
Abstract
Transplantation of photoreceptor precursor cells (PPCs) differentiated from human embryonic stem cells (hESCs) is a promising approach to treat common blinding diseases such as age-related macular degeneration and retinitis pigmentosa. However, existing PPC generation methods are inefficient. To enhance differentiation protocols for rapid and high-yield production of PPCs, we focused on optimizing the handling of the cells by including feeder-independent growth of hESCs, using size-controlled embryoid bodies (EBs), and addition of triiodothyronine (T3) and taurine to the differentiation medium, with subsequent removal of undifferentiated cells via negative cell-selection. Our novel protocol produces higher yields of PPCs than previously reported while reducing the time required for differentiation, which will help understand retinal diseases and facilitate large-scale preclinical trials.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Keywords:
1 Introduction
Retinal degenerations resulting in irreversible vision loss, such as age-related macular degeneration (AMD) and retinitis pigmentosa (RP), affect millions of people around the world. These and other retinal degenerations lead to loss of cells in the retinal pigment epithelium and/or neurosensory retina, which can ultimately lead to blindness. Although there are some effective therapies for “wet” AMD (e.g., anti-VEGF agents and photodynamic therapy), there are no available treatments for most retinal degenerations including “dry” AMD, RP, and other hereditary retinal dystrophies.
Cell transplantation is a promising approach for replacing damaged, degenerating cells of the neural retina. However, finding the optimal source of transplantable cells and achieving efficient production of sufficient amounts of the desired cells remain challenging. It has been proposed that the appropriate cells for transplantation are ones that have been partially differentiated towards the retinal fate (1) although the optimal stage has yet to be determined.
In the past several years, significant advances have been made in obtaining and generating cells derived from fetal retinal tissue (1), human adult stem cells (2, 3), human embryonic stem cells (hESCs; (4)), and induced pluripotent stem cells (iPSCs; (5)). Most work has been focused on hESCs as these stem cells have a remarkable capacity for self-renewal and can differentiate into any cell type in the body under the right conditions. These characteristics make them an ideal source for cell replacement therapies.
Exposing hESCs to growth factors and modulators of developmental signaling pathways that mimic normal retinal development has successfully directed undifferentiated hESCs to both retinal pigment epithelium (6, 7) and neural retina cell fate (8, 9). Most of the reported protocols begin with the formation of embryoid bodies (EBs), which are sphere-like structures derived from undifferentiated hESC colonies (10, 11). These EBs were formed by enzymatically detaching hESC colonies and allowing them to “curl” and form a sphere. Using these techniques resulted in a differentiated cell population that consisted of a mixture of most cell types of the neural retina (e.g. ganglion cells, amacrine cells, bipolar cells, horizontal cells, and photoreceptor cells) (6–9).
One of the limiting factors in the production of meaningful amounts of retinal precursor cells for preclinical and clinical studies has been the time-consuming nature of the differentiation protocols (up to 150 days) that often result in low yields of the desired cells (8, 9, 12). As a consequence, the differentiation process can also be very expensive.
Our goal was to develop a method to efficiently produce large amounts of retinal precursor cells. We focused on developing a protocol for just one cell type, photoreceptor precursor cells (PPCs), rather than the production of a range of retinal cells because that is the cell type most in demand for preclinical trials. We modified information from several previous studies and experimented with EBs of variable sizes.
The resulting novel protocol begins with the feeder-free growth and the use of chemically defined media for hESC maintenance, reducing the variability associated with growth on feeder cells. The second stage is the use of size-selected EBs with a diameter of ~200 μm, containing 1,000 cells, to synchronize the differentiation process and finally, the addition of taurine and T3 during the differentiation period. This method produces, in 17 days, a cell population enriched for PPCs that express BLIMP1 (a protein expressed in early photoreceptor development) (13, 14), CRX, recoverin, and s-opsin.
The protocol outlined in this chapter describes in detail the maintenance and expansion of undifferentiated hESCs grown in feeder-free systems and the multistep generation of PPCs over the course of 17 days. The protocol then summarizes a method we use to confirm the identity of the PPCs: RT-qPCR and flow cytometry. We also outline the procedure to further enrich PPC purity using magnetic separation to remove undifferentiated cells still expressing the hESC marker SSEA-4.
