
Abstract
The last 100 years have seen a dramatic alteration in the treatment of cancer. Aside from small molecule inhibitors of protein tyrosine kinases, monoclonal antibodies have also been found to provide valuable therapeutic approaches for modulating tumour pathophysiology. As our knowledge of cancer biology improves, the specificity of this new generation of drugs is generally delivering an improved therapeutic ratio compared to traditional cytotoxic agents. However, patient selection through the use of biomarkers is key in optimising efficacy and improving cost-effectiveness. The most recent wave of revolutionary new systemic therapy approaches to cancer has arrived in recent years in the form of immune checkpoint inhibitors, now clinically validated as modulators of immune-regulatory pathways. The future of oncology therapeutics includes a combination of cytotoxic agents, targeted therapies and immunotherapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Checkpoint inhibition
- Cytotoxic
- Drug resistance
- Immunotherapy
- Monoclonal antibody
- Oncogene
- Signalling
- Tyrosine kinase
1 Biology of Cancer
1.1 Cancer as a Genetic Disease
The malignant phenotype of cancer is driven by a series of genetic aberrations in a cell clone that evolves in Darwinian fashion to form a tumour. The first recognition of a cellular origin for viral oncogenes was made in 1970. Proto-oncogenes exist in the normal genome and generally encode proteins that have an important role to play in regulating normal cell growth, proliferation and development. If these genes are dysregulated, they have the potential to contribute to tumourigenesis (Croce 2008). Another important group of genetic contributors to initiating and maintaining malignancy are tumour suppressor genes, with an anti-proliferative effect in the normal cell. Tumour suppressor genes undergo genetic change such as deletion and missense mutation, resulting in loss of function in cancer cells. MicroRNAs are the products of genes that do not encode any protein. These short RNA sequences contribute to tumourigenesis by complementing the sequence of specific mRNAs and so preventing their translation.
The hallmarks of cancer can be considered within a conceptual framework entailing the fundamental aspects of neoplastic biology. Genetic insults to oncogenes and tumour suppressor genes contribute to tumour formation by affecting key aspects of this biology (Fig. 2) (Hanahan and Weinberg 2011). For example, the action of oncogenes may lead to abnormal growth and proliferation in the absence of appropriate signals, failure of programmed cell death, upregulated angiogenesis or unconstrained replication potential. Loss of tumour suppressor gene function can result in absence of normal signals controlling cell division.
There may be hundreds of genetic changes to the germline genome in a single cancer cell. The mechanisms giving rise to these mutations are only partially understood. In some cancers there is clearly a role for chemical carcinogens (cigarette smoke in lung cancer). In others, oncogenic viral genes act to inactivate tumour suppressors in infected cells, exemplified by E6 antigen expressed by human papilloma virus. E6 inactivates p53 and contributes to the increased incidence of cervical carcinoma in individuals infected by this virus.
In some cases, there is inherited susceptibility to genetic events in families. For example, a defective allele of the tumour suppressor genes BRCA1 or BRCA2 is inherited by some patients with breast, ovarian or prostate cancer. More commonly, these events occur in the somatic genome. Multistep tumourigenesis refers to a serial accumulation of insults and partly accounts for the fact that cancer is more common in a large long-lived organism such as man. Between 1,000 and 10,000 mutations have been implicated as contributing to a single human cancer, the majority of which affect dominantly acting oncogenes (Stratton 2011). Defects in a critical subset of genes, known as driver mutations, must arise in a single cell in order to give rise to a malignant phenotype. These are thought to be critical to providing a survival advantage to the cancer clone, whilst a larger number are likely to be passenger molecular events arising in an increasingly unstable genome. This genetic instability gives rise to the multiple and heterogeneous clone characteristic of a mature cancer. An exponential expansion in understanding of these aspects of cancer biology has defined potential targets for new therapies, a number of which has been approved in the past two decades.
1.2 Signalling in Health and Malignant Disease
Cellular signalling pathways composed of extracellular soluble ligands, transmembrane receptors and intricate intracellular kinase cascades are ubiquitous in nature. The ErbB receptor family has been more extensively studied than any other signal transduction network. EGFR is a receptor tyrosine kinase in this family, which consists of four members: EGFR (HER1/ErbB1), HER2 (ErbB2), HER3 (ErbB3) and HER4 (ErbB4) (Salomon et al. 1995). Ligand binding results in rapid receptor dimerisation, phosphorylation and activation of intracellular signalling pathways, which in turn leads to cell growth, proliferation and differentiation (Yarden and Sliwkowski 2001). ErbB receptors undergo various types of alteration and dysregulation in human tumours including gene amplification, receptor overexpression, activating mutations, overexpression of receptor ligands and/or loss of negative regulatory controls (Fig. 3) (Krause and Van Etten 2005). These tyrosine kinases can be targeted both by inhibitors of the intracellular signalling domain and by monoclonal antibodies specific for the extracellular ligand-binding domain.
1.3 Tumour Microenvironment and Host Immunity
Tumours evolve through multistep tumourigenesis to form complex ‘organs’. It is not only the individual carcinogenic cells that define its properties but also the microenvironment which nurtures its development and progression. The tumour microenvironment is composed of multiple cell types including lymphoid, myeloid, stromal and endothelial cells. The microenvironment is a hostile environment therapeutically due to low pH, necrosis, hypoxia, shortage of nutrient and the presence of immunosuppressive host immune components (Riviere and Sadelain 2017; Sadelain et al. 2017). Between and within patients, there is tumour heterogeneity which is reflected in histopathology showing varying degrees of differentiation, invasion, inflammation and vascularity. During tumour evolution there are progressive changes within the microenvironment (Hanahan and Weinberg 2011). Advances in immunotherapy have arisen from the current understanding of the ability of a tumour to circumvent host immunity.
Genetic alterations within a tumour cell result in the expression of neoantigens, which are processed into peptides that can bind to the major histocompatibility class I (MCHI) molecules on the surface of cancer cells, differentiating them from normal cells. These cancer-specific peptide-MHCI complexes can be recognised by host CD8+ T-cells. However, through evolutionary deletion of this complex, known as immune editing, cancer cells can avoid host attack (Chen and Mellman 2013). Expression on the tumour cell surface of ligands for inhibitory T-cell receptors such as PD1 provides another mechanism for immune evasion. Immune-mediated tumour cell death releases neoantigens, to be captured by dendritic cells in regional lymph nodes, which in turn prime and activate effector T-cells by presenting antigens in the context of MHCI and MHC II molecules. T-cells may subsequently traffic to and infiltrate the tumour, where they recognise and bind to cancer cells through interaction with the T-cell receptor and co-stimulatory pathways to cause further tumour cell death, completing the so-called cancer-immunity cycle (Chen and Mellman 2013).
The tumour microenvironment plays a critical role in the modulation of immune activity. In a microenvironment which is otherwise not responsive to immunotherapy, chemotherapeutics can be used to sensitise the tumour to become immunogenic. In vitro studies have demonstrated that chemotherapeutics induce a systemic host response including adaptive immunity. They influence tumour-host interactions, stimulating CD8+ T-cell activation and infiltration into the tumour microenvironment in otherwise T-cell-naive tumours. Immunogenic chemotherapeutics also have direct actions on the tumour bed. Collectively, these processes can sensitise tumours to immunotherapy. Together, chemotherapeutics and checkpoint inhibitors can provide a synergistic treatment option for tumours resistant to checkpoint blockade therapy alone (Chen and Mellman 2013).
2 Introduction
2.1 History
The introduction of both surgery and radiotherapy for the treatment of cancer predate the advent of drug therapy by many years. First used clinically more than a century ago, radiotherapy was used either alone or in conjunction with surgery as the only available treatment modality until the first trials with cancer-targeting drugs in the 1940s. Approximately two thirds of patients will require radiotherapy during their cancer treatment. X-rays were first used diagnostically by Wilhelm Conrad Rontgen in 1895. Subsequently, skin cancers were treated with x-rays due to low tissue penetration. In the early days dosimetry was unsophisticated, and toxicities often outweighed the benefits of treatment. However, by the 1920s the radiobiological properties of electromagnetic radiation (x-rays and gamma rays), particles (electrons, protons and neutrons) and radioactive isotopes (particularly radium) were better understood. Radiotherapy either directly damages cellular DNA or causes indirect damage through the production of free radicals, thereby damaging the genome of clonogenic tumour cells. This in turn leads to mitotic arrest and cell death when the cells enter mitosis without repair of this DNA damage. However, normal cells, particularly those that are rapidly dividing, may also be damaged. Therefore, radiation oncology clinicians and physicists have to plan accurately focused radiation beams, with fractionation of the total treatment dose to allow for maximum dose delivery to the tumour, whilst sparing normal tissue and allowing sufficient repair and recovery of non-tumour regions unavoidably included in the treatment field.
By the 1980s devices used to deliver proton beams were established, particularly to treat benign diseases such as keloid scarring. The following decades saw the marriage of machines delivering x-rays and advanced computer software allowing for three-dimensional conformal imaging. As computer systems became more sophisticated, intensity-modulated radiation therapy and stereotactic radiotherapy were introduced. In many centres, these techniques form the mainstay of radical treatment. In addition, we are now able to add a fourth dimension of time to accommodate for real-time motion as a result of the breathing cycle for treatment of tumours in the lungs and upper abdomen. Currently under trial is ‘adaptive radiotherapy’ that allows for repeating imaging in between fractions to account for alterations in the size and motion of tumours during radiotherapy, which is particularly useful for rapidly responding tumours. Radiotherapy continues to remain an exceptionally important mode of treatment in both a radical and palliative context (Gianfaldoni et al. 2017). Coupled with surgery, radiotherapy remains the mainstay of treatment for tumours localised at the time of diagnosis. However, in many cases tumours are metastatic at the time of presentation. A detailed discussion of surgery and radiotherapy in the treatment of cancer is beyond the scope of this chapter which focusses on the diverse systemic therapies developed since the dawn of medical oncology 75 years ago.
During the First World War, nitrogen mustards were deployed as a chemical weapon. Soldiers that survived exposure to nitrogen mustards were noted to have reversible leucopenia and mucocutaneous blistering. In the 1940s, drugs in the same class were first used in clinical trials for the treatment of haematological malignancies (Gilman and Philips 1946) with promising outcomes. By the 1960s newer cytotoxic drugs were made available so that diseases such as leukaemia and some solid organ tumours, most notably germ cell malignancies, could be controlled by halting the dividing cell and, in cases such as testicular cancer, cured.
