
Abstract
Two-pore domain potassium channels are formed by subunits that each contain two pore-loops moieties. Whether the channels are expressed in yeast or the human central nervous system, two subunits come together to form a single potassium selective pore. TOK1, the first two-domain channel was cloned from Saccharomyces cerevisiae in 1995 and soon thereafter, 15 distinct K2P subunits were identified in the human genome. The human K2P channels are stratified into six K2P subfamilies based on sequence as well as physiological or pharmacological similarities. Functional K2P channels pass background (or “leak”) K+ currents that shape the membrane potential and excitability of cells in a broad range of tissues. In the years since they were first described, classical functional assays, latterly coupled with state-of-the-art structural and computational studies have revealed the mechanistic basis of K2P channel gating in response to specific physicochemical or pharmacological stimuli. The growing appreciation that K2P channels can play a pivotal role in the pathophysiology of a growing spectrum of diseases makes a compelling case for K2P channels as targets for drug discovery. Here, we summarize recent advances in unraveling the structure, function, and pharmacology of the K2P channels.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 An Introduction to Two-Pore Domain Potassium Channels
Potassium (K+) channels are a superfamily of multi-subunit membrane proteins that are fundamental for physiology throughout the tree of life. K+ channels are complex protein machines with a simple purpose: they open and close (gate) in a coordinated manner that allows the conduction of K+ ions down their electrochemical gradient, typically from the intracellular to extracellular space in mammalian tissues. Gating occurs in response to a panoply of stimuli and shapes the resting membrane potential and the dynamics of cellular excitability by regulating the flux of K+ ions. Thus, K+ channels are essential for many biological processes including neuronal, muscular, and cardiac function (Enyedi and Czirjak 2010).
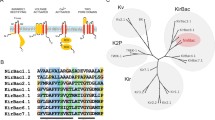
The superfamily of K+ channels is stratified into distinct subfamilies based on structural similarities, namely the number of transmembrane domains and pore forming domains present in each subunit. The largest subfamily includes the voltage (KV) channels and calcium activated (KCa) channels which are characterized by one reentrant pore loop (P-loop) and (typically) six transmembrane domains per subunit; holo-channels are tetramers. The inwardly rectifying K+ channels (KIR) also form as tetramers in which each subunit consists of a single P-loop and two transmembrane domains. The notion that all K+ channels are tetramers changed in 1995 when TOK1 was cloned from Saccharomyces cerevisiae (Ketchum et al. 1995). TOK1 channels have a distinct architecture: Functional channels are dimers of subunits with eight transmembrane domains (M1-M8), intracellular amino- and carboxy-terminal tails, and two reentrant P-loops located between transmembrane domains M5-M6 and M7-M8 (Fig. 1) (Ketchum et al. 1995). Although TOK channels are not found beyond fungi, K+ channel subunits with two P-loops from higher organisms were described soon after.

An overview of the two-pore domain potassium channels. (a) A phylogenic tree created using MEGA software to show the relatedness of the K2P subunits expressed in humans. The IUPHAR-standardized names of each subunit are in black. The phylogenic divisions of K2P subunits fall into the different subfamilies indicated, these are named for their physiological or pharmacological properties. (b) A cartoon depicting how the transmembrane domains and P-loops of human K2P channels are organized to create a single channel pore from a dimer of subunits. (c) A topological cartoon to show the organization of the 8-transmembrane domains (M1-M8) that comprise a TOK subunit. Note the two pore-loops (P1 and P2) between M5-M6 and M7-M8. (d) A topological cartoon to show the organization of the 4-transmembrane domains (M1–M4) that comprise a K2P subunit. Note the two pore-loops (P1 and P2) between M1-M2 and M3-M4
Unlike the KV and KIR channel subfamilies, discovery of the K2P channels was made possible using genome database mining rather than by a molecular cloning strategy (Goldstein et al. 1996; Lesage et al. 1996b; Yang and Jan 2013). In 1996, K2PØ (also called KCNKØ, or dORK) was cloned from Drosophila melanogaster and K2P1 (also called KCNK1 or TWIK1) was cloned from human kidney (Lesage et al. 1996b). Holo two-pore domain K+ (K2P) channels are dimers of subunits, with each subunit contributing two-P loops and four transmembrane domains to the structure (Feliciangeli et al. 2015; Goldstein et al. 2001; Guyenet et al. 2019; Kollewe et al. 2009; Medhurst et al. 2001; Yang and Jan 2013). In general, heterologous expression of K2P channels produces outward K+ currents under physiological conditions. The phenomenon of “background’” or “leak” K+ currents has been appreciated since the 1940s and was ratified in the membrane equations of Hodgkin and Huxley (Enyedi and Czirjak 2010; Goldman 1943; Goldstein et al. 1996, 2001; Hodgkin and Huxley 1952; Lesage et al. 1996b). In the last ~25 years, numerous studies have confirmed the central role that K2P channels play in determining the membrane potential in a broad range of excitable and non-excitable cell types (Goldstein et al. 2001) (Fig. 1).
K+ channel subunits are identified by a common sequence of amino acid residues that comprise the selectivity filter for K+ ions within the conduction pore of the channel (see also chapter “Comparison of K+ Channel Families”). This evolutionarily conserved structural domain is constructed from four P-loops that are held in position in the membrane between two transmembrane helixes that form the channel corpus. The surrounding architecture of the channel is comprised of transmembrane domains that correlate with their unique physiological functions. This architectural arrangement controls when the channels open and for how long (open probability), allowing the conductance of K+ ions down their electrochemical gradient through the selectivity filter of the pore with high fidelity (Doyle et al. 1998). Gating of K2P channels is regulated by a plethora of physicochemical and mechanical stimuli including stretch, temperature, pH, and various cell signaling, and second-messenger pathways (Chemin et al. 2007; Honore 2007; Lotshaw 2007). Despite significant progress, the mechanistic basis by which each of these stimuli influences the gating machinery, and in turn the activity of K2P channels, remains a matter of ongoing research.
A growing body of work, first using a classical structure-function approach, and more recently via snapshots of channel structures paired with molecular dynamics simulations has revealed that extrinsic regulators typically influence the open probability of K2P channels via allosteric pathways and via c-type gating in particular (Bagriantsev et al. 2011, 2012; Cohen et al. 2008; Lolicato et al. 2014, 2020; Piechotta et al. 2011; Schewe et al. 2016; Zilberberg et al. 2001). Compelling evidence supports that this mode of gating results from constriction of the extracellular region of the channel, occluding the conduction pathway for K+ ions (Hoshi et al. 1991; Yellen 1998). In common with data from other types of K+ channel, the c-type gating of K2P channels occurs at the selectivity (SF) (Bagriantsev et al. 2011; Cohen et al. 2008; Piechotta et al. 2011). For example, binding of high affinity quaternary ammonium (QA) deep within the K2P2 channel selectivity filter revealed that the gating process was occurring at the SF (Piechotta et al. 2011; Schewe et al. 2016). Furthermore, c-type gating works in concert with the carboxy-terminal tail of the channel to mediate the response to physicochemical stimuli such as temperature and mechanical force (Bagriantsev et al. 2011, 2012), indicating that allosteric interactions can transcend the channel corpus (Bagriantsev et al. 2011, 2012; Zilberberg et al. 2001).
K2P channels assemble as dimers with each subunit composed of four transmembrane domains and two P-loops, one between the M1 and M2 helices and one between the M3 and M4 helices (Fig. 1b, c) (Brohawn et al. 2012; Goldstein et al. 2001; Kollewe et al. 2009; Lolicato et al. 2017; Miller and Long 2012). In addition, the first extracellular loop of each K2P subunit (linking the M1 to M2 helices) contributes to a “cap-domain” located above the axis of the K+ selectivity filter. This structure bifurcates the pathway for K+ ions and is proposed to render K2P channels insensitive to many classical K+ channel blockers (such as protein toxins) by shielding the extracellular mouth of the pore via steric hindrance (Fig. 2) (Lolicato et al. 2017; Miller and Long 2012; Piechotta et al. 2011; Zuniga and Zuniga 2016). The extracellular cap-domain is formed when the two extracellular helices (E1 and E2) assemble (Şterbuleac 2019). The cap-domain has not been observed in other K+ channels and was first revealed upon elucidation of the structure of K2P1 (TWIK1) and K2P4 (TRAAK) by X-ray crystallography. The placement and movement of the transmembrane helices allow the channel to adopt the two unique states, “up” and “down” (Brohawn et al. 2012; Miller and Long 2012; Şterbuleac 2019). Transitioning from the “up” to the “down” states reveals fenestrations which allow molecules to interact with the channel’s inner pore (Feliciangeli et al. 2015; Şterbuleac 2019). The cap-domain has been observed on all K2P channel structures solved to date, including K2P1, K2P2, K2P3, K2P4, and K2P10 (Brohawn et al. 2012; Dong et al. 2015; Lolicato et al. 2017; Miller and Long 2012; Pope et al. 2020; Rödström et al. 2020).

