
Abstract
Modeling tissues and organs using conventional 2D cell cultures is problematic as the cells rapidly lose their in vivo phenotype. In microfluidic bioreactors the cells reside in microstructures that are continuously perfused with cell culture medium to provide a dynamic environment mimicking the cells natural habitat. These micro scale bioreactors are sometimes referred to as organs-on-chips and are developed in order to improve and extend cell culture experiments. Here, we describe the two manufacturing techniques photolithography and soft lithography that are used in order to easily produce microfluidic bioreactors. The use of these bioreactors is exemplified by a toxicity assessment on 3D clustered human pluripotent stem cells (hPSC)-derived cardiomyocytes by beating frequency imaging.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Keywords:
- Microfluidics
- Photolithography
- Soft lithography
- Human pluripotent stem cells (hPSCs)
- Organ-on-a-chip
- Cardiomyocytes
- Cardiac bodies (CBs)
- 3D cell culture models
- Drug assessment
1 Introduction
The technology to manufacture bioreactors, with features at the scale of tens to hundreds of micrometers, originates from the production methods for microelectronics and microelectromechanical systems [1]. The ability of the technology to adapt to the scale and structure of biological systems and their applications is especially suitable for replicating the functions of living tissues and organs. Mimicking the in vivo environment is of major interest for establishing better prediction models during pharmaceutical development. With such models the assessment of efficacy and safety of drug candidates can be significantly improved, leading to better decisions for continuing drug development, than if conventional static 2D cell culture models are used [2–5]. Microfluidic bioreactors that consist of one or several cell types, and that are continuously perfused with cell culture media, providing nutrients and removing toxic wastes as well as inducing shear stress, are also known as organs-on-chips [6]. Important indicators of cellular state, such as essential proteins and metabolite markers, otherwise lost shortly after isolation, become easier to detect and monitor over extended time periods using these advanced cell culture platforms. Incorporating iPSCs with disease specific genotypes into organs-on-chips is expected to further improve the predictability of clinical trials [2, 7].
The most commonly applied fabrication method for microfluidic bioreactors is photolithography, followed by soft lithography. In photolithography, a master is created by spin coating a photoresist onto a silicon wafer that is polymerized by UV-light under a mask with transparent patterns. Soft lithography is then used for replica molding of the master in polydimethylsiloxane (PDMS), an optically transparent and gas permeable silicone elastomer. The photolithographic and soft lithographic fabrication methods are described in this chapter as well as the use of a microfluidic bioreactor with defined niches for drug assessment of beating clusters of hPSC-derived cardiomyocytes, known as cardiac bodies (CBs) [8–11].
2 Materials
Photolithography and soft lithography fabrication as well as cell culture require clean environments. Make sure that the laboratory is suitable for the production of the lithographic masters of the bioreactor and that all equipment used for culture of cells is sterile.
2.1 Photolithography
-
1.
Silicon wafer, 4″ (SWI, Taiwan).
-
2.
Washing solution (TL1): H2O, H2O2 (VWR International, PA, USA) and NH3 (Merck, NJ, USA) (5:1:1 ratio by volume).
-
3.
Negative photo resists SU-8 10 and SU-8 3035 (MicroChem Corp., MA, USA).
-
4.
Spin coater (Spin150, APT, Germany).
-
5.
Photolithographic mask (Acreo, Sweden): The patterns of the bioreactor are created using any CAD software and printed on a plastic film. The desired structures should be transparent and the rest black.
-
6.
Mask aligner with UV-light source (Kari Süss, Germany).
-
7.
Developer (Micro Resist Technology, Germany).
-
8.
Isopropanol (VWR International).
-
9.
Silanization solution: 3 % tetramethylsilane (Sigma-Aldrich, MO, USA) in 99.5 % ethanol.
2.2 Soft Lithography
-
1.
Silicon wafer patterned by photolithography (master).
-
2.
Microscope glass slides (VWR International).
-
3.
PDMS pre-polymer and curing agent (Sylgard 184, Dow Corning, MI, USA).
-
4.
Punches (Syneo, TX, USA).
-
5.
Bioreactor connections: stainless steel tubes (0.64 mm OD, New England Small Tube Corp., NH, USA), Tygon tubing (0.5 mm ID, 1.52 mm OD, VWR International), PEEK tubing (0.36 mm OD, LabSmith Inc., CA, USA).