Over the next decades, the spectrum of cytotoxic agents expanded further with candidate drugs exhibiting antimitotic activity through a variety of mechanisms. The landmark discovery of platinum conjugates, particularly cisplatin (Rosenberg et al. 1969), allowed the first curative treatment for patients even with advanced testicular cancer. The phenomenon of tumour resistance to anticancer therapeutics was overcome in some contexts with the ability to safely combine multiple cytotoxic agents. Drug combinations have been particularly effective in haematological malignancies, especially aggressive lymphomas and acute lymphoblastic leukaemia. The potential for the more common tumours of epithelial origin such as breast, colorectal and lung cancer to benefit from cytotoxic drugs led to the development of further drug classes including taxanes and antimetabolites in the last two decades of the twentieth century. However, although these drugs demonstrate useful palliative and adjuvant efficacy in various settings, they failed to deliver the hoped-for outcome of cure in common advanced-stage solid tumours, partly due to evolving tumour cell resistance.
In response to a perceived stalling of progress with newer cytotoxics used in more complex and toxic combinations, drug discovery and clinical development in oncology began to focus at the end of the twentieth century on new classes of drugs, driven by progress in the molecular understanding of tumour biology. For example, transtuzumab, a monoclonal antibody targeting the oncogenic HER2 receptor, was licensed for the treatment of breast cancer in 1998. More recently, research into the host immune system’s response to cancer has led to the development of immune checkpoint inhibitors and adoptive T-cell therapy.
2.2 Roles for Systemic Therapies
A common characteristic of cancers is a high mitotic index reflecting rapid cell proliferation. Antimitotic drugs, targeted agents and immunotherapies are each now widely used in the treatment of cancer patients, with either radical or palliative intent. In the radical setting, they are often used following surgery, commonly known as adjuvant therapy. In some rapidly proliferating lymphomas, acute leukaemia and germ cell tumours, chemotherapy alone may be curative. However, in more common metastatic tumours of epithelial origin such as breast, lung and colorectal, cure is almost never achieved. In these situations, the aim is improvement in quality of life through symptom control and extension of survival time.
With the use of novel targeted therapies, combination cytotoxic regimens and immune checkpoint inhibitors, a pivotal decision is often the selection of optimal therapy and sequence of treatment for each individual. Even where cure cannot be offered with any certainty, cancer may now be considered a chronic disease in some tumour types. Throughout treatment the aim remains to prolong life and maintain a reasonable quality of life.
Systemic therapies are also used in the neoadjuvant and adjuvant setting. In the former, the goal is to reduce the size of the tumour, facilitating a successful outcome with radical surgery or radiotherapy. Adjuvant therapies make a meaningful reduction in the risk of relapse after radical treatment, thereby extending overall survival following surgical treatment in many common cancers including colorectal, breast and lung. The rationale for adjuvant therapies is that despite locally confined disease macroscopically, and using the most sensitive imaging techniques, it is apparent in retrospect that many patients had micrometastases at the time of radical local treatment. Studies showing circulating tumour cells and epithelial cells in the bone marrow even in patients with early stage cancers support this hypothesis.
Systemic therapies are used concurrently or sequentially with radiotherapy, particularly for locally advanced head and neck, lung, breast and some gastrointestinal cancers. Together, both modalities provide an efficacy not achievable by either modality alone.
2.3 Cytotoxic Drugs
Cytotoxic agents interact with the cellular machinery of mitosis, the cellular process critical to malignant proliferation. This machinery includes DNA synthesis, DNA structure, and tubulin-based cytoskeletal mitotic structures. Box 1 summarises the broad classes of cytotoxic agents. The inevitable consequences of targeting proliferating cells is that all organs with rapid rates of healthy cell turnover, such as hair follicles, mucosal surfaces in the gastrointestinal tract and bone marrow, are potentially affected. Haematopoietic lineages are affected in chronological sequences dictated by their normal circulating half-life. In this way, leucocytes are depopulated first, followed by platelets and then red cells. After the administration of chemotherapy, leucocytes take approximately 3 weeks to recover in the absence of exogenous growth factors. Therefore, chemotherapy is generally administered every 3 weeks.
Box 1 Summary of Broad Classes of Cytotoxic Drugs
Agents targeting DNA structure | |
Alkylation | Cyclophosphamide CCNU Melphalan |
Platinum coordination cross-linking | Cisplatin |
Double-stranded cleavage via topoisomerase 2 antibiotics | Doxorubicin Daunorubicin Podophyllotoxins Etoposide Teniposide |
Single-stranded cleavage via topoisomerase 1 | Topotecan Irinotecan |
Intercalation blocking RNA synthesis | Dactinomycin |
Uncertain mechanism | Bleomycin |
Agents targeting DNA synthesis: antimetabolites | |
Pyrimidine analogues | 5-flurouracil Capecitabine (fluoropyrimidines) |
Antifolates | Methotrexate (DHFR) Pemetrexed (TS;DHFR) |
Agents targeting tubulin | |
Taxanes (stabilise microtubules) | Paclitaxel Docetaxel Novel taxanes |
Vinca alkaloids (inhibit tubular polymerisation) | Vinorelbine Vincristine Vinblastine |
The primary dose-limiting factor for cytotoxic treatments is unwanted toxicities on normal tissues. It is regarded desirable to administer as high a dose as tolerable, both to maximise anticancer efficacy, but to limit toxicities affecting quality of life. Phase I trials are traditionally designed to reach a maximum tolerated dose, with the result that at approved doses, cytotoxic drugs and their combinations are usually associated with a narrow therapeutic index. In routine clinical practice, this translates to the necessity for a thorough assessment of fitness and comorbidities as an essential precursor to prescribing chemotherapy. In the context of clinical drug development, early-phase trials are generally conducted in populations of cancer patients, rather than in healthy volunteers, because some compensation for toxicity by clinical benefit is at least a possibility.
2.4 Targeted Therapies
By the first decade of the twenty-first century, a new era of cancer therapeutics was born with the exponential advent of many classes of drugs that mediated anticancer effects through targets other than the mitotic machinery. These have become generally known as ‘targeted therapies’.
Targeted therapies can be divided into two broad categories: therapeutic monoclonal antibodies or small molecules. The latter penetrate the cell membrane and can interact with their cellular targets. Unlike cytotoxics, these agents tend to have more intrinsic specificity for cancer cells, and so they are associated with a higher therapeutic ratio than cytotoxic drugs. Unlike cytotoxic therapies they are seldom associated with significant myelosuppression. However, on-target toxicities are still observed because the targets for these agents often have a physiological role to play, in addition to their aberrant function in the cancer cell. In many cases significant efficacy has been observed at well-tolerated doses. Biological markers such as cell surface expression of antigens or hormone receptors, or molecular features in the cancer genome, are used to select patients who would benefit from these agents, thereby personalising the approach to cancer treatment.
2.5 Immunotherapy
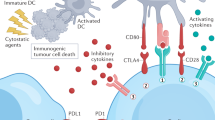
Clinical validation of immunotherapy in the treatment of cancer has only recently been achieved, but the proposed concept that the patient’s immune system might be induced to control tumour cells is over a century old. Neoantigens, resulting from tumourigenic mutations in the cancer genome, are expressed or presented on the cancer cell surface, potentially rendering these cells recognisable as non-self by host cytotoxic CD8+ T-cells. However, cancer cells can evade immune surveillance by expressing proteins such as PDL1, which can engage with its receptor, the inhibitory molecule PD1, on the T-cell surface. Inhibiting the PDL1/PD-1 interaction can restore antitumour T-cell activity (Fig. 1). The concept of immune evasion has been established as a biological hallmark of tumour capabilities (Fig. 2) (Hanahan and Weinberg 2011).

Targeting the immune checkpoint. Neoantigens in cancer cells potentially render them recognisable as non-self. However, cancer cells can evade immune surveillance by expressing proteins such as PD-L1 recognised by the negative regulatory T-cell receptor PD1. Inhibiting the PD-L1/PD1 interaction can restore T-cell cytotoxic activity

Therapeutic targeting of the Hallmarks of Cancer. Extracted with permission (from (Hanahan and Weinberg 2011)) Properties recognised to be responsible for tumour evolution, and how respective cancer therapeutics are developed to target tumours, are depicted below
So-called checkpoint inhibitor monoclonal antibodies target the suppressive mechanisms at the interface between T-cell and tumour or between T-cells and antigen-presenting cells. Aside from PD1 and PDL1, these targets include CTLA-4, and the first of these checkpoint inhibitors gained marketing approval in 2013. This approach has shown unprecedented clinical benefit across multiple tumour groups, and in many indications is better tolerated than cytotoxic treatment, or offers increased efficacy alone or in combination. However, there remains a large proportion of patients that fail to respond to the current early generation checkpoint inhibitors. Measurement of tumour PDL1 expression has been used as a clinical biomarker to enrich the patient population for those that may respond to treatment. However, in practice, PD1 and PDL1 expression correlates poorly with clinical response. Laboratory studies show that the proportion of cancer cells responding to checkpoint inhibitors may be increased by combining them with immunogenic drugs which alter cell expression of proteins and/or the tumour microenvironment (Havel et al. 2019).
3 Clinical Trials in Oncology
3.1 Phase 1 Trials
Traditionally, Phase 1 oncology trials are conducted in patients with advanced cancer as opposed to healthy volunteers. This has been because of the low therapeutic ratio expected for cytotoxic drugs. More recently, with the advent of better-tolerated targeted agents, initial dosing in healthy volunteers is more commonly undertaken, especially where the toxicity profile is predictable from other drugs in the same class. For example, single-dose administration to explore initial pharmacokinetics can often be best conducted in this way. This strategy reduces the risk of exposing cancer patients to subtherapeutic doses.
The commonest dose escalation scheme remains the traditional ‘3 + 3’ design, in which three patients are first treated at a given dose, with a further three added if a single dose-limiting toxicity (DLT; as predefined for each study) occurs. If no DLT is seen in the first three patients, or in no more than one of six patients in an expanded cohort, then dose may be escalated for the next cohort. A dose at which two or more DLTs occur is regarded as intolerable, and a dose level below this is likely to be explored as the maximum tolerated dose (MTD). There is no statistical basis to this trial design, but it has proved practical and informative in the development of countless drugs for the treatment of cancer. Nevertheless, newer strategies including accelerated dose escalation and the continuous reassessment method (CRM) are becoming more widely adopted (Piantadosi et al. 1998). CRM, based on Bayesian statistics, aims to adjust dose increments and cohort sizes by taking into account emergent toxicity data and has the potential for reducing the time and sample number required in a dose escalation trial.
The usual pharmacokinetic parameters such as Cmax, t1/2 and AUC are collected in cancer Phase 1 studies and may inform key decisions including dose escalation and dosing schedule. These data answer the fundamental question of whether drug can circulate at adequate concentrations to allow therapeutic effect, as predicted from target plasma concentration established in preclinical models.
