
Abstract
With the development of fast transportation and cavalry, the horse represents the domestic animal that most influenced human history. Yet, the evolutionary history of the horse was not limited to the last 5,500 years since it was first domesticated. It is rooted within a 55 million-year-long time span, where a large number of lineages radiated and became extinct. Together with zebras, hemiones, and donkeys, the horse belongs to the genus Equus, the only remaining equine lineage living in the planet. Even though the survival of exploitable ancient DNA molecules is at best limited to the last million years, the sequencing of short mitochondrial and nuclear DNA fragments, as well as of complete genome sequence from archaeological and paleontological material, has illuminated our understanding of the evolutionary history of the horse family. Such work has not only revisited the evolutionary tempo of Equus and the phylogenetic relationships within and outside the genus but also revealed how past climates and human activities have shaped the genetic makeup of the horse species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The history of ancient DNA (aDNA) is intimately linked to the horse family as the first aDNA sequence ever characterized was obtained from the quagga zebra (Equus quagga quagga), an extinct member of this family, closely related to the plains zebra (Higuchi et al. 1984). With a short stretch of the mitochondrial DNA (mtDNA) sequence, a new research area was born, which makes use of the tiny pieces of aDNA preserved in museum specimens and subfossil material to recover direct genetic information from the past (see Ermini et al. (2015), Hofreiter et al. (2015), Pedersen et al. (2015), and Llamas et al. (2017) for reviews). Within no more than three decades and thanks to the developments of high-throughput DNA sequencing, aDNA research has moved from the sequencing of mainly minute amounts of maternally transmitted mitochondrial markers to complete genome sequencing [see Stoneking and Krause (2011) and Orlando et al. (2015) for reviews], first of the woolly mammoth (Miller et al. 2008), ancient human individuals (Rasmussen et al. 2010, 2011), and archaic hominins (Green et al. 2010; Reich et al. 2010; Meyer et al. 2012) but, soon enough, of members of the horse family. The complete genome of the extinct quagga was released in December 2014 and was obtained from only ~50 mg of hair shaft (Jónsson et al. 2014). A year earlier, a bone preserved in the oldest permafrost reported in the planet delivered enough DNA molecules to reconstruct a first draft of the nuclear genome of a Middle Pleistocene horse (Orlando et al. 2011, 2013). It was dated to 560,000–780,000 years (kyrs) ago (Froese et al. 2008) and demonstrated the last million years as a credible temporal limit for genome sequencing in frozen conditions. Still in 2014, the genome sequence of animals that lived prior to domestication times was characterized, enabling the first direct comparison with the genomes of present-day domesticates. Perhaps not surprisingly given their importance for human history, horses provided the first such comparisons (Schubert et al. 2014) and have been followed by others since, including aurochs/cattle (Park et al. 2015; Braud et al. 2017), wolves/dogs (Skoglund et al. 2015; Frantz et al. 2016), and maize (Da Fonseca et al. 2015; Ramos-Madrigal et al. 2016) (see MacHugh et al. 2017; Scheu 2017 for reviews).
Ancient DNA studies applied to equine subfossil material have greatly improved our understanding of the evolutionary history of the horse family. These have addressed a broad range of topics, including phylogenetics, extinction, and population dynamics, as well as domestication, conservation, and human management. The present chapter highlights a broad, complementary panel of the literature within these research areas.
2 Phylogenetics and Taxonomy
2.1 The Rise of Equus
The horse belongs to the genus Equus, a genus defined by a series of cranial and postcranial morphological features (Eisenmann and Baylac 2000; Franzen 2010). Equus comprises three extant species of zebras (the Grévy’s zebra, E. grevyi; the plains zebra, E. quagga; and the mountains zebra, E. zebra) and three extant species of asses (the hemione, E. hemionus; the Tibetan kiang, E. kiang; and the African ass, E. africanus) (Fig. 1; see Orlando 2015 for a review), the latter of which has been domesticated some 5,500 years ago to form the domestic donkey (E. asinus, Rossel et al. 2008). Molecular phylogenies based on partial mtDNA (Weinstock et al. 2005; Orlando et al. 2009) and/or nuclear genes (Steiner et al. 2012), complete mitogenomes (Vilstrup et al. 2013), and whole genome sequences (Jónsson et al. 2014) support a deep split within Equus, where the horse (E. caballus) and other extinct lineages showing horse-like morphological features, the so-called caballines, form a first clade and where both zebras and asses form a second group, the so-called stenonines.

