
Abstract
Chronic obstructive pulmonary disease (COPD) and asthma are both common respiratory diseases that are associated with airflow reduction/obstruction and pulmonary inflammation. Whilst drug therapies offer adequate symptom control for many mild to moderate asthmatic patients, severe asthmatics and COPD patients symptoms are often not controlled, and in these cases, irreversible structural damage occurs with disease progression over time. Transient receptor potential (TRP) channels, in particular TRPV1, TRPA1, TRPV4 and TRPM8, have been implicated with roles in the regulation of inflammation and autonomic nervous control of the lungs. Evidence suggests that inflammation elevates levels of activators and sensitisers of TRP channels and additionally that TRP channel expression may be increased, resulting in excessive channel activation. The enhanced activity of these channels is thought to then play a key role in the propagation and maintenance of the inflammatory disease state and neuronal symptoms such as bronchoconstriction and cough. For TRPM8 the evidence is less clear, but as with TRPV1, TRPA1 and TRPV4, antagonists are being developed by multiple companies for indications including asthma and COPD, which will help in elucidating their role in respiratory disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- ‘Chronic obstructive pulmonary disease’ (COPD)
- ‘Transient receptor potential’ (TRP)
- Asthma
- Cough
- TRPA1
- TRPM8
- TRPV1
- TRPV4
1 Transient Receptor Potential Channels
Transient receptor potential channels are a superfamily of 28 transmembrane cation permeable channels that can be subdivided into seven families – namely, TRP ankyrin (TRPA), canonical (TRPC), melastatin (TRPM), mucolipin (TRPML), NOMPC (TRPN), polycystin (TRPP) and vanilloid (TRPV) – on the basis of sequence homology. With the exception of TRPN channels, which have only been detected in fish, 27 of these ion channels spanning the other six TRP families are expressed in mammals.
In general, TRP channels share certain properties, namely, they are generally Ca2+-preferring cation channels (albeit with varying selectivities), and possess six transmembrane domains with a pore region between the fifth and sixth transmembrane regions (Clapham 2005; Szallasi et al. 2007). Collectively, the TRP ion channel family form an array of cellular sensors for a huge range of endogenous and exogenous chemical and physical stimuli, and their ability to coordinate and integrate a spectrum of physiological stimuli has implicated them with key roles in the pathogenesis of many mammalian diseases. For the purposes of this chapter, we will focus on those TRP channels that have been heavily implicated with roles in asthma and chronic obstructive pulmonary disease (COPD), namely, TRPA1, TRPV1, TRPV4 and TRPM8. We will first briefly summarise the characteristics of COPD and asthma. The structural and functional properties of each TRP channel will then be briefly discussed, along with the evidence for the channels involvement in asthma and/or COPD. Then the drugs affecting that channel will be highlighted, with a particular emphasis on those drugs currently in, or aimed for, clinical trials. Finally the fortunes of current TRP channel drug development will be summarised, and the future merits and prospects of TRP channel drug development will be considered.
1.1 COPD
Chronic obstructive pulmonary disease (COPD) is a prevalent and debilitating respiratory disease with associated systemic comorbidities. It is a leading cause of death and disability worldwide, and disease incidence is predicted to continue increasing, such that COPD is predicted to be the third leading cause of death by 2020 (Vestbo et al. 2013). COPD is characterised by irreversible and progressive reduction of airflow, measured as a decline in lung function through spirometry (Barnes and Stockley 2005; Rabe et al. 2007; Paredi et al. 2010). Current treatments provide essentially only moderate symptomatic relief and do not halt progression of the disease (Barnes 2013). The airflow limitation in COPD is accompanied by an abnormal inflammatory response to noxious inhaled particulates or gases, for example, tobacco smoke, which is thought to be one of the primary causative agents for the initiation of COPD (Barnes and Stockley 2005; Rabe et al. 2007; Salvi and Barnes 2009). Typically such exposures must take place over a long duration before the symptoms of COPD appear – hence most diagnoses of COPD are made when patients are in middle age.
Chronic cough is often one of the first complaints that patients present with prior to a diagnosis of COPD: such that the recent GOLD strategy for diagnosis, management and treatment of COPD notes that patients presenting with chronic cough accompanied by a decline in actual compared to predicted spirometry values should be considered for a diagnosis of COPD (Vestbo et al. 2013). Indeed, cough was found to be the most commonly experienced symptom, as reported by 70% of 3,265 COPD sufferers interviewed, and occurring daily in 46% of the same population (Rennard et al. 2002).
One of the primary characteristics of COPD is an abnormal inflammation of the airways that is unresponsive to standard anti-inflammatories, including the gold-standard treatment of corticosteroids (Barnes 2013). It is thought that chronic exposure to noxious gases/particles drives inflammation in the lungs (Decramer et al. 2012). Cigarette smoke (CS), for example, stimulates multiple pulmonary immune cells (in particular macrophages, but also neutrophils, and CD4+ Th1 and CD8+ Tc lymphocytes) to release many inflammatory mediators, which recruits further inflammatory cells (in particular neutrophils, but also macrophages and CD4+ Th1 and CD8+ Tc lymphocytes) to the airways (Barnes 2008). It is thought that this positive feedback loop, along with repeated stimulation provided by chronic CS exposures, drives a persistent and progressive inflammation. This inflammation, along with exposure to the damaging components of CS, causes destruction of the alveolar structure and enhances mucus production (via goblet cell hyperplasia and hypertrophy) and fibrosis, resulting in reduction in the surface area for gas exchange, reduced elastic recoil and increased airflow obstruction (Hogg et al. 2004; Barnes 2008; Lai and Rogers 2010). The exact proportion of these structural changes may vary between patients, as COPD is an umbrella term that covers several interrelated lung diseases, including chronic bronchitis, small airway disease (SAD) and emphysema (Barnes 2004; Sturton et al. 2008). Whatever the proportion in an individual patient, these inflammatory-driven structural changes contribute to the reduction in airflow, as assessed by spirometry.
1.2 Asthma
Defining asthma is somewhat difficult, as there is no single definitive genetic or environmental cause or trigger for development of this disease (Hargreave and Nair 2009). However, broadly speaking, asthma is a chronic inflammatory disease of the airways, characterised by sudden but transient decreases in airflow associated with dyspnea, cough and wheeze, which are generally fully reversible by bronchodilator treatment (Morosco and Kiley 2007). It has been estimated that 300 million people worldwide may suffer from asthma, with the highest incidences in the Americas, Europe and Australia of 5–10% of the population (Masoli et al. 2004).
Sudden bronchospasm of airway smooth muscle in asthmatics (‘asthma attack’) causes breathlessness and wheeze. Bronchospasm may be triggered by many stimuli, which in normal subjects would be innocuous, including dust and other allergens, pollen, air pollution, exercise and cold air (Eder et al. 2006). This airway hyperresponsiveness (AHR) is driven either by chronic airway inflammation and/or structural changes to the airways induced by this inflammation (Lommatzsch 2012). Whilst the chronic inflammation in asthma also involves an abnormal activation of multiple immune cells, it is different from the inflammation observed in COPD in that in the majority of asthmatics it is suppressed by anti-inflammatory therapy and involves different cell types, including CD4+ Th2 cells, mast cells and eosinophils (Barnes 2008).
2 TRPV1
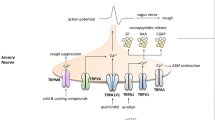
TRPV1 is well known as the receptor responsible for the perception of heat, particularly so for mediating the ‘spicy hot’ effects of capsaicin, the active constituent of chilli peppers from piquant Capsicum spp. plants, which activates TRPV1 on sensory nerves (Caterina et al. 1997). However, TRPV1 is a polymodal receptor, responding to capsaicin, its ultra-potent structural analog resiniferatoxin, and also xenobiotics, noxious heat (>42°C), acidic conditions/protons (low pH), as well as endogenous agents such as anandamide and inflammatory eicosanoids such as bradykinin and PGE2 (Fig. 1) (Caterina et al. 1997; Vriens et al. 2009; Grace et al. 2012). Some of these agents, such as capsaicin, heat, acid and anandamide, activate TRPV1 via a direct interaction with the channel to cause a lowering of its voltage dependency, leading to opening of the pore domain (Caterina et al. 1997; Zygmunt et al. 1999; Jordt et al. 2000; Vriens et al. 2004). By contrast, other activators of the channel, such as PGE2 and bradykinin, act indirectly by second messengers, in these cases released subsequent to activation of G-protein-coupled receptors (respectively the B2 and EP3 receptors) (Maher et al. 2009; Grace et al. 2012).

Diagram showing domain topology and residues important in activation of TRPV1, along with selected endogenous/exogenous TRPV1 activators and TRPV1 antagonists in clinical trials for respiratory indications (Szolcsányi and Sándor 2012)
2.1 TRPV1 Roles in Airway Disease
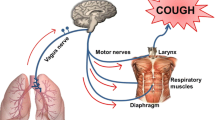
TRPV1 receptors are predominantly expressed in the peripheral nervous system and, relevant to the control of airway functions, in a subset of vagal (both jugular and nodose origin) ganglia sensory neurons, as well as pulmonary innervating dorsal root and nasal trigeminal ganglia neurons (Banner et al. 2011). TRPV1-positive fibres innervate multiple tissue types, including the nose, trachea, parenchyma, alveoli and vessels, throughout the respiratory tract (Grace et al. 2014). Classically, TRPV1 is thought to be expressed on a capsaicin-sensitive subset of slow-conducting unmyelinated C-fibres (Coleridge and Coleridge 1984); however it is now acknowledged that there is a wider population of TRPV1-expressing neurons which includes the fast-conducting myelinated Aδ-fibres (Adcock et al. 2014). Due to their expression in these nerve fibres innervating the lung, TRPV1 receptors in the airway have received particular attention for their ability to provoke cough in both animal species and human subjects. Indeed, the threshold for cough provocation by capsaicin has been found to have been lowered in various populations of asthmatics and COPD patients who have chronic cough compared to healthy control subjects (Choudry and Fuller 1992; Wong and Morice 1999; Doherty et al. 2000; Weinfeld et al. 2002; Blanc et al. 2009; Belvisi et al. 2016). Furthermore, a lowered capsaicin cough threshold, or rather increased sensitivity to capsaicin, is one of the key clinical measures which define a population of patients proposed to have cough hypersensitivity syndrome (Chung 2011; Millqvist 2011).