2 Materials
2.1 Culture of Undifferentiated hESCs
-
1.
The hESC line used: WA-09 (WiCell Research Institute).
-
2.
hESC growth media: Complete TeSR™2 (STEMCELL Technologies).
-
3.
Dispase (1 mg/ml in DMEM/F-12; STEMCELL Technologies).
-
4.
Serological glass pipettes.
2.2 Surface Coating for Adherent Cell Growth of hESCs and Differentiating PPCs
-
1.
Growth Factor Reduced Matrigel™ (BD Biosciences).
-
2.
DMEM/F-12 (Life Technologies).
-
3.
Six-well plates with Nunclon-treated surface (Nunc).
2.3 Generation of PPCs from Undifferentiated hESCs
-
1.
Accutase™ (STEMCELL Technologies).
-
2.
Dispase.
-
3.
AggreWell™ 400 plate (STEMCELL Technologies).
-
4.
Cell strainers (40 and 100 μm pores; Fisher or STEMCELL Technologies).
-
5.
Ultra-Low Attachment plates (Corning).
-
6.
EB formation medium: TeSR™2 supplemented with 10 μM Rho-Associated Coil Kinase (ROCK) inhibitor (STEMCELL Technologies).
-
7.
EB resuspension medium: DMEM/F12, 10 % knockout serum replacement, custom B-27 and N-2 supplements (Life Technologies), 1 ng/ml mouse noggin, 1 ng/ml recombinant human Dkk-1 and 5 ng/ml recombinant human IGF-1 (R&D Systems).
-
8.
PPC differentiation medium: DMEM/F12, custom B-27 and N-2 supplements, 10 ng/ml mouse noggin, 10 ng/ml recombinant human Dkk-1, 10 ng/ml recombinant human IGF-1 and 5 ng/ml recombinant human basic FGF (Life Technologies). When indicated, 20 mM taurine (Sigma) and 40 ng/ml triiodothyronine (T3; Sigma) are added to the differentiation medium as specified below.
-
9.
The factors: Noggin, Dkk-1 and basic FGF are dissolved in Dulbecco’s PBS (DPBS; Life Technologies) containing 0.1 % bovine serum albumin (BSA) at a concentration of 10, 100 or 2 μg/ml, respectively. IGF-1 was dissolved in DPBS at a concentration of 200 μg/ml. All factors should be stored in single-use aliquots at −80 °C.
-
10.
T3 is first dissolved in 1 N NaOH at 100 mg/ml and then diluted in DMEM to 40 μg/ml. Stored in single-use aliquots at −80 °C. Taurine powder can be directly dissolved in PPC differentiation medium and filter-sterilized before use.
2.4 Magnetic Removal of Undifferentiated Cells
-
1.
SSEA-4 Positive Selection kit (STEMCELL Technologies).
-
2.
EasySep® Magnet (STEMCELL Technologies).
-
3.
Accutase.
-
4.
Polystyene 5 ml round-bottom tubes (Falcon).
-
5.
Selection buffer: DPBS, 2 % Fetal Bovine Serum (FBS) and 1 mM EDTA.
2.5 Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
-
1.
Aurum™ Total RNA Fatty and Fibrous Tissue kit (Bio-Rad).
-
2.
iScript™ cDNA Synthesis kit (Bio-Rad).
-
3.
UltraPure™ DNase/RNase-free Distilled water (Life Technologies).
-
4.
FAM- or VIC-labeled TaqMan® Gene Expression primers (Life Technologies).
-
5.
TaqMan® Universal Master Mix (Life Technologies).
-
6.
MicroAmp® 96-well reaction plate and the corresponding Optical Adhesive Covers (Applied Biosystems).
-
7.
Real Time PCR System, such as the ViiA™ 7 (Applied Biosystems).
2.6 Flow Cytometry
2.6.1 Cell Surface Labeling
-
1.
Accutase.
-
2.
Blocking buffer: DPBS with 10 % human Serum (Sigma).
-
3.
Washing and resuspension buffer: DPBS with 2 % FBS.
-
4.