Whether drug delivered to tumour can accumulate there and exert a biological effect requires on-treatment tumour biopsy, allowing for improved pharmacodynamic analysis. As an example, western blotting of phosphoproteins in a study of a kinase inhibitor may help to demonstrate successful target modulation and corresponding downstream effects. Serial sampling of tumours is preferred to collection of surrogate tissues such as skin or peripheral blood mononuclear cells. This is because drug penetration of normal tissues may differ from that in tumour, where abnormal vasculature, altered pH and hypoxia may give rise to very different localised effects. In general, patients participating in Phase 1 cancer trials are perhaps surprisingly willing to undergo these procedures. Further insight into the effect of a drug on tumours may be provided by functional imaging, for example, evaluating tumour perfusion or vessel permeability using MRI techniques. However, few such imaging endpoints are validated for making go/no-go drug development decisions.
As in other areas of experimental therapeutics, the primary objectives of a Phase 1 oncology trial are to study the safety profile of the drug and to establish a recommended Phase 2 dose (RP2D). Traditionally, dose is escalated to the maximum tolerated dose (MTD), but for drugs with known mechanism of action and available assays to demonstrate target inhibition, an alternative is to determine an optimal biological dose (Adjei 2006). This endpoint may appear better suited to trials of rationally designed targeted therapies where activity might be expected below the MTD, but nevertheless it has not become established as a standard (Parulekar and Eisenhauer 2004). This is because of concern that a reliance on the demonstration of PD effect in tumours may increase the risk of selecting a Phase 2 dose below maximal clinical activity, which might occur if the drug’s mechanism of action is incompletely understood, or biomarkers of efficacy are misleading in samples from heterogeneous tumours (Yap et al. 2010). Ideally, randomised Phase 2 trials should compare biologically active doses with the higher MTD. If escalation to MTD is not possible, as with some new well-tolerated non-cytotoxic agents, RP2D may be selected based on PK and PD parameters.
The likelihood of efficacy is clearly an important metric when discussing trial participation with patients. Assessment of response is never a primary objective in a Phase 1 trial, but where seen this is of course extremely encouraging. There is evidence, in the current era of rationally designed drugs and with many trials combining novel agents with more established therapies, that on average 11% of Phase 1 patients experience a partial beneficial response (Horstmann et al. 2005). Protocols should preferably be written to allow enrichment with patients whose tumours express biomarkers believed to be predictive of response, and this can accelerate progression into late-phase clinical trials in an appropriately targeted population (Kwak et al. 2010).
3.2 Phase 2 Trials
Compared with other disciplines, Phase 2 trials in oncology have often been conducted as single-arm (non-randomised) studies with response rate as the primary endpoint. Two-stage designs incorporating early stopping rules in the event of lack of efficacy are widely used for ethical purposes to minimise the numbers of patients treated on an ineffective agent (Simon 1989). Lack of randomisation in cancer studies may have arisen from a reluctance to allocate patients with a life-threatening diagnosis to placebo, or to no treatment, in indications where there is no standard of care. Response rate (RR) was an obvious endpoint to focus on when most agents studied were cytotoxic and, if active, were expected to shrink tumours. RR in these Phase 2 trials is usually compared to historical controls, if available. However, multiple experiences of promising Phase 2 activity followed by a negative Phase 3 trial, as well as a shift to studies of targeted agents, have led to a renewed emphasis on randomisation in Phase 2 trials (Eisenhauer et al. 2009). Evidence-based treatment options in many cancers have expanded over recent years, so comparators for control arms are more likely to exist, although in some settings a placebo control arm may still be appropriate. Strategies for reducing exposure to placebo are discussed below.
Another endpoint commonly used for efficacy assessment in Phase 2 trials of cancer drugs is progression-free survival (PFS, time from randomisation to disease progression), which may be a more meaningful surrogate of clinical benefit. PFS is also likely to allow a more appropriate assessment of efficacy of newer drug classes with mechanisms of action likely to block proliferation rather than induce apoptosis and tumour shrinkage.
Assessment of disease status in solid tumours is generally performed using CT scanning, and reproducible quantification of this is essential for determination of RR and PFS. An arbitrary but widely accepted technique for evaluating disease status is provided by the response evaluation criteria in solid tumours (RECIST) in which the long axis of selected target lesions is summed to provide a total measurement (Eisenhauer et al. 2009). Progression of disease is defined as an enlargement of the RECIST measurement by more than 20%, and conversely reduction by more than 30% represents a partial response (complete response if no assessable disease remains). RECIST disease assessment has, however, been widely criticised as being cumbersome and misleading, and some have argued for the use of RECIST to be ‘resisted’ (Ratain and Eckhardt 2004). Nevertheless, RECIST criteria have led to a useful international standard.
Additional imaging modalities such as PET and functional MRI are also frequently used in Phase 2 trials to assess efficacy and explore mechanism of action (Josephs et al. 2009). In some patients, measurable disease by radiological imaging is not present, and other measures of tumour burden are being evaluated as intermediate endpoints of clinical benefit, for example, circulating tumour-secreted proteins (tumour markers), tumour cell counts and circulating plasma nucleic acids. Prostate-specific antigen (PSA) is shed into the plasma in proportion to tumour burden, and criteria for PSA change in response to trial therapies have been agreed (Small and Roach 2002). Phase 2 trials provide an opportunity for development and initial validation of novel biomarkers to inform patient selection for future studies. This is especially important for therapies with a defined target where marketing approval may not be granted in the absence of a companion diagnostic to maximise efficacy in a defined patient population.
3.3 Phase 3 Trials
As in other disciplines, Phase 3 trials in oncology are randomised studies and wherever possible are designed so that both patient and investigator are blind to the treatment allocation (Booth and Tannock 2008). The control arm may be placebo, if there is no currently available evidence-based active treatment, or may be a standard treatment. Blinding may not be practical if, for example, an oral therapy is being compared to another administered parenterally. Key eligibility criteria include histological diagnosis, stage, prior therapies and performance status (an important measure of fitness and symptom burden) (Oken et al. 1982). Upper age limits are rarely appropriate, but older patients have historically been significantly under-represented in Phase 3 oncology trials, clearly an undesirable situation when most common cancers are more common in older patients. An accepted primary endpoint for Phase 3 oncology trials is overall survival, which has the advantages of a lack of ambiguity or bias. However, as the treatment armamentarium expands in many tumour types, this endpoint is increasingly likely to be confounded by post-study therapies. As a result, PFS is increasingly accepted for registration trials. RECIST measurements in serial CT scans are generally used to assess this endpoint. PFS has clinical relevance in many cases because disease progression in metastatic cancer often causes worsening symptoms and deterioration in quality of life (QOL).
Prospective assessment of QOL is desirable in Phase 3 trials. This is especially the case in oncology where any improvements in symptoms, OS or PFS need to be counterbalanced by consideration of potentially considerable toxicity. The UK National Institute for Health and Care Excellence (NICE) analyses measures of efficacy including QOL and takes into account drug pricing when evaluating cost-effectiveness for use of new therapies in the UK National Health Service. NICE uses a measure of benefit that corrects survival improvement for QOL, called a QALY (quality-adjusted life year), so that greater value is attached to a year’s extra survival at a perfect level of fitness than to the same period at an impaired level of function (Faden and Chalkidou 2011).
Phase 3 trials are large undertakings including sometimes thousands of patients treated at hundreds of centres and are therefore costly to conduct. It is self-evident that measures should be taken to maximise the chances of success, but in the era of targeted therapies, this has not always occurred. In fact oncology drugs are less likely to progress successfully through clinical development than most other clinical disciplines, with only 5% of cancer drugs awarded Investigational New Drug status going on to gain marketing approval (Adjei et al. 2009). By contrast, some of the most important Phase 3 results with novel agents have been obtained through careful selection of patients with tumours expressing the target, as in trials of trastuzumab in HER2+ breast cancer, or EGFR inhibition in EGFR-mutated non-small-cell lung cancer (Mok et al. 2009; Slamon and Pegram 2001). It is important to note that the predictive value of biomarkers such as HER2 amplification or EGFR mutation can only be definitively confirmed in a randomised trial because this is the only way to exclude a purely prognostic effect of these markers.
The inclusion of a placebo arm in an oncology trial can be problematic and may impair recruitment because of patients’ negative perceptions of this design. A number of strategies have been proposed for minimising exposure to placebo, including weighted randomisation and crossover design. Crossover is particularly suitable if OS is not the primary endpoint and allows patients on the placebo arm to receive experimental treatment upon progression. Interim analyses conducted by a robust data monitoring committee help to keep the sample number to a minimum and so minimise exposure to placebo in the control arm.
4 Rationally Designed Therapies
Advances in the understanding of cancer biology have led to the identification of new targets and driven the development of specific therapies directed against them. Examples include drug classes directed against ligands, receptors, intracellular signalling components and cellular machinery such as the proteasome and chromatin (Box 2).
Box 2 Targets for Anticancer Therapies: Examples of Approved and Investigational Drugs
Ligands | |
Steroid Hormones | AIs Abiraterone Apalutamide Enzalutamide |
VEGF | Bevacizumab |
Receptors | |
Oestrogen | Tamoxifen |
erbB | Cetuximab Trastuzumab |
Receptor tyrosine kinases | |
erbB | Erlotinib Gefitinib Afatinib Osimertinib |
VEGFR | Sunitinib Sorafenib Axitinib |
MET | Hh |
Intracellular kinases | |
mTOR | Everolimus |
BRAF | Vemurafenib |
MEK | Trametinib Cobimetinib Binimetinib |
bcr-abl | Imatinib |
EML4-ALK | Crizotinib Brigatinib Alectinib |
Proteasome | |
Bortezomib | |
Chromatin | |
HDACs Demethylase inhibitors PARP inhibitors – olaparib | |
Immunomodulating antibodies | |
PD1 | Pembrolizumab Nivolumab |
PDL1 | Atezolizumab |
CTLA4 | Ipilimumab |
Haematological targets | |
CD20 | Rituximab |
CD52 | Alemtuzumab |
CD20 | Ofatumumab |
4.1 Ligands as a Target
4.1.1 Oestrogen
Oestrogen is crucial for the growth and propagation of hormone-sensitive breast cancer. The circulating oestrogen ligand binds to and activates cytosolic receptors in tumour cells. These receptors are expressed in approximately 75% of all breast cancers, suggesting that these tumours may respond to oestrogen deprivation. Removing sites of oestrogen production (oophorectomy or irradiation), antagonising oestrogen activity or blocking oestrogen synthesis can all reduce available oestrogen. Tamoxifen is a selective oestrogen-receptor antagonist that blocks ligand binding, thereby blocking tumour cell proliferation. In premenopausal women with tumours strongly expressing oestrogen receptor (ER+), 5 years of adjuvant tamoxifen reduced the risk of recurrent disease and reduced the risk of death by 34% (Early Breast Cancer Trialists’ Collaborative 2005). More recently a comprehensive statistical model, PREDICT 2.0, has been developed to assess the survival benefit of adjuvant hormone treatment over a 5- and 10-year period, based on numerous clinical factors. This model is widely used in clinical practice to select patients that are likely to benefit from adjuvant hormone, targeted or cytotoxic treatment (Punglia et al. 2018).
