
Abstract
Xanthine oxidase inhibitors are primarily used in the clinical prevention and treatment of gout associated with hyperuricemia. The archetypal xanthine oxidase inhibitor, Allopurinol has been shown to have other beneficial effects such as a reduction in vascular reactive oxygen species and mechano-energetic uncoupling. This chapter discusses these properties and their relevance to human pathophysiology with a focus on Allopurinol as well as newer xanthine oxidase inhibitors such as Febuxostat and Topiroxostat.
Graphical Abstract

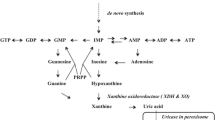
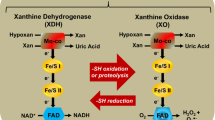
Xanthine oxidase (XO) and xanthine dehydrogenase (XDH) are collectively referred to as xanthine oxidoreductase (XOR). XDH is initially synthesised as a 150-kDa protein from which XO is derived, e.g. under conditions of ischemia/hypoxia either reversibly by conformational changes (calcium or SH oxidation) or irreversibly by proteolysis, the latter leading to formation of a 130-kDa form of XO. Both, XO and XDH, catalyse the conversion of hypoxanthine via xanthine to uric acid, the former by using oxygen forming superoxide and hydrogen peroxide and the latter NAD+. However, XDH is in principle also able to generate ROS.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Urate and Xanthine Oxidoreductase
Urate is a heterocyclic purine derivative. In humans and some primates, it is the final product of purine breakdown. The majority of urate is filtered through the kidney (60–70%) and at least 90% of this is re-absorbed through the GLUT9 and URAT1 anion transporters. Humans lack a functional uricase gene that is present in most other lower mammals. Uricase further oxidises urate into water-soluble allantoin (Chen et al. 2016).
Urate is thought to exert opposing actions on ROS extracellularly versus intracellularly. Circulating urate is thought to contribute to 70% of all free radical scavenging capability of plasma. It is an effective scavenger of carbon-centred radicals and peroxylradicals (Waring 2002). For example, it reacts with peroxynitrite (ONOO−) to release nitric oxide (NO) and therefore induce vasodilatation (Skinner et al. 1998). However, under hydrophobic conditions, it can accelerate the oxidation of LDL, increased monocyte-chemoattractant protein (MCP)-1, high sensitivity C-reactive protein and inflammatory interleukins (Bagnati et al. 1999). Therefore, it is thought that overall, hyperuricaemia contributes to the progression of CV disease because of the overwhelming oxidant property of urate (Chen et al. 2016) as well as the free radical by-products of its formation (George and Struthers 2008) (see below).
Xanthine oxidoreductase (XOR) is part of a group of enzymes known as the molybdenum iron-sulphur flavin hydroxylases. It was first discovered in milk by Schardinger in 1902 (Berry and Hare 2004) and is thought to be involved in reactions that produce reactive oxygen species (ROS) such as nitrite which enable newborn infants to overcome gut-associated bacterial gastroenteritis (Hancock et al. 2002; Stevens et al. 2000). XOR is widely distributed throughout various organs including the liver, gut, lung, kidney, heart, brain and plasma (Pacher et al. 2006) with the highest levels being found in the gut and the liver (Parks and Granger 1986). In the myocardium, it is localised to the capillary endothelial cells (Cicoira et al. 2002). The gene encoding for XOR is located at the short arm of chromosome 2 (Ichida et al. 1993). It exists in two interconvertible forms known as xanthine oxidase (EC 1.1.3.22) and xanthine dehydrogenase (EC 1.17.1.4) (Della Corte et al. 1969). Both enzymes consist of two identical subunits of 145 kDa.
The mechanism by which XOR catalyses hypoxanthine and xanthine conversion is complex and has been previously described in detail (Berry and Hare 2004; Hille and Massey 1981). A fully reduced XO contains six electrons, and its re-oxidation involves electron transfer to oxygen molecules which generates two H2O2 and two O2− species (Hille and Massey 1981) for every fully reduced XO molecule. It is interesting to note that XDH can theoretically produce more O2− per mole of oxygen during NADH oxidisation than XO. Along with NADPH oxidase, it is a major generator of ROS in the human.
2 Allopurinol
Allopurinol (C5H4N4O) is the archetypal and longest established XO inhibitor in clinical use. It is a weak acid with a dissociation constant (pKa) of 9.4. It has a molecular mass of 136.11 g/mol. It is rapidly converted to oxypurinol by aldehyde oxidoreductase. While allopurinol is an analogue of hypoxanthine, oxypurinol (or alloxanthine) is an analogue of xanthine (Day et al. 2007). Oxypurinol is much more lipid soluble than allopurinol (octanol/water partition coefficient 14 vs. 0.28 for allopurinol) (Day et al. 2007).
2.1 Pharmacokinetics
Approximately 90% of allopurinol is absorbed from the gastrointestinal tract. It is rapidly absorbed and reaches peak plasma concentrations within 30–60 min (Pea 2005) following oral administration. It is rapidly metabolised to its active metabolite oxypurinol. For every 100 mg oral dose of allopurinol, 90 mg of oxypurinol is formed (Day et al. 2007). When given orally, oxypurinol has a lower bioavailability than allopurinol. An early pharmacokinetics study showed that 300 mg of allopurinol produced a slightly higher plasma levels than 600 mg of oral oxypurinol (Elion et al. 1968). Allopurinol has a short half-life in plasma between 2 and 3 h and has negligible protein binding. It has an apparent volume of distribution of 1.2 to 2.2 L/kg in healthy volunteers (Day et al. 2007). The renal clearance of allopurinol is 1.54 mL/min/kg (Day et al. 2007). Oxypurinol is detected in plasma 15–20 min after an oral dose of allopurinol, reaches peak plasma levels in 3–4 h (Guerra et al. 2001) and has a much longer plasma half-life between 14 and 30 h because it is reabsorbed in the proximal tubule of the kidney (Pea 2005). It is responsible for much of the hyperuricaemic action of allopurinol. It is excreted almost entirely unchanged in urine. Therefore the renal clearance of oxypurinol is therefore the most important aspect of the clinical pharmacokinetics of allopurinol (Day et al. 2007). Patients treated with allopurinol excrete 70% of the dose as oxypurinol and only 10% as allopurinol which indicates that the vast majority of allopurinol is converted to oxypurinol (Elion et al. 1966). Thus, the major route of allopurinol elimination is via oxidation to oxypurinol (Turnheim 1999). Turnheim et al. showed that although allopurinol elimination is not reduced in the aged, the elimination of its metabolite oxypurinol is reduced due to age-related reduction in renal function (Turnheim 1999).
