 6 h post-injection [24]. The thermoregulatory effects caused by a single administration of methylone did not differ from the outcomes observed after repeated administration of the drug. Piao and colleagues evaluated the acute effects of methylone using 5-HT transporter (SERT) and dopamine (DA) transporter (DAT) knockout (KO) mice and observed a slight diminution in the hyperthermic effects of methylone in DAT KO mice, whereas a slight enhancement of these effects was seen in SERT KO mice. Administration of selective D1 and D2 receptor antagonists reduced methylone-induced hyperthermia, but these drugs also had hypothermic effects of their own in saline-treated mice, which complicates interpretation of the findings [41].
MDPV exhibits a few important differences in altering body temperature by comparison to other β-ketoamphetamines. Acute exposure to MDPV produces hyperthermia [47] at elevated temperatures but not at normal ambient temperatures, which contrasts with what is observed for MDMA and methamphetamine [60]. MDPV has been shown to elevate body temperature in some cases of human medical emergency and fatal overdose deaths [62]. In rats, treatment with 1.0, 5.6, and 10.0 mg/kg of MDPV elicited a significant hypothermia when compared with the vehicle condition [44]. The effects were dose dependent and lasted up to 3 h after dosing [44]. However, an unexpected lack of dose dependence was characterized when MDPV was administered in the 20°C ambient environment. At that cool ambient temperature, MDPV doses from 1 to 30 mg/kg each induced a rise in core temperature of approximately 1.5°C, which was not different from that observed following saline administration, and the time course of this effect was also similar across all tested doses [46]. Besides affecting body temperature, MDPV has been found to induce brain hyperthermia through an increase in peripheral vasoconstriction [40]. Furthermore, an age-dependent effect of MDPV on thermoregulation was documented in rats. While adolescent rats increased their body temperature following MDPV administration, adults showed a decrease in temperature [48].
The β-ketoamphetamines mephedrone, methylone, and MDPV differentially affect the temperature effects of their non-β-ketoamphetamine counterparts [19]. In vivo studies demonstrated that the mephedrone-induced hyperthermia is not enhanced by concomitant administration of methamphetamine [23]. When given in two-drug combinations, mephedrone, methylone and MDPV caused significant increases in body temperature [19]. When methylone or MDPV are given with mephedrone, the initial hypothermic effect of mephedrone was retained and slightly exaggerated. Combined treatment with MDPV and methylone results in a steady 1–2°C increase in core body temperature that becomes evident within 15 min of treatment and persists for at least 8–9 h [19]. Neither MDPV nor methylone attenuated the hyperthermic effects of methamphetamine in mice [19].
3 Neurotransmitters
Alterations in monoaminergic systems are one of the hallmarks of the most studied non-keto-amphetamines. Methamphetamine is perhaps best known for its toxic effects on DA nerve terminals of the striatum [63, 64]. MDMA has also been shown to cause long-term deficits in DA and 5-HT nerve ending in both laboratory animals and humans [65–67]. Even amphetamine has been linked to nerve terminal damage [66]. Amphetamine neurotoxicity manifests as long-term depletion of DA and 5-HT, inhibition of their biosynthetic enzymes tyrosine hydroxylase (TH) and tryptophan hydroxylase 2 (TPH-2), inactivation of DAT and SERT, reduction in function of the vesicle monoamine transporter (VMAT), degeneration of fine, unmyelinated axons, and apoptosis [3]. Reductions in DA, TH, and DAT have been documented in the postmortem striatum of chronic methamphetamine users [68]. Studies of the effects of β-ketoamphetamines on neurotransmitter systems are summarized in Table 1.
Methcathinone is a potent releaser of DA but not 5-HT, similar to amphetamine and methamphetamine [15]. Methcathinone has previously been shown to release radiolabeled DA [16] and 5-HT from rat brain preparations with similar DA versus 5-HT selectivity to amphetamine and methamphetamine, but with two- to threefold lower potency [15]. Multiple administrations of methcathinone caused persistent deficits in monoaminergic systems [11], reflected by decreases in DA and 5-HT uptake capacity, tissue content and associated TH and TPH-2 activities in frontal cortex, hippocampus, and neostriatum after four doses of 30 mg/kg in rats [8, 9]. However, the effects of this drug seemed to be more accentuated in rats compared to mice. While in mice methcathinone produced long lasting depletions of striatal DA, in rats it caused significant depletions of both DA and 5-HT [11]. A single high-dose administration of methcathinone increased striatal DA release, as measured by microdialysis in conscious rats [8]. Methcathinone was also found to increase the metabolites homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA) levels in striatum [7].
Mephedrone alone did not cause persistent reductions in the levels of DA, 5-HT, or TPH-2 [18, 19, 22, 28, 69] aside from a small decrease in the DA metabolite HVA in the mouse striatum [28]. Mephedrone is considered a more potent releaser of DA than MDMA [15, 70]. An in vivo microdialysis study in rats showed that mephedrone produced a rapid and pronounced increase in DA levels in the nucleus accumbens that was comparable with amphetamine and greater than MDMA, which only elevates DA levels moderately [71]. While both mephedrone and MDMA also produced strong increases in extracellular 5-HT, amphetamine had only a moderate effect on 5-HT levels [71]. Self-administration of mephedrone was shown to decrease the levels of striatal 5-HIAA in rats [21]. Mephedrone administered in a binge of 3 doses of 10 mg/kg every 2 h showed that the extracellular increase in striatal DA seen after the first mephedrone injection was similar in magnitude and time course to those following the second and third injections. However, the extracellular overflow of striatal 5-HT was more variable but was enhanced when second and third injections were given when compared with the first response [26]. Other microdialysis studies in the rat nucleus accumbens showed that mephedrone elevated extracellular DA and 5-HT levels, with relatively higher effects on 5-HT levels [24, 27], similar to MDMA and unlike methamphetamine, which preferentially increases DA [24]. Thus, mephedrone shares some of the DA-releasing properties of amphetamine and methamphetamine and the 5-HT-releasing property of MDMA [15]. Repeated administration of mephedrone in rats showed no significant effect on tissue concentrations of DA, 5-HT, or their metabolites in the striatum or frontal cortex and hippocampus 7 days post-administration although the concentration of DOPAC was significantly increased in this region following mephedrone [29]. The same study further evaluated the acute effects of mephedrone in comparison with MDMA, and reported reductions in hippocampal 5-HT and 5-HIAA 2 h after a single injection of MDMA but not following acute mephedrone [29]. The expression of TH, a biochemical marker of neuronal integrity in DA neurons, was found to be decreased in frontal cortex but not in the striatum after a binge regimen of 25 mg/kg of mephedrone [20]. The overall effects of mephedrone do not involve long-lasting depletions of DA but they seem to affect 5-HT. Repeated administration of mephedrone in rats caused persistent decreases in hippocampal 5-HT levels but no changes were observed in striatal DA after 7 days of treatment [70]. Another study reported decreases in TH and TPH-2 after a binge of mephedrone for 2 consecutive days [25]. The fact that mephedrone has DA-releasing capability resembling methamphetamine and yet does not cause DA deficits is of significant interest for studying the differential mechanisms of damage induced by stimulants.