2.3 Cell Culture
2.3.1 Production of Cardiac Bodies
-
1.
Human pluripotent stem cells (hPSC) including the following human induced pluripotent stem cell (hiPSC) and human embryonic stem cell (hESC) lines: HES3 NKX2-5eGFP/w [12], hCBiPS2 [13], and hHSC_F1285T_iPS2 [14].
-
2.
hPSC culture medium: mTeSR (Stem Cell Technologies, Vancouver, Canada) supplemented with 10 μM Y-27632 (Tocris, UK) for 24 h post seeding.
-
3.
Cardiomyogenic differentiation medium RPMI1640 (Life Technologies, CA, USA) supplemented with B27 minus insulin (Gibco, USA) supplemented with the chemical WNT pathway agonist CHIR99021 (Millipore, MA, USA) and the WNT antagonist IWP2 or IWR1 (Tocris)
-
4.
hPSC expansion and cardiomyogenic differentiation in suspension culture was performed [9, 11]. In brief: For the inoculation of suspension culture, dissociation into single cells is performed by accutase treatment for 5 min at 37 °C followed by dilution to 3.3 × 105 cells/ml in mTeSR1 plus 10 μM Y-27632 (Tocris) and seeding into 12-well suspension plates (Greiner-BioOne, Austria) or Erlenmeyer flasks (125 ml scale; VWR-International) in 1.5 ml/well or 20 ml/flask, respectively. Flasks are agitated at 75 rpm (orbital shaker, Infors-HT, Switzerland).
-
5.
Differentiation of resulting aggregates is induced on day 0 (4 days after single cell inoculation) using 7.5 μM CHIR99021 (Millipore) for 24 h. On day 3, IWP2 or IWR1 (Tocris) is added at 5 and 4 μM for 48 h, respectively. During the differentiation, cells are kept in RPMI1640 (Life Technologies) supplemented with B27 (minus insulin). Medium is entirely replaced on days 0 (+CHIR 99021), 1, 3 (+IWP2 or IWR1), and 5 applying 3 or 20 mL depending on the respective culture format, i.e. 12-well dish and Erlenmeyer flask. Aggregates are cultured in RPMI1640 supplemented with B27 from day 7 onwards and used from day 10–30 for experiments.
-
6.
Resulting cardiac bodies (CBs) are monitored by light microscopy; images are captured (AxiovertA1; Zeiss, Germany) and processed via the software AxioVision (Zeiss) to define diameter and size distribution ranging from ~200–400 μm.
2.3.2 Cardiac Body Monitoring in a Microfluidic Bioreactor
-
1.
Microfluidic bioreactor.
-
2.
Cardiac bodies.
-
3.
RPMI1640 supplemented with B27 minus insulin.
-
4.
Petri dish (90 mm, VWR International).
-
5.
Video microscope (SVM340, LabSmith Inc., CA, USA).
-
6.
Syringes pump (Fusion 100, Chemyx Inc., TX, USA).
-
7.
Three-port valve (MV201, LabSmith).
-
8.
1 ml syringe (BD Plastipak, NJ, USA).
-
9.
20 ml syringe (BD Plastipak).
-
10.
Blunt ended cannulas (Sterican, B. Braun, Germany).
-
11.
Crocodile clips.
-
12.
Clamps.
3 Methods
3.1 Master Fabrication by Photolithography
Using the protocol presented here creates structures with thicknesses of 20 and 200 μm (Fig. 1). For other thicknesses check the specification of the photoresist and adjust spin times, baking times and UV-light exposure accordingly.

Overview of the process of photolithography. A silicon wafer is covered with photoresist by spin coating. The photoresist is exposed to UV-light under a mask having a defined transparent pattern. UV-exposed photoresist is polymerized while unexposed photoresist is washed away
-
1.
Immerse the silicon wafer in washing solution for 10 min at 85 °C to remove organic residues. Rinse the wafer in deionized water and let dry in an oven at 100 °C for 10 min.
-
2.
Spin coat SU-8 10 on the wafer at 500 rpm for 10 s, then ramped at 150 rpm/s at 1500 rpm for 35 s to create the first layer of photoresist with a thickness of 20 μm (see Notes 1 and 2 ).