As well as playing a role in eliciting cough, TRPV1-expressing C-fibres have been demonstrated to release pro-inflammatory neuropeptides such as substance P (SP) and calcitonin gene-related peptide (CGRP) which mediate neurogenic inflammation via their retrograde release from peripheral terminals in rodent airways, although it is unclear if this phenomenon occurs in humans (Belvisi 2003). Indeed, capsaicin inhalation is also associated with parasympathetic bronchoconstriction, mucus hypersecretion, vasodilatation and the sensation of dyspnea (Couto et al. 2013), further implicating the TRPV1 receptor a role in symptoms other than cough in asthma and COPD. Furthermore, administration of the TRPV1 antagonist SB-704498 was found to reduce subsequent airway hyperresponsiveness to histamine in an ovalbumin-sensitised guinea pig model of allergic asthma (Delescluse et al. 2012).
In respiratory disease settings, TRPV1 function in the lung is thought to be modulated by some or all of three main mechanisms: elevated levels of direct TRPV1 agonists or channel openers; sensitisation of the channel by, for example, phosphorylation to induce activation to otherwise innocuous stimuli; and the increase in expression or de novo expression of TRPV1 in individual cells.
In the case of the first two mechanisms, the progressive and persistent pulmonary inflammation observed in asthmatics and COPD patients elevates the levels of multiple TRPV1 activators and sensitisers, including, e.g. inflammatory prostanoids and eicosanoids, neurotrophins, lowered pH and proteases (PAR2 agonists) (Adcock 2009; Grace et al. 2014; Veldhuis et al. 2015).
It should be noted that it is difficult in some cases to make a distinction between a ‘sensitiser’ and an ‘activator’ of TRPV1, with some GPCR agonists, having been shown to both sensitise TRPV1 to subsequent stimuli and to cause activation of TRPV1 (Fischer and Reeh 2007; Maher et al. 2009). Especially in the context of the inflammatory milieu, it is difficult to determine the exact role a single constituent plays. It is clear however that modulation of TRPV1 by GPCRs is an important factor in inflammatory diseases given that TRPV1 contains domains for binding of proteins, such as A-kinase-anchoring protein (AKAP), that can influence its activity, by, for instance, facilitating interactions with the signalling cascade kinases PKA and PKC (Veldhuis et al. 2015). Interestingly it is thought that the subcellular localisation of TRPV1 along with accessory proteins such as AKAP facilitates the regulation of TRPV1 by a multitude of signalling pathways, allowing TRPV1 to integrate signals to a wide range of stimuli – recently reviewed in Nilius and Szallasi (2014). Sensitisers are generally thought to enhance the activity of TRPV1 via phosphorylation of specific residues, enabling the channel to respond to normally innocuous stimuli, or even to become activated spontaneously (Nilius and Szallasi 2014).
In addition to the increase in TRPV1 activation currents via elevated levels of activators/sensitisers, expression of TRPV1 may be increased under inflammatory conditions. This may occur in cells that had previously expressed TRPV1, in which case the increased surface expression would be expected to contribute, along with increased levels of activators/sensitisers to increased TRPV1 activation. However, intriguingly Lieu et al. have recently shown in guinea pigs that ovalbumin sensitisation/challenge or neurotrophin administration increases the number of TRPV1-expressing neurons, particularly of Aδ-type fibres, suggesting that the neural pathways innervating the airways are plastic and may be moulded by the inflammatory environment seen in asthma and COPD (Lieu et al. 2012). This data is corroborated by two other studies demonstrating increases in the proportion of TRPV1-expressing neurons in nodose ganglia in both a rat ovalbumin sensitisation/challenge model of allergic asthma and a guinea pig cigarette smoke exposure model of COPD (Zhang et al. 2008; Wortley et al. 2014a). It seems likely therefore that TRPV1 expression can be induced in neurons that previously did not express TRPV1, increasing the number of peripheral sensory inputs which may cause cough or airway hyperresponsiveness. In human subjects with chronic cough and severe asthma, capsaicin cough hypersensitivity has been reported (Doherty et al. 2000; Belvisi et al. 2016), and increased expression of TRPV1-like immunoreactivity has been detected in bronchial biopsies (Groneberg 2004; Mitchell et al. 2005; McGarvey et al. 2013).
By contrast, less evidence has been published regarding the expression of non-neuronal TRPV1. However, preliminary data has suggested that TRPV1 mRNA expression in the whole lung homogenate of emphysema patients is increased compared with healthy nonsmokers and nonsmokers, suggesting that TRPV1 expression may be increased in other tissues as well as neuronal cells (Grace et al. 2014). In addition, the TRPV1 inhibitor JNJ17203212 reduced the release of ATP from human bronchial epithelial cells (HBEC), and in vivo, TRPV1−/− mice exhibited less cigarette smoke-induced ATP release and subsequent neutrophilic inflammation in bronchoalveolar lavage fluid (Baxter et al. 2014). Despite these intriguing data, the role of TRPV1 in other lung tissues and cells in respiratory health and diseases such as asthma and COPD is relatively poorly understood (Gharat 2007).
There are no current firm descriptions of TRPV1 variant channelopathies per se (Banner et al. 2011). However Smit et al. more recently described associations between several TRPV1 SNPs and increased risk of usual and nocturnal cough in non-asthmatics, which was also correlated with cigarette smoking and occupational irritant exposures (Smit et al. 2012). Whilst the functional effects of SNPs associated with increased risk of cough are unknown, another SNP variant TRPV1-I585V, which – by contrast – is associated with a reduced risk of current cough or wheeze in asthmatics, is reported to decrease channel activity by 20–30% (Cantero-Recasens et al. 2010). The associations of these SNPs with either increased risk or protection from cough and wheeze may suggest potential benefit could be derived from the antagonism of TRPV1 in disease, particularly in asthmatics and COPD patients who suffer from these symptoms.
2.2 TRPV1 Channel Antagonists
Of all the TRP channels, antagonists for TRPV1 are the most advanced in terms of drug development and clinical trials. However, development of these compounds has not been as straightforward as was initially hoped. The state of development of TRPV1 antagonists has been well documented in recent reviews (Preti et al. 2012; Nilius and Szallasi 2014), and here we will focus on selected compounds to describe the difficulties in development of TRPV1 antagonists, as well as updating with the latest reports of the most promising compounds, especially those targeted for respiratory conditions.
The main difficulties with TRPV1 antagonist development have been related to its function as a thermosensor for hot temperatures, and in particular this has meant that adverse effects such as increased body temperature and latent withdrawal to noxious hot stimuli have dogged development. For instance, the TRPV1 antagonist AMG 517 was entered into phase 1 and 1b clinical trials, where administration in patients following molar extraction caused significant and long-lasting increases in body temperature to above 40°C (Gavva et al. 2008). Due to safety concerns, these trials were terminated before the analgesic effect could be determined.
Another compound, MK-2295 (or NGD-8243), a Merck/Neurogen compound, entered a phase 2A POC trial also against dental pain, with 182 subjects receiving either drug or placebo (clinicaltrials.gov identifier NCT00387140). In this study the compound was similarly reported to have undesired effects, including causing an increase in body temperature and altering noxious heat sensation threshold (Xia et al. 2011). These adverse effects were correlated with target engagement, and reportedly meant a dose regimen could not be established that would allow efficacy whilst avoiding risk for burn injury, with some subjects reportedly unable to detect potentially harmful hot temperatures (Xia et al. 2011; Moran et al. 2011).
By contrast, the compound PHE377 has completed a phase 1b PoP trial, with the developing company PharmEste reporting that the compound was ‘well tolerated’, has ‘on-target activity’ and ‘does not increase body temperature’ (PharmEste website 2012). Unfortunately we could not find a peer-reviewed publication presenting this data; however the company is reportedly seeking partners for further development in a ‘phase 2 clinical study in chronic pain with different aetiologies’. It seems, therefore, that the development of TRPV1 antagonists which avoid the problem of hyperthermia may be possible, an idea substantiated by the preclinical development of BCTP, which is highly efficacious at inhibiting responses to capsaicin, RTX and heat, yet exhibits only mild effects on body temperature in rats (0.6°C increase) (Nash et al. 2012; Ferrer-Montiel et al. 2012).