The architecture of a K2P channel. An overview of the three-dimensional architecture of K2P2 (Crystal structure, PDB ID: 6CQ6) showing views from the side, the top (extracellular), and the bottom (intracellular) of the channel. The helices of one subunit are colored to reflect the segments of a single subunit: four transmembrane domains (M1–M4); two portions of extracellular loop1 that contribute to the cap-domain (EC1 and EC2); two selectivity filter helices (SFH1 and SFH2), one contributes to each P-loops. Images were rendered from the PDB files indicated using UCSF Chimera software (https://www.rbvi.ucsf.edu/chimera)
The unique topology of K2P channels is shared among 15 human genes designated “KCNK” by the Human Gene Organization nomenclature (Lesage and Barhanin 2011; Yang and Jan 2013) (Table 1). These genes encode 15 K2P channel subunits that are classified into six subfamilies based on similarities in structural and functional properties: tandem of pore domains in a weak inward rectifying K+ channel (TWIK); TWIK-related K+ channel (TREK); TWIK-related acid sensitive K+ channel (TASK); TWIK-related alkaline pH-activated K+ channel (TALK); TWIK-related spinal cord K+ channel (TRESK); and tandem pore domain halothane-inhibited K+ channel (THIK) (Table 1). To mitigate the variance in the pharmacological and physiological attributes that were subsequently associated with different members of each subfamily the nomenclature of the K2P channels “K2PX” was designated by the International Union of Basic and Clinical Pharmacology (IUPHAR) (Table 1). However, the descriptive names of these channels have utility and remain in common use.
2 The Role of K2P Channels in Pathology and Pain Signaling
Numerous studies have linked K2P channels to cardiac and neuronal diseases. In this section we highlight examples. K2P channels have also been linked to neurodevelopmental disorders including Birk-Barel syndrome. K2P channels have also been linked to neurodevelopmental disorders including Birk-Barel syndrome. This rare genetic disease is associated with mutation of the glycine residue at position 236 (Gly236) to arginine (a positively charged residue) in the KCNK9 gene (encodes K2P9, also called TASK3) and is characterized by intellectual disability, hypotonia and hyperactivity. Two-electrode voltage-clamp (TEVC) studies of WT and mutant channels expressed in Xenopus oocytes revealed that while wild type (WT) channels passed measurable currents, mutant channels had no measurable current. In addition, co-expression of mutant channel with either WT or K2P3 channels (which form functional heterodimers with K2P9) resulted in decreased current (Barel et al. 2008). Using the bacterial K+ channel KcsA to generate a homology model structure, Barel and colleagues determined that the expected location of the Gly236 residue was in the ion conduction pathway. It was therefore postulated that a mutation to arginine may result in the disruption of physical and electrostatic interactions in the pore that would diminish current by impeding the conduction of K+ ions.
KCNK18 gene encodes for K2P18 or the TRESK channel and is primarily expressed in trigeminal root ganglion (TRG) and dorsal root ganglion (DRG). Truncations and other mutations in KCNK18 have been associated with familial migraine (Lafrenière et al. 2010). Expression of mutant K2P18 channels resulted in decreased current density when expressed in oocytes. This observation led Lafrenière and colleagues to propose that an increase in the functional expression of WT K2P18 could protect against migraines and that as yet unidentified mutations in KCNK18 could lead to an increase in migraine risk (Lafrenière et al. 2010).
Following whole exome sequencing (WES) studies conducted on patients with arrhythmic disorders, Decher and colleagues identified a heterozygous K2P2 mutation (Ile267Thr) in a patient with right ventricular outflow tract ventricular tachycardia (RVOT-VT). When expressed in Xenopus oocytes, K2P2 Ile267Thr channels have decreased current compared to WT channels. Further, co-expression of WT and mutant channel resulted in reduced current density in what is known as a “dominant-negative” behavior (Decher et al. 2017). It was found that the mutant channel was more permeable to sodium (Na+) ions, unlike WT channels. This change in ion selectivity of the channel was attributed to the mutation of the isoleucine residue in the second pore loop to threonine. A change in the selectivity of K2P channels that permits an increase in the conductance of sodium has previously been observed for development-related alternative-translation initiated truncation variants of K2P2 and for mutation in K2P1 (Thomas et al. 2008). Following their observations, Decher and colleagues sought to reverse this defect in ion selectivity by finding drugs that would “rescue” the channel. Incubation of the channel with the following K2P2 blockers verapamil (62 μM) and fluoxetine (80 μM) and activators 2-APB (50 μM) and riluzole (500 μM) did not alter the selectivity of the channel (Decher et al. 2017). In contrast 5 μM of BL-1249 rescued channel function. Authors hypothesized that BL-1249 may be binding at a unique site that differs from the other compounds.
K2P channels are expressed ubiquitously across excitable and non-excitable tissues (Lesage 2003; Lesage and Lazdunski 2000). Several K2P channels are expressed in the TRG and DRG (Mathie and Veale 2015). The DRG and TRG somatosensory neurons give rise to the peripheral axonal fibers that innervate various tissues including the skin, muscle, and viscera and ascend to the spinal cord (DRG) or brainstem (TRG) (Mathie and Veale 2015; Plant 2012). Damage-sensing (nociceptive) somatosensory neurons detect and respond to noxious stimuli through activation of Aδ fibers which are lightly myelinated neurons that respond to localized pain, and via C-fibers which are unmyelinated neurons that are activated by a range of noxious stimuli (Plant 2012). Aα and Aβ fibers are myelinated fibers that respond to innocuous, mainly mechanical stimuli (Plant 2012). K2P channels expressed in the DRG and TRG modulate neuronal excitability and response to noxious and innocuous mechanical stimuli.
Using a rat neuropathic pain model, Pollema and colleagues demonstrated that following spared nerve injury (SNI) levels of mRNA for KCNK3 and KCNK9 (that encode for K2P3 and K2P9 channels, respectively), were downregulated compared to sham controls. Downregulation of these K2P channels following SNI implicates these channels in neuropathic pain phenotypes. Interestingly, four weeks post SNI, only mRNA for KCNK1 (which encodes for K2P1) remained downregulated hinting at the importance of this channel in maintaining the neuropathic pain phenotype (Pollema-Mays et al. 2013). Contrary to this study, another group found that while still using the SNI model, intrathecal delivery of K2P18 in an adenovirus vector reduced the response of rats to neuropathic pain (Zhou et al. 2013).
2.1 K2P Channel Pharmacology
Although multiple lines of evidence support a role for K2P channels in pain physiology, pharmacological options that target these proteins remain elusive. Given that present pharmacophores lack the ability to selectively inhibit K2P channels, development of selective pharmacological agents is therefore imperative in order to study distinct characteristics of each channel. Intensive efforts to identify selective, potent, and efficacious pharmacophores are in progress. For example, Bagriantsev and colleagues utilized a 384 well plate yeast-based screening assay to identify K2P blockers and activators in a high-throughput fashion. They began by screening a library containing 106,281 small molecules for their ability to inhibit the growth of yeast expressing K2P2. From this screen the library of small molecules was narrowed to 320 compounds that were selected for their ability to inhibit 44–99% of growth (Bagriantsev et al. 2013). A dose-response screen revealed 61 compounds that successfully prevented the growth of yeast expressing K2P2. TEVC experiments conducted in Xenopus oocytes revealed that 2 inhibitors ML45, ML58 and 3 activators ML12, ML42, and ML67 altered K2P2 channel activity (Bagriantsev et al. 2013). Bagriantsev et al. selected the activator ML67 which caused an ~11 fold (EC50 213 ± 1.2 μM) increase in K2P2 channel current for further characterization. Through TEVC experiments it was found that the compound activated closely related channels (K2P10, EC50 ~ 250 μM) but not the more distantly related K2P3 channel. Substitution of a tricyclic ring to the ML67 compound yielded the compound ML67–33 which was 5 times more potent than the other ML-67 derivatives (Bagriantsev et al. 2013). Mutations at the P1 pore helix (Gly1371) and M4 (Trp275) of K2P2 resulted in decreased channel activity. Conversely, triple glycine mutations at the C-terminal lead to channels that could be activated by the compound. As a result, the authors postulated that ML-67-33 mediates its effects on K2P2 activity by modulating the C-type gate. Compounds such as ML-67-33, a selective and potent activator of K2P2 channels, provide an approach by which similar compounds could be developed and assayed. In this chapter we provide a concise summary of the pharmacology and regulation of K2P channels in that they may be explored further toward the development of novel pharmacophores.