Tamoxifen, an oestrogen receptor antagonist, has long been the gold standard of endocrine treatment in ER+ breast cancer, but its use is associated with some significant (but uncommon) adverse effects including endometrial cancer and thromboembolism. Furthermore, a significant number of women receiving tamoxifen experience disease recurrence or progression, whether they are treated in the adjuvant or metastatic setting. There is therefore a need for further agents to treat tamoxifen-resistant disease.
Other drugs in the aromatase inhibitor category, such as letrozole and anastrozole, block the production of oestrogen by preventing the last step of oestrogen synthesis. They are aromatase-specific and thus have little effect on the synthesis of other steroids or on the adrenal axis (Choueiri et al. 2004). Anastrozole, letrozole and exemestane have all been compared with tamoxifen in randomised studies in the metastatic setting. In a large Phase 3 randomised trial, letrozole demonstrated a superior outcome when compared with tamoxifen. Based upon these results, studies of aromatase inhibitors in the adjuvant setting were also undertaken comparing efficacy and toxicity with that of tamoxifen. In addition to replacing tamoxifen with an aromatase inhibitor as an initial adjuvant therapy, other strategies that have been investigated are switching between tamoxifen and an aromatase inhibitor or the addition of extended adjuvant aromatase inhibition after 5 years of tamoxifen. In practice, letrozole is used more commonly in women with postmenopausal status.
4.1.2 Androgen
In 1941 Charles Huggins demonstrated that at initial presentation, prostate cancer is an androgen-dependent cancer that responds to either surgical or hormonal withdrawal of circulating androgen (Huggins and Hodges 2002). Twenty-five years later he received the Nobel Prize for his observation. This led to the development of first-generation anti-androgens, for example, bicalutamide, a partial agonist of the androgen receptor. Inevitably, the disease enters a castration-refractory phase. There is good evidence that this phase is driven by upregulation of androgen receptors, leading to increased sensitivity to even low levels of circulating and intratumoural ligand. Second-generation anti-androgens, such as abiraterone, specifically inhibit the adrenal synthetic enzymes 17 alpha-hydroxylase and C17,20-lyase, significantly decreasing testosterone production in castration-refractory prostate cancer. Abiraterone is associated with marked progression-free and overall survival benefit (de Bono et al. 2011). Enzalutamide blocks testosterone from binding to cytosolic androgen receptor, which impedes receptor migration to the nucleus and thereby inhibits androgen-dependent gene expression. The SPARTAN trial showed that the third-generation anti-androgen, apalutamide, which has a high affinity for the androgen receptor, showed superior disease-free survival in cases of non-metastatic castrate-resistant prostate cancer (Smith et al. 2018). As a result, apalutamide has recently been licensed for this indication.
4.2 Targeting Receptors
4.2.1 Vascular Endothelial Growth Factor (VEGF) and Its Receptors
Vascular endothelial growth factor (VEGF) is a key component of a pathway regulating tumour angiogenesis. Tumour-derived VEGF is the ligand for a group of three receptors, VEGFR-1, 2 and 3 (also known as Flt-1, KDR and Flt-4, respectively) expressed on endothelial cells. Both the ligand and its receptor family are the target of rationally designed drugs (Ferrara 2005). Agents targeting VEGF include the monoclonal antibody bevacizumab and the soluble VEGF-binding protein aflibercept. Bevacizumab has been approved for the treatment of a range of solid tumours including ovarian and lung cancer. Aflibercept is an engineered soluble receptor from extracellular domains of VEGFR-1 and VEGFR-2. It binds to all isoforms of VEGF and has a higher affinity than bevacizumab for VEGF A and B.
The VEGF receptor is a target for validated small molecular inhibitors (Rhee and Hoff 2005). These so-called multi-targeted inhibitors include vandetanib, which inhibits EGFR and VEGFR-2, as well as sorafenib, sunitinib and cediranib which have broad specificity for receptor tyrosine kinases including members of the VEGFR family. Vandetanib is licensed for use in medullary thyroid cancer. Sunitinib and sorafenib are both approved for use in advanced renal cell cancer, and sorafenib is also active in hepatocellular and thyroid carcinoma. Unsurprisingly, a prominent toxicity of these agents is hypertension, because of the involvement of VEGF in blood pressure homeostasis, although this toxicity is readily managed with careful blood pressure monitoring and early introduction of antihypertensives.
4.2.2 Epidermal Growth Factor Receptor
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase (RTK) and a member of the ErbB receptor superfamily. Whilst it was first discovered in 1962, its role in tumourigenesis was only understood in the 1980s. EGFR overexpression is associated with poorer outcomes in various human malignancies; pathways involved in EGFR signal transduction therefore represent promising therapeutic targets. Binding of extracellular growth factor ligands to the ErbB receptor family causes dimerisation of the receptors, forming homo- or heterodimers that stimulate tyrosine kinase activity, initiating intracellular signalling cascades. The considerable clinical impact of therapies targeting EGFR and HER2 explains the central role these two receptors play in driving human cancer. The past three decades have seen the development of both monoclonal antibodies and small molecule tyrosine kinase inhibitors specific for ErbB family members.
Erlotinib and gefitinib are small molecule reversible inhibitors selective for the intracellular tyrosine kinase domain of EGFR (Baselga 2002). These orally bioavailable drugs prevent ATP binding and autophosphorylation of the EGFR tyrosine kinase. Trials in unselected patient populations resulted in response rates of 10–19% (reviewed in (Spicer and Rudman 2010)). Modest improvement in overall survival was observed in comparison with placebo in randomised trials with these agents. Further analyses from these studies reported variations in efficacy according to clinical characteristics and activating mutations in the EGFR gene were eventually identified as a potent predictive biomarker.
Second-generation tyrosine kinase inhibitors (TKIs) such as afatinib and dacomatinib irreversibly bind to EGFR and HER2. Dacomatinib showed a statistically superior overall survival compared with gefitinib (34 versus 27 months), in patients with an EGFR mutation in exon 19 or 21 (Wu et al. 2019). Osimertinib is a third-generation drug, less potently active against wild-type EGFR, and, importantly, able to bind avidly to the target even when bearing the T790M point mutation characteristic of resistance to earlier-generation inhibitors (Soria et al. 2018).
Cetuximab is a chimeric IgG monoclonal antibody against the extracellular domain of EGFR approved for use in colorectal, head and neck and lung cancers. The use of receptor-targeted antibody therapies offers two potential mechanisms of action, adding the potential for activation of immune mechanisms to the signalling inhibition also seen with small molecules. All antibodies approved for cancer therapy belong to the IgG immunoglobulin subclass and as such are able to recruit cells such as NK expressing Fcγ receptor, which in turn can have cytotoxic or phagocytic effects on the tumour cell. Thus, therapeutic monoclonal antibodies may have antitumour effects mediated by both signalling inhibition and by antibody-directed cellular cytotoxicity. Several current and future strategies in the development of antibody therapies are directed at improving or broadening the affinity of these molecules for their receptors on immune effector cells.
Genomic analysis is routinely carried out on diagnostic samples of adenocarcinomas in lung and colorectal cancer. Patients with advanced-stage disease are selected to receive primary treatment with EGFR-targeted agents. TKIs are routinely used first line in the case of EGFR-mutated lung cancer, and monoclonal antibodies are selected in the case of colorectal cancer lacking activating mutation of KRAS, the product of which signals downstream of EGFR.
4.2.3 HER2
Binding of ligand to the extracellular domain of RTKs induces receptor dimerisation, both between the same and different (heterodimerisation) receptor subtypes. Heterodimerisation is assumed to be of particular significance for HER2 (Klapper et al. 1999), for which no endogenous ligand has been identified. HER2 amplification can lead to constitutive proliferative signalling in the absence of ligand and has been detected in a wide range of tumour types including those originating from breast and stomach. The efficacy of the anti-HER2 monoclonal antibody trastuzumab appears to depend on HER2 overexpression in the targeted tumour, and this drug is approved in both these diseases where HER2 is upregulated. In patients with HER2-positive breast tumours, trastuzumab is associated with marked survival superiority in both the metastatic (Slamon et al. 2001) and adjuvant settings.
Even in the context of resistance to prior therapy with trastuzumab, the tumour can be effectively targeted, and systemic toxicities limited, using an antibody-drug conjugate (ADC). Trastuzumab emtansine (T-DM1) combines humanised antibody trastuzumab and the potent microtubule polymerisation inhibitor DM1, through a stable thioether linker. The latter component was found to have activity against breast cancer in the 1970s. However, as a single agent its toxicity far outweighed its benefits. Delivered via an ADC combination, the cytotoxic agent is internalised and delivered directly into the target cancer cells resulting in apoptosis. A randomised study demonstrated an impressive delay in progression-free survival in HER2-positive breast cancer (Hurvitz et al. 2013).
4.2.4 CD20
An early advance in the antibody therapy of human cancers was the development of rituximab, an IgG antibody specific for CD20. This target is ubiquitously expressed in lymphocytes of B-cell lineage. The addition of rituximab significantly improves the efficacy of chemotherapy of non-Hodgkin’s lymphomas (Coiffier 2005) and has also found a role in the therapy of chronic lymphocytic leukaemia.
4.3 Other Targets
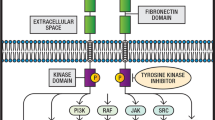
A growing knowledge of the diversity and complexity of signalling networks in malignant cells is reflected in the number of targeted therapies tested in clinical trials. In addition to the ligands and receptors outlined above, many others have been studied including inhibitors and antagonists of RTKs and other receptors such as MET, RET and FGFR (Jiang and Ji 2019). Targets of small molecules inhibiting intracellular kinases include mTOR, Akt, PI3K, BRAF, ALK and MEK (Fig. 3).