Indeed, the GSK candidate SB-705498 recently completed phase 2 clinical trials for chronic refractory cough and was reported to be well tolerated, with no significant increases in tympanic temperature. This was the first TRPV1 antagonist to be examined clinically as an antitussive, but disappointingly, SB-705498 lacked efficacy in improving 24 h cough counts (Belvisi et al. 2014). However, the shift in objectively measured capsaicin-evoked cough, whilst statistically significant, appeared to be slight, and TRPV1 occupancy was estimated to be approximately only 40% (±20% confidence intervals). The targeting of chronic idiopathic cough was based on the observation by Belvisi et al. that cough responses to capsaicin were differential across different disease groups, with COPD and chronic cough patients in particular exhibiting increased cough responses to capsaicin inhalation and additional studies showing that chronic cough patients show high spontaneous cough frequency over 24 h of ambulatory recording compared to other patient groups (Decalmer et al. 2007; Belvisi et al. 2016; Sumner et al. 2013). However, it remains to be seen if higher potency compounds that have greater efficacy at inhibiting capsaicin-evoked cough can be effective at reducing increased cough frequency in chronic idiopathic cough patients or chronic cough of other aetiologies.
Of note, another TRPV1 antagonist, XEN-D0501, has recently been reported to cause increases of only 0.74°C in a phase 1 clinical trial after a single dose, and following twice-daily repeated dosing, this increase above placebo was reduced to 0.3°C (Round et al. 2011). What is more, Wortley et al. have recently shown that XEN-D0501 was approximately 1,000 times more potent than SB-705498 at inhibiting capsaicin depolarisation of human vagus nerve in vitro, and 100 times more potent at inhibiting capsaicin-evoked cough in conscious guinea pigs (Wortley et al. 2014b). XEN-D0501 is currently in ongoing phase 2 clinical trials for both chronic idiopathic cough (clinicaltrials.gov identifier NCT02233699) and cough in COPD (clinicaltrials.gov identifier NCT02233686), which are expected to conclude in 2015.
2.2.1 Other Notable Clinical Candidates Now Discontinued
The sensory neural pathways underlying pain disorders share many similarities with those that cause abnormal cough, and other TRPV1 antagonists which have been in development as analgesics include AZD1386 and GRC-6211 which both were trialled in patients with dental pain following molar extraction and appeared efficacious. It appears however that subsequently both compounds have been discontinued, in the former case due to liver enzyme elevations (Bonney and Carr 2013) and the latter for unspecified reasons following the sale of the compound by Glenmark to Lilly (Lilly press release 2007; Kym et al. 2009; Xia et al. 2011).
3 TRPA1
TRPA1, formerly known as ANKTM1, is currently the only mammalian-expressed member of the TRP ankyrin family and is named for the large number of ankyrin repeat motifs (14–19) on its N-terminus (Nilius et al. 2012). Near this ankyrin repeat domain are residues that enable TRPA1 to be activated by electrophilic compounds, which include a wide range of endogenous and exogenous reactive chemicals and irritants, so implicating TRPA1 with a key role as a noxious chemosensor. This list of TRPA1 activators includes environmental irritants (constituents of cigarette smoke, air pollution and vehicle exhaust fumes), pungent components of foods, hypochlorite (produced endogenously by immune cells and exogenous constituent of warfare agents) and endogenous agents, commonly produced in inflammatory processes (Nilius and Szallasi 2014). The newest addition to the list of TRPA1 activators are components of bacterial cell walls, an intriguing finding that suggests that the peripheral sensory nervous system (which expresses TRPA1 and responds to LPS) can detect and respond to bacterial infections independently of the immune system (Meseguer et al. 2014). A selected list of some of the wide range of the compounds activating TRPA1 is illustrated in Fig. 2. However as well as chemical ligands, TRPA1 was also initially discovered to be a sensor of physical stimuli, responding to noxious cold temperatures (below 17°C) (Story et al. 2003) and contributing to cellular responses to mechanical stresses (Brierley et al. 2011). However there is some controversy surrounding its supposed cold sensitivity, with one group suggesting that rat and mouse TRPA1 expressed in heterologous expression systems are activated at cold temperatures (Chen et al. 2013), but that in vivo a TRPA1 antagonist does not affect paw withdrawal time to noxious cold (Chen et al. 2011). In support of this latter finding, Zhou et al. report that TRPA1 does not play a role in cold sensation in afferent bronchopulmonary C-fibres (Zhou et al. 2011). This confusion was furthered by two recent contradictory reports, with Chen et al. suggesting that human TRPA1 (albeit in heterologous expression systems) is unresponsive to cold temperatures, whereas Moparthi et al. suggest that human TRPA1 is intrinsically cold sensitive (Chen et al. 2013; Moparthi et al. 2014). It seems therefore that the thermoTRP status of TRPA1 as a cold sensor is far from definitively proven.

3.1 TRPA1 in Asthma and COPD
Although TRPA1 was first identified in human cultured fibroblasts (Jaquemar et al. 1999), expression studies have shown that TRPA1 is predominantly expressed in sensory nociceptive neurons, including, in the respiratory tract, those of vagal and dorsal root ganglia, as well as those of nasal trigeminal ganglia origin (Story et al. 2003; Bandell et al. 2004; Bautista 2005; Nassenstein et al. 2008; Jang et al. 2012).
Recently, however, TRPA1 expression has also been demonstrated in immune cells involved in the inflammatory response in asthma and COPD – such as B cells, CD4+ and CD8+ T cells and mast cells (Prasad et al. 2008; Banner et al. 2011). Additionally, TRPA1 expression has been observed in human lung bronchial epithelial cell lines, and these observations have recently been extended to native pulmonary epithelial cells (Mukhopadhyay et al. 2011; Büch et al. 2013).
Activation of TRPA1 channels has been shown to depolarise vagal pulmonary C-fibres and Aδ-nociceptors in rodent species (Bessac et al. 2008; Taylor-Clark et al. 2008; Nassenstein et al. 2008; Birrell et al. 2009; Andrè et al. 2009; Grace et al. 2012; Adcock et al. 2014), and by extension TRPA1 receptors on these fibre types are thought to mediate cough provoked by TRPA1 agonists in human subjects (Birrell et al. 2009). Indeed, the wide range of exogenous irritants and noxious agents that activate TRPA1, along with its ability to provoke cough, has indicated TRPA1 with a key role as a key defensive noxious sensor in the lungs (Geppetti et al. 2009; Grace and Belvisi 2011).
Interestingly, in pulmonary vagal ganglia neurons, TRPA1 is expressed almost exclusively in a subpopulation of TRPV1-expressing cells, with up to 98% of TRPA1-expressing cells also expressing TRPV1 (Hondoh et al. 2010). TRPA1 subunits have been demonstrated to form functional TRPA1/V1 heterodimers with TRPV1, although there is no dependency between the two for the formation of functional channels (Akopian 2011). What is more, a recent screen of compounds acting on TRPV1 and TRPA1 suggests that the responsiveness to various agonists may be modulated depending on whether these channels are either expressed individually or together (Sadofsky et al. 2014). It will be particularly interesting and relevant to future clinical development to discover whether native cells that co-express TRPV1/A1 differ in their functional responses to agonists compared to cell populations expressing TRPV1 or TRPA1 alone.
As well as evoking cough, many TRPA1-activating irritants cause asthma-like symptoms, including cough, wheeze and dyspnea, hinting at a role for TRPA1 in asthma (Grace et al. 2014). Indeed, TRPA1 has been linked with a key role in the airway hyperresponsiveness (AHR) and bronchoconstriction characteristic of asthma, with a TRPA1 antagonist (HC030031) reversing the AHR to acetylcholine in an ovalbumin-sensitised mouse model (Caceres et al. 2009) and abolishing the late asthmatic response observed following ovalbumin sensitisation/challenge in rat and murine models of asthma (Raemdonck et al. 2012). Additionally, Trankner et al. recently demonstrated that TRPV1-expressing nerves are essential for the development of allergic AHR in a murine model, with selective ablation of TRPV1-expressing nerve fibres abolishing AHR, and direct optogenetic stimulation of these same fibres induces AHR in non-challenged but sensitised control mice (Tränkner et al. 2014). Given that a TRPV1 antagonist did not block this effect and that TRPA1 is almost only expressed in TRPV1-expressing nerve fibres, this supports the concept that TRPA1 plays a key role in the development of AHR in asthma. Furthermore, recent work by Hox et al. has demonstrated that nonallergic AHR can be induced by a single exposure of hypochlorite (TRPA1 agonist) + ovalbumin in wild-type, but not TRPA1−/− mice (Hox et al. 2013).
It is thought that the excessive activation of TRPA1 on sensory nerves in asthma and COPD is due to the increased levels of both endogenous pro-inflammatory signalling molecules (the ‘inflammatory soup’) and the exogenous stimuli which are implicated in the development of COPD – and to some extent asthma – such as cigarette smoke (Andrè and Campi 2008; Simon and Liedtke 2008; Lin et al. 2010; Kanezaki et al. 2012). However, as well as being excessively activated by pro-inflammatory stimuli, it is also thought that TRPA1 activation itself causes the release of pro-inflammatory agents that help to sustain the persistent inflammation observed in asthma and COPD. COPD-relevant TRPA1 agonist sources include cigarette smoke constituents (including acrolein, crotonaldehyde and nicotine), other potential COPD-causative agents such as wood smoke and ozone as well as endogenous aldehydes produced following oxidative stress exposure, such as 4-hydroxynonenal (Taylor-Clark et al. 2007; Trevisani et al. 2007; Shapiro et al. 2013). Recent studies have suggested the expression of TRPA1 in non-neuronal pulmonary cell types, including fibroblasts, epithelial cells and smooth muscle cells, and that expression and release of pro-inflammatory cytokines can be induced from these cell types (Nassini et al. 2012).