3 The THIK Channels: K2P12 and K2P13
The THIK subfamily is composed of THIK2 (K2P12, KCNK12) and THIK1 (K2P13, KCNK13) channels (Girard et al. 2001; Rajan et al. 2001). The mammalian K2P12 and K2P13 channels share 64% homology as well as a similar pore region structure (Renigunta et al. 2014). While K2P13 channels are expressed ubiquitously, K2P12 channels are expressed in the lungs, spleen, and brain (Rajan et al. 2001). When expressed heterologously in Xenopus oocytes, only K2P13 channel activity can be measured while K2P12 channel activity is largely undetectable. K2P13 currents are activated by arachidonic acid and inhibited by halothane, quinidine, and weakly by hypoxia (a ~ 13% reduction compared to control when Po2 is decreased to 20 mmHg) (Table 2) (Campanucci et al. 2005; Enyedi and Czirjak 2010; Feliciangeli et al. 2015; Renigunta et al. 2014).
K2P12 channels are one of five channels: K2P1 (TWIK1), K2P6 (TWIK2), K2P7 (kcnk8), and K2P15 (TASK5) that are classified as electrically silent channels because they do not pass measurable K+ current in either native cells or in heterologous expression systems (Renigunta et al. 2014). Two groups reported that lack of detectable K2P12 channel activity was a result of the channel possibly being sequestered in the endoplasmic reticulum (ER) and thus resulting in low expression of the channel at the cellular membrane (Blin et al. 2014; Chatelain et al. 2013). However, detection of K2P12 channel activity is possible under specific circumstances. Thus, it was found that substitution of a proline residue within M2 helix or deletion of 18 to 19 AA found in the N-terminus (corresponding to an ER retention/retrieval signaling motif) results in the appearance of macroscopic K2P12 activity that is comparable to K2P13 (* Chatelain et al. 2013; Renigunta et al. 2014). Removal of the AAs from the N-terminus however prevented the channel from being activated by arachidonic acid even though it could still be inhibited by both halothane and quinidine (Renigunta et al. 2014). Of great physiological relevance, heterodimerization of K2Ps 12 and 13 results in functional channels presumably because K2P13 masks the ER retention motif on the K2P12 subunit (Bayliss et al. 2019).
4 The TRESK Subfamily: K2P18
The TRESK subfamily contains only the K2P18 channel, encoded by KCNK18 (Sano et al. 2003). Discovery of K2P18 in 2003 was made possible following the completion of the human genome project (Sano et al. 2003). Sano and colleagues utilized the human draft sequencing data to clone the K2P18 subunit from the complementary DNA of the spinal cord. Subsequence expression analysis found mRNA transcript for KCNK18 throughout the central and peripheral nervous systems (Bayliss et al. 2019; Enyedi et al. 2012; Enyedi and Czirjak 2015; Gada and Plant 2019; Tulleuda et al. 2011; Weir et al. 2019). In rodents, expression of K2P18 has also been detected in the spleen, thymus, and testis (Enyedi and Czirjak 2010). K2P18 channels contribute to the leak or background K+ current which plays an important role in the regulation of neuronal excitability (Hwang et al. 2015). When studied using symmetrical K+ solutions, K2P18 channels displayed outward rectification (Lengyel et al. 2018; Sano et al. 2003). Tulleuda and colleagues reported a decrease in channel activity following neuronal injury, which alters neuronal excitability and thus changes “pain pathways.”
K2P18 shares ~19% sequence homology with other members of the K2P family (Lengyel et al. 2018; Sano et al. 2003). Despite this, human K2P18 is predicted to be structurally like the rest of the K2P channels. It however differs in that its intracellular loop found between the second and third transmembrane domains is longer (>120 amino acids (AA) compared to the 20–30 AA in the other K2P channels) and its C-terminal is shorter (30 AA long compared to the ≥120 AA in the other K2P channels) (Enyedi and Czirjak 2015; Sano et al. 2003). In contrast to most other K2P channels, K2P18 has a short C-terminal tail. This structural difference may indicate differential regulation of K2P18, including how regulatory events might allosterically influence the activity of the channel. (Braun et al. 2015).
The activity of K2P18 channels is enhanced by volatile (inhaled) anesthetics (e.g., isoflurane, sevoflurane, halothane, desflurane) but is inhibited by local anesthetics, including bupivacaine, tetracaine, ropivacaine, mepivacaine, lidocaine, as well as unsaturated fatty acids (Table 3) (Czirjak et al. 2004; Liu et al. 2004). Like most K2P channels, K2P18 channels are sensitive to differences in extracellular and intracellular pH, however the degree of sensitivity differs in the human ortholog compared to rodent orthologs (Lotshaw 2007). In contrast to other K2P channels, K2P18 is modulated by the cytosolic concentration of Ca2+ ions. Thus, K2P18 channels are regulated by activation of Gαq-coupled receptors, which lead to downstream release of Ca2+ from intracellular stores (Table 3). However, a series of elegant studies by Czirják et al. showed that the direct application of Ca2+ ions to the inside of the membrane was insufficient to stimulate K2P18 in off-cell patches, suggesting that additional cytoplasmic factors are required to activate the channels (Czirjak et al. 2004). Subsequent studies found that the Ca2+-dependent activation of K2P18 is mediated by the calmodulin-dependent protein phosphatase, calcineurin, which interacts with the C-terminal tail of the channel (Czirjak et al. 2004). This regulatory mechanism that activates K2P18 channels can be inhibited by pharmacological inhibitors of calcineurin such as cyclosporine. In addition, mutant channels that lack the calcineurin binding site are still subject to regulation by a novel-type of protein kinase C (Pergel et al. 2019).
5 The TALK Subfamily: K2P5, K2P16, and K2P17
The TALK family includes the K2P5, (TASK2, KCNK5), K2P16 (TALK1, KCNK16), and K2P17 (TALK2, TASK4, KCNK17) channels (Decher et al. 2001; Girard et al. 2001; Reyes et al. 1998). K2P16 and K2P17 channels share 37% homology (Lotshaw 2007). When K2P5 was first cloned from human kidney it was assigned to the TASK subfamily. However, it was later reassigned to the TALK subfamily because it had more sequence similarity (~30%) to K2P16 and K2P17 and, in addition, its pH sensitivity was in the alkaline range, similar to that of K2P16 and K2P17 (Enyedi and Czirjak 2010; Lotshaw 2007; Reyes et al. 1998). In humans, K2P5 expression has been detected in the kidneys, pancreas, and liver. Transcripts for KCNK5 were also detected in DRG and spinal cord (Medhurst et al. 2001) (Enyedi and Czirjak 2010). In humans, mRNA for KCNK17 has been found in the liver, heart, pancreas, and lungs while K2P16 channels appear to be expressed exclusively in the pancreas (Duprat et al. 2005; Girard et al. 2001; Lotshaw 2007).
All TALK subfamily channels are activated by extracellular and intracellular alkalinization and inhibited by extracellular acidification (Cid et al. 2013) (Table 4). The pH-sensing of K2P5 requires Arg244; substitution of this amino acid with neutral residues abolishes the response of the channel to changes in alkalization of the extracellular pH (pHo) (Niemeyer et al. 2007). Protonation of Arg244 residue lowers K+ occupancy of the selectivity filter resulting in pore-blockade (Cid et al. 2013).
TALK channels are also sensitive to changes in the intracellular pH (pHi) (Niemeyer et al. 2010). It is postulated that lys245, located on the C-terminus of K2P5, acts as a sensor for pHi (Cid et al. 2013). Given the findings, it may be that the regulation of K2P5 channel activity by pHo and pHi occurs via effects on independent gates (Cid et al. 2013; Niemeyer et al. 2010); however, the mechanistic details that subserve this idea are yet to be elucidated.
K2P5 activity can be inhibited by Gβγ subunits of the heterotrimeric G protein (Anazco et al. 2013) (Table 4). Añazco and colleagues suggested that Gβγ modulation plays a role in the channel’s ability to react to changes in cell volume (this is a result of neutralization of a lysine residue in the C-terminus that is important for inhibition by Gβγ). Although modulation of K2P5 by Gβγ is possible, it remains an open question in the field. Evidence to support Gβγ-modulation of K2P channel activity can be found in the K2P2 channels (Woo et al. 2012). Finally, Duprat and colleagues demonstrated that both K2P16 and K2P17 channels can be activated by nitric oxide (NO) and reactive oxygen species (ROS) (Table 4) (Duprat et al. 2005).