Polystyene 5 ml round-bottom tubes.
-
5.
Primary antibody, as desired (see note 1).
-
6.
Alexa Fluorophore-conjugated secondary antibody.
-
7.
Propidium iodide (Life Technologies).
2.6.2 Intracellular Labeling
-
1.
Accutase.
-
2.
DPBS with 2 % FBS.
-
3.
DPBS with 2 % paraformaldehyde (PFA).
-
4.
Saponin Permeabilization Buffer (SPB): DPBS, 0.1 % Bovine Serum Albumin (BSA) and 0.2 % saponin (Sigma).
-
5.
Polystyene 5 ml round-bottom tubes.
-
6.
Primary antibody, as desired (see note 2).
-
7.
Alexa Fluorophore-conjugated secondary antibody.
3 Methods
3.1 Coating Plates with Matrigel™
Both human embryonic stem cells (hESCs) and differentiating PPCs grow on tissue culture plates coated with Matrigel. Matrigel is supplied as a frozen solution. Thaw on ice overnight in a 4 °C refrigerator and make single-use aliquots into precooled 1.5 ml centrifuge tubes using precooled pipette tips (see note 3). It is important to keep all the solutions cold prior to coating, as Matrigel will solidify rapidly at room temperature.
-
1.
For coating a 6-well plate, remove an aliquot of 0.5 mg Matrigel from the −80 °C freezer and place on ice (see note 3).
-
2.
Dispense 5 ml of cold DMEM/F-12 into a 15 ml conical tube.
-
3.
Add 1 ml cold DMEM/F-12 to the frozen Matrigel aliquot and pipette up and down to dissolve. Quickly transfer the Matrigel solution to the conical tube and mix well.
-
4.
Distribute 1 ml of diluted Matrigel per well, swirl the plate to ensure uniform coating and incubate at room temperature for at least 1 h.
-
5.
If the coated plate is to be used more than 1 h later, add 1 ml of DMEM/F-12 to prevent the surface from drying (see note 4).
3.2 Culture of Undifferentiated Human Embryonic Stem Cells
-
1.
Culture the hESC line WA09 in TeSR™2 growth medium in a 37 °C incubator in 5 % CO2. Change medium daily and manually remove spontaneously differentiating cells, as needed.
-
2.
Passage cells every 4–6 days by incubating the cells with 1 ml Dispase per well. When colonies start to “curl” and detach from the surface (typically 5–10 min), remove the Dispase solution and carefully wash colonies two times with DMEM/F12. Using a glass pipette and 2 ml of fresh DMEM/F-12, the colonies should be gently scraped off the surface and collected in a 15 ml conical tube. Use 2 ml fresh DMEM/F-12 to collect the remaining colonies and add to the conical tube. Centrifuge the cell suspension for 5 min at 300 × g and resuspend the cell pellet in fresh TeSR™2 by slowly pipetting up and down to break the colonies to smaller pieces. Distribute the colonies on Matrigel coated plates, according to the split ratio required, and slowly move the plates side to side and front to back to uniformly disperse the colonies.
3.3 Generation of PPCs from hESCs
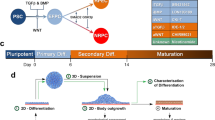
The differentiation of hESCs to PPCs is a multistep procedure (Fig. 1). The first step is the generation of size-controlled embryoid bodies (EBs), which we have recently shown can be used to enhance the yield of PPCs (15). The generation of size-controlled EBs can be performed in two ways; manual selection of medium-sized EBs, which result in the highest proportion of PPCs when separated from a mixed-size EB population, or by using the commercially available AggreWell 400 plate to generate 1,000-cell EBs (see note 5). The protocol below outlines both methods. Representative images of the EBs generated by this protocol can be seen in Fig. 2.

Schematic representation of the photoreceptor precursor cells differentiation protocol

Representative images of the EB sizes produced in this protocol. Scale bar is 400 μm
3.3.1 Generation and Manual Selection of Medium-Size EBs
-
1.