Mechanisms of receptor tyrosine kinase activation in cancer. (a) Binding of upregulated ligand (L) to the extracellular domain, or presence of an activating kinase domain mutation (jagged arrow), leads to receptor dimerisation and autophosphorylation (P) of the intracellular kinase domain. Activation of downstream signalling events (open brown arrows) results in proliferation. (b) Overexpression of the receptor itself, for example, as a result of gene amplification in the tumour genome, results in inappropriate extracellular domain proximity and again activates downstream signalling. Erlotinib and gefitinib are small molecule reversible inhibitors of the intracellular tyrosine kinase domain of the EGFR receptor
Translocations in the cancer genome can result in unique fusion kinases that can be the driver for some cancers. Some of these are the target for approved inhibitors. Examples include inhibition by imatinib and other drugs of the fusion kinase encoded by BCR-ABL on the Philadelphia chromosome resulting from a balanced translocation in most cases of chronic myeloid leukaemia (CML; see below) and targeting with crizotinib, alectinib and other molecules of the EML4-ALK fusion gene product present in about 5% of non-small-cell lung cancers.
Proteasomes which degrade tumour suppressor gene products are appealing targets for cancer therapy. Bortezomib has established activity in multiple myeloma (Richardson et al. 2005). The structure of chromatin, and hence the expression of genes controlling the malignant phenotype, can be modulated by histone deacetylase or demethylase inhibition (Piekarz and Bates 2009; Turner et al. 2004). Therapy-targeting mechanisms maintaining the integrity of the cancer genome itself, especially poly-ADP-ribose polymerase (PARP), have proven clinically effective especially in tumours occurring on a background of germline heterozygosity for DNA repair genes such as BRCA1 and BRCA2, discussed further below (Fong et al. 2009). Such tumours become more dependent on parallel DNA repair pathways than are surrounding normal cells, allowing targeting of DNA repair that is highly selective for tumour cells through a process that has become known as synthetic lethality.
Other promising approaches include antisense technology, oncolytic viral therapy, vaccines, immune checkpoint inhibition and adoptive T-cell therapies.
4.4 Targeting the T-Cell
4.4.1 Checkpoint Inhibition
The introduction of immunotherapy using checkpoint inhibition has provided unprecedented improvement in outcomes for some cancers with largely manageable toxicities. Immune checkpoint inhibitors targeting molecules such as CTLA-4 and PD1 enhance the activity of endogenous T-cells against tumour antigens. However, a proportion of patients exhibit incomplete efficacy (intrinsic resistance), and others will experience loss of tumour control in due course (acquired resistance) (Havel et al. 2019).
Blockade of PD1 or its ligand PDL1, through use of antibodies including pembrolizumab, has been validated as a therapeutic strategy as outlined above. These drugs have demonstrated a survival benefit across several malignancies including lung, melanoma, lymphoma, renal cell carcinoma, head and neck squamous cell carcinoma and bladder cancer. PDL1 is often expressed on tumours or within the microenvironment. PD1 or PDL1 directed targeted therapeutics can stimulate exhausted T-cells by blocking this inhibitory T-cell signalling interaction.
Observations support the prediction that checkpoint blockade is only effective in tumours that are infiltrated with T-cells (‘hot’ tumours) and in those with a high burden of somatic mutations. Identifying biomarkers to select patients that are likely to respond to checkpoint inhibition has generally been unsuccessful in many tumour groups such as melanoma and renal cancer. In lung cancer, higher expression of PD1 expression is used to select patients that are more likely to benefit from immunotherapy, but even here the association between PD1 expression and activity is incomplete (Havel et al. 2019).
4.4.2 Adoptive T-Cell Therapy
T-cells are an essential part of adaptive immunity and central to pathogen clearance, and their physiological function also includes tumour surveillance and rejection. During the early stages of development in the thymus, T-cells mature the specificity of their T-cell receptor (TCR), which recognises processed antigens in an MHC context. Some tumours have significant populations of host T-cells present in their microenvironment, and in some circumstances, it is possible to extract this population of tumour-infiltrating lymphocytes (TILs) from a surgically resected tumour, culture them ex vivo and reinfuse into the patient to generate a durable clinical response (Yang and Rosenberg 2016).
The artificial genetic transfer of TCR genes, or chimeric antigen receptors (CAR), to naive T-cells, which originally do not have any antitumour specificity, is a compelling concept which has shown promising results in several tumour types. This autologous approach entails T-cell extraction from patient blood and manipulating the expression of tumour-targeting receptors through genetic engineering. A period of cytotoxic conditioning which results in depletion of endogenous T-cells may be important prior to introducing primed T-cells, in part to address the population of inhibitory lymphocytes already present in the tumour microenvironment. Frequently observed toxicities following administration of adoptive T-cell therapies include cytokine release syndrome, central neurotoxicity and infections which can be fatal. However, major responses have been achieved with CAR-T-cell therapy in the treatment of acute leukaemia, although the treatment of solid tumours is proving more difficult, presumably because of the presence of a hostile microenvironment in solid, but not liquid tumours (Sadelain et al. 2017).
5 Nuclear Medicine Therapies
A further option for targeted treatment of metastatic disease is the use of radioactive pharmaceuticals to combine diagnostic imaging and therapy. A single agent is formed by combining a diagnostic and therapeutic radioisotope with a binding molecule to allow for diagnosis, drug delivery and treatment response monitoring. Broadly this is referred to as the theranostic approach. The radiopharmaceutical component is identical or a similar molecule that is radiolabelled differently or administered at different dosages. For example, iodine-123 is a gamma emitter, and iodine-131 is a gamma and beta emitter, both of which can be used for theranostic purposes (Yordanova et al. 2017) Desirable properties of therapeutic radionuclide include emission characteristics proportional to the tumour volume, minimising local toxicity. This is a particularly attractive option for patients with multiple comorbidities that are unfit for cytotoxic treatments.
5.1 Radio-Iodine in Thyroid Cancer
Iodine is used in the formation of the thyroid hormones thyroxine (T4) and triiodothyronine (T3), in the thyroid gland. Physiologically, these are vital in human development and metabolism. In 1946, the first radiopharmaceutical was developed from the neutron bombardment of tellurium-131 forming the radionuclide 131I. 131I combines beta and gamma emitters to irradiate cancerous cells, thyroid remnant or distant metastatic disease. It is licensed for particular cases of papillary and follicular carcinoma. Iodine is taken up by the follicular cells of the thyroid gland, whilst some is directly excreted renally. Beta emission, which penetrates up to 1 mm, is therapeutic, whilst simultaneous gamma emission can image the target lesion using SPECT or a gamma camera, allowing for real-time visualisation. Physiologically the salivary glands take up some iodine, and therefore a common toxicity is sialadenitis. Strict precautions for patients receiving radioactive treatment, including isolation, should be adhered to. Successful ablation of the thyroid gland will often require subsequent long-term thyroxine replacement.
5.2 Somatostatin Analogues
Somatostatin receptors (SSTRs) have important physiological roles including inhibition of hormones secreted by the pituitary gland, inhibition of pancreatic exocrine hormones (insulin and glucagon), inhibition of motility and exocrine secretions in the GI tract and central nervous system regulation. They are overexpressed in neuroendocrine tumours where somatostatin plays a critical role in secretion and growth (Reubi and Schonbrunn 2013). Neuroendocrine tumours originate from endocrine organs or neuroendocrine cells within an organ such as the gastrointestinal tract. They often have a high density of somatostatin receptors, making diagnostic and targeted therapy an attractive treatment option.
Three somatostatin analog tracers known as DOTA-TATE, DOTA-TOC and DOTA-NOC are labelled with gamma-emitting gallium-68 to specifically target SSTRs for diagnostic purposes. By targeting tumours with alpha- or beta-emitting isotopes such as 90Y or 177Lu, selective radiotherapy can be delivered using these peptides to the primary tumour and metastatic sites with high specificity and sensitivity. This is known as peptide receptor radionuclide therapy (PRRT) (Yordanova et al. 2017).
Octreotide and lanreotide are synthetic somatostatin analogues, specific for SSTR2, one of the five known receptor subtypes, and can be used for the treatment of symptoms caused by hormone overproduction including non-malignant conditions such as acromegaly. Targeting the SSRTs inhibits downstream signalling, halting cell growth and stimulating apoptosis.
5.3 Radium-223
Radium-223 is an alpha-emitting isotope which selectively binds to areas of increased bone turnover, through its property of mimicking calcium. It is used in cases of metastatic castrate-resistant prostate cancer with bone-only metastasis, to prevent morbidity associated with skeletal metastasis. Alpha particles travel a short range, and radium-223 has low emission of gamma photons, optimising safety of administration and minimizing concerns about close patient contact afterwards. Studies have shown a delay in time to first symptomatic skeletal event and improved overall survival (Parker et al. 2018).
5.4 PSMA Ligand: Lutetium
Prostate-specific membrane antigen (PSMA) is a transmembrane protein which plays a critical role in cell migration, survival and proliferation through a receptor-internalisation process. It is overexpressed on the surface of prostate cancer cells, particularly in patients with high-grade and castrate-refractory disease, allowing for the development of specific diagnostic and therapeutic ligands. The latter often uses a small beta-emitting molecule, lutetium 177 (177Lu), which binds to PSMA with high affinity.
Lutetium has a long half-life, and preliminary studies have shown promising outcomes (Yordanova et al. 2017). Whilst radionuclide therapy manipulates tumour-specific receptors, the radioactive component emits radiation which can disseminate causing local toxicity. As such, use of this therapy is dependent on the sites of metastases. PSMA has a physiological role in normal intestinal, renal and salivary gland, where it is expressed albeit to a lesser degree compared to cancer cells, driving the toxicity profile of this targeted treatment, with dry mouth being a common side effect. Radionuclides are often excreted by the kidneys, and therefore these remain as the most pertinent organs at risk. Overall, radionuclides are better tolerated than cytotoxics.
6 Pharmacogenetics, Pharmacogenomics and Patient Selection for Treatment
An understanding of somatic mutations in the cancer genome has led to the development of targeted therapies. These mutations can serve as biomarkers predicting clinical benefit. Retrospectively, they may seem predictable given a drug’s mechanism of action, an example being the use of the HER2-specific trastuzumab only in those patients with HER2 amplification on their tumour. Other predictive somatic genetic events include the BCR-ABL chromosomal translocation in CML sensitive to imatinib, EGFR mutations responding to EGFR inhibitors (erlotinib, gefitinib, afatinib, osimertinib; see elsewhere in this chapter) and ALK mutations in non-small-cell cancer responding to crizotinib, alectinib and other members of a growing class of tyrosine kinase inhibitors. Other biomarkers predictive of toxicity, rather than benefit, are polymorphisms in the patient’s somatic genome (Wang et al. 2011). Predicting clinical benefit from immune checkpoint blockade appears to be more complex than simple reference to tumour PDL1 expression, and other factors such as tumour mutational burden are being investigated.