Phylogenetic relationships within living members of the equine family [adapted from Jónsson et al. (2014)]
The time to the most recent common ancestor (tMRCA) of Equus remained controversial, with estimates ranging between ~2 and over 6 million years (myrs) depending on the molecular clock calibration methods considered (see Steiner et al. 2012; Orlando et al. 2013, references therein). Sequencing a draft genome of a Middle Pleistocene horse preserved in the permafrost of the Yukon territory, Orlando and colleagues found that the MRCA of caballines and stenonines lived at least some ~4–4.5 myrs ago (Orlando et al. 2013). The authors used coalescence simulations under a divergence model to predict, in the absence of gene flow, the credible distribution range of the F summary statistics between the ancient and present-day horse genomes. This statistics was originally introduced by the Neanderthal Genome Consortium (Green et al. 2010; Reich et al. 2010) and represents the probability to sample a derived allele in the ancient genome at a site where present-day genomes are heterozygous. The F-statistics increases for younger population divergence times as the derived neutral allele is then highly likely to not be lost in any descending population. Conversely, for older population divergences, the persistence of the neutral derived allele in both descending populations is highly unlikely, and the F-statistics decreases. Carefully accounting for demographic changes within the population leading to present-day horses, Orlando and colleagues found that coalescence simulations, where the genome-wide mutation rate was calibrated assuming ~4–4.5 myrs for the tMRCA of caballines and stenonines and where the divergence between ancient and present-day populations was constrained by the age of the Middle Pleistocene fossil, could reproduce the F-statistics observed between the ancient and present-day horse genomes. This suggested that the MRCA of all living Equus lived ~4–4.5 myrs ago, in line with the oldest dates of the monodactyle Plesippus simplicidens, one candidate for the earliest fossil of Equus (MacFadden and Carranza-Castaneda 2002). Further work based on the diploid genome sequence of all living members of the genus and exploiting the CoalHMM statistical model (Mailund et al. 2012) revealed that the caballine and stenonine lineages coexisted in sympatry in North America and remained genetically connected until 2.9–3.8 myrs ago, before they finally became reproductively fully isolated (Jónsson et al. 2014).
2.2 Paleontological Over-splitting
The relatively limited number of equine species living today is in sharp contrast to the large number of Pleistocene species described in the fossil record. Over 50 equine species have been named in the Pleistocene of the Americas, and genetic analyses of short DNA fragments of the mitochondrial hypervariable region have revealed this as a typical case of paleontological over-splitting (Weinstock et al. 2005). In fact, most of the morphological diversity found in the Pleistocene of the Americas can be lumped into three main genetic clusters. The first is the caballine lineage leading to the horse, which only survived in the Old World before it was reintroduced in the Americas following the Spanish conquest. The second is referred to as New World stilt-legged (NWSL) horses as they were endemic to North America and shared hemione-like gracile limbs. They disappeared from the fossil record of Alaska and the Yukon territory approximately ~31 kyrs ago (Guthrie 2003, 2006). Their mtDNA sequences, based on partial fragments (Weinstock et al. 2005), or on the whole mitogenome (Vilstrup et al. 2013), were clearly divergent to those of hemiones (and any extant stenonine), contradicting scenarios positing their origin following the expansion of the hemione distribution range through Beringia during past glacial periods. Finally, a third group includes species endemic to South America, the so-called hippidiforms for their characteristic morphological features, including robust limbs and a prominent nasal notch (Orlando et al. 2003, 2009; Der Sarkissian et al. 2015a).