Incubate hESCs with Dispase at 37 °C until colonies begin to peel off the plate (typically 20–30 min). Gently wash the colonies with the Dispase solution, transfer to a 15 ml conical tube, and collect residual colonies with 2 ml of DMEM/F-12 per well. Let the colonies settle to the bottom of the tube for ~5 min, aspirate the supernatant, and wash the colonies with 4–6 ml of fresh DMEM/F-12. After colonies have settled down and the supernatant aspirated the second time, resuspend the colonies in EB resuspension buffer and place on Ultra-Low Attachment plates in a volume of 5 ml per well for 3 days in the 37 °C incubator (see note 6). Move the plates slowly from side to side and front to back 3–4 times daily, to prevent sticking of EBs.
-
2.
At day 4 of incubation, place the plate in the sterile tissue culture hood and manually remove the largest EBs (diameter of ~400 μm or more) using a 200 μl pipette tip, and pass the remaining suspension over 100 μm nylon cell strainer (see note 7). Single cells and small EBs will flow through. Turn the cell strainer so that the EBs are facing a new 50 ml conical tube and elute the “trapped” EBs in 2–4 ml PPC differentiation medium. Distribute the eluted EBs on Matrigel-coated plates and move the plate from side to side and front to back to evenly distribute the EBs (see note 8).
3.3.2 Manufacture of 1,000-Cell EBs Using AggreWell Plates
-
1.
Wash one or more wells of hESCs once with DMEM/F-12, aspirate and replace with 0.75 ml of room temperature Accutase per well. Incubate the cells at 37 ° C for 5–10 min, until most cells have dislodged. With a 1 ml tip, pipette the cells several times to dissociate any remaining clumps and collect the cell suspension of all wells into a conical tube. Collect the remaining cells with 4 ml DMEM/F-12 per well into the same tube and pellet cells by centrifugation for 5 min at 300 × g. Resuspend the cell pellet in 1 ml EB formation media and count the viable cells with trypan blue. To generate 1,200 EBs with 1,000 cells per EB, add ~1.2 × 106 cells to one well of the AggreWell400 and centrifuge the AggreWell plate for 3 min at 100 × g. Place the AggreWell plate in a 37 ° C incubator for 24 h.
-
2.
Harvest EBs by pipetting the medium in the well up and down several times with a 1 ml tip to dislodge most of the EBs. Pass the EB suspension through a 40 μm cell strainer and wash the AggreWell five more times with 1 ml of DMEM/F-12 to dislodge the remaining EBs. Pass all the suspensions through the same cell strainer.
-
3.
Invert the cell strainer so that the EBs are facing a 50 ml conical tube. Release the EBs from the cell strainer in 5 ml of EB resuspension buffer and distribute evenly to two or more wells (see note 9). The final volume in each well should be 5 ml of EB resuspension buffer.
-
4.
Incubate the EBs for 3 days in the 37 °C incubator. To prevent sticking of EBs, move the plate slowly from side to side and front to back 3–4 times daily. At day 4 of incubation, collect all the EBs in a 50 ml conical tube and centrifuge for 5 min at 100 × g. Aspirate the medium and carefully resuspend the EBs in PPC differentiation medium (2 ml per well, see note 10). Evenly distribute the EBs on Matrigel coated plates by moving the plate from side to side and front to back.
3.3.3 Adherent Growth of PPCs
-
1.
Culture the cells, originating from EBs generated by both manual size selection and AggreWell plates, in PPC differentiation medium for 2 weeks without passaging. Change the culture medium every other day. On the 7th day of adherent growth, supplement the PPC differentiation medium with taurine and T3 (20 mM and 40 ng/ml, respectively) and continue with the same feeding schedule.
-
2.
After 2 weeks of adherent growth, cells may be frozen in PPC differentiation medium containing 10 % DMSO for future use, or they may be harvested for analysis of differentiation stage.
3.4 Analysis of Differentiation Stage Using RT-qPCR
Relative gene expression can be quantified with the TaqMan® primer/probe system. Use FAM-labeled primers for the gene of interest and VIC-labeled primers for an internal control (such as GAPDH). All the details described below refer to the use of the TaqMan® system and the ViiA™ 7 Real Time PCR system.
-
1.