6 The TWIK Subfamily: K2P1, K2P6, and K2P7
Following its initial description in 1996, K2P1 (TWIK1, KCNK1) was observed to have low channel activity in heterologous expression systems (Goldstein et al. 1998; Lesage et al. 1996b; Pountney et al. 1999). However, since mRNA transcripts for KCNK1, the gene that encodes for the K2P1 subunit, are found in the kidney, placenta, lungs, heart, and the brain (Gaborit et al. 2007; Lesage et al. 1996b; Talley et al. 2001), several groups pursued potential cellular and biophysical mechanisms that would limit the activity of K2P1 channels. Data to support three hypotheses have been presented: SUMOylation of K2P1 channels at the plasma membrane; rapid endocytosis of K2P1 channels from the plasma membrane, and hydrophobic dewetting of the channel pore.
SUMOylation is an enzyme-mediated post-translational modification pathway that links a ~100 amino acid Small Ubiquitin-like MOdifier (SUMO) protein to the epsilon amine-group of lysine residues in specific motifs (Hay 2005). Although SUMOylation was not thought to occur at the plasma membrane, the process was shown to inhibit the activity of K2P1 channels because K+ selective currents were observed when SUMO was removed from the channel by a SUMO-specific proteases (SENPs), or when the SUMOylation site (K2P1-Lys274) was mutated to prevent SUMO-binding (Plant et al. 2010; Rajan et al. 2005). SUMOylation is now known to regulate the activity of an array of ion channels in multiple tissues. The process is rapid, reversible, and dynamic and is often challenging to capture biochemically. In keeping with observations of numerous soluble SUMO substrates, such as transcriptional regulators, SUMOylation of K2P1 channels is labile and is often not observed when cells and tissues are studied after detergent purification (Feliciangeli et al. 2007; Hay 2005). Therefore, SUMOylation is typically studied in live cells using real-time electrophysiology, spectroscopy, and microscopy (Plant et al. 2010).
Studies in MDCK and HEK293 cells found that the low activity of K2P1 could be attributed to rapid, endocytic recycling of the channel from the plasma membrane (Feliciangeli et al. 2010, 2015). The process is dynamin-dependent based on analysis of a di-isoleucine motif: mutation of Ile293 and Ile294) resulted in measurable currents upon heterologous expression. Further, K2P1 was found to associate with ARF6, a small G protein that modulates endocytosis at the apical surface of epithelial cell (Decressac et al. 2004).
Following the resolution of the crystal structure of human K2P1, molecular dynamic simulations (MDS) of ion permeation identified a “hydrophobic cuff” in the inner vestibule of the channel, below the selectivity filter, comprised of four residues: Leu146 on M2 and Leu261 on M4, from each subunit (Aryal et al. 2014; Miller and Long 2012). MDS revealed that stochastic motion of the cuff restricted the access of water molecules to the internal entrance of the pore, creating an energetic barrier to the permeation of K+. Based on this model, substitution of Leu146 with hydrophilic residues resulted in a K2P1 channel variant that passed robust currents in Xenopus oocytes (Aryal et al. 2014; Chatelain et al. 2012).
Determining how SUMOylation, the hydrophobic gating barrier, and rapid endocytosis contribute individually or together to the regulation of K2P1 in native cells remains an area of active study that is spurred on by the observation that K2P1 knockout mice exhibit altered physiology in several tissues, including pancreatic β cells and the kidney (Chatelain et al. 2012; Nie et al. 2005). Similarly, K2P1 has been shown to play a key physiological and developmental role in the atria of transgenic zebrafish (Christensen et al. 2016). K2P1 has also been shown to mediate arrhythmogenic depolarization of cardiac myocytes exposed to low concentrations of K+ associated with hypokalemia (Gotter et al. 2011). A part of the enigmatic character of K2P1 can be attributed to heterodimerization with K2P3 and K2P9 subunits in rat neurons and with K2P2 in rat astrocytes (Hwang et al. 2014; Plant et al. 2012). The resultant heteromeric channels have distinct properties. For example, the activity of K2P1-K2P3 and K2P1-K2P9 channels is increased by volatile, halogenated ester-based anesthetics and is subject to regulation by the SUMO pathway (Plant et al. 2012).
The TWIK subfamily is also composed of K2P6 (TWIK2, KCNK6) and the K2P7 (Kcnk8, KCNK7) channels. K2P6 was described by two independent groups (Chavez et al. 1999; Pountney et al. 1999) and shares 34% sequence identity with K2P1. In contrast, K2P7 is more closely related to K2P6 (94% homology) (Lesage and Lazdunski 2000; Lotshaw 2007). K2P6 and K2P7 are expressed in peripheral tissues and peripheral blood leukocytes, respectively (Lesage and Lazdunski 2000; Medhurst et al. 2001).
In native cells all TWIK channels have low channel activity and as a result they are sometimes considered to be electrically silent (Bockenhauer et al. 2000; Renigunta et al. 2014), limiting functional characterization of the channels as well as the development of selective pharmacological tools (Lotshaw 2007). When active, K2P1 and K2P6 currents are inhibited by barium, quinine, or quinidine (Table 5) (Lesage et al. 1996b). Separately, K2P1 channels can also be inhibited by intracellular (Lesage et al. 1996b) as well as extracellular acidification (Plant et al. 2010). K2P1 is also regulated by PKC activation by phorbol esters such as PMA, which enhances channel activity (Table 5) (Lesage et al. 1996b).
7 The TREK Subfamily: K2P2, K2P10, and K2P4
The TREK subfamily is composed of K2P2 (TREK1, KCNK2), K2P10 (TREK2, KCNK10), and K2P4 (TWIK-related arachidonic-acid-stimulated K+ channel or TRAAK, K2P4, KCNK4) channels (Bang et al. 2000; Fink et al. 1998). In humans K2P2 and K2P10 tissue expression overlaps in the CNS and periphery tissues while K2P4 expression is most notable in the neurons (Lesage et al. 2000; Meadows et al. 2000; Medhurst et al. 2001). K2P10 channel shares 65% sequence similarity to K2P2 and 45% similarity to K2P4 (Bang et al. 2000; Ozaita and Vega-Saenz de Miera 2002). The K2P2 and K2P10 channels exhibit similar outward rectification (Lesage et al. 2000; Maingret et al. 1999; Medhurst et al. 2001). The differences between K2P2 and K2P10 currents can be seen when comparing unitary currents of the two channels under high extracellular concentration of K+. Under this condition K2P10 exhibits inward rectification (Lesage et al. 2000; Maingret et al. 1999; Medhurst et al. 2001) while both K2P2 and K2P4 exhibit Goldman-Hodgkin-Katz (GHK) rectification (Fink et al. 1998).
The TREK subfamily of K2P channels are noted for their sensitivity to mechanical stimuli. These mechanosensitive channels are modulated by numerous physicochemical stimuli including pH, temperature, mechanical stress (stretch, shear, and swelling), polyunsaturated fatty acids (PUFAs), anesthetics (volatile), and protein phosphorylation (Table 6) (Lotshaw 2007; Maingret et al. 1999). K2P2 channels are also activated by an acidic pHi (Maingret et al. 2000), likely due to protonation of a glutamic acid residue at position 306 (Glu306). Protonation of this residue is an important regulator of the response of K2P2 channels to mechanical stimulation (Honore et al. 2002).
TREK channels are also activated by heat (Kang et al. 2005; Maingret et al. 2000). Thus, at 37 °C K2P2 channels exhibit outward rectification (Kang et al. 2005; Maingret et al. 2000) that is lost upon cooling (Kang et al. 2005; Maingret et al. 2000). K2P2 and K2P10 are also activated by halogenated volatile anesthetics such as chloroform, ether, halothane, isoflurane (Table 6) (Lesage et al. 2000; Maingret et al. 2000). Halothane is a more effective activator of K2P10 while chloroform is a more efficacious activator of K2P2 (Lesage et al. 2000). All the TREK subfamily channels are activated by riluzole, a neuroprotective drug that transiently activates K2P2 and K2P10 but permanently activates K2P4 (Lesage et al. 2000). The mechanism by which riluzole exerts its effect is believed to be a result of PKA inhibition as a result of cAMP accumulation (Lesage et al. 2000). In 2001, Bockenhauer and colleagues demonstrated that PKA phosphorylation of serine-348 (Ser348) results in an altered voltage-dependence of K2P2 channels, effectively reducing the open probability and thereby the channel activity (Bockenhauer et al. 2001).