The mechanism of XO inactivation by oxypurinol was determined by Massey et al. (Massey et al. 1970). Oxypurinol strongly binds at the active site of XO, resulting in the reduction of MoVI to MoIV. Its inhibition is time-dependent, and it is important to maintain an effective concentration of the inhibitor as spontaneous oxidation back from MoIV to MoVI will result in concomitant recovery of enzyme activity. It is an excellent substrate with a Vmax which is sixfold faster than xanthine. The mechanism of oxypurinol inhibition of its own production has been termed “suicidal” (Spector 1977). In a healthy individual with a creatinine clearance of 120 mL/min, the clearance of oxypurinol is 23 mL/min. As oxypurinol is a small molecule that is not bound to plasma proteins, it is freely filtered at the glomerulus (Elion et al. 1968)
2.2 Dose-Response Studies
Dose-response studies of the hyperuricaemic effect of allopurinol suggest that this increases relatively little with increasing doses of the drug (Day et al. 2007). The EC50 for allopurinol has been calculated as 5.6 ± 1.3 mg/L, which is identical to the trough level for the 300 mg/day dose (5.6 ± 0.6 mg/L). The steady-state oxypurinol concentration over a dose range of 50-900 mg allopurinol/day is shown in Table 1.
The oxypurinol concentration in steady-state was found to increase in a linear fashion up to the 600 mg/day dose of allopurinol. The concentration of oxypurinol did not increase proportionally between 600 mg/day and 900 mg/day (Graham et al. 1996) suggesting that tubular reabsorption of oxypurinol may be saturated at higher doses. The other possible explanation could be the saturation of xanthine oxidase, but this is unlikely as Spiekermann et al. (Spiekermann et al. 2003) have recently shown that complete inhibition of plasma XO activity in vivo requires an oxypurinol concentration of 1 mmol/L. Beneficial effects seen with higher dose could then be related to other effects of allopurinol. Although it is tempting to relate the efficacy of allopurinol to the degree of urate lowering it produces, there are many more factors that contribute to urate levels such as exogenous contributions from diet, endogenous production as well as renal function.
2.3 Pharmacodynamics
Allopurinol is generally safe and well tolerated since it was introduced into clinical practice 40 years ago. By the end of the 1980s, more than five million patient years of treatment and over 240 million doses had been prescribed (Vazquez-Mellado et al. 2001). Common adverse effects of allopurinol are gastrointestinal disturbance, hypersensitivity reactions (up to 8% of patients, sometimes occurring months to years after commencing medication) and skin rash (Committee 2007). In a study by McInnes et al., 6.2% of hospitalised patients monitored in a drug surveillance programme received allopurinol. After the exclusion of skin reactions, the most frequent reactions found were haematological abnormalities (0.6%), diarrhoea (0.3%) and pyrexia (0.3%). These adverse effects were found to be dose-related (McInnes et al. 1981). The rare allopurinol hypersensitivity syndrome (fever, rash, vasculitis, eosinophilia and renal failure) occurs in 0.4% of patients but can have a mortality of up to 25% (Gutierrez-Macias et al. 2005). It has been recently discovered that the HLA-B*5801 allele is a very significant risk factor for the allopurinol hypersensitivity syndrome (Hung et al. 2005) in Chinese patients. Whether or not this allele confers the same risk to other populations is yet to be known.
Clinically significant interactions between allopurinol and the endogenous purines mercaptopurine and azathioprine have been reported. As mentioned earlier, the initial discovery of allopurinol was as an agent to potentiate the anti-tumour effects of mercaptopurine so it is unsurprising that mercaptopurine levels are augmented by allopurinol because it is metabolised by XO into inactive metabolites (Pea 2005). Allopurinol enhances the anticoagulant effect of warfarin and increases the plasma concentration of didanosine, ciclosporin and theophylline (Committee 2007). The risk of allopurinol hypersensitivity syndrome is increased in elderly patients on thiazide diuretics (Schlesinger 2004) and ampicillin (Vazquez-Mellado et al. 2001) Both allopurinol and urate are removed by dialysis (Day et al. 2007).
2.4 Indirect Antioxidant Action (XO-Inhibition Mediated)
Allopurinol has also been shown to normalise endothelial dysfunction in individuals with Type 2 diabetes with mild hypertension and reduced plasma malondialdehyde (MDA) levels (Butler et al. 2000). MDA results from acid hydrolysis of lipid peroxides which are formed by free radical attack on plasma lipoproteins. It is therefore used as an indirect measure of oxidised LDL.
In the experimental murine myocardial infarction model, allopurinol significantly attenuated LV dilatation, hypertrophy, fibrosis and dysfunction. Once again, XO expression (as determined by electron spin resonance spectroscopy) and myocardial ROS generation were markedly increased in the post-MI ischemic model (Engberding et al. 2004). This suggests a role for allopurinol in LV remodelling, a possibility that we are investigating at present in our unit. Allopurinol has also been shown to be beneficial in conditions such as post coronary artery bypass surgery where it reduced ischemic events and produced less ST segment depression (Sisto et al. 1995) as well as in hypercholesterolaemic patients (Cardillo et al. 1997). Animal studies in other conditions such as diabetic retinopathy have yielded similar results both in terms of indirect and direct (see below) action of allopurinol. Allopurinol significantly improved the b-wave amplitude on electroretinography as well as 8-isoprostanes, a biomarker of ROS formation. Despite lowering urate to a similar degree, Benzobromarone did not result in any beneficial effect (Goharinia et al. 2017).