Whilst there is some evidence that TRPV1 expression is increased under disease conditions and specifically in asthma and COPD (as discussed previously), there is very limited evidence that TRPA1 expression is similarly modulated in inflammatory states. What little information is available indicates that mechanisms of enhanced TRPA1 expression are likely to be tissue-type specific, and to date TRPA1 up-regulation has not been investigated in tissue types or disease models of relevance to COPD (Malin et al. 2011; Bautista et al. 2013).
Again, by comparison to TRPV1, where Smit et al. found an association between TRPV1 SNPs and increased risk of cough and wheeze, the same authors could find no association between 29 TRPA1 SNPs and cough or wheeze in the same population (Smit et al. 2012).
The only known TRPA1 channelopathy is familial episodic pain syndrome, which is associated with functional activation of the channel at normal resting potentials, causing debilitating upper body pain; however there is no data available concerning how this syndrome impacts on the respiratory tract (Kremeyer et al. 2010; Nilius and Szallasi 2014). There are currently no reported associations between SNPs in TRPA1 and susceptibility to airway diseases (Nilius and Szallasi 2014).
3.2 TRPA1 Channel Antagonists
In contrast to TRPV1, TRPA1 antagonists have had less development time (due to the more recent identification of the TRPA1 receptor), and therefore with far fewer candidate compounds, only two candidate compounds have reached clinical trial stages. A recent (and thorough) review of the patent literature however suggests that several companies possess patented TRPA1 antagonists in various states of preclinical/biological testing, including Abbott (two compounds), Merck Sharp & Dohme, Scripps Research Institute, Janssen (two compounds), Glenmark Pharmaceuticals (seven compounds) and Hydra Biosciences (four compounds), with some indicated for asthma and respiratory conditions (Preti et al. 2012).
One of those compounds that has reached clinical trials however is CB-189,625, which was developed by Cubist Pharmaceuticals and Hydra Biosciences. Indicated for acute (perioperative) pain and certain inflammatory conditions, CB-189,625 completed phase 1 clinical trials, with no adverse effects reported apart from those attributed to vehicle (Bokesch et al. 2012; Preti et al. 2012). However, according to a publicly available quarterly financial report in 2013, the compound was discontinued due to ‘solubility concerns’ (Cubist Pharmaceuticals 2013).
By contrast, the Glenmark Pharmaceuticals candidate GRC-17536 appears to have fared better, having completed phase 1 and, recently, phase 2 trials in subjects with painful diabetic peripheral neuropathy (clinicaltrials.gov identifier NCT01726413). There are to the best of our knowledge no published works on the results of these trials; however a press release indicates that the compound was well tolerated in phase 2, with no adverse effects reported and a statistically significant and clinically relevant positive response observed (Evaluate Group press release 2014).
In addition, and following preclinical data with this compound showing an antitussive effect against CA-provoked cough (Mukhopadhyay et al. 2014), GRC-17536 is currently in another phase 2 clinical trial for chronic refractory cough (clinicaltrialsregister.eu identifier 2013-002728-17). The trial is described as a double-blind crossover placebo-controlled trial of the effect of GRC-17536 on 24 h cough counts in chronic cough patients refractory to treatment, with target engagement judged by GRC-17536 inhibition of CA-induced cough. Interestingly, the compound has been formulated as a dry powder for inhalation, although the reason for this change of formulation from previous clinical trials is unknown. Whilst it is unclear when it is due to conclude, the results of this first clinical trial of a TRPA1 antagonist in a respiratory condition are highly anticipated.
4 TRPV4
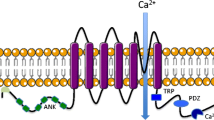
TRPV4 was initially discovered to be expressed in rat kidney and was originally identified as a putative osmosensor with sequence similarities to TRPV1 and TRPV2; hence its original designation of VR-OAC, or vanilloid receptor-related osmotically activated channel (Liedtke et al. 2000). TRPV4 is a Ca2+- and Mg2+-permeable nonselective cation channel composed of 871 amino acids with three ankyrin repeats near the NH2-terminus (Fig. 3). Like TRPA1 and TRPV1, TRPV4 is a thermosensor, although unlike the former two, TRPV4 is thought to be involved in the sensation of tepid to warm temperatures, imbuing TRPV4 with constitutive activity in basal conditions as its activation range of 24–42°C overlaps with normal body temperatures (Nilius et al. 2005; Belmonte and Viana 2008). Again in similarity to TRPV1 and TRPA1, TRPV4 is a polymodal sensor which responds to a range of endogenous and exogenous chemical and physical stimuli including arachidonic acid and derivatives, endocannabinoids, synthetic α-phorbols and mechanical stimuli, such as changes in osmolarity (Watanabe et al. 2003; Vriens et al. 2004; Willette et al. 2008).

4.1 TRPV4 Role in Airway Disease
Its wide tissue expression in tissues all over the body (including heart, lung, kidney, CNS, skin and sweat glands) and variously in multiple neuronal and non-neuronal cell types aligns with the many different cellular functions in which TRPV4 is involved (Vincent and Duncton 2011; Grace et al. 2014).
Unlike that of the TRPA1 and TRPV1 channels discussed previously, the role of neuronally expressed TRPV4 in the lungs is not well characterised (Grace et al. 2014). However, recently it has been demonstrated that airway sensory nerves can be activated by TRPV4 agonists (Bonvini et al. 2016). In this study, TRPV4 agonists evoked cough in guinea pigs and also induced depolarisation or calcium entry into vagal nerves and neurons – specifically nodose but not jugular neurons. This finding means it is possible that TRPV4 could play a role in the abnormal cough response in COPD and asthmatic patients, where the osmolarity of the pulmonary surfaces is altered, and levels of endogenous activators (such as arachidonic acid) are increased, although this idea requires validation with further work (Grace et al. 2014).
Whilst neuronal pulmonary TRPV4 has not been widely investigated, more is known about TRPV4 for its expression and functional role in a wide range of non-neuronal lung tissues, including structural cells such as airway smooth muscle, epithelial cells, pulmonary vessels as well as in inflammatory cells such as alveolar macrophages and neutrophils (Liedtke et al. 2000; Jia et al. 2004; Yang 2006; Alvarez et al. 2006; Dietrich et al. 2006; Banner et al. 2011). Due to the postulated increased levels of TRPV4 activators present, it is thought that excessive activation of TRPV4 could play multiple roles in the pathology of asthma and COPD, dependent on the specific tissues in which it is expressed.
For example, it has been demonstrated that TRPV4 is expressed in human airway smooth muscle cells (Jia et al. 2004), and a specific TRPV4 agonist elicits significant contraction of both guinea pig and human smooth muscle (via the release of cysteinyl leukotrienes), which is blocked using a selective TRPV4 antagonist, implicating TRPV4 with a role in the variable airflow obstruction observed in asthma (Bonvini et al. 2013; McAlexander et al. 2014). Furthermore, TRPV4 activation in both human airway smooth muscle and human airway epithelial cells has been shown to cause the enhanced release of ATP (Seminario-Vidal et al. 2011; Takahara et al. 2014; Baxter et al. 2014), TRPV4−/− mice had reduced pulmonary ATP release and neutrophilic inflammation following CS exposure and TRPV4 expression was upregulated in whole lung tissues from COPD patients (Baxter et al. 2014). What is more, TRPV4 activation has also been suggested to contribute to both neurogenic inflammation via the release of neuropeptides and the stimulation of alveolar macrophages to release reactive oxygen and nitrogen species (Vergnolle et al. 2010; Hamanaka et al. 2010). These data collectively suggest TRPV4 plays a role in the inflammatory processes underlying asthma and COPD pathologies (Esther et al. 2008; Willart and Lambrecht 2009; Mortaz et al. 2010; Riteau et al. 2010; Eltom et al. 2014). What is more, in rodent and murine models TRPV4 activation has been implicated with a role in the formation of pulmonary oedema (Thorneloe et al. 2012), which is thought to be due to its role as a regulator of endothelial permeability (Jian et al. 2008).
Similarly to TRPV1 and TRPA1, TRPV4 can be activated by multiple stimuli including endogenous inflammatory lipids, changes in airway surface osmolarity and mucus production (Vincent and Duncton 2011; Grace et al. 2014). In addition to the evidence that levels of TRPV4 activators are upregulated in asthma and COPD, there is also the suggestion that TRPV4 may be sensitised to subsequent stimulation via phosphorylation of the channel, for example via PKA and PKC activity (Grant et al. 2007; Poole et al. 2013).
Two known diseases are caused by genetic changes resulting in abnormal TRPV4 function: Charcot-Marie-Tooth disease type 2C and scapuloperoneal spinal muscular atrophy (Wu et al. 2010). Whilst there are serious systemic repercussions of both channelopathies (bone dysplasia and peripheral nervous degeneration), there is no evidence linking these channelopathies of TRPV4 with asthma, COPD or other respiratory diseases, although it is unclear whether a gain or loss of function is encoded by the TRPV4 mutant protein (Nilius and Owsianik 2010). As Nilius and Owsianik point out when discussing TRPV4 channelopathies, there appears to be a disconnect between the anticipated problems (osmoregulation, pulmonary hypertension or endothelial dysfunction) and those observed in TRPV4-related channelopathies (Nilius and Owsianik 2010).
By contrast, however, several SNPs of TRPV4 have been found to confer increased susceptibility to COPD pathologies, indicating that the increased activation of TRPV4 may contribute to disease progression in COPD patients (Zhu et al. 2009). Interestingly, the study by Cantero-Recasens et al., which found an association between TRPV1 SNPs and cough/wheeze in asthma, found no similar associations with known TRPV4 SNPs and asthma symptom risk (Cantero-Recasens et al. 2010). It may be, however, that there are other asthma risks (e.g. for exacerbation) that are associated with TRPV4 SNPs that were not considered in the design of this study.