The cranial and postcranial morphologies of hippidiforms and Equus are so different that these lineages have been modeled as two independent branches in the equine tree, possibly diverging some ~10–12 myrs ago (Alberdi and Prado 1993, 1998; MacFadden 1997). The first partial mitochondrial sequences obtained for three Patagonian remains yet indicated hippidiforms nested within Equus, as a sister group to caballine horses (Orlando et al. 2003). This first suggested that these specimens belonged to Equus that were misclassified within Hippidion, a typical hippidiform genus. Further morphological analysis of the specimens rejected the possibility of sample misassignment (Alberdi et al. 2005), and additional partial sequences of the mitochondrial hypervariable region, including specimens from the montane Peruvian range, confirmed the phylogenetic placement within Equus (Weinstock et al. 2005). However, the closest living relative of Equus – the rhinos and tapirs – diverged some ~55 myrs ago from the equine lineage, which makes the location of the equine root particularly difficult based on partial mitochondrial sequences only. Despite supporting a root placement nesting hippidiforms within Equus, as a sister group to caballine horses, a further study indicated possible alternative roots more in line with classical evolutionary models based on paleontological evidence, resulting in hippidiforms and Equus in two reciprocally monophyletic separate lineages (Orlando et al. 2009). The latter was finally confirmed using whole mitogenome sequences of nine specimens from two Hippidion species (Der Sarkissian et al. 2015a). The divergence time between hippidiforms and Equus was, however, estimated to be ~5.6–6.5 myrs ago, which likely roots the deep origins of hippidiforms in North America as the South American Hippidion is not known prior to ~3 myrs ago.
Another example of marked morphological plasticity within a single genetic group is provided by the genetic analyses of specimens revealed to be conspecific to South African plains zebras. These included the extinct quagga (Leonard et al. 2005; Jónsson et al. 2014), which showed a range of coat-color morphotypes along its South African range, and the extinct giant Cape zebra, which was both larger and bigger than present-day plains zebras and became extinct by the end of the late Pleistocene (Orlando et al. 2009).
2.3 Identifying Species and Hybrids
Complete skulls are classically considered to provide the best morphological characters to discriminate extant equine species and reconstruct their phylogenetic relationships to fossil specimens [Groves and Willoughby 1981; Eisenman 1998; see, however, Cucchi et al. 2017 for recent methodological developments applying geometric morphometrics, GMM (see Lawling and Polly 2010 for a review), approaches to occlusal enamel folding patterns of cheek teeth]. Complete skulls are, however, relatively rare in the fossil record, which often consists of fragmentary remains, limiting the identification of species and possible hybrid forms. In contrast, limited amounts of genetic data can supply archaeologists with such information. For example, a short minibarcode (<90 bp) within the mitochondrial hypervariable region has been found to show limited variation within species but large differences between species (Orlando et al. 2009). Amplifying and sequencing such fragments can, thus, provide a first candidate for the taxonomy of the remains analyzed. The sequencing of the whole mitogenome, instead of the sole minibarcode, can offer a complementary approach (although more work-intensive and less cost-effective), especially now that simple, robust target-enrichment methods for the mitogenome are available (Maricic et al. 2010). Applying this methodology, Cardoso and colleagues could identify donkeys within the bone assemblages of a Chalcolithic fortified site from Portugal (Cardoso et al. 2013). This site predated by more than one millennium the supposed introduction of the donkey in the region by the Phoenicians, revealing that even minute amounts of genetic information can significantly change our understanding of the past species dynamics.
However, as mtDNA is maternally inherited, the approach above cannot address whether the specimen analyzed was a purebred individual or a hybrid. Yet, mules – the offspring of a jack and a mare – have been extensively used in the Roman Army for transportation (Johnstone 2004), and their identification requires the analysis of markers that are both maternally and paternally inherited. Leveraging the whole genome sequence data now available for all living members of the horse family (Jónsson et al. 2014), the Zonkey pipeline provides a fast and cost-effective method for a molecular identification of the species, the sex, and the hybrid status of equine remains in the archaeological record. The methodology is based on shotgun sequencing of raw extracts and requires no more than 10,000 endogenous reads (Schubert et al. 2017). F1 hybrids are detected based on both ADMIXTURE profiles (Alexander et al. 2009) and TreeMix phylogenetic reconstruction (Pickrell and Pritchard 2012), allowing one migration edge, whereas the molecular sex is estimated from the proportion of high-quality reads aligned against the X chromosome and autosomes. Applied to 18 archaeological remains, the approach revealed the presence of mules in situations where morphological evidence remained inconclusive (Schubert et al. 2017). This was, for example, the case of six genetically identified mules from the Byzantine site of Yenikapi (Turkey) and one genetically identified mule from the Roman site of Dangstetten (Germany). The approach proved also useful for the identification of species in Southwest Iran, where no less than four equine species coexisted in sympatry and where identifying which among the horse, the donkey, the hemione, and the now extinct European ass, Equus hydruntinus, was present in the Chalcolithic faunal assemblage of Mehr Ali based on tooth morphology alone was impossible.