A recent placebo-controlled clinical trial (n = 100) in patients with acute coronary syndrome (ACS) showed early (1 month) reduction of markers of oxidative stress malondialdehyde (MDA), oxidised LDL. This reduction was sustained for up to 2 years (Huang et al. 2017).
Allopurinol in chronic heart failure (CHF) was assessed by Doehner et al. (Doehner et al. 2002) and by Farquharson et al. (Farquharson et al. 2002). Doehner et al. showed that the degree of improvement in forearm blood flow correlated with the degree of urate lowering. Interestingly, they also measured allantoin, a marker of oxygen free radical generation, which was reduced by 20% following 300 mg/day allopurinol. Farquharson et al. (Farquharson et al. 2002) from our unit found a 181% change in forearm blood flow with 300 mg allopurinol. They also found a 33% reduction in plasma MDA levels in patients treated with 300 mg allopurinol suggesting that the improvement in endothelial function and NO bioavailability seen was due (at least in part) to a reduction of ROS. Allopurinol also reduced B-type Natriuretic peptide (BNP) in stable CHF patients, although the reduction did not correlate with the fall in urate (Gavin and Struthers 2005). Uric acid also directly inactivates NO (Gersch et al. 2008), and therefore allopurinol may increase NO bioavailability through this indirect pathway also.
Our group demonstrated, for the first time, the antioxidant effect of high-dose allopurinol in reducing vascular oxidative stress. We studied patients with chronic heart failure and found that the effect of allopurinol on endothelial vascular function was due to xanthine oxidase inhibition and not urate lowering (George et al. 2006). We also demonstrated that there was a steep dose-response curve with high dose allopurinol (600 mg/day) significantly better than standard dose (300 mg/day) in this respect. At high dose, allopurinol completely negated the benefits seen with high dose intra-arterial vitamin C infusions (Fig. 1). This is further strengthened by evidence that the beneficial effect of vitamin C co-infusion in patients with CHF was greatest in patients with the highest levels of oxidative stress as measured by extracellular SOD (ecSOD) (Landmesser et al. 2002) and XO activity.

Absolute forearm blood flow data for acetylcholine (50, 100 nmol/min) + vitamin C 25 mg/mL vs acetylcholine alone for – placebo, 300 mg allopurinol and 600 mg allopurinol (mean ± SEM)
This finding has been further confirmed by two other studies in coronary artery disease (Rajendra et al. 2011) and heart failure (Ogino et al. 2009) and is now widely accepted as a possible mechanism for the benefits seen with allopurinol. Our group has also previously demonstrated that high-dose (but not low-dose) allopurinol reduced oxidised LDL, further confirming allopurinol’s antioxidant impact. However, as xanthine oxidase is significantly upregulated in acute ischemia or inflammation (Spiekermann et al. 2003) and otherwise constitutionally is expressed at low levels (Panus et al. 1992), there remains doubt that treating high-risk stable patients with long-term high-dose allopurinol will provide any benefit at all. Large ongoing trials such as ALL-HEART are seeking to address this question (Mackenzie et al. 2016). The biology of XO suggests that allopurinol is most beneficial in the acute ischaemia/reperfusion/inflammation setting rather than the chronic stable setting.
The other possible explanation which also relates to urate and superoxide formation is that in patients with low baseline oxidative stress, there are proportionately more urate (a known antioxidant) molecules to combat oxidative stress. In patients with high background or ischemia-induced oxidative stress however, inhibition of XO will reduce proportionately more superoxide (due to the cascade formation of superoxide). The reduction in urate with allopurinol may be an unfortunate price to pay, and the system may already be overwhelmed at this stage. This is supported by evidence in rat myocardium where the magnitude of functional improvement seen with XO inhibitors was dependent on the initial level of XO activity (Kogler et al. 2003). In chronic diseases such as CHF, sustained high levels of ROS may exceed the capacity of cellular enzymatic and non-enzymatic antioxidants (Deanfield et al. 2007) to counter its effects. Using electron spin resonance, Spiekermann et al. demonstrated that both NADPH oxidase and xanthine oxidase are upregulated in patients with coronary artery disease (Spiekermann et al. 2003). Others have demonstrated increased levels in CHF (Landmesser et al. 2002; Amado et al. 2005).
2.5 Direct Antioxidant Action
Allopurinol directly scavenges free radicals as demonstrated by Das et al. and others (Das et al. 1987; Hoey et al. 1988; Ricardo et al. 1995) in in vitro hearts where evidence of free radical scavenging occurred in the absence of XO activity. Further evidence for a possible direct antioxidant effect of allopurinol comes from models of experimental colitis where tungsten (a potent XO inhibitor) failed to improve symptoms whereas allopurinol did (Keshavarzian et al. 1990). Augustin et al. suggested that this direct effect was only seen at higher doses (Augustin et al. 1994). This was also seen in mice paracetamol toxicity models where lower doses (sufficient to block XO activity) of allopurinol failed to show antioxidant protection but higher doses did (Knight et al. 2001). There have been other non-XO effects of allopurinol suggested such as copper chelation, preventing LDL oxidation as described above (Malkiel et al. 1993), inhibition of heat shock protein (hsp) expression (Nishizawa et al. 1999) and calcium sensitisation (below). Allopurinol treatment reduces early changes in inflammation such as leukocyte activation by reducing adherence, rolling and extravasation (Granger et al. 1989). Similarly, animal studies in global cerebral ischemia-reperfusion have demonstrated that the ROS lowering effect of allopurinol was not related to its XO inhibition activity but rather due to its direct free radical scavenging activity. This was not evident with febuxostat (Yamaguchi et al. 2015).