Whilst much work remains to be done to highlight directly the role of TRPV4 in asthma and COPD in the specific context of these diseases, there is evidence that activators of TRPV4 are elevated in disease and that activation of TRPV4 leads to the development of asthma and COPD pathologies in animal models. When taken together with the associations between TRPV4 SNPs and risk for COPD, there seems to be a role for TRPV4 activation in the development of these diseases, providing a rationale for the clinical development of specific antagonists of TRPV4.
4.2 TRPV4 Antagonists
There are few TRPV4 antagonist clinical candidates in advanced stages of development. Currently, the GlaxoSmithKline compound GSK2798745 is in a phase 1 trial to assess safety and tolerance in healthy subjects and stable heart failure patients (clinicaltrials.gov identifier NCT02119260). Otherwise it is unclear whether existing compounds in preclinical testing for respiratory indications will be advanced to clinical trials. Given the channelopathies related to systemic change of function of this channel, and its wide expression profile, it may be that the lungs are ideal target tissues for antagonists of TRPV4, with the potential for compounds to be targeted relatively selectively to the lung using inhaled formulations.
5 TRPM8
TRPM8 is another ‘thermoTRP’, in that, like TRPA1, it is thought to respond to cold – or more accurately cool – temperatures in the range 15–28°C (Peier et al. 2002; McKemy 2013). In this way, TRPM8 is thought to be more a physiological sensor of innocuous cool temperatures, rather than a sensor for the detection of painful noxious cold temperatures (McKemy 2013).
Whilst it is a cold thermosensor, TRPM8 shares little sequence homology with TRPA1 and in particular lacks the N-terminus ankyrin repeat sequence structure of TRPA1, as well its responsiveness to electrophilic noxious compounds (Story et al. 2003; Latorre et al. 2007). Instead, chemical TRPM8 activators, such as menthol, icilin and eucalyptol, in general seem to have the shared characteristic of eliciting a cooling sensation (Peier et al. 2002; Zhou et al. 2011). Menthol itself has been variously reported to be analgesic, to have antitussive properties, to be a bronchodilator and to reduce airway irritation and inflammation caused by cigarette smoke irritants (Morice et al. 1994; Wright et al. 1997; Galeotti et al. 2002; Willis et al. 2011; Millqvist et al. 2012). However, whilst menthol is commonly thought of as a TRPM8 agonist, it acts as a TRPA1 agonist at higher concentrations, and may well also activate a host of other targets, including possibly opioid receptors (Galeotti et al. 2002; Karashima et al. 2007). Indeed, data presented recently indicates that the beneficial (bronchodilator/antitussive) effects of menthol in the airways are not due to TRPM8 agonism (Maher et al. 2014). This highlights the particular difficulty in interpreting the literature on TRPM8 due to the use of nonselective ligands (Fig. 4).

Diagram showing domain topology and residues important in activation of TRPM8, along with selected endogenous/exogenous TRPM8 activators and TRPM8 ligands in clinical trials for respiratory indications (Latorre et al. 2011)
5.1 TRPM8 in Asthma and COPD
TRPM8 is primarily expressed in neurons, particularly in subpopulations of neurons originating in the dorsal root and trigeminal ganglia (Clapham et al. 2001; Peier et al. 2002; Story et al. 2003). Interestingly, the subpopulation of TRPM8-expressing neurons appears to be mostly distinct from those subpopulations that express TRPA1, which could imply a distinction in the physiological responses to ‘cool’ versus ‘noxious cold’ sensing (Clapham et al. 2001; Peier et al. 2002; Story et al. 2003; Hondoh et al. 2010). It may therefore be relevant to note that whilst TRPA1 expression almost completely overlaps with the well-known pain/cough receptor TRPV1, only about 30% of TRPM8 also express TRPV1 in DRG neurons (Okazawa et al. 2004). Limited TRPM8 expression in vagal ganglia neurons has also been observed in some studies (Xing et al. 2008; Nassenstein et al. 2008), and whilst TRPM8 mRNA has been detected in retrograde-stained airway jugular neurons, it is thought that the proportion of TRPM8-expressing vagal ganglia neurons is much lower than the reported 60% of TRPM8-expressing nasal trigeminal neurons (Hondoh et al. 2010; Plevkova et al. 2013).
Inhalation of cold air can cause bronchoconstriction and cough and induce plasma protein extravasation and mucus production (Yoshihara et al. 1996; Peier et al. 2002; Carlsen and Carlsen 2002; Xing et al. 2008). However, it is unclear whether this is due to the activation of TRPM8, or the noxious cold sensor TRPA1, or some relative proportion of the two channels. Furthermore, contradictory data exists from multiple studies indicating that activation of TRPM8 (mostly via menthol inhalation/application) inhibits cough and bronchoconstriction (Morice et al. 1994; Kenia et al. 2008; Ito et al. 2008; Millqvist et al. 2012; Wise et al. 2012).
There is also the suggestion that activation of TRPM8 by menthol in nasal trigeminal neurons can inhibit the cough reflex, although this may tell us more about the neurological integration of peripheral nervous signals in the CNS than it does about the role of TRPM8 in disease conditions (Buday et al. 2012).
A truncated TRPM8 splice variant has been detected in human bronchial epithelial cells, and this activation of this variant has been shown to induce pro-inflammatory cytokine transcription (Sabnis et al. 2008a, b). Furthermore, TRPM8 activation has been shown to play a key role in mucus production and mast cell activation (Cho et al. 2010; Li et al. 2011; Grace et al. 2014).
It is unknown whether the expression of TRPM8 is altered in disease states. What is more, whilst it is thought that modulators (usually activators) of TRPV1, TRPA1 and TRPV4 are increased in disease states or inflammatory conditions, it is unclear if in a similar manner TRPM8 activity is modulated by the inflammatory milieu, with no known endogenous activators of TRPM8 having been described (Grace et al. 2014).
To the best of our knowledge there are no SNPs of TRPM8 with relevance to asthma or COPD, and likewise there are no known channelopathies that can offer insight into the function of TRPM8 in the airways.
The various apparently conflicting findings of the consequences of TRPM8 activation make it hard to predict whether TRPM8 agonism or antagonism would be of more benefit in asthma or COPD. Perhaps future work with more specific agonists would help to elucidate the role of this channel in various disease states and tissues. Therefore the next section of this chapter will discuss the development and use of both TRPM8 agonists and antagonists.
5.2 Drugs Affecting TRPM8 Channels
Menthol is already currently available and widely used in OTC lozenges, nasal sprays and cough syrups; usually indicated as cough and cold remedies (Morice et al. 1994; Laude et al. 1994; Kenia et al. 2008; Preti et al. 2012). In these OTC remedies, menthol is often ascribed with antitussive properties and increasing airflow (reducing dyspnea). However, whilst clinical studies have found that menthol does inhibit the cough response to inhaled capsaicin and citric acid (Morice et al. 1994; Laude et al. 1994; Wise et al. 2012), appropriate controls for inhalation of menthol have not been established. This makes these data hard to interpret, given the conscious control of the cough reflex, and the demonstrated effect of mindfulness on the cough reflex (Young et al. 2009). What is more, recently menthol has been shown to increase the perception of nasal patency, but no effect on nasal airflow (Eccles 2003; Kenia et al. 2008).
Interestingly, menthol can attenuate respiratory irritation induced by TRPA1 and TRPV1 agonist constituents of cigarette smoke (Willis et al. 2011). However, as menthol is known to activate several receptors besides TRPM8 (as discussed previously), it is hard to know whether to attribute any clinical benefits to TRPM8 agonism.
To the best of our knowledge, currently no TRPM8 antagonists have reached clinical development stages for asthma and COPD. However, the Pfizer candidate PF-05105679 has completed a phase 1 trial (clinicaltrials.gov identifier NCT01393652) and was reported to be generally well tolerated whilst reducing inhibition of pain induced by the cold pressor test (Winchester et al. 2014). It was also reported that around a third of participants receiving PF-05105679 experienced a sensation of ‘feeling hot’; however no effect on core temperature was observed – although the reason for this observation was not established.
Furthermore, antagonists are in development and biological testing by Bayer HealthCare, Glenmark Pharmaceuticals, RaQualia Pharma Inc., Amgen, and Janssen (for more details, see the excellent patent review by Preti et al. 2012). Of note, Janssen and Amgen have specifically mentioned COPD and asthma (respectively) in their patent applications, although Janssen has multiple compounds that are also indicated for ‘respiratory conditions’, although to date there are no published validations of these compounds in preclinical models of asthma or COPD (Preti et al. 2012).
It seems likely that future preclinical biological work to understand the mechanisms of how TRPM8 modulation affects the course of disease is required to ‘pathfind’ for the preclinical and clinical development of TRPM8 drugs.
6 Discussion
Targeting TRP channels with antagonist drugs may be an effective strategy for reducing the elevated activity of these channels and consequent adverse effects/symptoms in asthma and COPD. Whilst it is apparent from the first generation of antagonists that TRP channels play a role in homeostasis as well as as gatekeepers of these adverse effects, it may be possible to design compounds that can avoid these adverse effects. For example, hot temperature gating of TRPV1 is structurally separate to ligand-gating site(s), and this has allowed the development of specific antagonists that block activation of the ligand-gating site to prevent chemical activators, but allow activation by hot temperatures (it is thought). Such antagonists hold promise that they may allow the desired blockade of adverse effects caused by TRP activation whilst still allowing the activation of the TRP channel by normal endogenous stimuli such as hot temperatures. This offers an optimal clinical profile for patients, especially in the case of TRPV1, for example, allowing the reflexive sensing of dangerous heat for the avoidance of burning.