Inhibition of K2P2 and K2P10 but not K2P4 was demonstrated to be mediated by activators of protein kinases (Table 6). Lesage and colleagues found that co-expression of K2P10 and Gαs-coupled receptor 5HT4 resulted in decreased channel activity when the receptors were activated by 5-hydroxytryptamine. In contrast, co-expression K2P10 and Gαi-coupled mGluR2 receptors increased channel activity upon stimulation by glutamate (Lesage et al. 2000). Lastly, co-expression of K2P10 and the Gαq-coupled receptor mGluR1 resulted in inhibition of channel activity upon stimulation of mGluR1 by glutamate (Lesage et al. 2000). Signaling through Gαq results in activation of phosholipase C (PLC) which results in the hydrolysis of PIP2 into diacylglcerol (DAG) and inositol 1,4,5-triphosphate (IP3) production. Lesage et al. postulated that inhibition of the channel may be a result of activation of protein kinase C (PKC) by DAG (Fig. 3).

Interactions between K2P channels and pharmacophores. A comparison of four pharmacophores that have been captured in complex with K2P channels. For each channel, one subunit is shown in light blue and the adjacent subunit is shown in yellow. In each case, the structure of the pharmacophore is given above a zoomed-in view of how the molecule interacts with the channel protein. Right-hand column: ML402 (top) or ML335 (bottom) interacting with K2P2 channel (PDB ID: 6CQ9, 6CQ8) (Lolicato et al. 2017). Left-hand column: Brominated fluoxetine derivative or Norfluoxetine binding to K2P10 (PDB ID: 4XDL, 4XDK (Dong et al. 2015)
8 The TASK Subfamily: K2P3, K2P9, and K2P15
The TASK subfamily is composed of K2P3 (TASK1, KCNK3), K2P9, (TASK3, KCNK9), and K2P15 (TASK5, KCNK9) channels (Duprat et al. 1997; Kim and Gnatenco 2001; Kim et al. 2000). The K2P3 channel was first isolated based on its sequence homology to K2P1 and K2P2 (Duprat et al. 1997). In general, the TASK channels share low sequence similarity with other K2P channels (<30%) however, amongst each other TASK channels share relatively high sequence similarity (>50%) (Ashmole et al. 2001; Duprat et al. 1997, 2007; Kim et al. 2000). TASK channels are expressed in most tissues with notable expression in the placenta and pancreas (Ashmole et al. 2001; Duprat et al. 1997; Kim et al. 2000; Rajan et al. 2000). While K2P3 and K2P9 can form functional homodimers or heterodimers, K2P15 channels are electrically silent when expressed alone or with other TASK channels (Ashmole et al. 2001; Bayliss and Barrett 2008; Czirják and Enyedi 2002; Duprat et al. 2007). Under physiological conditions activation of TASK1 and TASK3 channels occurs instantaneously and the channels exhibit outward rectification (Duprat et al. 2007; Kim et al. 2000).
The sine qua non of TASK channels is inhibition of the channel activity by extracellular acidification (Table 7) (Czirják and Enyedi 2002; Duprat et al. 1997; Kim et al. 2000; Rajan et al. 2000). In mutational studies of Guinea pig K2P9 (62.3% and 88.3% homology to human K2P3 and K2P9, respectively) Rajan and colleagues found that the histidine at position 98 (His98) conferred pH sensitivity to the channel (Lopes et al. 2000, 2001; Rajan et al. 2000). Similarly, Lopes and colleagues found that protonation of the equivalent residue in K2P3 conferred pH-sensitivity to that channel (Lopes et al. 2000, 2001). Of note, K2P3 and K2P9 heterodimers are also inhibited by by extracellular acidification (Czirják and Enyedi 2002).
Two groups have found that K2P9 homodimers are inhibited by Ruthenium Red (RR) while micromolar concentrations of RR were also unable to inhibit K2P3 homodimers in both Xenopus oocytes and COS-7 cells (Table 7). Interestingly, K2P3-K2P9 heterodimers are minimally inhibited by RR (Czirják and Enyedi 2002; Kang et al. 2004). RR appears to inhibit K2P9 homodimers by binding to Glutamate 70 (Glu70) on both subunits (Czirjak and Enyedi 2003). With K2P3-K2P9 heterodimers there is only one subunit with Glu70 for RR to bind which is likely insufficient to cause inhibition (Czirjak and Enyedi 2003).
K2P3 and K2P9 are both inhibited by Gαq (Chen et al. 2006) although whether this result is secondary to hydrolysis of PIP2 remains an area of active debate. Both K2P3 and K2P9 are activated by volatile anesthetics (halothane and isoflurane) (Kang et al. 2004; Patel et al. 1999).
9 TOK Channels
K+ channel subunits with two pore domains are not limited to expression in higher order eukaryotes but have also been identified in fungi. The transient outward current (TOK) channels were first cloned and described in Saccharomyces cerevisiae following a genome search that identified a P domain peptide sequence homologous to those of other K+ channels (Ketchum et al. 1995). In contrast to the K2P subunits discussed above, TOK channels are dimers of subunits with eight transmembrane domains with intracellular amino- and carboxy-terminal tails (M1-M8), with two reentrant P-loops located between transmembrane domains composed with two P-loops regions located between M5 and M6 and M7 and M8 (Fig. 3) (Ketchum et al. 1995; Lesage et al. 1996a; Zhou et al. 1995). Expression of the S. cerevisiae TOK (ScTOK) channels in Xenopus oocytes revealed K+-selective channels with outward rectification that were activated by depolarizing voltages (Ketchum et al. 1995; Lesage et al. 1996a; Zhou et al. 1995). Activation of ScTOK channels is coupled to the K+ equilibrium potential (EK) in that changes in the external concentration of K+ results in loss of outward rectification (Bertl et al. 1998; Ketchum et al. 1995; Lesage et al. 1996a; Zhou et al. 1995). ScTOK currents are inhibited by barium ions, quinine, or tetraethylammonium (TEA) (Ketchum et al. 1995; Lesage et al. 1996a; Zhou et al. 1995).
TOK channels have now been identified in a range of fungi, including strains that are pathogenic to humans. A comparative study of four pathogenic fungi, Aspergillus fumigatus (AfTOK1), Candida albicans (CaTOK), and two strains of Cryptococcus neoformans (CnTOK and H99TOK), by Lewis and colleagues revealed that the TOK subfamily of K+ channels share similar biophysical characteristics as ScTOK (Lewis et al. 2020). Their unique distribution in only fungi suggests that these TOK channels could be important therapeutic targets for anti-fungal pharmaceutics. This intriguing proposal is supported by data showing that extracellular K1 killer toxin kills Saccharomyces yeast by increasing the open probability of ScTOK and perturbing K+ homeostasis (Ahmed et al. 1999). In contrast, infection with killer toxin virus protects against the effects of the external toxin, allowing virus-positive cells to propagate (Sesti et al. 2001). Thus, selective, small molecule activators of TOK channels are potential anti-fungal agents.
10 Conclusion and Future Perspectives
The K+ channels comprise a large, diverse, and ubiquitous superfamily of membrane proteins that regulate various biological processes in both excitable and non-excitable cells (Kuang et al. 2015; Tian et al. 2014). The two-pore domain K+ channels constitute a subfamily of K+ channels that are categorized based on structural and sequence similarity. Since the discovery of these channels more than 20 years ago much has been revealed about these channel’s physiology and pharmacology. The expression of K2P channels is widespread across various tissues and organ systems. This broad distribution and expression highlight their importance in the biology of many tissues and suggest that K2P channels will continue to emerge as important potential druggable targets for the treatment of diverse diseases. Given the fundamental role that K2P channels play in physiology, it is not surprising that their activity is tightly regulated and modulated by diverse physicochemical and mechanical stimuli including temperature, mechanical stress, pHi, pHo, second-messenger pathways, PUFAs, and phosphoinositides.