Methylone administration does not result in damage to DA nerve endings in mice [19]. Binge administration of methylone to single-housed rats (3 or 10 mg/kg, 3 doses) has no long-lasting effects on brain tissue monoamines [24] but produced significant elevations on extracellular levels of DA and 5-HT [24, 34, 42]. There seem to be species differences in the sensitivity to long-term neurochemical effects of methylone [28]. The effects of drug treatments on mouse brain monoamine levels in the frontal cortex, striatum, and hippocampus indicate that methylone did not cause any significant changes in neurotransmitter levels. However, in the rat brain methylone had a profound impact on 5-HT levels, causing a decrease in 5-HT levels in the frontal cortex, striatum, and hippocampus comparable to that induced by amphetamine. Additionally, 5-HIAA levels were reduced in the striatum and hippocampus [28].
In vivo microdialysis studies indicate that MDPV increases extracellular concentrations of DA in the nucleus accumbens in a dose-related manner similar to cocaine. However, MDPV was tenfold more potent than cocaine in its ability to increase extracellular dopamine [49]. This robust stimulation of DA transmission by MDPV predicts serious potential for abuse [49]. Despite the high potency to block the DAT, MDPV did not produce DA efflux. Thus, this cathinone is thought to be a pure transporter uptake inhibitor [15].
Studies in mice indicate that combined treatment with mephedrone and methamphetamine or MDMA did not change the status of 5-HT nerve endings to an extent that was different from either drug alone [18]. Methamphetamine and MDMA alone caused mild reductions in 5-HT but did not change SERT and TPH2 levels [23]. While mephedrone did not produce changes in the 5-HT system, it enhanced the DA and TH depletions induced by methamphetamine, amphetamine, and MDMA in striatum [23].
In mice, none of the β-ketoamphetamines mephedrone, methylone, or MDPV in combination with each other resulted in changes in striatal DA or TH, but mephedrone and methylone potentiated the depletions of DA and TH induced by administration of methamphetamine [19, 23]. On the other hand, MDPV was able to prevent the striatal decreases in DA and TH observed after administering methamphetamine, MDMA, and MPTP [19]. Consistent with this study, in vitro data showed that MDPV blocked methamphetamine-induced DA release with high potency reflecting its elevated efficiency as an uptake inhibitor. The finding suggests that the more potently a drug antagonizes the DA release produced by methamphetamine, the more potently it also blocks DA uptake [51].
4 Biochemical Mechanisms: Oxidative Stress and Cytotoxicity
Evidence indicates that reactive oxygen species (ROS) are responsible for amphetamine-related damage but neither the manner by which these drugs cause oxidative stress nor the cellular source of the reactant species is known [4, 68]. Oxidative stress is believed to be a prominent factor in methamphetamine-induced cellular toxicity [72]. By increasing DA release, amphetamines increase the available DA for oxidation and its metabolism into ROS [73]. Methamphetamine’s ability to flood the intracellular medium with DA is thought to be the first step in a cascade that leads to mitochondrial dysfunction, enhanced excitatory neurotransmission, increases in oxidative stress, nerve ending damage, and apoptosis [74]. Similar to the other amphetamines, metabolism of MDMA also results in the formation of ROS, which ultimately induce long-term neurotoxic effects [2]. However, none of the β-ketoamphetamines (methcathinone, mephedrone, methylone, and MDPV) showed cytotoxicity at the highest concentrations tested in functional assays [36]. Studies of the effects of β-ketoamphetamines on oxidative stress and cytotoxicity are summarized in Table 1.
Methcathinone is manufactured by a clandestine process that involves potassium permanganate oxidation of ephedrine and pseudoephedrine contained in readily available pharmaceuticals. Intravenous injections of such methcathinone preparations expose users to a high manganese load because the resultant methcathinone is not purified [75]. Although studies of methcathinone abusers have described movement disorders similar to Parkinson’s disease attributed to the manganese toxicity, the syndrome lacks typical features of this condition such as resting tremor and gait initiation failure [75]. The accumulation of manganese can lead to the development of encephalopathy and might trigger secondary pathogenic mechanisms, such as mitochondrial dysfunction and oxidative stress [76].
Animal studies showed that mephedrone induced an increase in the expression of the antioxidant enzymes superoxide dismutase, catalase, and glutathione peroxidase in the hippocampus, striatum, and frontal cortex in rats. Along with these enzyme protein increases, treatment with mephedrone caused a significant increase in the levels of lipid peroxidation in the frontal cortex [20]. In mice, a single injection of mephedrone at 2.5 mg/kg caused both an increase in lipid peroxidation levels and a decrease of catalase activity in the hippocampus and prefrontal cortex [37] whereas at a dose of 25 mg/kg, mephedrone induced a significant increase of glutathione peroxidase in striatum [30].
The exposure of mouse cultured cortical cells to various concentrations of mephedrone for 24 h or 48 h caused a concentration-dependent decrease in metabolically active cells. The calculated LD50 value for mephedrone after 24 h of incubation was significantly higher to that obtained after 48 h of drug exposure [25]. In addition, neuroblastoma cells exposed to mephedrone showed an increase in cytotoxic damage only at high concentrations [31]. Cell culture studies documented the cytotoxicity of methylone and methamphetamine in CHO cells stably expressing the rat transporters DAT, NET, and SERT. Methylone was not cytotoxic to any cell line except that expressing the SERT [43], indicating higher specificity for the 5-HT system.
The cytotoxic effects of MDPV have only been evaluated in a developing brain mouse model, where a single administration of the drug caused a prominent increase in the number of apoptotic cells in the piriform cortex, retrosplenial area, hippocampus CA1, and nucleus accumbens, without an overall change in the density of cells. The neurons of the nucleus accumbens appeared to be especially sensitive to MDPV as they showed an increase of apoptotic cells in the core and shell regions of the accumbens. However, this effect of MDPV was not observed in the brain of adult mice [45]. While methylone alone was reported to be cytotoxic in cell lines expressing the rat SERT, the combination of methamphetamine with methylone caused a significant increase in the toxicity in the cells stably expressing the rat monoamine transporters DAT, NET, and SERT but not in the control CHO cells [43].
5 Neurotransmitter Transporters and Receptors
The non-keto-amphetamines including amphetamine, methamphetamine, and MDMA cause neurotoxic effects to monoaminergic systems in part through alterations in DA and 5-HT transporters and receptors [66]. Methamphetamine causes acute increases in both DA and 5-HT release that result from the direct and indirect actions on the DAT and SERT. Amphetamines can disrupt the vesicle proton gradient to cause an increase in cytoplasmic DA and 5-HT from vesicular compartments by altering the function of the vesicular monoamine transporter-2 (VMAT-2). Both methamphetamine and MDMA also increase 5-HT release through similar transporter mediated mechanisms, though MDMA has a preferential affinity for SERT over DAT and consequently more pronounced effects on the 5-HT system [74]. Studies of the effects of β-ketoamphetamines on neurotransmitter transporters and receptors are summarized in Table 1.