Following 17 days of differentiation, isolate total RNA from cell cultures using the Aurum™ Total RNA Fatty and Fibrous Tissue kit according to the manufacturer’s instructions.
-
2.
For each sample to be processed, use the iScript™ cDNA Synthesis kit to reverse transcribe 1 μg of total RNA according to the manufacturer’s instructions.
-
3.
Dilute the resulting cDNA 1:10 by adding 180 μl UltraPure™ distilled water. Further dilute a portion of the 10x diluted sample 1:2 to make a “working stock” of 20× (see note 11). All samples should be stored at −20 °C.
-
4.
The RT-qPCR reaction mix is as follows (scale up according to the number of samples required):
-
6 μl UltraPure™ distilled water
-
10 μl TaqMan® Universal Master Mix
-
1 μl FAM-labeled TaqMan® Gene Expression assay (gene of interest; see note 12)
-
1 μl VIC-labeled TaqMan® Gene Expression assay (internal control)
-
2 μl cDNA
-
20 μl total volume per reaction
-
-
5.
Load all samples into the 96-well plate, seal the plate with an optical adhesive film, centrifuge the plate briefly and load the plate into the instrument. Thermal cycling conditions should be as follows: initial incubation consisting of 2 min at 50 °C and then 10 min at 95 °C, followed by 40 cycles of: 15 s at 95 °C and 1 min at 60 °C.
-
6.
Analyze the data using the comparative C T method to calculate the fold change in the expression of the gene of interest compared to a calibrator (such as undifferentiated cells).
3.5 Flow Cytometry
The protocols detailed below have been optimized for the antibodies we used to detect specific cell surface and intracellular antigens (SSEA-4 and CRX, respectively; see notes 1 and 2).
3.5.1 Cell Surface Labeling
-
1.
To collect and make a single cell suspension, use Accutase as described in Sect. 3.3.2. Note that differentiated cells detach quicker with Accutase than undifferentiated hESCs (less than 5 min). Use ice-cold DPBS to resuspend the resulting cell pellet.
-
2.
Count the cells and distribute ~2 × 105 cells per sample into a 1.5 ml conical tube.
-
3.
Centrifuge the tubes at 300 × g for 5 min and resuspend the cell pellet in 100 μl DPBS/10 % human serum. Block on ice for ~10 min.
-
4.
Add 1 μl of anti-SSEA-4 antibody directly to each sample, vortex briefly, and incubate on ice for 30 min.
-
5.
Add 1 ml of DPBS/2%FBS, vortex briefly, and centrifuge the tubes at 300 × g for 5 min.
-
6.
Aspirate the supernatant and resuspend each cell pellet in 100 μl of Alexa-488 goat anti-mouse IgG diluted 1:400 in DPBS/2 % FBS.
-
7.
Vortex briefly and incubate on ice in the dark for 15 min.
-
8.
Aspirate the supernatant, add 1 ml of DPBS/2%FBS, vortex briefly, and centrifuge the tubes at 300 × g for 5 min.
-
9.
Aspirate the supernatant and resuspend the cell pellets in 300 μl of DPBS/2%FBS supplemented with propidium iodide (diluted 1:1,000). Transfer the cell suspension into a 5 ml round-bottom tube.
-
10.
Analyze on a flow cytometer instrument, such as the BD Biosciences FACSCalibur.
3.5.2 Intracellular Labeling
-
1.
Prepare a single-cell suspension as described above, but resuspend the cell pellet in ice-cold DPBS/2 % FBS.
-
2.
Count the cells and distribute ~8 × 105 cells per sample into a 1.5 ml conical tube.
-
3.
Centrifuge the tubes at 300 × g for 5 min and fix the cells by resuspending the pellet in 250 μl DPBS/2 % PFA and incubating for 15 min on ice.
-
4.
Add 1 ml of DPBS/2%FBS, mix carefully, and centrifuge the tubes at 300 × g for 5 min.
-
5.
Aspirate the supernatant and permeabilize the cells by resuspending the pellet with 0.5 ml Saponin Permeabilization Buffer (SPB) and then incubate for 15 min at room temperature.
-
6.
Centrifuge the tubes at 300 × g for 5 min, aspirate the supernatant and resuspend each cell pellet in 100 μl CRX antibody diluted 1:200 in SPB.