Despite the growing body of work which has implicated K2P channels in various cardiac and neuronal diseases, there is much that is yet to be learned about K2P physiology and its role in pathophysiology. A present obstacle in attaining this knowledge is the lack of channel selective pharmacophores although this landscape is starting to evolve, particularly for the TREK subfamily of K2P channels. Following the elucidation of several K2P structures we now appreciate that a part of the delay in identifying selective pharmacophores comes from the cap-domain of the K2P channels. This structural feature, seemingly unique amongst K+ channels, protects the outer mouth of the channel pore from infiltration by classical K+ channel blockers, particularly protein toxins. However, the same structural revolution that identified the problems has also helped to initiate solutions. Using computational approaches to understand the dynamics of K2P channels, researchers have started to identify druggable pockets and binding sites within the channel corpus. Of note, Lolicato and colleagues identified a cryptic binding pocket behind the pore of the K2P2 channel that can co-ordinate the newly identified channel activators ML335 and ML402 (Lolicato et al. 2017). Bagriantsev and colleagues demonstrated that selective and potent compounds of K2P channels can also be identified using high-throughput screens (Bagriantsev et al. 2013). These powerful approaches promise to break the gridlock in the development of selective new K2P channel modulators in the future.
References
Ahmed A, Sesti F, Ilan N, Shih TM, Sturley SL, Goldstein SA (1999) A molecular target for viral killer toxin: TOK1 potassium channels. Cell 99:283–291. https://doi.org/10.1016/s0092-8674(00)81659-1
Anazco C, Pena-Munzenmayer G, Araya C, Cid LP, Sepulveda FV, Niemeyer MI (2013) G protein modulation of K2P potassium channel TASK-2: a role of basic residues in the C terminus domain. Pflugers Arch 465:1715–1726. https://doi.org/10.1007/s00424-013-1314-0
Aryal P, Abd-Wahab F, Bucci G, Sansom MS, Tucker SJ (2014) A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat Commun 5:1–9
Ashmole I, Goodwin PA, Stanfield PR (2001) TASK-5, a novel member of the tandem pore K+ channel family. Pflugers Arch 442:828–833. https://doi.org/10.1007/s004240100620
Bagriantsev SN, Peyronnet R, Clark KA, Honore E, Minor DL Jr (2011) Multiple modalities converge on a common gate to control K2P channel function. EMBO J 30:3594–3606. https://doi.org/10.1038/emboj.2011.230
Bagriantsev SN, Clark KA, Minor DL Jr (2012) Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J 31:3297–3308. https://doi.org/10.1038/emboj.2012.171
Bagriantsev SN, Ang K-H, Gallardo-Godoy A, Clark KA, Arkin MR, Renslo AR, Minor DL Jr (2013) A high-throughput functional screen identifies small molecule regulators of temperature-and mechano-sensitive K2P channels. ACS Chem Biol 8:1841–1851
Bang H, Kim Y, Kim D (2000) TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem 275:17412–17419. https://doi.org/10.1074/jbc.M000445200
Barel O, Shalev SA, Ofir R, Cohen A, Zlotogora J, Shorer Z, Mazor G, Finer G, Khateeb S, Zilberberg N (2008) Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am J Hum Genet 83:193–199
Bayliss DA, Barrett PQ (2008) Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci 29:566–575. https://doi.org/10.1016/j.tips.2008.07.013
Bayliss DA, Czirják G, Enyedi P, Goldstein SA, Lesage F, Minor DL Jr, Plant LD, Sepúlveda F, Winn BT (2019) Two P domain potassium channels (version 2019.4) in the IUPHAR/BPS guide to pharmacology database. IUPHAR/BPS Guide Pharmacol. https://doi.org/10.2218/gtopdb/F79/2019.4
Bertl A, Bihler H, Reid J, Kettner C, Slayman CL (1998) Physiological characterization of the yeast plasma membrane outward rectifying K+ channel, DUK1 (TOK1), in situ. J Membr Biol 162:67–80
Blin S, Chatelain FC, Feliciangeli S, Kang D, Lesage F, Bichet D (2014) Tandem pore domain halothane-inhibited K+ channel subunits THIK1 and THIK2 assemble and form active channels. J Biol Chem 289:28202–28212. https://doi.org/10.1074/jbc.M114.600437
Bockenhauer D, Nimmakayalu MA, Ward DC, Goldstein SA, Gallagher PG (2000) Genomic organization and chromosomal localization of the murine 2 P domain potassium channel gene Kcnk8: conservation of gene structure in 2 P domain potassium channels. Gene 261:365–372
Bockenhauer D, Zilberberg N, Goldstein SA (2001) KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat Neurosci 4:486–491. https://doi.org/10.1038/87434
Braun G, Lengyel M, Enyedi P, Czirjak G (2015) Differential sensitivity of TREK-1, TREK-2 and TRAAK background potassium channels to the polycationic dye ruthenium red. Br J Pharmacol 172:1728–1738. https://doi.org/10.1111/bph.13019
Brohawn SG, del Mármol J, MacKinnon R (2012) Crystal structure of the human K2P TRAAK, a lipid-and mechano-sensitive K+ ion channel. Science 335:436–441
Campanucci V, Brown S, Hudasek K, O’kelly I, Nurse C, Fearon I (2005) O2 sensing by recombinant TWIK-related halothane-inhibitable K+ channel-1 background K+ channels heterologously expressed in human embryonic kidney cells. Neuroscience 135:1087–1094
Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, Warth R, Barhanin J, Lesage F (2012) TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci 109:5499–5504
Chatelain FC, Bichet D, Feliciangeli S, Larroque MM, Braud VM, Douguet D, Lesage F (2013) Silencing of the tandem pore domain halothane-inhibited K+ channel 2 (THIK2) relies on combined intracellular retention and low intrinsic activity at the plasma membrane. J Biol Chem 288:35081–35092. https://doi.org/10.1074/jbc.M113.503318
Chavez RA, Gray AT, Zhao BB, Kindler CH, Mazurek MJ, Mehta Y, Forsayeth JR, Yost CS (1999) TWIK-2, a new weak inward rectifying member of the tandem pore domain potassium channel family. J Biol Chem 274:7887–7892. https://doi.org/10.1074/jbc.274.12.7887
Chemin J, Patel AJ, Delmas P, Sachs F, Lazdunski M, Honore E (2007) Regulation of the Mechano-gated K2P channel TREK-1 by membrane phospholipids. Curr Top Membr 59:155–170. https://doi.org/10.1016/S1063-5823(06)59007-6
Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, Viana F, Garrison JC, Bayliss DA (2006) Inhibition of a background potassium channel by Gq protein alpha-subunits. Proc Natl Acad Sci U S A 103:3422–3427. https://doi.org/10.1073/pnas.0507710103
Christensen AH, Chatelain FC, Huttner IG, Olesen MS, Soka M, Feliciangeli S, Horvat C, Santiago CF, Vandenberg JI, Schmitt N (2016) The two-pore domain potassium channel, TWIK-1, has a role in the regulation of heart rate and atrial size. J Mol Cell Cardiol 97:24–35
Cid LP, Roa-Rojas HA, Niemeyer MI, Gonzalez W, Araki M, Araki K, Sepulveda FV (2013) TASK-2: a K2P K(+) channel with complex regulation and diverse physiological functions. Front Physiol 4:198. https://doi.org/10.3389/fphys.2013.00198
Cohen A, Ben-Abu Y, Hen S, Zilberberg N (2008) A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J Biol Chem 283:19448–19455. https://doi.org/10.1074/jbc.M801273200
Czirják G, Enyedi P (2002) Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem 277:5426–5432
Czirjak G, Enyedi P (2003) Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol Pharmacol 63:646–652. https://doi.org/10.1124/mol.63.3.646
Czirjak G, Toth ZE, Enyedi P (2004) The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J Biol Chem 279:18550–18558. https://doi.org/10.1074/jbc.M312229200
Decher N, Maier M, Dittrich W, Gassenhuber J, Bruggemann A, Busch AE, Steinmeyer K (2001) Characterization of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett 492:84–89. https://doi.org/10.1016/s0014-5793(01)02222-0
Decher N, Ortiz-Bonnin B, Friedrich C, Schewe M, Kiper AK, Rinne S, Seemann G, Peyronnet R, Zumhagen S, Bustos D, Kockskamper J, Kohl P, Just S, Gonzalez W, Baukrowitz T, Stallmeyer B, Schulze-Bahr E (2017) Sodium permeable and "hypersensitive" TREK-1 channels cause ventricular tachycardia. EMBO Mol Med 9:403–414. https://doi.org/10.15252/emmm.201606690
Decressac S, Franco M, Bendahhou S, Warth R, Knauer S, Barhanin J, Lazdunski M, Lesage F (2004) ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GTP exchange factor for ARF6. EMBO Rep 5:1171–1175
Dong YY, Pike AC, Mackenzie A, McClenaghan C, Aryal P, Dong L, Quigley A, Grieben M, Goubin S, Mukhopadhyay S (2015) K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 347:1256–1259