In animals, repeated administration of methcathinone was shown to reduce the content of DA and 5-HT, the number of transporter sites, as well as the activity of TPH-2 and TH [9]. In humans, persistent reductions of DAT density have been reported using PET in methcathinone users and are suggestive of loss of DAT or loss of DA terminals [10, 77]. Methcathinone exhibited a monoamine transporter inhibition profile that was very similar to that of the non-keto analogs amphetamine and methamphetamine, with high inhibitory potencies at the DAT and low potencies at the SERT [13, 15, 78]. It is believed that the deficits in DAT and SERT produced by methcathinone may reflect potential long-term damage to DA and 5-HT neurons. Nevertheless, to become evident, these neural deficits require massive, multiple doses of methcathinone over several days. Such doses are 10 to 100 times higher than behaviorally active doses [12]. Deficits in DA function induced by methcathinone were prevented by pretreatment with dopamine D1 or D2 receptor antagonists, whereas 5-HT changes were prevented with a depletion of striatal DA by lesioning with 6-hydroxydopamine [8]. Apparently the serotonergic neurotoxicity of methcathinone is promoted by the presence of the N-methyl group on the drug molecule as it was earlier reported that no long-term changes in 5-HT levels were observed with repeated high doses of the desmethyl parent compound, cathinone [12]. In a cell study, methcathinone was less efficacious in releasing preloaded radiolabeled neurotransmitter via VMAT-2 than methamphetamine [33]. Methcathinone also exhibited low μM potency at 5-HT2A receptor binding [15].
Mephedrone administration alone did not cause persistent reductions in the levels of SERT [18]. Uptake inhibition studies using rat synaptosomes found that mephedrone potently inhibits DAT and SERT [35, 70], and the drug is a substrate for DAT, SERT, and the norepinephrine transporter (NET) [24]. Similar to methcathinone, mephedrone can bind to 5-HT2A receptors and stimulation of these receptors has also been shown to enhance DA release potentially increasing abuse liability [79]. Mephedrone and methcathinone also exhibited affinity for α1A adrenoceptors, which have been implicated in stimulant-induced vasoconstriction, hyperthermia, and euphoria [15] and methcathinone has been found to be a low potency partial agonist at the 5-HT1A receptors [33].
In mice, repeated administration of mephedrone induced a significant loss in DA and 5-HT reuptake sites in striatum, hippocampus, and frontal cortex [25]. In addition, mephedrone decreased the number of D2 receptors in striatum and the number of 5-HT2A receptors in frontal cortex and hippocampus of treated mice [25]. In adolescent mice, mephedrone elicited an increase in D3 receptors in the striatum [30]. In rats, a binge of mephedrone induced a significant loss in DAT in frontal cortex and a decrease in the density of SERT in striatum, cortex, and hippocampus. This effect was accompanied by decreased TPH-2 expression in all the three brain areas and a moderate decrease in the concentration of D2 receptors in the striatum [20]. The effects of mephedrone on the human monoamine transporters were studied using cell lines stably expressing the human NET, DAT, and SERT. These data indicate that mephedrone and MDMA were equally potent in inhibiting noradrenaline uptake at NET. Compared to their NET inhibition potency, both drugs were weaker uptake inhibitors at DAT and SERT, with mephedrone being more potent than MDMA at DAT and less potent than MDMA at SERT. Nonetheless, mephedrone and MDMA differed most in their inhibition of DA uptake by synaptic vesicles isolated from human striatum, with MDMA being tenfold more potent than mephedrone, and their ability to release DA from human VMAT expressing cells [80]. In general, the in vitro releasing capabilities of mephedrone resemble those of MDMA. With regard to selectivity ratios, mephedrone displayed NET/DAT ratios and DAT/SERT ratios close to unity, similar to MDMA [24]. Interestingly, a recent report suggests that the para ring-substitution of the methyl group in mephedrone left-shifted the SERT inhibition curves over the DAT inhibition curves (DAT:SERT inhibition ratios <1), resulting in monoamine transporter inhibition profiles that were more similar to MDMA and less similar to the parent compound methcathinone [36]. Similarly, in vitro and in vivo studies have shown that methcathinone para ring-substituted analogs increase monoamine release via SERT relative to DAT, and that this shift in selectivity markedly reduces the abuse-related effects of the drugs as assessed by intracranial self-stimulation [81, 82].
Methylone was reported to act somewhat more potently in inhibiting DAT than SERT at the human transporter [15], but equally potent for DAT and SERT inhibition in rat synaptosomes [83]. Methylone was a substrate for NET and DAT, with slightly lower potency at SERT, displaying a selectivity profile similar to mephedrone but about half as potent. In general, the in vitro releasing capabilities of methylone resembled those of MDMA. With regard to selectivity ratios, methylone displayed NET/DAT ratios and DAT/SERT ratios close to unity, similar to MDMA [24]. In cells expressing the VMAT-2, methylone elicited less than 35% of methamphetamine maximal efficacy to stimulate release of neurotransmitter via the VMAT-2 [33]. Similar to mephedrone, methylone is also a low potency partial agonist at the 5-HT1A receptors, and an antagonist with very low potency at the 5-HT2A receptor [33]. While methamphetamine and MDMA are likely to be substrates for VMAT-2, methcathinone and methylone are not. Therefore, the behavioral effects of methcathinone and methylone arise largely from the drugs’ effects at the plasma membrane transporters, not VMAT-2. In summary, due to the large decrease in potency at VMAT-2, methcathinone and methylone are highly selective for the plasma membrane catecholamine transporters and moderately selective for SERT. As a result of its greater potency at the SERT, methylone is somewhat less discriminating than methcathinone at the plasma membrane [13].
MDPV exhibited very high affinity for the DAT and NET in the low nanomolar range (<10 nM) in vitro, consistent with its high potency as a DAT and NET inhibitor [15, 33, 49]. MDPV exhibited the most potent DAT inhibition [15], being at least tenfold more potent than cocaine and methamphetamine [15]. In contrast, MDPV is a weak inhibitor of the SERT, resulting in high DAT selectivity, with DAT/SERT inhibition ratios >100. MDPV is also one of the most potent NET inhibitors [15]. Studies using fast-scan cyclic voltammetry in mouse striatal slices indicate that MDPV is more potent than cocaine at inhibiting DA clearance [49]. In contrast to mephedrone, MDPV is a very potent NET and DAT inhibitor with very low 5-HT activity, reflected by high DAT:SERT inhibition ratios. The 3,4-methylenedioxy ring substitution that is shared by MDMA and MDPV would be predicted to increase serotonergic activity compared with the non-substituted compound methamphetamine. However, in the case of MDPV, the SERT inhibition potency is very low despite the presence of this substitution [36]. In this regard, data have shown that the carbonyl and the extended alpha alkyl groups in MDPV have greater contributions to this drug’s affinity for DAT than the methylenedioxy group [84]. In addition, in vitro findings revealed that the presence of a pyrrolidine ring in any cathinone-like compound such as MDPV confers potent blocking properties at DAT and NET [85].