-
3.
Remove solvents from the photoresist by baking the wafer at 65 °C for 1 min on a hot plate, followed by 10 min at 95 °C.
-
4.
Expose the photoresist to UV-light at 187 mJ/cm2 under the photolithographic mask.
-
5.
Move the wafer to a hot plate at 100 °C for 15 min. The patterns should be visible after this polymerization step. Let the wafer cool down for 5 min before the next step.
-
6.
Continue by spin coating SU-8 3035 on the wafer at 500 rpm for 10 s, then ramped at 150 rpm/s at 1500 rpm for 35 s to create a second layer of photoresist with a thickness of 100 μm. Bake the wafer at 65 °C for 1 min on a hot plate, followed by 100 °C for 65 °C.
-
7.
Repeat step 6 to create a third layer with a thickness of 100 μm (see Note 1 and 2 ).
-
8.
Align the second photolithographic mask according to the patterns visible from the first exposure. Expose the photoresist to UV-light at 400 mJ/cm2 (see Note 3 ).
-
9.
Remove unexposed photoresist by immersing the wafer in developer for 10 min followed by rinsing in isopropanol, then immerse for another 2 min in developer, followed by rinsing in isopropanol and water.
-
10.
Use a DekTak to check the height of the structures.
-
11.
Put the wafer in a silanization solution for 10 min, rinse in 99.5 % ethanol and incubate at 120 °C for 30 min.
3.2 Microfluidic Bioreactor Fabrication by Soft Lithography
-
1.
Mix PDMS pre-polymer and curing agent thoroughly at a 10:1 ratio (w/w) for at least 2 min. Degas with vacuum in a desiccator to remove bubbles (see Note 4 ).
-
2.
PDMS membrane: Pour PDMS between two flat sheets of Plexiglas separated by a 200 μm diameter copper wire. Clamp the Plexiglas and let cure at room temperature overnight. Put in the oven at 60 °C for 10 min for final curing.
-
3.
PDMS bubble trap: Pour 20 g of PDMS into a 90 mm petri dish and let cure overnight. Put in the oven at 60 °C for 10 min for final curing. Cut out a 5 × 5 × 4 mm cuboid and punch a hole through it. Cut a square of the plasma membrane and bond to the cuboid by exposing the surfaces to oxygen plasma for 1 min. Put the bubble trap in the oven for 10 min for irreversible bonding (see Note 5 ).
-
4.
Clean the master in water and ethanol two times and blow dry. Pour the mixture of PDMS pre-polymer and curing agent over the master and cure in the oven at 60 °C for 2 h (Fig. 2).
Fig. 2 
Overview of the bioreactor production by soft lithography. PDMS is cured on the master, peeled off, and bonded to a microscope glass slide. Tubing is connected to the channel for cell seeding and cell culture medium perfusion by punching
-
5.
Cut rectangles of PDMS from the master leaving at least 0.5 cm between the patterns and the edge.
-
6.
Use tape to remove residues from the PDMS piece, rinse in water and ethanol and blow dry.
-
7.
Use a 0.4 mm ID punch to make holes for the medium inlet.
-
8.
Use a 0.64 mm ID punch to make holes for medium outlet, cell inlet and connection to the bubble trap.
-
9.
Remove residues from the PDMS piece.
-
10.
Oxidize the surfaces of the PDMS piece and a clean glass slide in oxygen plasma for 1 min. Bond the PDMS to the glass slide and make sure no air bubbles are trapped in between. Put in the oven at 60 °C for 10 min for irreversible bonding.
-
11.
Remove residues from both the PDMS piece and the bubble trap membrane and oxidize in oxygen plasma for 1 min. Bond the bubble trap to the designated hole in the PDMS piece.
-
12.
Attach the tubing to the medium inlet and outlet, the cell inlet and the bubble trap. Seal with PDMS and cure in the oven at 60 °C for 10 min (see Note 6 ).
-
13.
Autoclave.

3.3 Beating Frequency Monitoring of 3D CBs in a Microfluidic Bioreactor
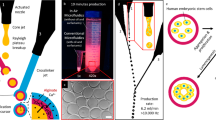
This step should be performed in a LAF hood. Place the device on top of a LabSmith inverted microscope and use it to detect bubbles, to monitor the CB seeding, and to record the beating frequency of CBs (Fig. 3).