Animal studies in experimentally induced uveitis show that at very high doses (up to 50 mg/kg), allopurinol behaves as a free radical scavenger with intrinsic antioxidant properties. Crucially, this was only achieved far beyond the XO inhibition dose of 10 mg/kg and not at that dose itself.
2.6 Mechano-energetic Uncoupling
This phenomenon refers to an imbalance between left ventricular performance and myocardial energy consumption (Kittleson and Hare 2005). The role of XO inhibition may either be to maintain cardiac output while reducing myocardial oxygen consumption or even to increase cardiac output without increasing myocardial oxygen consumption. In dogs with pacing-induced heart failure, allopurinol improved myocardial contractility, efficiency in oxygen utilisation, prevented increases in systemic vasoconstriction and ameliorates reductions in myocardial contractility (Amado et al. 2005; Ekelund et al. 1999; Saavedra et al. 2002). In murine post-ischaemic cardiomyopathy models, allopurinol attenuated the increase in end-systolic and end-diastolic volumes (Naumova et al. 2006), increased survival, augmented ventricular function as well as reduced products of lipid peroxidation (Stull et al. 2004).
Khan et al. found a direct protein-protein interaction between XO and neuronal NOS in the sarcoplasmic reticulum of cardiac myocytes (Khan et al. 2004). Allopurinol improved myofilament calcium sensitivity as contraction force increases without a concomitant rise in systolic Ca2+ influx. The effects were not seen in endothelial NOS-deficient mice suggesting a role for neuronal NOS preventing XO inhibition of cardiac excitation-contraction coupling (Khan et al. 2004). The finding that allopurinol is a potent myofilament Ca2+ sensitizer, particularly in the setting of ischaemia, is thought to be due to the inhibition of basal XO production. As with the previous study by Khan et al., Perez et al. found an almost exclusive increase in force generation without a lowering of inward transient Ca2+ (Perez et al. 1998).
Despite the small sample size (n = 9), Cappola et al. showed using cardiac catheterisation that direct intra-coronary infusions of allopurinol in these patients resulted in a marked decrease in myocardial oxygen consumption (MVO2) with no decrease in the rate of left ventricular pressure rise (dP/dT), stroke work or ventricular load (Cappola et al. 2001). Patients post-CABG given allopurinol have also been shown to require less inotropic support (Sisto et al. 1995).
As alluded to earlier, the most potent ROS-generating systems are the NADPH oxidase and xanthine oxidase enzymes, and angiotensin II is the most potent inducer of NADPH oxidase (Griendling et al. 1994; Harrison et al. 2003). However, as we have previously demonstrated, patients already on an ACE inhibitor or an AT1 receptor blocker still derive improvement in vascular function from XO inhibition suggesting that there is still a significant level of oxidative stress present even in patients who are optimally treated with current evidence-based treatments (George et al. 2006). These actions are summarised in Fig. 2.

Summary of effects of xanthine oxidase inhibitors on hyperuricaemia, cardiac function and cardiovascyular haemodynamics
3 Febuxostat
3.1 Pharmacokinetics
Febuxostat (2-(3-cyano-4-[2-methylpropoxyl]phenyl)-4-methylthiazole-5-carboxylic acid) is a thiazolecarboxylic acid derivative, selective for inhibition of both the oxidised and reduced forms of xanthine oxidase, and does not resemble a purine or pyrimidine (Ernst and Fravel 2009). Febuxostat has selective affinity for both the oxidised and reduced forms of xanthine oxidase, with an in vitro inhibition (Ki) value of <1 nM (mean [SD], 1.2 [0.05] × 10−10) (Takano et al. 2005). The drug has an oral bioavailability of 85% (Kamel et al. 2017), achieves maximum plasma concentration in approximately 1.5 h and has a mean elimination half-life varying between 1.3 and 15.8 h (Khosravan et al. 2006). Febuxostat is mainly metabolised via glucuronidation (22–44% of the dose) and oxidation (2–8%) with only 1–6% of the dose being excreted unchanged via the kidneys (Khosravan et al. 2006). Therefore renal function is not a key determinant in its use. It is now recognised that Febuxostat is at least as effective as allopurinol in urate reduction (Faruque et al. 2013).
Data on the antioxidant effects of febuxostat are conflicting. Theoretically, it is a better antioxidant agent as inflammatory/hypoxic conditions upregulate tissue XO expression which results in sequestration and immobilisation of XO by endothelial glycosaminoglycans (GAG) . Immobilised GAG-bound XO is resistant to allopurinol but not febuxostat (Malik et al. 2011). Detailed crystallography studies revealed that febuxostat reaction with XO is confined to critical amino acid residues in the tunnel leading to the Mo cofactor, where it effectively blocks substrate access to the active site (Okamoto and Nishino 2008). Thus, febuxostat should not be affected by enzyme redox state and interaction with XO should not induce ROS formation (Malik et al. 2011).
Animal models of renal ischemia-reperfusion have demonstrated amelioration of ROS and therefore tubular injury and interstitial fibrosis by febuxostat (Tsuda et al. 2012). In studies using streptozocin-induced diabetic rat model, febuxostat reduced both urinary 8-OHdG, significantly decreased renal infiltration of macrophages resulting in reduced oxidative stress, transcription levels of inflammatory genes (E-selectin and VCAM), inflammation-induced enzymes (COX-2), inflammatory mediators (NF-kB) and renal cortical nitrotyrosine. This suggests a possible therapeutic effect for febuxostat in slowing deterioration of kidney function in the setting of diabetic nephropathy (Lee et al. 2014).