6.1 Lung Cancer and EGFR Mutations
Approximately 90% of non-small-cell lung carcinoma cases are associated with tobacco exposure. Cancers in the remaining 10% tend to occur with relatively greater frequency in younger, female, non-smoking patients, most often with a particular histology. It is now understood that these clinical characteristics correlate with a discrete underlying biology that drives the malignant phenotype. Specifically, this is an upregulation of EGFR signalling and in particular mutations in the tyrosine kinase domain of this receptor. Sensitivity to EGFR inhibition with TKIs such as geftinib and erlotinib is associated with activating EGFR mutations (Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004). NREGFR kinase domain mutations are found in four exons (Klapper et al. 1999; Slamon et al. 2001; Coiffier 2005; Richardson et al. 2005) which are in close proximity to the ATP-binding pocket. In-frame deletions in exon 19, and an exon 21 substitution (L858R), are the most common mutations, together representing 85–90% of all EGFR mutations found in NSCLC. The location of these mutations leads to an alteration in the catalytic site, resulting in enhanced affinity for the competitive TKI relative to ATP substrate. Retrospective analyses show superior outcomes including response rates of up to 75% in patients with activating mutations treated with EGFR-specific TKIs.
Trials comparing first-line TKI treatment (gefitinib, erlotinib and afatinib) versus the previous gold standard of platinum-based chemotherapy in patients with EGFR-mutated lung adenocarcinoma showed a superior progression-free survival with TKIs (Mok et al. 2009; Rosell et al. 2012). Tailoring treatment of lung cancer according to mutation status has become the standard of care. Furthermore, 50% of patients that progress during or following first-line treatment have evidence of EGFR T790M point mutation (discussed further below). In these cases, osimertinib, an oral, third-generation, irreversible EGFR-TKI that selectively inhibits both EGFR-TKI–sensitizing and EGFR T790M resistance mutations, with lower activity against wild-type EGFR, is licensed globally. More recently, studies comparing first- and second-generation TKI versus third-generation TKIs in treatment-naive patients with EGFR mutations showed superior efficacy with the latter group (Soria et al. 2018).
6.2 BRCA1, BRCA2 Mutation and PARP Inhibition
Poly(ADP-ribose)polymerase (PARP) is an enzyme activated by damage to the genome and involved in DNA repair. PARP1 acts at DNA single-strand breaks via the mechanism of base excision repair (BER). PARP synthesises ADP-ribose polymers, which protect the strand break and provide a scaffold for assembly of the DNA repair complex.
BRCA1 and BRCA2 are tumour suppressor genes encoding proteins critical for DNA repair and genomic stability. BRCA-deficient cells are dependent on BER because alternative repair mechanisms are inactivated. PARP inhibition can induce synthetic lethality in cells where BRCA1 or BRCA2 are mutated, by inhibiting the alternative BER repair pathway. In patients with BRCA protein loss due to hereditary mutation of one BRCA allele, and somatic loss of the other allele in their tumour, inhibition of PARP function creates irreparable damage to tumour DNA. By contrast, normal tissues heterozygous for BRCA function are unaffected. The predicted combination of efficacy and tolerability has been confirmed in patients selected for genomic BRCA mutation.
PARP inhibition may also play a critical role in tumours presenting features of “BRCAness” (Turner et al. 2004), in which other genetic changes occur in sporadic tumours to create a phenotype similar to that of BRCA mutation carriers. These tumours may also be vulnerable to PARP inhibitors in combination with DNA-damaging agents. Biomarkers useful for patient selection in this setting are yet to be definitively identified.
6.3 Prediction of Toxicity
Anticancer drug therapy can be associated with significant toxicity. Variation in clinical outcome between individuals may partly be attributed to genomic polymorphism. For example, patients carrying one of four variants in the DPYD gene encoding dihydropyrimidine dehydrogenase, present in 5% of the UK population, experience significant toxicity to 5-fluoruracil (5-FU) (Diasio 2001). Similarly, the number of dinucleotide repeats in the promoter of UGT1A1 is associated with increased toxicity of irinotecan because of reduced metabolism. Testing for these predictive mutations prior to therapy is often performed, to guide dose reduction or offer alternative therapy. Such personalised strategies are already being successfully used in other health disciplines, for example, to optimise the efficacy of azathioprine treatment of patients with inflammatory bowel disease. Therapeutic drug monitoring, as used in the routine prescribing of oral anticoagulation and post-transplant immunosuppression, has been relatively underused in oncology but may now be gaining some traction.
7 Resistance Mechanisms
The variation in efficacy seen between patients with the same histological diagnosis can partly be explained by heterogeneity in resistance mechanisms. Broadly, these mechanisms can be classified as genetic or pharmacokinetic. Whilst drug resistance maybe de novo, it may also be acquired as a result of the selection pressure of the therapy itself.
It is widely appreciated that alterations in tumour vascularity can be responsible for tumour resistance. These can be altered by altering the structure of the drug to enhance delivery, such as with the case of liposomal doxorubicin or albumin-bound paclitaxel. Secondly, a number of pharmacokinetic resistance mechanisms are driven by membrane transporter proteins, especially members of the multidrug resistance family such as MDR1, also known as P-glycoprotein (Pgp). These proteins can drive ATP-dependent efflux of drugs from cancer cells. This has been seen following treatment with platinum- and anthracycline-based chemotherapy and can be overcome by co-administering with either small molecules, non-competitive inhibitors or competitive inhibitors. The third principle involves drug inactivation through gamma-glutamyl-cysteine synthetase or gluteihione-based enzymes. Some other resistance mechanisms arise from somatic genetic events in the cancer genome that alter the structure of the drug target or DNA damage and repair.
7.1 CML BCR-ABL Mutations and Resistance to Imatinib
CML pathogenesis is driven by a reciprocal translocation between chromosome 9 and 22 to give risk to a fusion protein kinase, BCR-ABL1. The first therapeutically successful small molecule tyrosine kinase inhibitor, imatinib, developed was against the BCR-ABL1 mutation. Imatinib has a high therapeutic ratio because the target is expressed in malignant cells only. Complete haematological response was seen in 53 of 54 patients treated at doses of >300 mg. The rational design and spectacular efficacy of this TKI ensured that imatinib became widely recognised as the paradigm for a new generation of targeted therapies.
Cytogenetic and/or molecular monitoring at 3, 6 and 12 months following initial TKI therapy can highlight patients in whom primary therapy with imatinib is likely to be ineffective. This is used to categorise the disease as being either BCR-ABL1 dependent versus independent. BCR-ABL1 dependence, whereby resistance occurs later in the course of the disease, suggests the mechanism of resistance is likely related to mutations in the kinase domain affecting the structure of BCR-ABL, which ultimately results in subtherapeutic delivery of drug to the target through impairment of binding or interference of biological and cellular processes. Over half of cases of BCR-ABL1 resistance are attributed to point mutations at the ATP-binding kinase domain (KD). In vitro studies show the T315I ‘gatekeeper’ point mutation causes steric hindrance preventing inhibitor binding and an active conformation of the fusion kinase, thereby promoting drug efflux. Second-generation inhibitors nilotinib and dasatinib retain activity against the majority of kinase domain mutations, aside from the T351I ‘gatekeeper’ mutation through tighter binding to a similar, inactive ABL1 conformation (Gorre et al. 2001). This allows for sequential treatment according to emergent resistance mutations. A further generation of agents with activity against T315I is in development.
Resistance due to dasantinib is a result of distinct mutations including VS299 and F317. All TKIs used in CML undergo hepatic first-pass metabolism via CYP3A4, strong inducers of which will lead to TKI resistance. In a fewer number of cases, intrinsic or primary resistance is observed and commonly associated with BCR-ABL1-independent mechanisms mediated through alternative survival pathways, of which numerous have been identified (Patel et al. 2017). For example, the activation of parallel integrin and/or growth factor receptor signalling pathways adds another dimension responsible for imatinib resistance. The latter pathway, named a dependence pathway, given its receptor dependence and ligand independence, is crucial for survival and anti-apoptotic signalling mediated via activation of the PI3K/Akt pathway in multiple cancers. Regardless of the numerous upstream pathways, they may converge onto the same downstream signals thereby allowing for therapeutic targets. Amongst the downstream pathways are STAT3, PIK3/AKT and RAF/MEK/ERK.
7.2 Molecular Markers of Resistance to EGFR Inhibition
Patients with activating EGFR mutations generally show an initial response to inhibition; however, inevitably acquired resistance develops to first- and second-generation EGFR-TKIs. Numerous mechanisms, including second EGFR mutations, are associated with the development of resistance to TKI therapy. Approximately 40–50% of acquired resistance to first-generation EGFR inhibitors can be accounted for by the T790M mutation, the commonest resistance event, in exon 20 of the EGFR kinase domain (Chen et al. 2008). This mutation results in the insertion of a bulky methionine residue at the active site and is analogous to the T315I gatekeeper mutations seen in CML. A molecular analysis of circulating tumour cells from TKI-naïve patients with metastatic NSCLC found the T790M mutation in 38% of patients. The presence of T790M even before patient exposure to TKI was associated with a significantly shorter progression-free survival compared with patients who did not have detectable levels of T790M (Maheswaran et al. 2008). Therefore, this resistance mechanism is naturally evolving and, in some cases, very likely to be propagated by TKI selection. More recently, the third-generation TKI osimertinib, with irreversible and covalent binding at the active site (cysteine-797 residue in the ATP-binding site), has been approved.
Other less common EGFR mutations can also lead to resistance. Additionally, alterations in parallel signalling pathways, such as MET amplification, may also overcome the effects of all three generations of TKI therapy (Sequist et al. 2011). The presence of mutations in other signalling components may be associated with intrinsic resistance and the lack of sensitivity to TKI therapy. Specifically, an activating KRAS mutation is present in 15–25% of lung adenocarcinomas and correlates with de novo lack of sensitivity to EGFR TKIs. Similarly, patient selection for treatment with monoclonal antibodies specific for EGFR, such as cetuximab, is informed by the presence of activating mutation in the KRAS oncogene, encoding a downstream signalling partner of EGFR, which perhaps unsurprisingly predicts lack of benefit in colorectal cancer (Karapetis et al. 2008).