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77. https://doi.org/10.1126/science.280.5360.69
Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M (1997) TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16:5464–5471. https://doi.org/10.1093/emboj/16.17.5464
Duprat F, Girard C, Jarretou G, Lazdunski M (2005) Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J Physiol 562:235–244. https://doi.org/10.1113/jphysiol.2004.071266
Duprat F, Lauritzen I, Patel A, Honore E (2007) The TASK background K2P channels: chemo- and nutrient sensors. Trends Neurosci 30:573–580. https://doi.org/10.1016/j.tins.2007.08.003
Enyedi P, Czirjak G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90:559–605. https://doi.org/10.1152/physrev.00029.2009
Enyedi P, Czirjak G (2015) Properties, regulation, pharmacology, and functions of the K(2)p channel, TRESK. Pflugers Arch 467:945–958. https://doi.org/10.1007/s00424-014-1634-8
Enyedi P, Braun G, Czirjak G (2012) TRESK: the lone ranger of two-pore domain potassium channels. Mol Cell Endocrinol 353:75–81. https://doi.org/10.1016/j.mce.2011.11.009
Feliciangeli S, Bendahhou S, Sandoz G, Gounon P, Reichold M, Warth R, Lazdunski M, Barhanin J, Lesage F (2007) Does sumoylation control K2P1/TWIK1 background K+ channels? Cell 130:563–569
Feliciangeli S, Tardy MP, Sandoz G, Chatelain FC, Warth R, Barhanin J, Bendahhou S, Lesage F (2010) Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J Biol Chem 285:4798–4805
Feliciangeli S, Chatelain FC, Bichet D, Lesage F (2015) The family of K2P channels: salient structural and functional properties. J Physiol 593:2587–2603
Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M (1998) A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 17:3297–3308. https://doi.org/10.1093/emboj/17.12.3297
Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S (2007) Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582:675–693. https://doi.org/10.1113/jphysiol.2006.126714
Gada K, Plant LD (2019) Two-pore domain potassium channels: emerging targets for novel analgesic drugs: IUPHAR review 26. Br J Pharmacol 176:256–266. https://doi.org/10.1111/bph.14518
Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, Lazdunski M, Lesage F (2001) Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem Biophys Res Commun 282:249–256. https://doi.org/10.1006/bbrc.2001.4562
Goldman DE (1943) Potential, impedance, and rectification in membranes. J Gen Physiol 27:37–60. https://doi.org/10.1085/jgp.27.1.37
Goldstein SA, Price LA, Rosenthal DN, Pausch MH (1996) ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae. Proc Natl Acad Sci 93:13256–13261
Goldstein SA, Wang KW, Ilan N, Pausch MH (1998) Sequence and function of the two P domain potassium channels: implications of an emerging superfamily. J Mol Med (Berl) 76:13–20. https://doi.org/10.1007/s001090050186
Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N (2001) Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2:175–184. https://doi.org/10.1038/35058574
Gotter AL, Santarelli VP, Doran SM, Tannenbaum PL, Kraus RL, Rosahl TW, Meziane H, Montial M, Reiss DR, Wessner K, McCampbell A, Stevens J, Brunner JI, Fox SV, Uebele VN, Bayliss DA, Winrow CJ, Renger JJ (2011) TASK-3 as a potential antidepressant target. Brain Res 1416:69–79. https://doi.org/10.1016/j.brainres.2011.08.021
Guyenet PG, Stornetta RL, Souza G, Abbott SBG, Shi Y, Bayliss DA (2019) The retrotrapezoid nucleus: central chemoreceptor and regulator of breathing automaticity. Trends Neurosci 42:807–824. https://doi.org/10.1016/j.tins.2019.09.002
Hay RT (2005) SUMO: a history of modification. Mol Cell 18:1–12. https://doi.org/10.1016/j.molcel.2005.03.012
Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117:500
Honore E (2007) The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci 8:251–261. https://doi.org/10.1038/nrn2117
Honore E, Maingret F, Lazdunski M, Patel AJ (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K(+) channel TREK-1. EMBO J 21:2968–2976. https://doi.org/10.1093/emboj/cdf288
Hoshi T, Zagotta WN, Aldrich RW (1991) Two types of inactivation in shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7:547–556
Hwang EM, Kim E, Yarishkin O, Woo DH, Han K-S, Park N, Bae Y, Woo J, Kim D, Park M (2014) A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat Commun 5:1–15
Hwang HY, Zhang E, Park S, Chung W, Lee S, Kim DW, Ko Y, Lee W (2015) TWIK-related spinal cord K+ channel expression is increased in the spinal dorsal horn after spinal nerve ligation. Yonsei Med J 56:1307–1315
Kang D, Han J, Talley EM, Bayliss DA, Kim D (2004) Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J Physiol 554:64–77. https://doi.org/10.1113/jphysiol.2003.054387
Kang D, Choe C, Kim D (2005) Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol 564:103–116. https://doi.org/10.1113/jphysiol.2004.081059
Ketchum KA, Joiner WJ, Sellers AJ, Kaczmarek LK, Goldstein SA (1995) A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 376:690–695
Kim D, Gnatenco C (2001) TASK-5, a new member of the tandem-pore K(+) channel family. Biochem Biophys Res Commun 284:923–930. https://doi.org/10.1006/bbrc.2001.5064
Kim Y, Bang H, Kim D (2000) TASK-3, a new member of the tandem pore K(+) channel family. J Biol Chem 275:9340–9347. https://doi.org/10.1074/jbc.275.13.9340
Koh SD, Monaghan K, Sergeant GP, Ro S, Walker RL, Sanders KM, Horowitz B (2001) TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J Biol Chem 276:44338–44346. https://doi.org/10.1074/jbc.M108125200
Kollewe A, Lau AY, Sullivan A, Roux B, Goldstein SA (2009) A structural model for K2P potassium channels based on 23 pairs of interacting sites and continuum electrostatics. J Gen Physiol 134:53–68. https://doi.org/10.1085/jgp.200910235
Kuang Q, Purhonen P, Hebert H (2015) Structure of potassium channels. Cell Mol Life Sci 72:3677–3693
Lafrenière RG, Cader MZ, Poulin J-F, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafrenière F, McLaughlan S, Dubé M-P (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16:1157–1160
Lengyel M, Czirjak G, Enyedi P (2018) TRESK background potassium channel is not gated at the helix bundle crossing near the cytoplasmic end of the pore. PLoS One 13:e0197622. https://doi.org/10.1371/journal.pone.0197622
Lesage F (2003) Pharmacology of neuronal background potassium channels. Neuropharmacology 44:1–7. https://doi.org/10.1016/s0028-3908(02)00339-8
Lesage F, Barhanin J (2011) Molecular physiology of pH-sensitive background K2P channels. Physiology 26:424–437
Lesage F, Lazdunski M (2000) Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279:F793–F801. https://doi.org/10.1152/ajprenal.2000.279.5.F793
Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J (1996a) A pH-sensitive yeast outward rectifier K channel with two pore domains and novel gating properties. J Biol Chem 271:4183–4187
Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J (1996b) TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15:1004–1011
Lesage F, Terrenoire C, Romey G, Lazdunski M (2000) Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem 275:28398–28405. https://doi.org/10.1074/jbc.M002822200
Lewis A, McCrossan ZA, Manville RW, Popa MO, Cuello LG, Goldstein SA (2020) TOK channels use the two gates in classical K+ channels to achieve outward rectification. FASEB J. https://doi.org/10.1096/fj.202000545R
Liu C, Au JD, Zou HL, Cotten JF, Yost CS (2004) Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth Analg 99:1715–1722. https://doi.org/10.1213/01.ANE.0000136849.07384.44
Lolicato M, Riegelhaupt PM, Arrigoni C, Clark KA, Minor DL Jr (2014) Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron 84:1198–1212. https://doi.org/10.1016/j.neuron.2014.11.017
Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, Minor DL Jr (2017) K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547:364–368. https://doi.org/10.1038/nature22988
Lolicato M, Natale A, Aberemane-Ali F, Crottès D, Capponi S, Duman R, Wagner A, Rosenberg JM, Grabe M, Minor DL (2020) K2P channel C-type gating involves asymmetric selectivity filter order-disorder transitions. bioRxiv 6(44):eabc9174
Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA (2000) Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem 275:16969–16978
Lopes CM, Zilberberg N, Goldstein SA (2001) Block of Kcnk3 by protons evidence that 2-P-domain potassium channel subunits function as homodimers. J Biol Chem 276:24449–24452