In small studies of patients with gout, febuxostat has been shown to have superior effects on oxidative stress and pulse wave velocity compared to low-moderate dose allopurinol (Tausche et al. 2014). In a study of haemodialysis patients, febuxostat was shown to significantly reduce high-sensitivity CRP and asymmetric dimethylarginine (ADMA) levels and improve endothelial dysfunction (reduced ADMA-mediated eNOS inhibition) compared to placebo (Alshahawey et al. 2017). Furthermore, the increase in ADMA levels and inhibition of nitric oxide production seen with proton pump inhibitors (PPI’s) has been shown to be blunted by febuxostat (Pinheiro et al. 2016). Febuxostat has also been shown to reduce other markers of oxidative stress such as oxidised LDL and EPA/AA (eicosapentaenoic acid/arachidonic acid) ratio (Sezai et al. 2013).
However when the impact of antioxidant defence is studied, febuxostat seems to reduce both biological antioxidant potential (BAP) and ROS metabolites (derivatives of reactive oxygen metabolites (d-ROMs) in equal measure (Fukui et al. 2015). In a study of obese adults with Type 2 diabetic nephropathy, febuxostat showed no effect on adipose tissue thiobarbituric acid reducing substances (TBARS) and adiponectin concentrations (Beddhu et al. 2016).
Recent large multi-centre trials of febuxostat have been reported showing mixed effects. The febuxostat for Cerebral and Cardiorenovascular Events Prevention Study (FREED) trial (Kojima et al. 2019) met its primary composite cardiovascular endpoint, but this was driven by a reduction in progression to renal dysfunction. There was no evidence of cardiovascular or cerebrovascular event rate reduction with febuxostat. This latter finding is consistent with the findings of the CARES (Cardiovascular Safety with Febuxostat vs Allopurinol) trial (White et al. 2018) which demonstrated no beneficial effect of febuxostat on cardiovascular events and in fact demonstrated that all-cause mortality and cardiovascular mortality were higher with febuxostat than with allopurinol (hazard ratio for death from any cause, 1.22 [95% CI, 1.01 to 1.47]; hazard ratio for cardiovascular death, 1.34 [95% CI, 1.03 to 1.73]). However, as Choi et al. point out, the use of non-XOI or placebo group is needed to determine whether the results of the CARES trial were due to the beneficial effects of allopurinol or the deleterious effects of febuxostat (Choi et al. 2018). Other ongoing trials such as Febuxostat versus Allopurinol Streamlined Trial (FAST) may provide some clarity to this issue.
3.2 Mechano-energetic Uncoupling
In a trial of hyperuricaemic patients undergoing cardiac surgery (NU-FLASH trial) comparing febuxostat with low-dose (300 mg) allopurinol, patients in the febuxostat arm showed significant reductions in systolic blood pressure, pulse-wave velocity and LV mass index compared to allopurinol (Sezai et al. 2013).
4 Topiroxostat
Topiroxostat [4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2- pyridinecarbonitrile] is a non-purine XOR inhibitor, approved in Japan in 2013 for the treatment of patients with hyperuricaemia. There is limited experience internationally with this agent. Topiroxostat behaves initially as a competitive type inhibitor to xanthine oxidoreductase before forming a strong covalent linkage to molybdenum via oxygen in the hydroxylation reaction intermediate (Chen et al. 2016). It also displays a potent non-covalent competitive type inhibition of XOR with a Ki value of 5.7 × 10−9 M (Matsumoto et al. 2011). Topiroxostat has good oral bioavailability with a half-life of up to 7.5 h after oral administration. It is predominantly eliminated in the urine. It is a strong inhibitor of Cyp 2C9 and has no inducing effect on CYP enzymes. Topiroxostat has a greater inhibitory effect on plasma XOR compared to tissue XOR (the opposite is observed with febuxostat) (Nakamura et al. 2016).
Mouse models of minimal change nephrotic syndrome demonstrated that nitrotyrosine and 8-hydroxy-2-deoxyguanosine (8-OHdG) were significantly ameliorated by topiroxostat (Kawamorita et al. 2017). The recently reported TROFEO trial (Sezai et al. 2017) in hyperuricaemic patients with cardiovascular disease comparing the effects of febuxostat and topiroxostat showed similar urate, antioxidant, anti-inflammatory and reno-protective effects for both drugs. The renoprotective effects of topiroxostat for hyperuricemic patients with overt diabetic nephropathy (ETUDE) study concluded that high-dose topiroxostat (160 mg/day) significantly reduced L-Fatty Acid Binding Protein (FABP), a validated biomarker of tubulointestitial damage and oxidative stress (Mizukoshi et al. 2018). There has not been any direct head-to-head antioxidant effect comparison between allopurinol and topiroxostat.
Table 2 summarises the current clinical trial evidence using XO inhibitors on ROS and other CV outcomes.