In up to 10% of cases where response to EGFR targeted therapy has failed, there is histological transformation of the tumour to a small-cell morphology known as epithelial-mesenchymal transition (Druker et al. 2001) with reduction in EGFR expression and subsequent alteration in the biochemical behaviour of the disease. Repeat biopsies to clarify the histology are necessary to guide treatment (Westover et al. 2018)
7.2.1 Multiple Targets
Cancer development and progression is driven by an array of complex biological processes. The molecular signalling pathways in a tumour are adaptable and redundant (Yarden and Sliwkowski 2001), exemplified by the ErbB receptor family members. This allows HER2, which has no identified ligand, and HER3, which has no kinase activity, to become actively involved in signalling. This combination of complexity and redundancy in the cancer cell implies that therapy focusing on a single target may be unlikely to achieve adequate, long-term disease control for many patients. Less than half of acquired EGFR TKI resistance is attributed to non-EGFR-centric adaptions, otherwise known as ‘bypass’ resistance mechanisms as they activate the same downstream signalling pathways resulting in tumour survival and growth. Most commonly, these pathways are related to the ErbB receptors through which IGF1 is activated and MET amplified (Fig. 4) (Westover et al. 2018; Ricordel et al. 2018). Perhaps, targeting multiple receptors with a single agent can potentially overcome resistance driven by molecular heterogeneity and hence improve efficacy. Lapatinib, a reversible inhibitor of both EGFR and HER2, is active in HER2-positive metastatic breast cancer, a disease in which inhibitors of EGFR alone are not active. It is approved for use in combination with capecitabine chemotherapy (Geyer et al. 2006). This approach is analogous to the class of multi-targeted ‘dirty’ drugs that owe efficacy in inhibiting angiogenesis to their ability to inhibit multiple pathways activated by VEGF and related ligands, including VEGFR-1 and -2, platelet-derived growth factor (PDGF)-α and -β, c-Kit and fms-like tyrosine kinase 3.

Some targets for novel anticancer therapies. Inhibitory effects are indicated in red. Solid black arrows indicate activating effects
7.2.2 Irreversible Binding
The acquisition of resistance mutations in the EGFR gene, such as T790M described above, interferes with reversible erlotinib and gefitinib binding at the active site and suppresses the inhibition of EGFR signalling in non-small-cell lung cancer. An attractive feature of a number of second-generation inhibitors of ErbB receptors is irreversible binding to the target receptor. At least in preclinical studies, these irreversible inhibitors effectively inhibit EGFR signalling even in gefitinib-resistant cell lines harbouring the T790M mutation. Prolonged suppression of EGFR kinase activity results from covalent elimination of kinase activity until the synthesis of new receptors. Third-generation EGFR inhibitors irreversibly bind to the ATP-binding site in the kinase domain of the receptor, with superior activity in the presence of T790M mutations compared to the earlier-generation drugs.
All patients eventually develop resistance to treatment with EGFR inhibitors. Preclinical studies and clinical evidence alike provide evidence that resistance mechanisms to first-, second- or third-generation treatment are similar. They may be due to a single or a combination category of resistance mechanism including: tertiary EGFR mutation, bypass signalling, downstream activation, or histological transformation (Ricordel et al. 2018; Spicer and Rudman 2010).
7.3 ALK Resistance
Small molecule anaplastic lymphoma kinase (ALK) inhibitors, such as crizotinib, alectinib and brigatinib, have good clinical effect in ALK-rearranged non-small-cell lung cancers. However, as with EGFR inhibition, secondary resistance inevitably develops. Similar mechanisms of resistance as those seen in EGFR-TKI resistance are seen in in vitro studies. In particular, inactivating kinase domain mutation is deemed responsible for some resistance to crizotonib, most commonly L1196M and G1269A mutations. Following treatment with first-generation ALK inhibitors, these tumours respond well to potent second-generation ALK inhibitors, alectinib and ceritinib. Alternatively, activation of separate oncogenes can override ALK to become the dominant driver mutation (Doebele et al. 2012). Newer ALK-targeting drugs show clinical benefit in treatment-naive ALK-positive cancers compared to crizotinib, suggesting that resistance develops due to inadequate suppression of ALK.
An isolated resistance mechanism is seldom the cause of loss of disease control but is rather a combination of mechanisms evolving to allow tumour survival. A complex combination of drug efflux, modulation of drug metabolism, secondary mutations of the target protein, induction of alternate signalling, induction of epigenetic mechanisms and selection of a drug-refractory cancer stem cell population may be involved.
8 Cancer Drug Discovery and Preclinical Development
Traditional cancer drug discovery has relied heavily on screening large compound libraries for activity against cancer cell lines in culture, initially murine leukaemias, and later human cancer cell lines. Many of these compounds were originally natural products, such as the vinca alkaloids (derived from the periwinkle) and taxanes (including paclitaxel, now synthetically manufactured, but originally available only by extraction from the pacific yew). More recently, small molecular weight drugs have been rationally designed with reference to target crystal structures and optimised via in silico modelling and combinatorial chemistry. Whilst structural modelling techniques may be rational, they offer no guarantee that a resulting lead compound will have desirable pharmaceutical properties, which may still require optimisation using traditional preclinical pharmacology and medicinal chemistry approaches.
Since the mid-twentieth century, cancer cell lines have been derived from common human cancers and are used as an early step in the validation of novel targets and therapy combinations. A range of in vivo preclinical models in rodents include xenografts (immunocompromised animals bearing tumours derived from other species including human), and transgenic animals are also widely used in preclinical drug development of novel treatments for cancer. Transgenics null for tumour suppressor genes can in some cases spontaneously develop tumours; the use of conditional knockouts can allow tissue-specific gene targeting and tumourigenesis.
Preclinical toxicity testing for modern rationally designed targeted drugs has been the subject of much debate, in large part because of the species-specificity of many modern biotherapeutics. Non-human toxicity data might be informative for a new cytotoxic which is expected to target mitosis in any mammalian cell, or even a novel molecular therapy where a drug candidate binds to both human and animal target, but the exploration in animals of on-target toxicity for precisely targeted agents, especially monoclonal antibodies, may be falsely reassuring (Chapman et al. 2007).
9 Current Issues in the Development of Drugs in Oncology
9.1 Improving the Odds of Success of Phase 3 Trials
There is evidence that the clinical development of new agents for the treatment of cancer is less efficient than for many other diseases (Adjei et al. 2009). Over the years this has at least in part been due to the failure of many Phase 3 trials. This might seem paradoxical in the era of drugs that have been rationally designed to hit targets known to drive human cancers, but it is only recently that patient selection has been implemented to optimise efficacy in a subset of patients. In the last 5 years, open-label Phase 3 studies have shown impressive outcomes. Brigatinib compared to crizotinib in patients with ALK-positive advanced lung cancer improved progression-free survival with an impressive hazard ratio of 0.49 (Camidge et al. 2018). The preferable strategy is to adopt a patient selection approach from late Phase 1, assuming an appropriate biomarker is available. This optimises the chance of demonstrating efficacy (or lack of it) early on in clinical development and of beginning the validation of a companion diagnostic alongside the new therapy.
The explosion of new knowledge in cancer biology, and the associated plethora of new potential therapeutic targets, places an additional pressure on the clinical drug development community, namely, how to prioritise and pick winners early. Careful design and conduct of studies in appropriate patient populations allows the possibility of establishing proof of mechanism, and evidence of efficacy, prior to initiation of costly Phase 3 trials. A successful early-phase trial is not just one which leads on to Phase 2 and 3 studies but also one allowing the early discontinuation of a development programme where an agent fails to meet early and rational go/no-go criteria.
9.2 Regulation of Cancer Drug Development
The standards of preclinical safety assessment required for the new generation of therapies are the subject of ongoing debate. Conventional toxicity studies in animals may provide limited information relevant to human use in the context of potent species-specific novel agents (Chapman et al. 2007). Indeed, preclinical data may be falsely reassuring as occurred in a Phase 1 study of an immunostimulatory anti-CD28 antibody agonist. Despite acceptable toxicity findings in non-human primates, the first six patients treated in a Phase 1 clinical trial all experienced a life-threatening cytokine storm resulting from uncontrolled T-cell activation (Suntharalingam et al. 2006). The best available preclinical exploration of safety should of course continue to be required, but for the development of highly specific agents such as antibodies, the use of conventional animal studies, especially those in non-human primates, should not be mandated where information relevant to human use is unlikely to be forthcoming.
Improvements in study design are required, as discussed above, to reduce late-stage failures of new drugs for cancer. There is also a pressing need to address the regulatory burden placed on those conducting clinical studies in cancer, as in other disciplines (Rawlins 2011). Such changes are likely to reduce delays and improve cost-effectiveness in the development of new treatments for diseases where a high level of unmet need remains, without materially compromising the safety of trial participants.
One aspect of new drug regulation that has evolved to optimise access to novel agents is provision of earlier patient access to new agents through accelerated approval (Kwak et al. 2010). Here, interim approval is granted on the basis of results of early clinical trials, on the understanding that post-approval studies will be completed. This approach allows for drug approval based on the use of surrogate endpoints ‘reasonably likely to predict clinical benefit’, response rate being a typical surrogate in oncology. Accelerated approval is especially appropriate where dramatic clinical benefit is demonstrated in early-phase trials conducted in patients selected for expression of the drug target, as has been the case for the ALK inhibitor crizotinib. Further work is required to encourage the development of new drugs to address unmet need in rare cancers, where small numbers of patients mean randomised trials are difficult or impossible to conduct and therefore any potential commercial market is limited. One approach has been the introduction of the orphan drug initiative which has relevance for the less common cancer diagnoses (Braun et al. 2010).
10 Conclusion and Future Perspectives
Continued progress in identifying the molecular targets that drive the biology of malignant cells, especially protein kinases which are readily druggable using small molecular inhibitors, represents a key trend in cancer drug development. These small molecules have contributed to the large class of target therapies also including an ever-expanding number of monoclonal antibody-based therapies. The utility of antibodies is driven not only by their intrinsic specificity for a single target but by the relatively recent focus on these agents as offering immune modulation in addition to signalling inhibition. Antibody conjugation, glyco-engineering and even class switching are ongoing and expected future trends in the development of antibody drugs.
Manipulation of the interplay between host immunity and tumourigenesis has affirmed its role as a therapeutic modality across several solid organ tumour groups. However, appropriate regulation of individual immunity to prevent self-reactive toxicity and autoimmunity is yet to be achieved. Understanding of the complex downstream signalling network, alteration in the tumour microenvironment and changes in the surface expression of HLA molecules will allow for more predictable use of highly specific immunomodulatory therapies. The future may lie in a selective combination of cancer vaccines, checkpoint inhibitors and immune cytokine modulation (Wraith 2017; de Aquino et al. 2015).