(a) Schematics of the CB seeding process, (b) micrograph of CBs in bioreactor niches, (c) drawing of the bioreactor and connections, (d) the experimental setup as arranged in the incubator (reproduced with permission from ref. [8])
-
1.
Maintain CBs in petri dishes with cell culture medium. Replace half of the medium every third day (see Note 7 ).
-
2.
Insert blunt-ended cannulas into the Tygon tubing at the bubble trap.
-
3.
Connect the PEEK tubing at the medium inlet to a LabSmith three-port valve.
-
4.
Connect the other two ports of the valve to 1 ml syringes.
-
5.
Punch a hole in two Eppendorf tubes and use as sample/waste at the end of the two outlet tubing.
-
6.
Insert a 1 ml pipette tip in the Tygon tubing at the cell inlet.
-
7.
Rinse the device with ethanol at 1 μl/min for 30 min. Purge any bubbles formed in the device by gently pushing the syringe (see Note 8 ).
-
8.
Switch the inlet syringe to sterile water and rinse the device at 1 μl/min for 30 min.
-
9.
Switch the inlet syringe to cell culture medium and rinse the device at 1 μl/min for 30 min.
-
10.
Change one of the inlet syringes to drug supplemented medium.
-
11.
Close the main medium outlet with a crocodile clip. Make sure that the medium only flows through the drainage outlet.
-
12.
Sieve or manually pick 10 beating CBs with a diameter between 50 and 200 μm and seed them into the pipette tip at the cell inlet. Wait for a few minutes until the CBs reach the cell inlet. The CBs should follow the flow into separate niches (see Note 9 ).
-
13.
Stop the pump and close the drain medium outlet and the CB inlet with crocodile clips. Cover the pipette tip with aluminum foil.
-
14.
Move the whole setup to the incubator. Wait until the temperature has recovered before carefully opening the main medium outlet followed by the medium inlet. Start the syringe pump at a perfusion rate of 0.1 μl/min.
-
15.
Record the beating frequency of the CBs.
-
16.
To introduce the drug supplemented medium stop the pump. Gently switch the valve to the position of the drug supplemented medium syringe. Resume the perfusion with the new syringe.
-
17.
Record again the beating frequency of the CBs.
4 Notes
-
1.
Dispense approximately 1 ml of SU8 per inch of wafer diameter, i.e., 4 ml for a 4″ wafer. The first step of the spin coating is to distribute the photoresist over the wafer. Look at the wafer during this step to make sure that the photoresist is correctly dispersed.
-
2.
The manufacturer’s instructions for spin coating are made for photoresist on silicon. Spin coating a photoresist onto photoresist can give other thicknesses because of the friction change. If the thickness on your master is not approximately 200 μm revise this step and adjust the spin speed.
-
3.
Alignment of the mask over the master can be difficult. A suggestion for increased precision is to make alignment marks of different sizes in the masks, for example crosses, spread over the mask.
-
4.
The speed of PDMS curing depends on the temperature. In room temperature curing is completed after approximately 48 h. Store uncured PDMS in the freezer for reuse.
-
5.
If many bioreactors should be made cut out a larger piece of the 4 mm thick PDMS, e.g., 5 × 80 × 4 mm in the petri dish. Punch holes approximately every 5 mm and bond a PDMS membrane to it. Cut out bubble traps when needed. This will save a lot of time.
-
6.
Use a few days old PDMS for this step. It is stickier and will not trickle down and block the channels.
-
7.
CBs have a tendency to stick together. Therefore, seed them sparsely and give the dish a swirl every day to keep properly sized material available.
-
8.
The risk of bubble formation in the device can be reduced by equilibrating the bioreactor, the cell culture medium, the tubing and the syringes in the incubator over night before starting the experiment.
-
9.
Be sure that the size of the CBs is smaller than the height of the bioreactor or they will be trapped. Gently tap the microscope if CBs stick together. Tilt the microscope if necessary to move the CBs.