TRPV4 and TRPM8 channel antagonists are in various stages of preclinical development, and their use in animal models has the potential to tell us much more about the role of the these channels in respiratory disease. By contrast, the clinical trials of TRPV1 and TRPA1 antagonists for the indication of chronic cough in various patient populations are currently ongoing, and the results of these trials may inform us much more about the nature of these channels in human respiratory disease.
References
Adcock JJ (2009) TRPV1 receptors in sensitisation of cough and pain reflexes. Pulm Pharmacol Ther 22:65–70
Adcock JJ, Birrell MA, Maher SA et al (2014) Making sense of sensory nerves: an in vivo characterisation of Aδ- and C-fibres innervating Guinea-Pig Airways. Am J Respir Crit Care Med 189:A3969
Akopian AN (2011) Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol 12:89–94
Alvarez DF, King JA, Weber D et al (2006) Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res 99:988–995
Andrè E, Campi B (2008) Cigarette smoke–induced neurogenic inflammation is mediated by α, β-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest 118:2574–2582
Andrè E, Gatti R, Trevisani M et al (2009) Transient receptor potential ankyrin receptor 1 is a novel target for protussive agents. Br J Pharmacol 158:1621–1628
Bandell M, Story GM, Hwang SW et al (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41:849–857
Bang S, Hwang SW (2009) Polymodal ligand sensitivity of TRPA1 and its modes of interactions. J Gen Physiol 133:257–262
Banner KH, Igney F, Poll C (2011) TRP channels: emerging targets for respiratory disease. Pharmacol Ther 130:371–384
Barnes PJ (2004) Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 56:515–548
Barnes PJ (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8:183–192
Barnes PJ (2013) Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 131:636–645
Barnes PJ, Stockley RA (2005) COPD: current therapeutic interventions and future approaches. Eur Respir J 25:1084–1106
Bautista DM (2005) Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci 102:12248–12252
Bautista DM, Pellegrino M, Tsunozaki M (2013) TRPA1: A gatekeeper for inflammation. Annu Rev Physiol 75:181–200
Baxter M, Eltom S, Dekkak B et al (2014) Role of transient receptor potential and pannexin channels in cigarette smoke-triggered ATP release in the lung. Thorax 69:1080–1089
Belmonte C, Viana F (2008) Molecular and cellular limits to somatosensory specificity. Mol Pain 4:14
Belvisi MG (2003) Sensory nerves and airway inflammation: role of Aδ and C-fibres. Pulm Pharmacol Ther 16:1–7
Belvisi MG, Birrell MA, Khalid S et al (2016) Neurophenotypes in airway diseases. Insights from translational cough studies. Am J Resp Crit Care Med 193(12):1364–1372
Bessac BF, Sivula M, von Hehn CA et al (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118:1899–1910
Birrell MA, Belvisi MG, Grace M et al (2009) TRPA1 agonists evoke coughing in guinea pig and human volunteers. Am J Respir Crit Care Med 180:1042–1047
Blanc F-X, Macedo P, Hew M, Chung KF (2009) Capsaicin cough sensitivity in smokers with and without airflow obstruction. Respir Med 103:786–790
Bokesch PM, Chandorkar G, van Lier JJ, Donovan J (2012) Safety and pharmacokinetics (PK) of novel first-in-class analgesic: TRPA1 antagonist CB-189,625 in healthy adult males. In: 31st Annu. Eur. Soc. Reg. Anaesth. Congr.
Bonney I, Carr D (2013) The future of pain pharmacotherapy. In: Deer TR, Leong MS, Buvanendran A, et al (eds) Comprehensive treatment of chronic pain by medical, interventional, and integrative approaches. Springer, New York, pp 199–209
Bonvini SJ, Adcock JJ, Grace MS et al (2013) Activation of TRPV4 causes bronchoconstriction: a possible role in respiratory disease? Eur Respir J 42:1759
Bonvini SJ, Birrell MA, Grace MS et al (2016) Transient receptor potential cation channel, subfamily V, member 4 and airway sensory afferent activation: role of adenosine triphosphate. J Allergy Clin Immunol 138:249–261
Brierley SM, Castro J, Harrington AM et al (2011) TRPA1 contributes to specific mechanically activated currents and sensory neuron mechanical hypersensitivity. J Physiol 589:3575–3593
Büch TRH, Schäfer EAM, Demmel M-T et al (2013) Functional expression of the transient receptor potential channel TRPA1, a sensor for toxic lung inhalants, in pulmonary epithelial cells. Chem Biol Interact 206:462–471
Buday T, Brozmanova M, Biringerova Z et al (2012) Modulation of cough response by sensory inputs from the nose – role of trigeminal TRPA1 versus TRPM8 channels. Cough 8:11
Caceres AI, Brackmann M, Elia MD et al (2009) A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci 106:9099–9104
Cantero-Recasens G, Gonzalez JR, Fandos C et al (2010) Loss of function of transient receptor potential vanilloid 1 (TRPV1) genetic variant is associated with lower risk of active childhood asthma. J Biol Chem 285:27532–27535
Carlsen K-H, Carlsen KCL (2002) Exercise-induced asthma. Paediatr Respir Rev 3:154–160
Caterina MJ, Schumacher MA, Tominaga M et al (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824
Chen J, Joshi SK, DiDomenico S et al (2011) Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152:1165–1172
Chen J, Kang D, Xu J et al (2013) Species differences and molecular determinant of TRPA1 cold sensitivity. Nat Commun 4:2501
Cho Y, Jang Y, Yang YD et al (2010) TRPM8 mediates cold and menthol allergies associated with mast cell activation. Cell Calcium 48:202–208
Choudry NB, Fuller RW (1992) Sensitivity of the cough reflex in patients with chronic cough. Eur Respir J 5:296–300
Chung KF (2011) Chronic “cough hypersensitivity syndrome”: a more precise label for chronic cough. Pulm Pharmacol Ther 24:267–271
Clapham DE (2005) International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev 57:427–450
Clapham DE, Runnels LW, Strübing C (2001) The TRP ion channel family. Nat Rev Neurosci 2:387–396
Coleridge JC, Coleridge HM (1984) Afferent vagal C fibre innervation of the lungs and airways and its functional significance. Rev Physiol Biochem Pharmacol 99:1–110
Couto M, de Diego A, Perpiñi M et al (2013) Cough reflex testing with inhaled capsaicin and TRPV1 activation in asthma and comorbid conditions. J Investig Allergol Clin Immunol 23:289–301
Cubist Pharmaceuticals (2013) Quarterly Report 31/03/13. In: OTC Mark. http://www.otcmarkets.com/edgar/GetFilingHtml?FilingID=9272339. Accessed 24 Nov 2014
D’hoedt D, Owsianik G, Prenen J et al (2008) Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J Biol Chem 283:6272–6280
Decalmer SC, Webster D, Kelsall AA et al (2007) Chronic cough: how do cough reflex sensitivity and subjective assessments correlate with objective cough counts during ambulatory monitoring? Thorax 62:329–334
Decramer M, Janssens W, Miravitlles M (2012) Chronic obstructive pulmonary disease. Lancet 379:1341–1351
Delescluse I, Mace H, Adcock JJ (2012) Inhibition of airway hyper-responsiveness by TRPV1 antagonists (SB-705498 and PF-04065463) in the unanaesthetized, ovalbumin-sensitized guinea pig. Br J Pharmacol 166:1822–1832
Dietrich A, Chubanov V, Kalwa H et al (2006) Cation channels of the transient receptor potential superfamily: their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol Ther 112:744–760
Doherty MJ, Mister R, Pearson MG, Calverley PM (2000) Capsaicin responsiveness and cough in asthma and chronic obstructive pulmonary disease. Thorax 55:643–649
Eccles R (2003) Menthol: effects on nasal sensation of airflow and the drive to breathe. Curr Allergy Asthma Rep 3:210–214
Eder W, Ege MJ, von Mutius E (2006) The asthma epidemic. N Engl J Med 355:2226–2235
Eltom S, Belvisi MG, Stevenson CS et al (2014) Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: an insight into the pathogenesis of COPD. PLoS One 9, e112829
Esther CR, Alexis NE, Clas ML et al (2008) Extracellular purines are biomarkers of neutrophilic airway inflammation. Eur Respir J 31:949–956
Evaluate Group press release (2014) Glenmark’s TRPA1 antagonist “GRC 17536” shows positive data in a proof of concept study. http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=530203. Accessed 15 Oct 2014
Everaerts W, Nilius B, Owsianik G (2010) The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 103:2–17
Ferrer-Montiel A, Fernández-Carvajal A, Planells-Cases R et al (2012) Advances in modulating thermosensory TRP channels. Expert Opin Ther Pat 22:999–1017
Fischer MJM, Reeh PW (2007) Sensitization to heat through G-protein-coupled receptor pathways in the isolated sciatic mouse nerve. Eur J Neurosci 25:3570–3575
Galeotti N, Di Cesare Mannelli L, Mazzanti G et al (2002) Menthol: a natural analgesic compound. Neurosci Lett 322:145–148
Gavva NR, Treanor JJS, Garami A et al (2008) Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136:202–210
Geppetti P, Patacchini R, Nassini R, Materazzi S (2009) Cough: the emerging role of the TRPA1 channel. Lung 188:63–68
Gharat L (2007) Medicinal chemistry of the vanilloid (Capsaicin) TRPV1 receptor: current knowledge and future perspectives. Drug Dev Res 68:477–497