Lotshaw DP (2007) Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem Biophys 47:209–256. https://doi.org/10.1007/s12013-007-0007-8
Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E (1999) Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274:26691–26696. https://doi.org/10.1074/jbc.274.38.26691
Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honore E (2000) TREK-1 is a heat-activated background K(+) channel. EMBO J 19:2483–2491. https://doi.org/10.1093/emboj/19.11.2483
Mathie A, Veale EL (2015) Two-pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pflugers Arch 467:931–943. https://doi.org/10.1007/s00424-014-1655-3
Meadows HJ, Benham CD, Cairns W, Gloger I, Jennings C, Medhurst AD, Murdock P, Chapman CG (2000) Cloning, localisation and functional expression of the human orthologue of the TREK-1 potassium channel. Pflugers Arch 439:714–722. https://doi.org/10.1007/s004249900235
Medhurst AD, Rennie G, Chapman CG, Meadows H, Duckworth MD, Kelsell RE, Gloger II, Pangalos MN (2001) Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res 86:101–114. https://doi.org/10.1016/s0169-328x(00)00263-1
Miller AN, Long SB (2012) Crystal structure of the human two-pore domain potassium channel K2P1. Science 335:432–436. https://doi.org/10.1126/science.1213274
Nie X, Arrighi I, Kaissling B, Pfaff I, Mann J, Barhanin J, Vallon V (2005) Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers Arch 451:479–488
Niemeyer MI, Gonzalez-Nilo FD, Zuniga L, Gonzalez W, Cid LP, Sepulveda FV (2007) Neutralization of a single arginine residue gates open a two-pore domain, alkali-activated K+ channel. Proc Natl Acad Sci U S A 104:666–671. https://doi.org/10.1073/pnas.0606173104
Niemeyer MI, Cid LP, Pena-Munzenmayer G, Sepulveda FV (2010) Separate gating mechanisms mediate the regulation of K2P potassium channel TASK-2 by intra- and extracellular pH. J Biol Chem 285:16467–16475. https://doi.org/10.1074/jbc.M110.107060
Ozaita A, Vega-Saenz de Miera E (2002) Cloning of two transcripts, HKT4.1a and HKT4.1b, from the human two-pore K+ channel gene KCNK4. Chromosomal localization, tissue distribution and functional expression. Brain Res Mol Brain Res 102:18–27. https://doi.org/10.1016/s0169-328x(02)00157-2
Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci 2:422–426
Pergel E, Lengyel M, Enyedi P, Czirják G (2019) TRESK (K2P18. 1) background potassium channel is activated by novel-type protein kinase C via dephosphorylation. Mol Pharmacol 95:661–672
Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Ehrlich G, Andres-Enguix I, Fritzenschaft H, Decher N, Sansom MS, Tucker SJ, Baukrowitz T (2011) The pore structure and gating mechanism of K2P channels. EMBO J 30:3607–3619. https://doi.org/10.1038/emboj.2011.268
Plant LD (2012) A role for K2P channels in the operation of somatosensory nociceptors. Front Mol Neurosci 5:21. https://doi.org/10.3389/fnmol.2012.00021
Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SA (2010) One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci 107:10743–10748
Plant LD, Zuniga L, Araki D, Marks JD, Goldstein SA (2012) SUMOylation silences heterodimeric TASK potassium channels containing K2P1 subunits in cerebellar granule neurons. Sci Signal 5:ra84
Pollema-Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M (2013) Expression of background potassium channels in rat DRG is cell-specific and down-regulated in a neuropathic pain model. Mol Cell Neurosci 57:1–9
Pope L, Lolicato M, Minor DL Jr (2020) Polynuclear ruthenium amines inhibit K2P channels via a “Finger in the Dam” mechanism. Cell Chem Biol 27(5):511–524.e4
Pountney DJ, Gulkarov I, Vega-Saenz de Miera E, Holmes D, Saganich M, Rudy B, Artman M, Coetzee WA (1999) Identification and cloning of TWIK-originated similarity sequence (TOSS): a novel human 2-pore K+ channel principal subunit. FEBS Lett 450:191–196
Rajan S, Wischmeyer E, Xin Liu G, Preisig-Muller R, Daut J, Karschin A, Derst C (2000) TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem 275:16650–16657. https://doi.org/10.1074/jbc.M000030200
Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, Karschin A, Derst C (2001) THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem 276:7302–7311. https://doi.org/10.1074/jbc.M008985200
Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA (2005) Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121:37–47
Renigunta V, Zou X, Kling S, Schlichthorl G, Daut J (2014) Breaking the silence: functional expression of the two-pore-domain potassium channel THIK-2. Pflugers Arch 466:1735–1745. https://doi.org/10.1007/s00424-013-1404-z
Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N, Lazdunski M (1998) Cloning and expression of a novel pH-sensitive two pore domain K+ channel from human kidney. J Biol Chem 273:30863–30869. https://doi.org/10.1074/jbc.273.47.30863
Rödström KE, Kiper AK, Zhang W, Rinné S, Pike AC, Goldstein M, Conrad LJ, Delbeck M, Hahn MG, Meier H (2020) A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature:1–5
Sano Y, Inamura K, Miyake A, Mochizuki S, Kitada C, Yokoi H, Nozawa K, Okada H, Matsushime H, Furuichi K (2003) A novel two-pore domain K+ channel, TRESK, is localized in the spinal cord. J Biol Chem 278:27406–27412
Schewe M, Nematian-Ardestani E, Sun H, Musinszki M, Cordeiro S, Bucci G, de Groot BL, Tucker SJ, Rapedius M, Baukrowitz T (2016) A non-canonical voltage-sensing mechanism controls gating in K2P K(+) channels. Cell 164:937–949. https://doi.org/10.1016/j.cell.2016.02.002
Sesti F, Shih TM, Nikolaeva N, Goldstein SA (2001) Immunity to K1 killer toxin: internal TOK1 blockade. Cell 105:637–644. https://doi.org/10.1016/s0092-8674(01)00376-2
Şterbuleac D (2019) Molecular determinants of chemical modulation of two-pore domain potassium channels. Chem Biol Drug Des 94:1596–1614
Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA (2001) Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci 21:7491–7505
Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SA (2008) Alternative translation initiation in rat brain yields K2P2. 1 potassium channels permeable to sodium. Neuron 58:859–870
Tian C, Zhu R, Zhu L, Qiu T, Cao Z, Kang T (2014) Potassium channels: structures, diseases, and modulators. Chem Biol Drug Des 83:1–26
Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X (2011) TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7:30. https://doi.org/10.1186/1744-8069-7-30
Weir GA, Pettingill P, Wu Y, Duggal G, Ilie AS, Akerman CJ, Cader MZ (2019) The role of TRESK in discrete sensory neuron populations and somatosensory processing. Front Mol Neurosci 12:170. https://doi.org/10.3389/fnmol.2019.00170
Woo DH, Han K-S, Shim JW, Yoon B-E, Kim E, Bae JY, Oh S-J, Hwang EM, Marmorstein AD, Bae YC (2012) TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151:25–40
Yang S-B, Jan LY (2013) Potassium channels: their physiological and molecular diversity. In: Roberts GCK (ed) Encyclopedia of biophysics. Springer, Berlin, pp 1933–1941
Yellen G (1998) The moving parts of voltage-gated ion channels. Q Rev Biophys 31:239–295
Zhou X-L, Vaillant B, Loukin SH, Kung C, Saimi Y (1995) YKC1 encodes the depolarization-activated K+ channel in the plasma membrane of yeast. FEBS Lett 373:170–176
Zhou J, Yang CX, Zhong JY, Wang HB (2013) Intrathecal TRESK gene recombinant adenovirus attenuates spared nerve injury-induced neuropathic pain in rats. Neuroreport 24:131–136. https://doi.org/10.1097/WNR.0b013e32835d8431
Zilberberg N, Ilan N, Goldstein SA (2001) KCNKO: opening and closing the 2-P-domain potassium leak channel entails "C-type" gating of the outer pore. Neuron 32:635–648. https://doi.org/10.1016/s0896-6273(01)00503-7
Zuniga L, Zuniga R (2016) Understanding the cap structure in K2P channels. Front Physiol 7:228. https://doi.org/10.3389/fphys.2016.00228
Conflict of Interest
The authors report no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kamuene, J.M., Xu, Y., Plant, L.D. (2021). The Pharmacology of Two-Pore Domain Potassium Channels. In: Gamper, N., Wang, K. (eds) Pharmacology of Potassium Channels. Handbook of Experimental Pharmacology, vol 267. Springer, Cham. https://doi.org/10.1007/164_2021_462
Download citation
DOI: https://doi.org/10.1007/164_2021_462
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84051-8
Online ISBN: 978-3-030-84052-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)