References
Alshahawey M, Shahin SM, Elsaid TW, Sabri NA (2017) Effect of febuxostat on the endothelial dysfunction in hemodialysis patients: a randomized, placebo-controlled, double-blinded study. Am J Nephrol 45:452–459
Amado LC, Saliaris AP, Raju SV, Lehrke S, St John M, Xie J, Stewart G, Fitton T, Minhas KM, Brawn J, Hare JM (2005) Xanthine oxidase inhibition ameliorates cardiovascular dysfunction in dogs with pacing-induced heart failure. J Mol Cell Cardiol 39:531–536
Augustin AJ, Boker T, Blumenroder SH, Lutz J, Spitznas M (1994) Free radical scavenging and antioxidant activity of allopurinol and oxypurinol in experimental lens-induced uveitis. Invest Ophthalmol Vis Sci 35:3897–3904
Bagnati M, Perugini C, Cau C, Bordone R, Albano E, Bellomo G (1999) When and why a water-soluble antioxidant becomes pro-oxidant during copper-induced low-density lipoprotein oxidation: a study using uric acid. Biochem J 340(Pt 1):143–152
Beddhu S, Filipowicz R, Wang B, Wei G, Chen X, Roy AC, DuVall SL, Farrukh H, Habib AN, Bjordahl T, Simmons DL, Munger M, Stoddard G, Kohan DE, Greene T, Huang Y (2016) A randomized controlled trial of the effects of febuxostat therapy on adipokines and markers of kidney fibrosis in asymptomatic hyperuricemic patients with diabetic nephropathy. Can J Kidney Health Dis 3:2054358116675343
Berry CE, Hare JM (2004) Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 555:589–606
Butler R, Morris AD, Belch JJ, Hill A, Struthers AD (2000) Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 35:746–751
Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, Hare JM (2001) Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104:2407–2411
Cardillo C, Kilcoyne CM, Cannon RO 3rd, Quyyumi AA, Panza JA (1997) Xanthine oxidase inhibition with oxypurinol improves endothelial vasodilator function in hypercholesterolemic but not in hypertensive patients. Hypertension 30:57–63
Chen C, Lu JM, Yao Q (2016) Hyperuricemia-related diseases and xanthine oxidoreductase (xor) inhibitors: an overview. Med Sci Monit 22:2501–2512
Choi H, Neogi T, Stamp L, Dalbeth N, Terkeltaub R (2018) New perspectives in rheumatology: implications of the cardiovascular safety of febuxostat and allopurinol in patients with gout and cardiovascular morbidities trial and the associated food and drug administration public safety alert. Arthritis Rheumatol 70:1702–1709
Cicoira M, Zanolla L, Rossi A, Golia G, Franceschini L, Brighetti G, Zeni P, Zardini P (2002) Elevated serum uric acid levels are associated with diastolic dysfunction in patients with dilated cardiomyopathy. Am Heart J 143:1107–1111
Committee JF (2007) British national formulary. British Medical Association and Royal Pharmaceutical Society of Great Britain, London
Das DK, Engelman RM, Clement R, Otani H, Prasad MR, Rao PS (1987) Role of xanthine oxidase inhibitor as free radical scavenger: a novel mechanism of action of allopurinol and oxypurinol in myocardial salvage. Biochem Biophys Res Commun 148:314–319
Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM (2007) Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet 46:623–644
Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and dysfunction: testing and clinical relevance. Circulation 115:1285–1295
Della Corte E, Gozzetti G, Novello F, Stirpe F (1969) Properties of the xanthine oxidase from human liver. Biochim Biophys Acta 191:164–166
Doehner W, Schoene N, Rauchhaus M, Leyva-Leon F, Pavitt DV, Reaveley DA, Schuler G, Coats AJS, Anker SD, Hambrecht R (2002) Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation 105:2619–2624
Ekelund UE, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, Kass DA, Marban E, Hare JM (1999) Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ Res 85:437–445
Elion GB, Kovensky A, Hitchings GH (1966) Metabolic studies of allopurinol, an inhibitor of xanthine oxidase. Biochem Pharmacol 15:863–880
Elion GB, Yu TF, Gutman AB, Hitchings GH (1968) Renal clearance of oxipurinol, the chief metabolite of allopurinol. Am J Med 45:69–77
Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, Fuchs M, Hilfiker-Kleiner D, Hornig B, Drexler H, Landmesser U (2004) Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation 110:2175–2179
Ernst ME, Fravel MA (2009) Febuxostat: a selective xanthine-oxidase/xanthine-dehydrogenase inhibitor for the management of hyperuricemia in adults with gout. Clin Ther 31:2503–2518
Farquharson CAJ, Butler R, Hill A, Belch JJF, Struthers AD (2002) Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 106:221–226
Faruque LI, Ehteshami-Afshar A, Wiebe N, Tjosvold L, Homik J, Tonelli M (2013) A systematic review and meta-analysis on the safety and efficacy of febuxostat versus allopurinol in chronic gout. Semin Arthritis Rheum 43:367–375
Fukui T, Maruyama M, Yamauchi K, Yoshitaka S, Yasuda T, Abe Y (2015) Effects of febuxostat on oxidative stress. Clin Ther 37:1396–1401
Gavin AD, Struthers AD (2005) Allopurinol reduces b-type natriuretic peptide concentrations and haemoglobin but does not alter exercise capacity in chronic heart failure. Heart 91:749–753
George J, Struthers AD (2008) The role of urate and xanthine oxidase inhibitors in cardiovascular disease. Cardiovasc Ther 26:59–64
George J, Carr E, Davies J, Belch JJ, Struthers A (2006) High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation 114:2508–2516
Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ, Henderson GN (2008) Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 27:967–978
Goharinia M, Zareei A, Rahimi M, Mirkhani H (2017) Can allopurinol improve retinopathy in diabetic rats? Oxidative stress or uric acid; which one is the culprit? Res Pharm Sci 12:401–408
Graham S, Day RO, Wong H, McLachlan AJ, Bergendal L, Miners JO, Birkett DJ (1996) Pharmacodynamics of oxypurinol after administration of allopurinol to healthy subjects. Br J Clin Pharmacol 41:299–304