Grace MS, Belvisi MG (2011) TRPA1 receptors in cough. Pulm Pharmacol Ther 24:286–288
Grace M, Birrell MA, Dubuis E et al (2012) Transient receptor potential channels mediate the tussive response to prostaglandin E2 and bradykinin. Thorax 67:891–900
Grace MS, Baxter M, Dubuis E et al (2014) Transient receptor potential (TRP) channels in the airway: role in airway disease. Br J Pharmacol 171:2593–2607
Grant AD, Cottrell GS, Amadesi S et al (2007) Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578:715–733
Groneberg DA (2004) Increased expression of transient receptor potential vanilloid-1 in airway nerves of chronic cough. Am J Respir Crit Care Med 170:1276–1280
Hamanaka K, Jian M-Y, Townsley MI et al (2010) TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 299:L353–L362
Hargreave FE, Nair P (2009) The definition and diagnosis of asthma. Clin Allergy 39:1652–1658
Hinman A, Chuang H-H, Bautista DM, Julius D (2006) TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103:19564–19568
Hogg JC, Chu F, Utokaparch S et al (2004) The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350:2645–2653
Hondoh A, Ishida Y, Ugawa S et al (2010) Distinct expression of cold receptors (TRPM8 and TRPA1) in the rat nodose–petrosal ganglion complex. Brain Res 1319:60–69
Hox V, Vanoirbeek JA, Alpizar YA et al (2013) Crucial role of transient receptor potential ankyrin 1 and mast cells in induction of nonallergic airway hyperreactivity in mice. Am J Respir Crit Care Med 187:486–493
Ito S, Kume H, Shiraki A et al (2008) Inhibition by the cold receptor agonists menthol and icilin of airway smooth muscle contraction. Pulm Pharmacol Ther 21:812–817
Jang Y, Lee Y, Kim SM et al (2012) Quantitative analysis of TRP channel genes in mouse organs. Arch Pharm Res 35:1823–1830
Jaquemar D, Schenker T, Trueb B (1999) An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J Biol Chem 274:7325–7333
Jia Y, Wang X, Varty L et al (2004) Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 287:L272–L278
Jian M-Y, King JA, Al-Mehdi AB et al (2008) High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am J Respir Cell Mol Biol 38:386–392
Jordt SE, Tominaga M, Julius D (2000) Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci U S A 97:8134–8139
Kanezaki M, Ebihara S, Gui P et al (2012) Effect of cigarette smoking on cough reflex induced by TRPV1 and TRPA1 stimulations. Respir Med 106:406–412
Karashima Y, Damann N, Prenen J et al (2007) Bimodal action of menthol on the transient receptor potential channel TRPA1. J Neurosci 27:9874–9884
Kenia P, Houghton T, Beardsmore C (2008) Does inhaling menthol affect nasal patency or cough? Pediatr Pulmonol 43:532–537
Khalid S, Murdoch R, Newlands A et al (2014) Transient receptor potential vanilloid 1 (TRPV1) antagonism in patients with refractory chronic cough: a double-blind randomized controlled trial. J Allergy Clin Immunol 134:56–62
Kremeyer B, Lopera F, Cox JJ et al (2010) A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 66:671–680
Kym PR, Kort ME, Hutchins CW (2009) Analgesic potential of TRPV1 antagonists. Biochem Pharmacol 78:211–216
Lai H, Rogers DF (2010) New pharmacotherapy for airway mucus hypersecretion in asthma and COPD: targeting intracellular signaling pathways. J Aerosol Med Pulm Drug Deliv 23:219–231
Latorre R, Brauchi S, Orta G et al (2007) ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium 42:427–438
Latorre R, Brauchi S, Madrid R, Orio P (2011) A cool channel in cold transduction. Physiology (Bethesda) 26:273–285
Laude EA, Morice AH, Grattan TJ (1994) The antitussive effects of menthol, camphor and cineole in conscious guinea-pigs. Pulm Pharmacol 7:179–184
Li M, Li Q, Yang G et al (2011) Cold temperature induces mucin hypersecretion from normal human bronchial epithelial cells in vitro through a transient receptor potential melastatin 8 (TRPM8)-mediated mechanism. J Allergy Clin Immunol 128:625–626
Liedtke W, Choe Y, Martí-Renom MA et al (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103:525–535
Lieu TM, Myers AC, Meeker S, Undem BJ (2012) TRPV1 induction in airway vagal low-threshold mechanosensory neurons by allergen challenge and neurotrophic factors. Am J Physiol Lung Cell Mol Physiol 302:L941–L948
Lilly press release (2007) Lilly press release. https://investor.lilly.com/releasedetail.cfm?releaseid=271993
Lin YS, Hsu CC, Bien MY et al (2010) Activations of TRPA1 and P2X receptors are important in ROS-mediated stimulation of capsaicin-sensitive lung vagal afferents by cigarette smoke in rats. J Appl Physiol 108:1293–1303
Lommatzsch M (2012) Airway hyperresponsiveness: new insights into the pathogenesis. Semin Respir Crit Care Med 33:579–587
Macpherson LJ, Dubin AE, Evans MJ et al (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445:541–545
Maher SA, Birrell MA, Belvisi MG (2009) Prostaglandin E2 mediates cough via the EP3 receptor: implications for future disease therapy. Am J Respir Crit Care Med 180:923–928
Maher S, Birrell M, Bonvini S et al (2014) P6 Menthol has beneficial effects in the airways through a Trpm8-independent mechanism. Thorax 69:A79–A80
Malin S, Molliver D, Christianson JA et al (2011) TRPV1 and TRPA1 function and modulation are target tissue dependent. J Neurosci 31:10516–10528
Masoli M, Fabian D, Holt S et al (2004) The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 59:469–478
McAlexander MA, Luttmann MA, Hunsberger GE, Undem BJ (2014) Transient receptor potential vanilloid 4 activation constricts the human bronchus via the release of cysteinyl leukotrienes. J Pharmacol Exp Ther 349:118–125
McGarvey LP, Butler CA, Stokesberry S et al (2013) Increased expression of bronchial epithelial transient receptor potential vanilloid 1 channels in patients with severe asthma. J Allergy Clin Immunol 133(3):704–12.e4
McKemy DD (2013) The molecular and cellular basis of cold sensation. ACS Chem Neurosci 4:238–247
Meseguer V, Alpizar YA, Luis E et al (2014) TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 5:3125
Millqvist E (2011) The airway sensory hyperreactivity syndrome. Pulm Pharmacol Ther 24:263–266
Millqvist E, Ternesten-Hasséus E, Bende M (2012) Inhalation of menthol reduces capsaicin cough sensitivity and influences inspiratory flows in chronic cough. Respir Med 107(3):433–438
Mitchell JE, Campbell AP, New NE et al (2005) Expression and characterization of the intracellular vanilloid receptor (TRPV1) in bronchi from patients with chronic cough. Exp Lung Res 31:295–306
Moparthi L, Survery S, Kreir M et al (2014) Human TRPA1 is intrinsically cold- and chemosensitive with and without its N-terminal ankyrin repeat domain. Proc Natl Acad Sci 111:16901–16906
Moran MM, McAlexander MA, Bíró T, Szallasi A (2011) Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov 10:601–620
Morice AH, Marshall AE, Higgins KS, Grattan TJ (1994) Effect of inhaled menthol on citric acid induced cough in normal subjects. Thorax 49:1024–1026
Morosco G, Kiley J (2007) Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma–Summary Report 2007. J Allergy Clin Immunol 120:S94–S138
Mortaz E, Folkerts G, Nijkamp FP, Henricks PAJ (2010) ATP and the pathogenesis of COPD. Eur J Pharmacol 638:1–4
Mukhopadhyay I, Gomes P, Aranake S et al (2011) Expression of functional TRPA1 receptor on human lung fibroblast and epithelial cells. J Recept Signal Transduct Res 31:350–358
Mukhopadhyay I, Kulkarni A, Aranake S et al (2014) Transient receptor potential ankyrin 1 receptor activation in vitro and in vivo by pro-tussive agents: GRC 17536 as a promising anti-tussive therapeutic. PLoS One 9:e97005
Nash MS, McIntyre P, Groarke A et al (2012) 7-tert-Butyl-6-(4-chloro-phenyl)-2-thioxo-2,3-dihydro-1H-pyrido[2,3-d]pyrimidin-4-one, a classic polymodal inhibitor of transient receptor potential vanilloid type 1 with a reduced liability for hyperthermia, is analgesic and ameliorates visceral hypersensitivity. J Pharmacol Exp Ther 342:389–398
Nassenstein C, Kwong K, Taylor-Clark T et al (2008) Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J Physiol 586:1595–1604
Nassini R, Pedretti P, Moretto N et al (2012) Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One 7:e42454
Nilius B, Owsianik G (2010) Channelopathies converge on TRPV4. Nat Genet 42:98–100
Nilius B, Szallasi A (2014) Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev 66:676–814
Nilius B, Voets T, Peters J (2005) TRP channels in disease. Sci STKE 2005:re8
Nilius B, Appendino G, Owsianik G (2012) The transient receptor potential channel TRPA1: from gene to pathophysiology. Pflugers Arch 464:425–458
Okazawa M, Inoue W, Hori A et al (2004) Noxious heat receptors present in cold-sensory cells in rats. Neurosci Lett 359:33–36
Paredi P, Goldman M, Alamen A et al (2010) Comparison of inspiratory and expiratory resistance and reactance in patients with asthma and chronic obstructive pulmonary disease. Thorax 65:263–267