The efficacy of the newer agents, both targeted and immune therapies, appears in many cases to be optimal in combination with other drugs, like the longer-established cytotoxic agents. The favourable therapeutic ratio of these newer agents facilitates their combination either with each other or with chemotherapy drugs. Rational pairings of agents targeted at either multiple components of a single signalling pathway, or at parallel pathways, are, respectively, likely to increase the chance of delivering optimal efficacy and of countering the development of resistance. Indeed a growing number of studies are showing improved outcomes with a combination of cytotoxics and checkpoint inhibition (Lisberg and Garon 2019).
The parallel development of biomarkers to inform patient selection will continue to be vital for the optimum use of cancer drugs. Targeted therapies are clearly not expected to be effective in tumours lacking expression of the target, but in many cases the presence of protein may not be enough. Additional molecular features of target activation (such as gene mutation, amplification and translocation) may exert a strong influence on driving oncogene addiction. For an individual patient, identifying which of these is present in their tumour will make increasing demands on cancer diagnostic services. Multiplex analysis for the presence of predictive biomarkers have now become the norm, and it may be that next-generation sequencing of patients’ cancer genome, or of a panel of selected genes, will some become routinely achievable as this technology becomes rapidly more cost-effective. Identification of the best biomarkers for selection of patients as candidates for checkpoint inhibitor immunotherapies remains work in progress. In some diseases and for some drugs, but not others, expression of the PDL1 protein in tumour tissue is associated with clinical benefit, but this correlation is not perfect. For example, current evidence suggests that lung cancer and melanoma patients may benefit from treatment with some of these drugs irrespective of the expression level of this target. Other selection parameters, such as tumour mutational burden (TMB) and tumour-infiltrating lymphocytes (TILs), are being investigated.
For reasons associated with the practicalities of clinical drug development, new anticancer agents are studied first in patients with advanced disease, and some of the significant advances made in the treatment of many of these patient groups have been described above. Whilst many of the targeted therapies have succeeded in delivering periods of good quality of life, long-term survival is still not expected in most metastatic cases of the common malignant diseases. This is largely due to resistance mechanisms, some of which are themselves becoming better understood at a molecular level. Similarly, the use of systemic therapies in the adjuvant setting is already proven to increase the chances of cure in several common cancers if treated when still at an early stage, and the clinical benefits of newer therapies will be amplified in this setting.
The cost of cancer care continues to escalate, and it has been projected that by 2020 spending in the United States will have risen in real terms by 600% in 30 years (Sullivan et al. 2011). This problem is amplified in less-developed nations, where already limited resources are challenged by rapid increases in cancer incidence across aging populations acquiring the risk factors associated with economic development. Initiatives such as careful patient selection based on predictive biomarkers, and changes to clinical trial design to allow earlier go/no-go decision-making, will become increasingly important in controlling drug costs. Rational therapy design has led directly to the development of molecular targeted therapies and immune checkpoint inhibitors. Combined with careful biomarker-driven patient selection, these newer treatment approaches provide the opportunity to make step changes in clinical outcomes to contrast with the modest increments made with many previous advances (Sobrero and Bruzzi 2009).
References
Adjei AA (2006) What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J Clin Oncol 24:4054–4055
Adjei AA, Christian M, Ivy P (2009) Novel designs and end points for phase II clinical trials. Clin Cancer Res 15:1866–1872
Baselga J (2002) Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist 7(Suppl 4):2–8
Booth CM, Tannock I (2008) Reflections on medical oncology: 25 years of clinical trials – where have we come and where are we going? J Clin Oncol 26:6–8
Braun MM et al (2010) Emergence of orphan drugs in the United States: a quantitative assessment of the first 25 years. Nat Rev Drug Discov 9:519–522
Camidge DR et al (2018) Brigatinib versus crizotinib in ALK-positive non-small-cell lung Cancer. N Engl J Med 379:2027–2039
Chapman K et al (2007) Preclinical safety testing of monoclonal antibodies: the significance of species relevance. Nat Rev Drug Discov 6:120–126
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10
Chen FL, Xia W, Spector NL (2008) Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res 14:6730–6734
Choueiri TK et al (2004) Role of aromatase inhibitors in the treatment of breast cancer. Clin Ther 26:1199–1214
Coiffier B (2005) State-of-the-art therapeutics: diffuse large B-cell lymphoma. J Clin Oncol 23:6387–6393
Croce CM (2008) Oncogenes and cancer. N Engl J Med 358:502–511
de Aquino MT et al (2015) Challenges and future perspectives of T cell immunotherapy in cancer. Immunol Lett 166:117–133
de Bono JS et al (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364:1995–2005
Diasio RB (2001) Clinical implications of dihydropyrimidine dehydrogenase on 5-FU pharmacology. Oncology (Williston Park) 15:21–26; discussion 27
Doebele RC et al (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18:1472–1482
Druker BJ et al (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344:1031–1037
Early Breast Cancer Trialists’ Collaborative, G (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365:1687–1717
Eisenhauer EA et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Faden RR, Chalkidou K (2011) Determining the value of drugs – the evolving British experience. N Engl J Med 364:1289–1291
Ferrara N (2005) The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS:209–231
Fong PC et al (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361:123–134
Geyer CE et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733–2743
Gianfaldoni S et al (2017) An overview on radiotherapy: from its history to its current applications in dermatology. Open Access Maced J Med Sci 5:521–525
Gilman A, Philips FS (1946) The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides. Science 103:409–415
Gorre ME et al (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293:876–880
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Havel JJ, Chowell D, Chan TA (2019) The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 19:133–150
Horstmann E et al (2005) Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med 352:895–904
Huggins C, Hodges CV (2002) Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol 167:948–951; discussion 952
Hurvitz SA et al (2013) Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 31:1157–1163
Jiang W, Ji M (2019) Receptor tyrosine kinases in PI3K signaling: the therapeutic targets in cancer. Semin Cancer Biol
Josephs D, Spicer J, O’Doherty M (2009) Molecular imaging in clinical trials. Target Oncol 4:151–168
Karapetis CS et al (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765
Klapper LN et al (1999) The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A 96:4995–5000
Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353:172–187
Kwak EL et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703
Lisberg A, Garon EB (2019) Does platinum-based chemotherapy still have a role in first-line treatment of advanced non-small-cell lung cancer? J Clin Oncol 37:529–536
Lynch TJ et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Maheswaran S et al (2008) Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 359:366–377
Mok TS et al (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361:947–957
Oken MM et al (1982) Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649–655
Paez JG et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Pao W et al (2004) EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 101:13306–13311
Parker C et al (2018) Current approaches to incorporation of radium-223 in clinical practice. Prostate Cancer Prostatic Dis 21:37–47
Parulekar WR, Eisenhauer EA (2004) Phase I trial design for solid tumor studies of targeted, non-cytotoxic agents: theory and practice. J Natl Cancer Inst 96:990–997
Patel AB, O’Hare T, Deininger MW (2017) Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am 31:589–612
Piantadosi S, Fisher JD, Grossman S (1998) Practical implementation of a modified continual reassessment method for dose-finding trials. Cancer Chemother Pharmacol 41:429–436
Piekarz RL, Bates SE (2009) Epigenetic modifiers: basic understanding and clinical development. Clin Cancer Res 15:3918–3926
Punglia RS et al (2018) Clinical risk score to predict likelihood of recurrence after ductal carcinoma in situ treated with breast-conserving surgery. Breast Cancer Res Treat 167:751–759
Ratain MJ, Eckhardt SG (2004) Phase II studies of modern drugs directed against new targets: if you are fazed, too, then resist RECIST. J Clin Oncol 22:4442–4445
Rawlins M (2011) A new era for UK clinical research? Lancet 377:190–192
Reubi JC, Schonbrunn A (2013) Illuminating somatostatin analog action at neuroendocrine tumor receptors. Trends Pharmacol Sci 34:676–688
Rhee J, Hoff PM (2005) Angiogenesis inhibitors in the treatment of cancer. Expert Opin Pharmacother 6:1701–1711
Richardson PG et al (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352:2487–2498
Ricordel C et al (2018) Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol 29:i28–i37
Riviere I, Sadelain M (2017) Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther 25:1117–1124
Rosell R et al (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13:239–246
Rosenberg B et al (1969) Platinum compounds: a new class of potent antitumour agents. Nature 222:385–386
Sadelain M, Riviere I, Riddell S (2017) Therapeutic T cell engineering. Nature 545:423–431
Salomon DS et al (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19:183–232
Sequist LV et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:75ra26
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10:1–10
Slamon D, Pegram M (2001) Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin Oncol 28:13–19
Slamon DJ et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Small EJ, Roach M 3rd (2002) Prostate-specific antigen in prostate cancer: a case study in the development of a tumor marker to monitor recurrence and assess response. Semin Oncol 29:264–273
Smith MR et al (2018) Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med 378:1408–1418
Sobrero A, Bruzzi P (2009) Incremental advance or seismic shift? The need to raise the bar of efficacy for drug approval. J Clin Oncol 27:5868–5873
Soria JC et al (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung Cancer. N Engl J Med 378:113–125
Spicer JF, Rudman SM (2010) EGFR inhibitors in non-small cell lung cancer (NSCLC): the emerging role of the dual irreversible EGFR/HER2 inhibitor BIBW 2992. Target Oncol 5:245–255
Stratton MR (2011) Exploring the genomes of cancer cells: progress and promise. Science 331:1553–1558
Sullivan R et al (2011) Delivering affordable cancer care in high-income countries. Lancet Oncol 12:933–980
Suntharalingam G et al (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355:1018–1028
Turner N, Tutt A, Ashworth A (2004) Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4:814–819
Wang L, McLeod HL, Weinshilboum RM (2011) Genomics and drug response. N Engl J Med 364:1144–1153
Westover D et al (2018) Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol 29:i10–i19
Wraith DC (2017) The future of immunotherapy: a 20-year perspective. Front Immunol 8:1668
Wu Z et al (2019) The international conference on intelligent biology and medicine (ICIBM) 2018: genomics with bigger data and wider applications. BMC Genomics 20:80
Yang JC, Rosenberg SA (2016) Adoptive T-cell therapy for cancer. Adv Immunol 130:279–294
Yap TA et al (2010) Envisioning the future of early anticancer drug development. Nat Rev Cancer 10:514–523
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Yordanova A et al (2017) Theranostics in nuclear medicine practice. Onco Targets Ther 10:4821–4828
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Khan, M., Spicer, J. (2019). The Evolving Landscape of Cancer Therapeutics. In: Barrett, J., Page, C., Michel, M. (eds) Concepts and Principles of Pharmacology. Handbook of Experimental Pharmacology, vol 260. Springer, Cham. https://doi.org/10.1007/164_2019_312
Download citation
DOI: https://doi.org/10.1007/164_2019_312
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-35361-2
Online ISBN: 978-3-030-35362-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)