An examination of methylone’s ability to influence the reverse transport of substrates through DAT, NET, and SERT was done in comparison with methamphetamine, since unlike cocaine, methamphetamine induces the release of monoamines via a reversal of transport. Similar to methamphetamine, methylone elicited the release of radiolabeled DA, NE, and 5-HT from CHO cells expressing the rat DAT, NET, and SERT. In addition, the combination of methylone with methamphetamine did not cause a further increase in the release of substrates [43]. None of the β-ketoamphetamines mephedrone, methylone, or MDPV in combination with each other resulted in changes in striatal DAT [19]. In combination with methamphetamine, mephedrone and methylone enhanced the reductions in DAT observed in mouse striatum [19, 23]. In contrast, administration of MDPV prevented the depletions in DAT observed after methamphetamine, amphetamine, MDMA, and MPTP [19].
6 Transendothelial Blood–Brain Barrier Dysfunction
The rate at which drugs reach the brain parenchyma depends not only on their route of administration but also on their ability to cross the cerebral endothelium, also called the blood–brain barrier (BBB), which constitutes the main brain interface modulating the exchange of compounds between the brain and blood [50]. Alterations in BBB function are likely involved in drug abuse neurotoxicity [1, 86]. Both MDMA and METH have been shown to produce disruption of the BBB as reflected by IgG extravasation and Evans Blue leakage [5, 87]. In fact, it was previously shown that METH compromises BBB function and its capacity to protect the brain against infection by the human immunodeficiency virus [88]. Studies of the effects of β-ketoamphetamines on BBB dysfunction are summarized in Table 1.
Methcathinone exhibited a brain permeability ratio ≥3, indicating high permeability. However, the apical to basolateral transport of methcathinone was not consistent with active transport by one of the blood-to-brain influx carriers [15]. No studies on the compromise of the BBB by methcathinone have been reported to date. The permeability ratio for mephedrone was >10, suggesting very high BBB permeability [15], and confirming that mephedrone readily enters the brain [15, 70]. Although highly permeable into the brain, mephedrone administration has not been linked to any BBB dysfunction. It is well recognized that compounds with a brain/plasma concentration ratio greater than 1 freely cross the blood–brain barrier and the obtained brain/plasma ratio for methylone of 1.42 demonstrates access to central nervous system [89]. As a reference, methamphetamine, amphetamine, and MDMA brain/plasma ratios are >3 [15].
Similar to mephedrone, the permeability ratio for MDPV was >10, suggesting a very high permeability [15]. The apical to basolateral transport of MDPV was significantly greater than basolateral to apical transport, consistent with active transport by one of the blood-to-brain influx carriers [15]. MDPV is a monoamine uptake inhibitor that is more lipophilic and potent than other cathinone derivatives. The high lipophilicity of this substance is caused by the pyrrolidine ring and the tertiary amino group creating a less polar molecule more able to cross the blood–brain barrier [90]. No combinations of the β-ketoamphetamines mephedrone, methylone, or MDPV with each other or any other amphetamine compound have been evaluated on BBB dysfunction to date.
7 Mechanisms of Action
Drugs that target monoamine transporters can be classified generally as either substrates, such as methamphetamine, or blockers, such as cocaine [83]. Both types of compounds elicit profound psychostimulant effects that render them liable for recreational abuse [91]. Substrates or blockers increase monoamine neurotransmitter concentrations in the synaptic cleft but this action can be the result of at least two distinct mechanisms [13]. One mechanism is through drug inhibition of plasma membrane transporter-mediated uptake of released neurotransmitters (i.e., for transporter blockers). The inhibition is believed to arise from competition by drugs for substrate binding sites in the monoamine uptake transporters, thereby reducing the effectiveness with which DA, 5-HT, and NE are cleared from the synapse following release. Typical DAT blockers are expected to fully inhibit DA uptake and to fully inhibit binding of another blocker, as well as release of substrate by reverse transport [92]. A second mechanism is through the drug-evoked release of the monoamine neurotransmitters, apparently by transporter-mediated exchange (i.e., for transporter substrates). The drug-evoked neurotransmitter release arises from two intracellular compartments. Methamphetamine and MDMA induce the release of newly synthesized, cytosolic pools of monoamines and also release monoamines from synaptic vesicle stores [13]. Typical DAT releasers are expected to fully release another substrate accumulated in cell or synaptosomes [92]. This mechanistic distinction is important to consider because transporter substrates and blockers display critical differences in their acute and long-term effects. Only substrates are translocated into cells where they could disrupt vesicular storage and stimulate non-exocytotic release of neurotransmitters by reversing the normal direction of transporter flux, and could produce persistent deficits in monoamine neurons, including depletion of neurotransmitters and loss of functional transporters [83]. The flux-coupled channel model suggests that whereas some cathinones, such as mephedrone, behave as DA-releasing agents (depolarizing current), some others such as MDPV act as DA-reuptake inhibitors (hyperpolarizing current) [93]. An “excitatory substrate” implies that in addition to the proposed transporter-mediated chemical effects of methamphetamine, mephedrone, and related cathinones, these substances have a depolarizing action that could itself promote exocytotic neurotransmitter release [32]. Structurally analogous MDPV, however, induces an outward hyperpolarizing current under similar conditions and therefore acting as an “inhibitory,” non-substrate blocker [93]. Results from release assays reveal that mephedrone and methylone function as substrates at monoamine transporters [33], thereby stimulating the release of radiolabeled substrates via DAT, NET, and SERT [83]. Mephedrone, methylone, and MDMA are non-selective transporter substrates (i.e., non-selective releasers), while methcathinone and amphetamine are selective substrates at DAT and NET. Mephedrone displays similar releasing potency at all three transporters and is about twice as potent as methylone [83]. While mephedrone, methylone, MDMA, and amphetamine are fully efficacious in the release assays, MDPV and cocaine are inactive as releasers [15]. MDPV displays a novel pharmacological profile when compared to other synthetic cathinones as it is a potent uptake blocker at DAT and NET with no measurable substrate activity [83]. When compared to the prototypical transporter blocker cocaine, MDPV was 50-fold more potent at DAT, 10-fold more potent at NET, and 10-fold less potent at SERT [94]. Taken together, the in vitro results indicate that mephedrone and methylone are non-selective transporter substrates, whereas MDPV is a pure catecholamine-selective transporter blocker [94]. This dichotomy of interaction with the DAT by mephedrone and methylone on one hand and by MDPV on the other can explain their opposing effects on methamphetamine-induced neurotoxicity [19]. While mephedrone and methylone enhanced the neurotoxic effects of methamphetamine, MDPV protected. It has previously shown that treatments resulting in an increase in the releasable pool of DA significantly accentuate methamphetamine-induced damage in DA nerve endings [95]. MDPV has an effect that is similar to more classical DAT blockers, including amphonelic acid and nomifensine, which also provide protection against methamphetamine-induced neurotoxicity [19]. By blocking DAT-mediated transport (inward or outward), MDPV blocks methamphetamine-induced efflux of DA [15]. Therefore, these properties as substrates or blockers represent an important mechanism by which synthetic cathinones influence the synaptic levels of monoamines but they do not explain why they lack neurotoxic properties on their own or how they enhance the neurotoxicity of the amphetamines.