-
7.
Incubate at room temperature for 30 min.
-
8.
Perform two washes by the addition of 1 ml of SPB to each sample, mix carefully and centrifuge at 300 × g for 5 min.
-
9.
After aspirating the supernatant for the second time, resuspend each cell pellet in 100 μl of Alexa-488 goat anti-rabbit IgG diluted 1:400 in SPB.
-
10.
Incubate on ice for 20 min and perform two washes as described above.
-
11.
Resuspend cell pellets in 300 μl of DPBS/2 % FBS and transfer the cell suspension into a 5 ml round-bottom tube.
-
12.
Analyze on a flow cytometer instrument, such as the BD Biosciences FACSCalibur.
3.6 Magnetic Removal of Residual Undifferentiated Cells Using EasySep® Magnetic Selection System
In order to reduce the risk of tumors resulting from residual undifferentiated cells, cells expressing hESC surface markers, such as stage-specific embryonic antigen-4 (SSEA-4), can be magnetically removed from the total cell population to enrich for the desired differentiated cells. The procedure below describes the depletion of SSEA-4 expressing cells using the EasySep® positive selection kit.
-
1.
Following 17 days of differentiation collect the total cell population with Accutase, as described above (Sect. 3.3.2) and resuspend in 100 μl of selection buffer (see note 13). Note that differentiated cells detach quicker with Accutase than undifferentiated hESCs (less than 5 min).
-
2.
Add 10 μl Selection Cocktail, mix well and incubate at room temperature for 15 min.
-
3.
Mix Magnetic nanoparticles by pipetting up and down several times and add 5 μl to the cell suspension. Mix well and incubate at room temperature for 15 min.
-
4.
Top up the cell suspension with selection buffer to a volume of 2.5 ml. Mix well, place the tube into the EasySep magnet, and set aside for 10 min.
-
5.
Pick up the magnet and invert the magnet and tube, pouring the desired fraction into a new 5 ml tube.
-
6.
Remove the tube with the unwanted cells and place the tube containing the desired fraction inside the magnet for a second round of magnetic separation. Repeat step 5.
-
7.
The enriched cells can now be used for further experiments.
4 Notes
-
1.
For cell surface labeling, we used the anti-human SSEA-4 monoclonal antibody, clone MC-817-70 (gift from Dr. Connie Eaves), which is now commercially available from several companies (e.g. STEMCELL Technologies and Millipore). We used this antibody at a 1:100 dilution.
-
2.
For intracellular labeling, we used the anti-CRX (cone rod homeobox) rabbit polyclonal antibody developed in our lab (16). When using the above protocol it is crucial to titrate the relevant antibody on a sample that is known to express the protein of interest.
-
3.
As mentioned above, Matrigel solidifies rapidly at room temperature. When preparing single-use aliquots for future use, make sure the tubes and tips are precooled in a −80 °C freezer a day before. One 6-well culture dish requires 0.5 mg Matrigel. It is useful to make aliquots of 0.5, 1, and 2 mg Matrigel to coat 1, 2, and 4 plates, respectively.
-
4.
Coated plates can be stored at 4 °C for up to 1 week but need to be warmed to room temperature for at least 30 min before use.
-
5.
We saw a significant improvement in the yield of PPCs when using the Aggrewell™400 to produce 1,000-cell EBs. However, if AggreWell plates are not available, medium sized EBs can be separated from a mixed size EB population to yield the highest percentage of PPCs in comparison to the mixed-size EB population and the small- and large-sized EBs. Refer to our 2013 paper (15) regarding size-controlled EBs.
-
6.
EBs generated from one confluent well of undifferentiated hESCs are transferred into one well of an Ultra-Low Attachment plate. This can be scaled up in the same ratio to generate larger amounts of PPCs.
-
7.
The largest EBs can be distinguished visually. Using a pipette with a 200 μl pipette tip, these EBs can be picked up and discarded.
-
8.
Typically, medium size EBs from several wells of mixed-size EBs were pooled together and evenly distributed onto Matrigel coated plates in a ratio of 1:1.5–1:2 (for example, from two wells of mixed-size EBs in suspension, three wells of medium size EBs were cultured).