The difficulty in assigning bone and tooth archaeological remains to a particular equine species is perhaps best illustrated in the case of the hydruntine, also known as the European wild ass, Equus hydruntinus. This species shows a mosaic of morphological characters, some of which are reminiscent of a stenonine species that lived prior to ~2 myrs ago, but others are present in donkeys, zebras, and hemiones (see references in Orlando et al. 2006), and others again are specific to this taxon. Depending on the material available, the species can thus be missed, or misidentified, and the discordance in the species’ assignment among archaeologists can become highly discordant. Geigl and Grange (2012) reported the results of a study where four archaeozoologists were asked to re-identify 23 bones and teeth originally attributed to E. hydruntinus. The archaeologists were unanimous for only one single sample, and for approximately a third of the samples investigated, E. hydruntinus could be confirmed by only a single archaeozoologist, while all the others disagreed. Luckily, early mitochondrial studies have established the close genetic proximity between hydruntines and hemiones (Orlando et al. 2006), which share a 28 bp deletion in the region covered by the mtDNA minibarcode (Orlando et al. 2009). The identification of horse specimens that were morphologically misassigned to hydruntines is thus genetically straightforward (Geigl and Grange 2012). In what is so far the largest genetic analysis of hydruntine specimens, Bennett and colleagues successfully recovered DNA information encompassing the mitochondrial barcode from 64 alleged hydruntine samples (Bennett et al. 2017). Approximately 40% of those turned out to be horses. Limiting their analyses to the remaining subset of specimens also showing reliable morphological identification, these authors revealed that the hydruntine range was structured into two clades. The first was represented by ~5–8-kyr-old material from Anatolia and the Balkans, while the second was present in France some ~100 kyrs ago and in Iran until the beginning of the twentieth century. The genetic distance observed between these clades and other groups of hemiones is somewhat comparable, suggesting the hydruntine as a conspecific member of hemiones. Hemiones were thus present in the Upper Paleolithic of Europe and were likely the inspirational source of some parietal paintings and engravings showing hemione-like equine silhouettes with particularly long ears.
The genetic analysis of other paleontological remains originally attributed to E. hydruntinus revealed the existence of a mitochondrial sequence both divergent to hemiones and all other present-day equine species (Orlando et al. 2009; Vilstrup et al. 2013). Its phylogenetic placement within stenonines still requires further clarification, but a careful morphological reanalysis of the specimens dismissed the original assignment to hydruntines (Eisenman 2010) and showed instead strong affinities with an equine group described in the Middle Pleistocene of Germany. These so-called Sussemiones were originally thought to have become extinct hundreds of thousand years earlier (Eisenman 2006, 2010) but likely survived in the Altai Siberian caves from Proskuriakov, Okladnikov, and Denisova until at least ~45 kyrs ago, as indicated by the age of the youngest sample genetically analyzed (Orlando et al. 2009). It thus seems that the strong morphological plasticity present in the equine paleontological record has not just resulted in situations similar to NWSL horses and hydruntines, where a single genetic species exhibited an entire range of morphotypes. It also caused situations where different genetic species (here, the Sussemione and the hydruntine) were lumped together within a single morphological species.