While the principal targets of amphetamines are plasmalemmal transporters, these drugs have concerted actions on other two important elements of the monoamine nerve ending: vesicular transporters and the degrading enzymes monoamine oxidase (MAO) and catechol-O-methyl transferase [91], both of which may contribute to their toxic properties. Amphetamine interactions with these three targets are the core tenet of the so-called weak base hypothesis [96]. Amphetamines enter nerve terminals via plasmalemmal transporters and disrupt vesicular storage as weak bases by dissipating the proton gradient across the membrane [96]. A reduction of the vesicular pH gradient promotes the reverse transport of DA into the cytosol. DA is then released into the synaptic space via reverse transport through the DAT. This flooding of the cytoplasm and synaptic space with the oxidatively labile DA is thought to be a critical first step in the neurotoxic cascade of the amphetamines [73]. These conditions of elevated concentrations of cytosolic monoamines could be further aggravated by inhibition of MAO [91]. Unlike amphetamines, mephedrone and methylone have little if any affinity for VMAT-2 [33]. Therefore, their lack of neurotoxicity could derive from an inability to promote the release of DA from storage vesicles into the cytoplasm.
If not toxic on their own, how can mephedrone and methylone increase the neurotoxicity of methamphetamine, MDMA, and amphetamine? We hypothesize that the enhancement of neurotoxicity elicited by the combination of methamphetamine plus either mephedrone or methylone could be explained by a reversal of greater numbers of DAT molecules than caused by either drug alone, resulting in heightened DA efflux into the cytoplasm (i.e., via methamphetamine actions on the VMAT) and synapse (i.e., via combined methamphetamine plus mephedrone actions on the DAT). This possibility is supported by the observation that amphetamine-induced DA release is greater when originating from both synaptic vesicles and cytoplasmic stores than from cytoplasmic stores only [97]. In addition, a possible inhibition of MAO could be speculated for mephedrone and methylone as it has been shown that MAO inhibitors increase significantly the DA depletion induced by methamphetamine [95]. However, a well-established mechanism to explain why some β-ketoamphetamines such as mephedrone and methylone are not neurotoxic on their own but are capable of potentiating the damage induced by amphetamines remains to be elucidated.
8 Conclusion
As β-keto analogs of amphetamines, synthetic cathinones may be expected to have amphetamine-like effects because of their structural similarity. However, β-ketoamphetamines are a diverse class of chemical compounds with differential neurotoxic properties on monoaminergic neurons. Some of the benchmarks used to gauge the neurotoxicity induced by amphetamines include inflammation, disruption of monoaminergic neurotransmitters, their transporters and receptors, alterations in thermoregulation, oxidative stress, and cytotoxicity. Compared to the effects induced by amphetamines on these parameters, the effects described for β-ketoamphetamines seem to be more moderate. Administration of synthetic cathinones is not consistently associated with long-term depletions in the levels of DA and 5-HT or with inhibition of these neurotransmitters biosynthetic enzymes. While hyperthermia has been established as one of the hallmark effects of amphetamines, synthetic cathinones elicit more complex responses that involve hypothermia and oscillations between hyper and hypothermia. Neuroinflammation markers such as microglial activation have not been documented after administration of synthetic cathinones and reports of increases in GFAP have been sparse. The evaluation of the effects of these cathinones on oxidative stress and cytotoxicity are limited and mostly circumscribed to in vitro studies, where concentrations are very high. Nonetheless, some studies in animals have described increases in lipid peroxidation and in the expression of antioxidant enzymes after mephedrone. Deficits in DAT and SERT were only observed after multiple doses that are several times higher than behaviorally active doses or with exacerbation of other factors such as high ambient temperature. Although only a few studies have reported the neurotoxic effects of β-ketoamphetamines alone or in combination with other drugs of the same group or with amphetamines, the overall outcome appears to be associated with their interaction with the vesicular and plasma transporters. The fact that mephedrone and methylone cause little or no toxicity themselves on the one hand, while being capable of enhancing amphetamines toxicity on the other hand, remains a provocative and open question that requires additional research. The role of these synthetic cathinones as weak bases to collapse the vesicular pH gradient necessary for monoamine storage, their capacity to increase the releasable pool of cytosolic monoamines, their potential to inhibit monoamine degrading enzymes, their ability to increase monoamine oxidation and metabolism into ROS, as well as their additive effects in recruiting DAT molecules along with amphetamines to enhance the DA efflux into the synapse, constitute some of the possible manners in which these β-ketoamphetamines may heighten the neurotoxicity induced by amphetamines.