-
9.
STEMCELL Technologies recommends incubating <1,000 EBs, generated in AggreWell plates, in one well of low attachment plate. When we generate EBs in one AggreWell we transfer the EBs into two low attachment wells, but when generating 2–3 AggreWells the resulting EBs are incubated in 3–4 low attachment wells, respectively.
-
10.
The plating density of AggreWell EBs should be 500–600 EBs per one well of a 6-well plate, assuming 1,200 EBs are harvested from each AggreWell used.
-
11.
cDNA was typically divided to a “working” sample (20×) and a “back up” sample (10×), to be used later on or in the case of an accidental contamination. The “working” stock is at a concentration of approximately 2.5 ng/μl and we use 2 μl (5 ng) for each RT-qPCR reaction.
-
12.
Each RT-qPCR reaction contains FAM-labeled primer for the gene of interest and a VIC-labeled primer to amplify GAPDH. This way we can normalize all reactions to one internal control in the same reaction.
-
13.
The manufacturer recommends using 100 μl selection buffer for samples containing up to 1.25 × 107 cells. In our laboratory we have not used samples that required a higher volume of selection buffer. However, this volume can be scaled up as required.
References
MacLaren RE, Pearson RA, MacNeil A et al (2006) Retinal repair by transplantation of photoreceptor precursors. Nature 444:203–207
Inoue T, Coles BL, Dorval K et al (2010) Maximizing functional photoreceptor differentiation from adult human retinal stem cells. Stem Cells 28:489–500
Wohl SG, Schmeer CW, Isenmann S (2012) Neurogenic potential of stem/progenitor-like cells in the adult mammalian eye. Prog Retin Eye Res 31:213–242
La TA, Lamba DA, Jayabalu A et al (2012) Production and transplantation of retinal cells from human and mouse embryonic stem cells. Methods Mol Biol 884:229–246
Phillips MJ, Wallace KA, Dickerson SJ et al (2012) Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest Ophthalmol Vis Sci 53:2007–2019
Idelson M, Alper R, Obolensky A et al (2009) Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 5:396–408
Lu B, Malcuit C, Wang S et al (2009) Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells 27:2126–2135
Lamba DA, Karl MO, Ware CB et al (2006) Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A 103:12769–12774
Osakada F, Ikeda H, Mandai M et al (2008) Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol 26:215–224
Bauwens CL, Peerani R, Niebruegge S et al (2008) Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells 26:2300–2310
Bratt-Leal AM, Carpenedo RL, McDevitt TC (2009) Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnol Prog 25:43–51
Mellough CB, Sernagor E, Moreno-Gimeno I et al (2012) Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells 30:673–686
Brzezinski JA, Lamba DA, Reh TA (2010) Blimp1 controls photoreceptor versus bipolar cell fate choice during retinal development. Development 137:619–629
Katoh K, Omori Y, Onishi A et al (2010) Blimp1 suppresses Chx10 expression in differentiating retinal photoreceptor precursors to ensure proper photoreceptor development. J Neurosci 30:6515–6526
Yanai A, Laver CR, Joe AW et al (2013) Differentiation of human embryonic stem cells using size-controlled embryoid bodies and negative cell selection in the production of photoreceptor precursor cells. Tissue Eng Part C Methods 19:755–764
Bibb LC, Holt JK, Tarttelin EE et al (2001) Temporal and spatial expression patterns of the CRX transcription factor and its downstream targets. Critical differences during human and mouse eye development. Hum Mol Genet 10:1571–1579
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this protocol
Cite this protocol
Yanai, A., Laver, C., Joe, A.W., Gregory-Evans, K. (2013). Efficient Production of Photoreceptor Precursor Cells from Human Embryonic Stem Cells. In: Turksen, K. (eds) Human Embryonic Stem Cell Protocols. Methods in Molecular Biology, vol 1307. Humana Press, New York, NY. https://doi.org/10.1007/7651_2013_57
Download citation
DOI: https://doi.org/10.1007/7651_2013_57
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2667-1
Online ISBN: 978-1-4939-2668-8
eBook Packages: Springer Protocols