3 Population Dynamics and Conservation
In addition to revisit the evolutionary tree of the horse family and second archaeozoologists in the identification of bone assemblages, aDNA studies have investigated the population dynamics of several equine species in the past, mostly horses. The two main questions addressed are: first, how did major climatic crises, in particular the Last Glacial Maximum (LGM), impact the horse demography (Lorenzen et al. 2011; Orlando et al. 2013; Schubert et al. 2014)? Second, did climate or human activities, including overhunting, drive the extinction of all equine species in the Americas at the end of the Late Pleistocene (Haile et al. 2009; Lorenzen et al. 2011; Willerslev et al. 2014)? The application of similar methodologies to both present-day individuals and museum specimens has also helped evaluate the impact of decades of captivity in a population generally considered as the last remaining truly wild horse on the planet (Der Sarkissian et al. 2015b). The main findings of these studies are presented below.
3.1 Extinction and Climate Change
Within the last 50 kyrs, a large fraction of the equine biodiversity became extinct. The exact extinction time of the Sussemiones is unknown but took place within the last ~45 kyrs (Orlando et al. 2009). The last NSWL horse remains were found in the Alaskan permafrost and were radiocarbon dated to ~31 kyrs cal. BP (Guthrie 2003). Hippidiform and horse populations vanished in the Americas around ~11–12 kyrs ago (Guthrie 2006), a time that also experienced the extinction of up to two thirds of the megafauna genera and four fifths of their species in this continent [see Stuart (2015) for a review]. The Giant Cape zebra from South Africa became extinct almost at the same time. The trace of the main mitochondrial lineage of the hydruntine is lost after ~3 kyrs ago (Orlando et al. 2006; Bennett et al. 2017), probably due to human-driven fragmentation of their habitat [claims of later survival, possibly up until the Middle Ages, have been dismissed as donkeys by genetic evidence (Orlando et al. 2009)]. The Atlas wild ass died out in Algeria probably due to overhunting by the Romans, some ~2 kyrs ago. By the late 1890s, no quagga zebras were roaming the South African savannahs, and the last captive specimen died in the early 1900s (Leonard et al. 2005).
While there is little doubt that human activities have driven most of these recent extinctions, several hypotheses have been proposed to account for those taking place around the Late Pleistocene–Holocene transition, some ~11 kyrs ago. In the Americas, this period overlaps not only with major climate changes, associated to the massive contraction of grasslands and tundra steppes, which are rich in nutrients, but also with human expansion. At one extreme, models posit climate as the main extinction driver, while at the other extreme, humans overkilled megafauna populations, including equids, possibly almost overnight (the Blietzkrieg hypothesis). Even though the latest horse macrofossils discovered north of the Cordilleran and Laurentide ice sheets dated to ~13–15 kyrs ago, traces of horse DNA have, however, been detected several millennia later in Alaskan permafrozen sediments (Haile et al. 2009). This demonstrated that horses survived at least until ~10.5 kyrs ago in the Americas. They thus overlapped with humans during almost three millennia before becoming extinct (Rasmussen et al. 2014), ruling out the Blitzkrieg extinction model. This does not rule out, however, humans as possible extinction drivers because a massive American potential range is predicted by climate niche modeling in the mid-Holocene, a time when horses already went extinct there (Lorenzen et al. 2011). Since climate niche modeling is purely trained on climatic variables, climate change in itself does not seem sufficient to have led horses to extinction. In Europe and Siberia, where extensive information on the presence of herbivores present in archaeological sites can be compiled, horses also represent the dominant species and show a massive overlap with humans from after the LGM (Lorenzen et al. 2011). Human activities might thus have contributed to shape the horse demographic trajectory within the last 10 kyrs, possibly including their extinction in the Americas.