Granger DN, Benoit JN, Suzuki M, Grisham MB (1989) Leukocyte adherence to venular endothelium during ischemia-reperfusion. Am J Phys 257:G683–G688
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994) Angiotensin ii stimulates nadh and nadph oxidase activity in cultured vascular smooth muscle cells. Circ Res 74:1141–1148
Guan W, Osanai T, Kamada T, Hanada H, Ishizaka H, Onodera H, Iwasa A, Fujita N, Kudo S, Ohkubo T, Okumura K (2003) Effect of allopurinol pretreatment on free radical generation after primary coronary angioplasty for acute myocardial infarction. J Cardiovasc Pharmacol 41(5):699–705
Guerra P, Frias J, Ruiz B, Soto A, Carcas A, Govantes C, Montuenga C, Fernandez A (2001) Bioequivalence of allopurinol and its metabolite oxipurinol in two tablet formulations. J Clin Pharm Ther 26:113–119
Gutierrez-Macias A, Lizarralde-Palacios E, Martinez-Odriozola P, Miguel-De la Villa F (2005) Fatal allopurinol hypersensitivity syndrome after treatment of asymptomatic hyperuricaemia. BMJ 331:623–624
Hancock JT, Salisbury V, Ovejero-Boglione MC, Cherry R, Hoare C, Eisenthal R, Harrison R (2002) Antimicrobial properties of milk: dependence on presence of xanthine oxidase and nitrite. Antimicrob Agents Chemother 46:3308–3310
Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H (2003) Role of oxidative stress in atherosclerosis. Am J Cardiol 91:7A–11A
Hille R, Massey V (1981) Studies on the oxidative half-reaction of xanthine oxidase. J Biol Chem 256:9090–9095
Hoey BM, Butler J, Halliwell B (1988) On the specificity of allopurinol and oxypurinol as inhibitors of xanthine oxidase. A pulse radiolysis determination of rate constants for reaction of allopurinol and oxypurinol with hydroxyl radicals. Free Radic Res Commun 4:259–263
Huang Y, Zhang C, Xu Z, Shen J, Zhang X, Du H, Zhang K, Zhang D (2017) Clinical study on efficacy of allopurinol in patients with acute coronary syndrome and its functional mechanism. Hell J Cardiol 58:360–365
Hung SI, Chung WH, Liou LB, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS, Chen MJ, Lai PC, Wu MS, Chu CY, Wang KH, Chen CH, Fann CS, Wu JY, Chen YT (2005) Hla-b*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 102:4134–4139
Ichida K, Amaya Y, Noda K, Minoshima S, Hosoya T, Sakai O, Shimizu N, Nishino T (1993) Cloning of the cdna encoding human xanthine dehydrogenase (oxidase): structural analysis of the protein and chromosomal location of the gene. Gene 133:279–284
Kamel B, Graham GG, Williams KM, Pile KD, Day RO (2017) Clinical pharmacokinetics and pharmacodynamics of febuxostat. Clin Pharmacokinet 56:459–475
Kawamorita Y, Shiraishi T, Tamura Y, Kumagai T, Shibata S, Fujigaki Y, Hosoyamada M, Nakagawa T, Uchida S (2017) Renoprotective effect of topiroxostat via antioxidant activity in puromycin aminonucleoside nephrosis rats. Physiol Rep 5(15):e13358
Keshavarzian A, Morgan G, Sedghi S, Gordon JH, Doria M (1990) Role of reactive oxygen metabolites in experimental colitis. Gut 31:786–790
Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM (2004) Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A 101:15944–15948
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L (2006) Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet 45:821–841
Kittleson MM, Hare JM (2005) Xanthine oxidase inhibitors: an emerging class of drugs for heart failure. Eur Heart J 26:1458–1460
Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H (2001) Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci 62(2):212–220
Kogler H, Fraser H, McCune S, Altschuld R, Marban E (2003) Disproportionate enhancement of myocardial contractility by the xanthine oxidase inhibitor oxypurinol in failing rat myocardium. Cardiovasc Res 59:582–592
Kojima S, Matsui K, Hiramitsu S, Hisatome I, Waki M, Uchiyama K et al (2019) Febuxostat for cerebral and cardiorenovascular events prevention study. Eur Heart J 40(22):1778–1786
Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, Harrison DG, Hornig B, Drexler H (2002) Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 106:3073–3078
Lee HJ, Jeong KH, Kim YG, Moon JY, Lee SH, Ihm CG, Sung JY, Lee TW (2014) Febuxostat ameliorates diabetic renal injury in a streptozotocin-induced diabetic rat model. Am J Nephrol 40:56–63
Mackenzie IS, Ford I, Walker A, Hawkey C, Begg A, Avery A, Taggar J, Wei L, Struthers AD, MacDonald TM (2016) Multicentre, prospective, randomised, open-label, blinded end point trial of the efficacy of allopurinol therapy in improving cardiovascular outcomes in patients with ischaemic heart disease: protocol of the all-heart study. BMJ Open 6:e013774
Malik UZ, Hundley NJ, Romero G, Radi R, Freeman BA, Tarpey MM, Kelley EE (2011) Febuxostat inhibition of endothelial-bound xo: implications for targeting vascular ros production. Free Radic Biol Med 51:179–184
Malkiel S, Har-el R, Schwalb H, Uretzky G, Borman JB, Chevion M (1993) Interaction between allopurinol and copper: possible role in myocardial protection. Free Radic Res Commun 18:7–15
Massey V, Komai H, Palmer G, Elion GB (1970) On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J Biol Chem 245:2837–2844
Matsumoto K, Okamoto K, Ashizawa N, Nishino T (2011) Fyx-051: a novel and potent hybrid-type inhibitor of xanthine oxidoreductase. J Pharmacol Exp Ther 336:95–103
McInnes GT, Lawson DH, Jick H (1981) Acute adverse reactions attributed to allopurinol in hospitalised patients. Ann Rheum Dis 40:245–249
Mizukoshi T, Kato S, Ando M, Sobajima H, Ohashi N, Naruse T, Saka Y, Shimizu H, Nagata T, Maruyama S (2018) Renoprotective effects of topiroxostat for hyperuricaemic patients with overt diabetic nephropathy study (ETUDE study): a prospective, randomized, multicentre clinical trial. Nephrology (Carlton, Vic) 23(11):1023–1030
Nakamura T, Murase T, Nampei M, Morimoto N, Ashizawa N, Iwanaga T, Sakamoto R (2016) Effects of topiroxostat and febuxostat on urinary albumin excretion and plasma xanthine oxidoreductase activity in db/db mice. Eur J Pharmacol 780:224–231