References
Whitesides GM (2006) The origins and the future of microfluidics. Nature 442(7101):368–373. doi:10.1038/nature05058
Alépée N (2014) State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. Altex. doi: 10.14573/altex1406111
Booth R, Kim H (2012) Characterization of a microfluidic in vitro model of the blood-brain barrier (muBBB). Lab Chip 12(10):1784–1792. doi:10.1039/c2lc40094d
Toh YC, Lim TC, Tai D, Xiao G, van Noort D, Yu H (2009) A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 9(14):2026–2035. doi:10.1039/b900912d
Kaneko T, Nomura F, Hamada T, Abe Y, Takamori H, Sakakura T, Takasuna K, Sanbuissho A, Hyllner J, Sartipy P, Yasuda K (2014) On-chip in vitro cell-network pre-clinical cardiac toxicity using spatiotemporal human cardiomyocyte measurement on a chip. Sci Rep 4:4670. doi:10.1038/srep04670
Bhatia SN, Ingber DE (2014) Microfluidic organs-on-chips. Nat Biotechnol 32(8):760–772. doi:10.1038/nbt.2989
Astashkina A, Mann B, Grainger DW (2012) A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol Ther 134(1):82–106. doi:10.1016/j.pharmthera.2012.01.001
Bergstrom G, Christoffersson J, Schwanke K, Zweigerdt R, Mandenius C-F (2015) Stem cell derived in vivo-like human cardiac bodies in a microfluidic device for toxicity testing by beating frequency imaging. Lab Chip 15(15):3242–3249. doi:10.1039/C5LC00449G
Kempf H, Olmer R, Kropp C, Ruckert M, Jara-Avaca M, Robles-Diaz D, Franke A, Elliott DA, Wojciechowski D, Fischer M, Roa Lara A, Kensah G, Gruh I, Haverich A, Martin U, Zweigerdt R (2014) Controlling expansion and cardiomyogenic differentiation of human pluripotent stem cells in scalable suspension culture. Stem Cell Rep 3(6):1132–1146. doi:10.1016/j.stemcr.2014.09.017
Kensah G, Roa Lara A, Dahlmann J, Zweigerdt R, Schwanke K, Hegermann J, Skvorc D, Gawol A, Azizian A, Wagner S, Maier LS, Krause A, Drager G, Ochs M, Haverich A, Gruh I, Martin U (2013) Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur Heart J 34(15):1134–1146. doi:10.1093/eurheartj/ehs349
Kempf H, Kropp C, Olmer R, Martin U, Zweigerdt R (2015) Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat Protoc 10(9):1345–1361. doi:10.1038/nprot.2015.089
Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL, Biben C, Hatzistavrou T, Hirst CE, Yu QC, Skelton RJ, Ward-van Oostwaard D, Lim SM, Khammy O, Li X, Hawes SM, Davis RP, Goulburn AL, Passier R, Prall OW, Haynes JM, Pouton CW, Kaye DM, Mummery CL, Elefanty AG, Stanley EG (2011) NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods 8(12):1037–1040. doi:10.1038/nmeth.1740
Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C, Zweigerdt R, Gruh I, Meyer J, Wagner S, Maier LS, Han DW, Glage S, Miller K, Fischer P, Scholer HR, Martin U (2009) Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5(4):434–441. doi:10.1016/j.stem.2009.08.021
Hartung S, Schwanke K, Haase A, David R, Franz WM, Martin U, Zweigerdt R (2013) Directing cardiomyogenic differentiation of human pluripotent stem cells by plasmid-based transient overexpression of cardiac transcription factors. Stem Cells Dev 22(7):1112–1125. doi:10.1089/scd.2012.0351
Acknowledgements
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 115439 (StemBANCC), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Christoffersson, J., Bergström, G., Schwanke, K., Kempf, H., Zweigerdt, R., Mandenius, CF. (2016). A Microfluidic Bioreactor for Toxicity Testing of Stem Cell Derived 3D Cardiac Bodies. In: Turksen, K. (eds) Bioreactors in Stem Cell Biology. Methods in Molecular Biology, vol 1502. Humana Press, New York, NY. https://doi.org/10.1007/7651_2016_340
Download citation
DOI: https://doi.org/10.1007/7651_2016_340
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6476-5
Online ISBN: 978-1-4939-6478-9
eBook Packages: Springer Protocols