References
Yamamoto BK, Moszczynska A, Gudelsky GA (2010) Amphetamine toxicities: classical and emerging mechanisms. Ann N Y Acad Sci 1187:101–121
Moratalla R, Khairnar A, Simola N, Granado N, Garcia-Montes JR, Porceddu PF, Tizabi Y, Costa G, Morelli M (2015) Amphetamine-related drugs neurotoxicity in humans and in experimental animals: main mechanisms. Prog Neurobiol
Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM (2004) Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther 311:1–7
Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM (2004) Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett 367:349–354
Rubio-Araiz A, Perez-Hernandez M, Urrutia A, Porcu F, Borcel E, Gutierrez-Lopez MD, O’Shea E, Colado MI (2014) 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy) disrupts blood-brain barrier integrity through a mechanism involving P2X7 receptors. Int J Neuropsychopharmacol 17:1243–1255
Rockhold RW, Carlton FB Jr, Corkern R, Derouen L, Bennett JG, Hume AS (1997) Methcathinone intoxication in the rat: abrogation by dextrorphan. Ann Emerg Med 29:383–391
Shortall SE, Green AR, Swift KM, Fone KC, King MV (2013) Differential effects of cathinone compounds and MDMA on body temperature in the rat, and pharmacological characterization of mephedrone-induced hypothermia. Br J Pharmacol 168:966–977
Gygi MP, Fleckenstein AE, Gibb JW, Hanson GR (1997) Role of endogenous dopamine in the neurochemical deficits induced by methcathinone. J Pharmacol Exp Ther 283:1350–1355
Gygi MP, Gibb JW, Hanson GR (1996) Methcathinone: an initial study of its effects on monoaminergic systems. J Pharmacol Exp Ther 276:1066–1072
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA (1998) Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 18:8417–8422
Sparago M, Wlos J, Yuan J, Hatzidimitriou G, Tolliver J, Dal Cason TA, Katz J, Ricaurte G (1996) Neurotoxic and pharmacologic studies on enantiomers of the N-methylated analog of cathinone (methcathinone): a new drug of abuse. J Pharmacol Exp Ther 279:1043–1052
Cozzi NV, Foley KF (2003) Methcathinone is a substrate for the serotonin uptake transporter. Pharmacol Toxicol 93:219–225
Cozzi NV, Sievert MK, Shulgin AT, Jacob P 3rd, Ruoho AE (1999) Inhibition of plasma membrane monoamine transporters by beta-ketoamphetamines. Eur J Pharmacol 381:63–69
Metzger RR, Haughey HM, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE (2000) Methamphetamine-induced rapid decrease in dopamine transporter function: role of dopamine and hyperthermia. J Pharmacol Exp Ther 295:1077–1085
Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME (2013) Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168:458–470
Glennon RA, Yousif M, Naiman N, Kalix P (1987) Methcathinone: a new and potent amphetamine-like agent. Pharmacol Biochem Behav 26:547–551
Angoa-Perez M, Kane MJ, Francescutti DM, Sykes KE, Shah MM, Mohammed AM, Thomas DM, Kuhn DM (2012) Mephedrone, an abused psychoactive component of ‘bath salts’ and methamphetamine congener, does not cause neurotoxicity to dopamine nerve endings of the striatum. J Neurochem 120:1097–1107
Angoa-Perez M, Kane MJ, Herrera-Mundo N, Francescutti DM, Kuhn DM (2014) Effects of combined treatment with mephedrone and methamphetamine or 3,4-methylenedioxymethamphetamine on serotonin nerve endings of the hippocampus. Life Sci 97:31–36
Anneken JH, Angoa-Perez M, Kuhn DM (2015) 3,4-Methylenedioxypyrovalerone prevents while methylone enhances methamphetamine-induced damage to dopamine nerve endings: beta-ketoamphetamine modulation of neurotoxicity by the dopamine transporter. J Neurochem 133:211–222
Lopez-Arnau R, Martinez-Clemente J, Rodrigo T, Pubill D, Camarasa J, Escubedo E (2015) Neuronal changes and oxidative stress in adolescent rats after repeated exposure to mephedrone. Toxicol Appl Pharmacol 286:27–35
Motbey CP, Clemens KJ, Apetz N, Winstock AR, Ramsey J, Li KM, Wyatt N, Callaghan PD, Bowen MT, Cornish JL, McGregor IS (2013) High levels of intravenous mephedrone (4-methylmethcathinone) self-administration in rats: neural consequences and comparison with methamphetamine. J Psychopharmacol 27:823–836
Motbey CP, Karanges E, Li KM, Wilkinson S, Winstock AR, Ramsay J, Hicks C, Kendig MD, Wyatt N, Callaghan PD, McGregor IS (2012) Mephedrone in adolescent rats: residual memory impairment and acute but not lasting 5-HT depletion. PLoS One 7, e45473
Angoa-Perez M, Kane MJ, Briggs DI, Francescutti DM, Sykes CE, Shah MM, Thomas DM, Kuhn DM (2013) Mephedrone does not damage dopamine nerve endings of the striatum, but enhances the neurotoxicity of methamphetamine, amphetamine, and MDMA. J Neurochem 125:102–110
Baumann MH, Ayestas MA Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203
Martinez-Clemente J, Lopez-Arnau R, Abad S, Pubill D, Escubedo E, Camarasa J (2014) Dose and time-dependent selective neurotoxicity induced by mephedrone in mice. PLoS One 9, e99002
Shortall SE, Spicer CH, Ebling FJ, Green AR, Fone KC, King MV (2015) Contribution of serotonin and dopamine to changes in core body temperature and locomotor activity in rats following repeated administration of mephedrone. Addict Biol
Wright MJ Jr, Angrish D, Aarde SM, Barlow DJ, Buczynski MW, Creehan KM, Vandewater SA, Parsons LH, Houseknecht KL, Dickerson TJ, Taffe MA (2012) Effect of ambient temperature on the thermoregulatory and locomotor stimulant effects of 4-methylmethcathinone in Wistar and Sprague-Dawley rats. PLoS One 7, e44652
den Hollander B, Rozov S, Linden AM, Uusi-Oukari M, Ojanpera I, Korpi ER (2013) Long-term cognitive and neurochemical effects of “bath salt” designer drugs methylone and mephedrone. Pharmacol Biochem Behav 103:501–509
Shortall SE, Macerola AE, Swaby RT, Jayson R, Korsah C, Pillidge KE, Wigmore PM, Ebling FJ, Richard Green A, Fone KC, King MV (2013) Behavioural and neurochemical comparison of chronic intermittent cathinone, mephedrone and MDMA administration to the rat. Eur Neuropsychopharmacol 23:1085–1095
Ciudad-Roberts A, Camarasa J, Ciudad CJ, Pubill D, Escubedo E (2015) Alcohol enhances the psychostimulant and conditioning effects of mephedrone in adolescent mice; postulation of unique roles of D3 receptors and BDNF in place preference acquisition. Br J Pharmacol 172:4970–4984
den Hollander B, Sundstrom M, Pelander A, Ojanpera I, Mervaala E, Korpi ER, Kankuri E (2014) Keto amphetamine toxicity-focus on the redox reactivity of the cathinone designer drug mephedrone. Toxicol Sci 141:120–131
Cameron KN, Kolanos R, Solis E Jr, Glennon RA, De Felice LJ (2013) Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. Br J Pharmacol 168:1750–1757
Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A (2013) Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85:1803–1815
Lopez-Arnau R, Martinez-Clemente J, Pubill D, Escubedo E, Camarasa J (2012) Comparative neuropharmacology of three psychostimulant cathinone derivatives: butylone, mephedrone and methylone. Br J Pharmacol 167:407–420
Martinez-Clemente J, Escubedo E, Pubill D, Camarasa J (2012) Interaction of mephedrone with dopamine and serotonin targets in rats. Eur Neuropsychopharmacol 22:231–236
Rickli A, Hoener MC, Liechti ME (2015) Monoamine transporter and receptor interaction profiles of novel psychoactive substances: para-halogenated amphetamines and pyrovalerone cathinones. Eur Neuropsychopharmacol 25:365–376
Budzynska B, Boguszewska-Czubara A, Kruk-Slomka M, Kurzepa J, Biala G (2015) Mephedrone and nicotine: oxidative stress and behavioral interactions in animal models. Neurochem Res 40:1083–1093
Lopez-Arnau R, Martinez-Clemente J, Abad S, Pubill D, Camarasa J, Escubedo E (2014) Repeated doses of methylone, a new drug of abuse, induce changes in serotonin and dopamine systems in the mouse. Psychopharmacology (Berl) 231:3119–3129
Lopez-Arnau R, Martinez-Clemente J, Pubill D, Escubedo E, Camarasa J (2014) Serotonergic impairment and memory deficits in adolescent rats after binge exposure of methylone. J Psychopharmacol 28:1053–1063
Kiyatkin EA, Kim AH, Wakabayashi KT, Baumann MH, Shaham Y (2015) Effects of social interaction and warm ambient temperature on brain hyperthermia induced by the designer drugs methylone and MDPV. Neuropsychopharmacology 40:436–445
Piao YS, Hall FS, Moriya Y, Ito M, Ohara A, Kikura-Hanajiri R, Goda Y, Lesch KP, Murphy DL, Uhl GR, Sora I (2015) Methylone-induced hyperthermia and lethal toxicity: role of the dopamine and serotonin transporters. Behav Pharmacol 26:345–352
Nagai F, Nonaka R, Satoh Hisashi Kamimura K (2007) The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur J Pharmacol 559:132–137
Sogawa C, Sogawa N, Ohyama K, Kikura-Hanajiri R, Goda Y, Sora I, Kitayama S (2011) Methylone and monoamine transporters: correlation with toxicity. Curr Neuropharmacol 9:58–62
Aarde SM, Creehan KM, Vandewater SA, Dickerson TJ, Taffe MA (2015) In vivo potency and efficacy of the novel cathinone alpha-pyrrolidinopentiophenone and 3,4-methylenedioxypyrovalerone: self-administration and locomotor stimulation in male rats. Psychopharmacology (Berl) 232:3045–3055
Adam A, Gerecsei LI, Lepesi N, Csillag A (2014) Apoptotic effects of the ‘designer drug’ methylenedioxypyrovalerone (MDPV) on the neonatal mouse brain. Neurotoxicology 44:231–236
Fantegrossi WE, Gannon BM, Zimmerman SM, Rice KC (2013) In vivo effects of abused ‘bath salt’ constituent 3,4-methylenedioxypyrovalerone (MDPV) in mice: drug discrimination, thermoregulation, and locomotor activity. Neuropsychopharmacology 38:563–573
King HE, Wetzell B, Rice KC, Riley AL (2014) 3,4-Methylenedioxypyrovalerone (MDPV)-induced conditioned taste avoidance in the F344/N and LEW rat strains. Pharmacol Biochem Behav 126:163–169
Merluzzi AP, Hurwitz ZE, Briscione MA, Cobuzzi JL, Wetzell B, Rice KC, Riley AL (2014) Age-dependent MDPV-induced taste aversions and thermoregulation in adolescent and adult rats. Dev Psychobiol 56:943–954
Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW (2013) Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology 38:552–562
Chapy H, Smirnova M, Andre P, Schlatter J, Chiadmi F, Couraud PO, Scherrmann JM, Decleves X, Cisternino S (2015) Carrier-mediated cocaine transport at the blood-brain barrier as a putative mechanism in addiction liability. Int J Neuropsychopharmacol 18
Simmler LD, Wandeler R, Liechti ME (2013) Bupropion, methylphenidate, and 3,4-methylenedioxypyrovalerone antagonize methamphetamine-induced efflux of dopamine according to their potencies as dopamine uptake inhibitors: implications for the treatment of methamphetamine dependence. BMC Res Notes 6:220
Numachi Y, Ohara A, Yamashita M, Fukushima S, Kobayashi H, Hata H, Watanabe H, Hall FS, Lesch KP, Murphy DL, Uhl GR, Sora I (2007) Methamphetamine-induced hyperthermia and lethal toxicity: role of the dopamine and serotonin transporters. Eur J Pharmacol 572:120–128
Crean RD, Davis SA, Von Huben SN, Lay CC, Katner SN, Taffe MA (2006) Effects of (+/-)3,4-methylenedioxymethamphetamine, (+/-)3,4-methylenedioxyamphetamine and methamphetamine on temperature and activity in rhesus macaques. Neuroscience 142:515–525
Cappon GD, Morford LL, Vorhees CV (1997) Ontogeny of methamphetamine-induced neurotoxicity and associated hyperthermic response. Brain Res Dev Brain Res 103:155–162
Dafters RI (1995) Hyperthermia following MDMA administration in rats: effects of ambient temperature, water consumption, and chronic dosing. Physiol Behav 58:877–882
Makisumi T, Yoshida K, Watanabe T, Tan N, Murakami N, Morimoto A (1998) Sympatho-adrenal involvement in methamphetamine-induced hyperthermia through skeletal muscle hypermetabolism. Eur J Pharmacol 363:107–112
Paulson PE, Robinson TE (1995) Amphetamine-induced time-dependent sensitization of dopamine neurotransmission in the dorsal and ventral striatum: a microdialysis study in behaving rats. Synapse 19:56–65
Yoshida K, Morimoto A, Makisumi T, Murakami N (1993) Cardiovascular, thermal and behavioral sensitization to methamphetamine in freely moving rats. J Pharmacol Exp Ther 267:1538–1543
Miller ML, Creehan KM, Angrish D, Barlow DJ, Houseknecht KL, Dickerson TJ, Taffe MA (2013) Changes in ambient temperature differentially alter the thermoregulatory, cardiac and locomotor stimulant effects of 4-methylmethcathinone (mephedrone). Drug Alcohol Depend 127:248–253
Gregg RA, Rawls SM (2014) Behavioral pharmacology of designer cathinones: a review of the preclinical literature. Life Sci 97:27–30
Aarde SM, Angrish D, Barlow DJ, Wright MJ Jr, Vandewater SA, Creehan KM, Houseknecht KL, Dickerson TJ, Taffe MA (2013) Mephedrone (4-methylmethcathinone) supports intravenous self-administration in Sprague-Dawley and Wistar rats. Addict Biol 18:786–799
Froberg BA, Levine M, Beuhler MC, Judge BS, Moore PW, Engebretsen KM, McKeown NJ, Rosenbaum CD, Young AC, Rusyniak DE (2015) Acute methylenedioxypyrovalerone toxicity. J Med Toxicol 11:185–194
Cadet JL, Jayanthi S, Deng X (2003) Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J 17:1775–1788
Kita T, Wagner GC, Nakashima T (2003) Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J Pharmacol Sci 92:178–195
Baumann MH, Wang X, Rothman RB (2007) 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacology (Berl) 189:407–424