Naumova AV, Chacko VP, Ouwerkerk R, Stull L, Marban E, Weiss RG (2006) Xanthine oxidase inhibitors improve energetics and function after infarction in failing mouse hearts. Am J Physiol Heart Circ Physiol 290:H837–H843
Nishizawa J, Nakai A, Matsuda K, Komeda M, Ban T, Nagata K (1999) Reactive oxygen species play an important role in the activation of heat shock factor 1 in ischemic-reperfused heart. Circulation 99:934–941
Ogino K, Kato M, Furuse Y, Kinugasa Y, Ishida K, Osaki S, Kinugawa T, Igawa O, Hisatome I, Shigemasa C, Anker SD, Doehner W (2009) Uric acid lowering treatment with benzbromarone in patients with heart failure: a double-blind placebo-controlled cross-over preliminary study. Circ Heart Fail 3:73–81
Okamoto K, Nishino T (2008) Crystal structures of mammalian xanthine oxidoreductase bound with various inhibitors: allopurinol, febuxostat, and fyx-051. J Nippon Med Sch 75:2–3
Pacher P, Nivorozhkin A, Szabo C (2006) Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 58:87–114
Panus PC, Wright SA, Chumley PH, Radi R, Freeman BA (1992) The contribution of vascular endothelial xanthine dehydrogenase/oxidase to oxygen-mediated cell injury. Arch Biochem Biophys 294:695–702
Parks DA, Granger DN (1986) Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl 548:87–99
Pea F (2005) Pharmacology of drugs for hyperuricemia. Mechanisms, kinetics and interactions. Contrib Nephrol 147:35–46
Perez NG, Gao WD, Marban E (1998) Novel myofilament ca2+-sensitizing property of xanthine oxidase inhibitors. Circ Res 83:423–430
Pinheiro LC, Oliveira-Paula GH, Portella RL, Guimaraes DA, de Angelis CD, Tanus-Santos JE (2016) Omeprazole impairs vascular redox biology and causes xanthine oxidoreductase-mediated endothelial dysfunction. Redox Biol 9:134–143
Rajendra NS, Ireland S, George J, Belch JJ, Lang CC, Struthers AD (2011) Mechanistic insights into the therapeutic use of high-dose allopurinol in angina pectoris. J Am Coll Cardiol 58:820–828
Ricardo SD, Bertram JF, Ryan GB (1995) Podocyte architecture in puromycin aminonucleoside-treated rats administered tungsten or allopurinol. Exp Nephrol 3:270–279
Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS, Harrison RW, Zeichner J, Mudrick D, Marban E, Kass DA, Hare JM (2002) Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res 90:297–304
Schlesinger N (2004) Management of acute and chronic gouty arthritis: present state-of-the-art. Drugs 64:2399–2416
Sezai A, Soma M, Nakata K, Hata M, Yoshitake I, Wakui S, Hata H, Shiono M (2013) Comparison of febuxostat and allopurinol for hyperuricemia in cardiac surgery patients (nu-flash trial). Circ J 77:2043–2049
Sezai A, Obata K, Abe K, Kanno S, Sekino H (2017) Cross-over trial of febuxostat and topiroxostat for hyperuricemia with cardiovascular disease (TROFEO trial). Circ J 81(11):1707–1712
Sisto T, Paajanen H, Metsa-Ketela T, Harmoinen A, Nordback I, Tarkka M (1995) Pretreatment with antioxidants and allopurinol diminishes cardiac onset events in coronary artery bypass grafting. Ann Thorac Surg 59:1519–1523
Skinner KA, White CR, Patel R, Tan S, Barnes S, Kirk M, Darley-Usmar V, Parks DA (1998) Nitrosation of uric acid by peroxynitrite: formation of a vasoactive nitric oxide donor. J Biol Chem 273:24491–24497
Spector T (1977) Inhibition of urate production by allopurinol. Biochem Pharmacol 26:355–358
Spiekermann S, Landmesser U, Dikalov S, Bredt M, Gamez G, Tatge H, Reepschlager N, Hornig B, Drexler H, Harrison DG (2003) Electron spin resonance characterization of vascular xanthine and nad(p)h oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation 107:1383–1389
Stevens CR, Millar TM, Clinch JG, Kanczler JM, Bodamyali T, Blake DR (2000) Antibacterial properties of xanthine oxidase in human milk. Lancet 356:829–830
Stull LB, Leppo MK, Szweda L, Gao WD, Marban E (2004) Chronic treatment with allopurinol boosts survival and cardiac contractility in murine postischemic cardiomyopathy. Circ Res 95:1005–1011
Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA (2005) Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci 76:1835–1847
Tausche AK, Christoph M, Forkmann M, Richter U, Kopprasch S, Bielitz C, Aringer M, Wunderlich C (2014) As compared to allopurinol, urate-lowering therapy with febuxostat has superior effects on oxidative stress and pulse wave velocity in patients with severe chronic tophaceous gout. Rheumatol Int 34:101–109
Tsuda H, Kawada N, Kaimori JY, Kitamura H, Moriyama T, Rakugi H, Takahara S, Isaka Y (2012) Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochem Biophys Res Commun 427:266–272
Turnheim K (1999) Oberbauer. Pharmacokinetics and pharmacodynamics of allopurinol in elderly and young subjects. Br J Clin Pharmacol 48:501–509
Vazquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R (2001) Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis 60:981–983
Waring WS (2002) Uric acid: an important antioxidant in acute ischaemic stroke. QJM 95:691–693
White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, Hunt B, Castillo M, Gunawardhana L, CARES Investigators (2018) Cardiovascular safety of febuxostat or allopurinol in patients with gout. N Engl J Med 378:1200–1210
Yamaguchi M, Okamoto K, Kusano T, Matsuda Y, Suzuki G, Fuse A, Yokota H (2015) The effects of xanthine oxidoreductase inhibitors on oxidative stress markers following global brain ischemia reperfusion injury in c57bl/6 mice. PLoS One 10:e0133980
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vickneson, K., George, J. (2020). Xanthine Oxidoreductase Inhibitors. In: Schmidt, H.H.H.W., Ghezzi, P., Cuadrado, A. (eds) Reactive Oxygen Species . Handbook of Experimental Pharmacology, vol 264. Springer, Cham. https://doi.org/10.1007/164_2020_383
Download citation
DOI: https://doi.org/10.1007/164_2020_383
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-68509-6
Online ISBN: 978-3-030-68510-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)