Peier AM, Moqrich A, Hergarden AC et al (2002) A TRP channel that senses cold stimuli and menthol. Cell 108:705–715
Pharmeste website (2012) PHE377: A novel clinical-stage TRPV1 antagonist devoid of “class” side effects for the treatment of chronic pain syndromes. http://www.pharmeste.com/repository/contenuti/paragrafi/file/PharmEste_Leaflet_2012.pdf. Accessed 14 Nov 2014
Plevkova J, Kollarik M, Poliacek I et al (2013) The role of trigeminal nasal TRPM8-expressing afferent neurons in the antitussive effects of menthol. J Appl Physiol 115:268–274
Poole DP, Amadesi S, Veldhuis NA et al (2013) Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem 288:5790–5802
Prasad P, Yanagihara AA, Small-Howard AL et al (2008) Secretogranin III directs secretory vesicle biogenesis in mast cells in a manner dependent upon interaction with chromogranin A. J Immunol 181:5024–5034
Preti D, Szallasi A, Patacchini R (2012) TRP channels as therapeutic targets in airway disorders: a patent review. Expert Opin Ther Pat 22:663–695
Rabe KF, Hurd S, Anzueto A et al (2007) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532–555
Raemdonck K, de Alba J, Birrell MA et al (2012) A role for sensory nerves in the late asthmatic response. Thorax 67:19–25
Rennard S, Decramer M, Calverley PMA et al (2002) Impact of COPD in North America and Europe in 2000: subjects’ perspective of Confronting COPD International Survey. Eur Respir J 20:799–805
Riteau N, Gasse P, Fauconnier L et al (2010) Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 182:774–783
Round P, Priestley A, Robinson J (2011) An investigation of the safety and pharmacokinetics of the novel TRPV1 antagonist XEN-D0501 in healthy subjects. Br J Clin Pharmacol 72:921–931
Sabnis AS, Reilly CA, Veranth JM, Yost GS (2008a) Increased transcription of cytokine genes in human lung epithelial cells through activation of a TRPM8 variant by cold temperatures. Am J Physiol Lung Cell Mol Physiol 295:L194–L200
Sabnis AS, Shadid M, Yost GS, Reilly CA (2008b) Human lung epithelial cells express a functional cold-sensing TRPM8 variant. Am J Respir Cell Mol Biol 39:466–474
Sadofsky LR, Sreekrishna KT, Lin Y et al (2014) Unique responses are observed in transient receptor potential ankyrin 1 and vanilloid 1 (TRPA1 and TRPV1) co-expressing cells. Cells 3:616–626
Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet 374:733–743
Seminario-Vidal L, Okada SF, Sesma JI et al (2011) Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem 286:26277–26286
Shapiro D, Deering-Rice CE, Romero EG et al (2013) Activation of transient receptor potential ankyrin-1 (TRPA1) in lung cells by wood smoke particulate material. Chem Res Toxicol 26:750–758
Simon SA, Liedtke W (2008) How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. J Clin Invest 118:2383–2386
Smit LAM, Kogevinas M, Antó JM et al (2012) Transient receptor potential genes, smoking, occupational exposures and cough in adults. Respir Res 13:26
Story GM, Peier AM, Reeve AJ et al (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829
Sturton G, Persson C, Barnes PJ (2008) Small airways: an important but neglected target in the treatment of obstructive airway diseases. Trends Pharmacol Sci 29:340–345
Sumner H, Woodcock A, Kolsum U et al (2013) Predictors of objective cough frequency in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187(9):943–949
Szallasi A, Cortright DN, Blum CA, Eid SR (2007) The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov 6:357–372
Szolcsányi J, Sándor Z (2012) Multisteric TRPV1 nocisensor: a target for analgesics. Trends Pharmacol Sci 33:646–655
Takahara N, Ito S, Furuya K et al (2014) Real-time imaging of ATP release induced by mechanical stretch in human airway smooth muscle cells. Am J Respir Cell Mol Biol 51(6):772–782
Taylor-Clark TE, Undem BJ, MacGlashan DW et al (2007) Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1). Mol Pharmacol 73:274–281
Taylor-Clark TE, McAlexander MA, Nassenstein C et al (2008) Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J Physiol 586:3447–3459
Thorneloe KS, Cheung M, Bao W et al (2012) An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4:159ra148
Tränkner D, Hahne N, Sugino K et al (2014) Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci 111:11515–11520
Trevisani M, Siemens J, Materazzi S et al (2007) 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci 104:13519–13524
Veldhuis NA, Poole DP, Grace M et al (2015) The G protein-coupled receptor-transient receptor potential channel axis: molecular insights for targeting disorders of sensation and inflammation. Pharmacol Rev 67:36–73
Vennekens R, Owsianik G, Nilius B (2008) Vanilloid transient receptor potential cation channels: an overview. Curr Pharm Des 14:18–31
Vergnolle N, Cenac N, Altier C et al (2010) A role for transient receptor potential vanilloid 4 in tonicity-induced neurogenic inflammation. Br J Pharmacol 159:1161–1173
Vestbo J, Hurd SS, Agustí AG et al (2013) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 187:347–365
Vincent F, Duncton MAJ (2011) TRPV4 agonists and antagonists. Curr Top Med Chem 11:2216–2226
Vriens J, Watanabe H, Janssens A et al (2004) Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A 101:396–401
Vriens J, Owsianik G, Janssens A et al (2007) Determinants of 4 alpha-phorbol sensitivity in transmembrane domains 3 and 4 of the cation channel TRPV4. J Biol Chem 282:12796–12803
Vriens J, Appendino G, Nilius B (2009) Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol 75:1262–1279
Watanabe H, Vriens J, Prenen J (2003) Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424:434–438
Weinfeld D, Ternesten-Hasséus E, Löwhagen O, Millqvist E (2002) Capsaicin cough sensitivity in allergic asthmatic patients increases during the birch pollen season. Ann Allergy Asthma Immunol 89:419–424
Willart MAM, Lambrecht BN (2009) The danger within: endogenous danger signals, atopy and asthma. Clin Exp Allergy 39:12–19
Willette RN, Bao W, Nerurkar S et al (2008) Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326:443–452
Willis DN, Liu B, Ha MA et al (2011) Menthol attenuates respiratory irritation responses to multiple cigarette smoke irritants. FASEB J 25:4434–4444
Winchester W, Gore K, Glatt S (2014) Inhibition of TRPM8 channels reduces pain in the cold pressor test in humans. J Pharmacol Exp Ther 351:259–269
Wise PM, Breslin PAS, Dalton P (2012) Sweet taste and menthol increase cough reflex thresholds. Pulm Pharmacol Ther 25:236–241
Wong CH, Morice AH (1999) Cough threshold in patients with chronic obstructive pulmonary disease. Thorax 54:62–64
Wortley M, Maher S, Bonvini S et al (2014a) P4 establishing a role for Trpv1 on sensory nerves in Copd associated chronic cough. Thorax 69:A78–A79
Wortley MA, Birrell MA, Maher SA et al (2014b) Profiling of XEN-D0501, a novel TRPV1 antagonist, in pre-clinical models of cough. Am J Respir Crit Care Med American Thoracic Society, A4979
Wright CE, Laude EA, Grattan TJ, Morice AH (1997) Capsaicin and neurokinin A-induced bronchoconstriction in the anaesthetised guinea-pig: evidence for a direct action of menthol on isolated bronchial smooth muscle. Br J Pharmacol 121:1645–1650
Wu L-J, Sweet T-B, Clapham DE (2010) International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 62:381–404
Xia R, Dekermendjian K, Lullau E, Dekker N (2011) TRPV1: a therapy target that attracts the pharmaceutical interests. Adv Exp Med Biol 704:637–665
Xing H, Ling JX, Chen M et al (2008) TRPM8 mechanism of autonomic nerve response to cold in respiratory airway. Mol Pain 4:22
Yang X-R (2006) Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol 290:L1267–L1276
Yoshihara S, Geppetti P, Hara M et al (1996) Cold air-induced bronchoconstriction is mediated by tachykinin and kinin release in guinea pigs. Eur J Pharmacol 296:291–296
Young EC, Brammer C, Owen E et al (2009) The effect of mindfulness meditation on cough reflex sensitivity. Thorax 64:993–998
Zhang G, Lin RL, Wiggers M et al (2008) Altered expression of TRPV1 and sensitivity to capsaicin in pulmonary myelinated afferents following chronic airway inflammation in the rat. J Physiol 586:5771–5786
Zhou Y, Sun B, Li Q et al (2011) Sensitivity of bronchopulmonary receptors to cold and heat mediated by transient receptor potential cation channel subtypes in an ex vivo rat lung preparation. Respir Physiol Neurobiol 177:327–332
Zhu G, Gulsvik A, Bakke P et al (2009) Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum Mol Genet 18:2053–2062
Zygmunt PM, Petersson J, Andersson DA et al (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Wortley, M.A., Birrell, M.A., Belvisi, M.G. (2016). Drugs Affecting TRP Channels. In: Page, C., Barnes, P. (eds) Pharmacology and Therapeutics of Asthma and COPD. Handbook of Experimental Pharmacology, vol 237. Springer, Cham. https://doi.org/10.1007/164_2016_63
Download citation
DOI: https://doi.org/10.1007/164_2016_63
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52173-2
Online ISBN: 978-3-319-52175-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)