Cadet JL, Krasnova IN, Jayanthi S, Lyles J (2007) Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res 11:183–202
Capela JP, Carmo H, Remiao F, Bastos ML, Meisel A, Carvalho F (2009) Molecular and cellular mechanisms of ecstasy-induced neurotoxicity: an overview. Mol Neurobiol 39:210–271
Imam SZ, el-Yazal J, Newport GD, Itzhak Y, Cadet JL, Slikker W Jr, Ali SF (2001) Methamphetamine-induced dopaminergic neurotoxicity: role of peroxynitrite and neuroprotective role of antioxidants and peroxynitrite decomposition catalysts. Ann N Y Acad Sci 939:366–380
Motbey CP, Hunt GE, Bowen MT, Artiss S, McGregor IS (2012) Mephedrone (4-methylmethcathinone, ‘meow’): acute behavioural effects and distribution of Fos expression in adolescent rats. Addict Biol 17:409–422
Hadlock GC, Webb KM, McFadden LM, Chu PW, Ellis JD, Allen SC, Andrenyak DM, Vieira-Brock PL, German CL, Conrad KM, Hoonakker AJ, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE (2011) 4-Methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J Pharmacol Exp Ther 339:530–536
Kehr J, Ichinose F, Yoshitake S, Goiny M, Sievertsson T, Nyberg F, Yoshitake T (2011) Mephedrone, compared with MDMA (ecstasy) and amphetamine, rapidly increases both dopamine and 5-HT levels in nucleus accumbens of awake rats. Br J Pharmacol 164:1949–1958
Toborek M, Seelbach MJ, Rashid CS, Andras IE, Chen L, Park M, Esser KA (2013) Voluntary exercise protects against methamphetamine-induced oxidative stress in brain microvasculature and disruption of the blood-brain barrier. Mol Neurodegener 8:22
Cadet JL, Brannock C (1998) Free radicals and the pathobiology of brain dopamine systems. Neurochem Int 32:117–131
Halpin LE, Collins SA, Yamamoto BK (2014) Neurotoxicity of methamphetamine and 3,4-methylenedioxymethamphetamine. Life Sci 97:37–44
Stepens A, Groma V, Skuja S, Platkajis A, Aldins P, Eksteina I, Martinsone I, Bricis R, Donaghy M (2014) The outcome of the movement disorder in methcathinone abusers: clinical, MRI and manganesemia changes, and neuropathology. Eur J Neurol 21:199–205
Levin OS (2005) “Ephedron” encephalopathy. Zh Nevrol Psikhiatr Im S S Korsakova 105:12–20
Kelly JP (2011) Cathinone derivatives: a review of their chemistry, pharmacology and toxicology. Drug Test Anal 3:439–453
Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA, Birkes J, Young R, Glennon RA (2003) In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther 307:138–145
Gudelsky GA, Yamamoto BK, Nash JF (1994) Potentiation of 3,4-methylenedioxymethamphetamine-induced dopamine release and serotonin neurotoxicity by 5-HT2 receptor agonists. Eur J Pharmacol 264:325–330
Pifl C, Reither H, Hornykiewicz O (2015) The profile of mephedrone on human monoamine transporters differs from 3,4-methylenedioxymethamphetamine primarily by lower potency at the vesicular monoamine transporter. Eur J Pharmacol 755:119–126
Bonano JS, Banks ML, Kolanos R, Sakloth F, Barnier ML, Glennon RA, Cozzi NV, Partilla JS, Baumann MH, Negus SS (2015) Quantitative structure-activity relationship analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 172:2433–2444
Sakloth F, Kolanos R, Mosier PD, Bonano JS, Banks ML, Partilla JS, Baumann MH, Negus SS, Glennon RA (2015) Steric parameters, molecular modeling and hydropathic interaction analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 172:2210–2218
Baumann MH, Partilla JS, Lehner KR (2013) Psychoactive “bath salts”: not so soothing. Eur J Pharmacol 698:1–5
Kolanos R, Solis E Jr, Sakloth F, De Felice LJ, Glennon RA (2013) Deconstruction of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS Chem Neurosci 12:1524–1529
Marusich JA, Antonazzo KR, Wiley JL, Blough BE, Partilla JS, Baumann MH (2014) Pharmacology of novel synthetic stimulants structurally related to the “bath salts” constituent 3,4-methylenedioxypyrovalerone (MDPV). Neuropharmacology 87:206–213
Kiyatkin EA, Sharma HS (2009) Acute methamphetamine intoxication: brain hyperthermia, blood-brain barrier, brain edema, and morphological cell abnormalities. Int Rev Neurobiol 88:65–100
O’Shea E, Urrutia A, Green AR, Colado MI (2014) Current preclinical studies on neuroinflammation and changes in blood-brain barrier integrity by MDMA and methamphetamine. Neuropharmacology 87:125–134
Coelho-Santos V, Leitao RA, Cardoso FL, Palmela I, Rito M, Barbosa M, Brito MA, Fontes-Ribeiro CA, Silva AP (2015) The TNF-alpha/NF-kappaB signaling pathway has a key role in methamphetamine-induced blood-brain barrier dysfunction. J Cereb Blood Flow Metab 35:1260–1271
Lopez-Arnau R, Martinez-Clemente J, Carbo M, Pubill D, Escubedo E, Camarasa J (2013) An integrated pharmacokinetic and pharmacodynamic study of a new drug of abuse, methylone, a synthetic cathinone sold as “bath salts”. Prog Neuropsychopharmacol Biol Psychiatry 45:64–72
Coppola M, Mondola R (2012) Synthetic cathinones: chemistry, pharmacology and toxicology of a new class of designer drugs of abuse marketed as “bath salts” or “plant food”. Toxicol Lett 211:144–149
Sitte HH, Freissmuth M (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci 36:41–50
Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL (2015) Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend 147:1–19
De Felice LJ, Glennon RA, Negus SS (2014) Synthetic cathinones: chemical phylogeny, physiology, and neuropharmacology. Life Sci 97:20–26
Araujo AM, Valente MJ, Carvalho M, Dias da Silva D, Gaspar H, Carvalho F, de Lourdes Bastos M, Guedes de Pinho P (2015) Raising awareness of new psychoactive substances: chemical analysis and in vitro toxicity screening of ‘legal high’ packages containing synthetic cathinones. Arch Toxicol 89:757–771
Thomas DM, Francescutti-Verbeem DM, Kuhn DM (2008) The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem 105:605–616
Sulzer D, Maidment NT, Rayport S (1993) Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem 60:527–535
Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA (1995) Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol Pharmacol 47:368–373
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Angoa-Pérez, M., Anneken, J.H., Kuhn, D.M. (2016). Neurotoxicology of Synthetic Cathinone Analogs. In: Baumann, M.H., Glennon, R.A., Wiley, J.L. (eds) Neuropharmacology of New Psychoactive Substances (NPS). Current Topics in Behavioral Neurosciences, vol 32. Springer, Cham. https://doi.org/10.1007/7854_2016_21
Download citation
DOI: https://doi.org/10.1007/7854_2016_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52442-9
Online ISBN: 978-3-319-52444-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)