Additionally, current evidence leaves no doubt that climate changes have also been a major driver of the horse demography during the last 50 kyrs. Bayesian skyline reconstructions based on the mitogenome diversity within present-day horses show a steady demography until their domestication where an exponential demographic increase is recovered (Lippold et al. 2011a, b; Achilli et al. 2012). This profile contrasts with those obtained when including ancient horse mitogenomes in the dataset (Orlando et al. 2013; Schubert et al. 2014) (Fig. 2), suggesting that the present-day mitogenome diversity only captures limited demographic information prior to domestication times. The demographic profile suggests a first expansion phase from ~100 kyrs ago, peaking right before the LGM, and followed by a massive decline. Interestingly, demographic reconstructions based on the pairwise sequential Markov chain (PSMC) model, which exploits patterns of heterozygosity variation along single diploid genomes (Li and Durbin 2011), are consistent with such Bayesian skyline profiles prior to the LGM (Orlando et al. 2013; Schubert et al. 2014; Librado et al. 2016) (Fig. 2). Leveraging partial hypervariable mtDNA sequences from over a hundred of radiocarbon dated horses, and the climate niche envelopes predicted at four time periods (42, 30, 21, and 6 kyrs ago), Lorenzen and colleagues found that the effective population size was significantly correlated to the predicted range size for the species, confirming climate, and in particular the transition to the cold, arid, and fully glacial marine isotope stage 2 some ~30 kyrs ago, as a major driver of the horse demography (Lorenzen et al. 2011). It is noteworthy that the PSMC profiles obtained from the single diploid genomes of present-day domesticates and wild individuals and Late Pleistocene animals all show similar trajectories (although with changes of a different magnitude), with two additional phases of expansion/collapse within the last 2 myrs (Orlando et al. 2013; Schubert et al. 2014; Librado et al. 2015). In particular, the Eemian period (~130 kyrs ago) also appeared associated with reduced effective sizes, suggesting an important role for glacial/interglacial transitions.

Horse demographic trajectories within the last 30–500 kyrs [adapted from Orlando et al. (2013), Schubert et al. (2014), Der Sarkissian et al. (2015b), and Librado et al. (2015, 2016, 2017)]. Complementary temporal resolution was obtained using Bayesian skyline modeling on patterns of mitochondrial DNA variation, using ancient DNA data (panel a; Orlando et al. 2013; Librado et al. 2017) or not (panel b; Lippold et al. 2011a), and PSMC reconstruction applied to the autosomal data underlying single diploid genomes of domestic horses (panel c; Orlando et al. 2013; Der Sarkissian et al. 2015b; Librado et al. 2015)
The climate changes underlying the global warming following the Late Pleistocene/Holocene transition might also have played a significant role in the horse extinction in North America. The sequencing of minibarcodes from permafrozen sediments spread across the whole Holarctic region indeed showed a complete ecological turnover in the post-LGM plant communities, with forbs no longer dominating the ecosystem but graminoids (Willerslev et al. 2014). The genetic analysis of pre-LGM coprolites from horses as well as other megafauna species, such as bisons, woolly rhinos, and woolly mammoths, yet suggested that these animals were predominantly forb feeders. Therefore, it is possible that the rise of graminoids associated with the post-LGM climate changes might not have provided conditions compatible with sustaining large horse (megafauna) populations. Whether the nutrient content of forbs helped sustain large herbivore populations, and/or the nitrogen input from such populations favored forbs in their competition with graminoids remains to be investigated.
3.2 Conservation
Except for the plains zebra and the Tibetan kiang, formally a species but recently proposed to represent a subspecies of hemiones (Bennett et al. 2017), all other wild equine species are presently considered vulnerable or (critically) endangered by the International Union for Conservation of Nature (IUCN) (Orlando 2015; see http://www.iucnredlist.org/). The situation is also concerning for horse domesticates, with approximately a quarter of breeds being considered endangered by the Food and Agriculture Organization (FAO; see http://www.fao.org/home/en/). It is thus essential that sound conservation programs are established before most of the equine diversity, be wild or domesticated, disappears. Ancient DNA studies of museum specimens can help toward the objective, as recently demonstrated for Przewalski’s horses (Der Sarkissian et al. 2015b).
Since the last tarpan died out in the Hellabrunn Zoo from Munich in 1887, the Przewalski’s horses have long been considered to represent the last surviving wild horses on the planet. Recent genomic work, however, demonstrated that these were the direct descendants of the earliest domestic horses known in the archaeological record (Gaunitz et al. 2018). They, thus, belong to a lineage that was once successfully domesticated but further returned feral.
Przewalski’s horses were first described as their own species but are now considered to be a subspecies of horses, despite their additional pair of chromosomes (Boyd and Houpt 1994). Following their discovery by the Western world in the second half of the nineteenth century, this population has experienced a massive demographic collapse until the last individual was caught in the wild in 1947, seen in 1969, and soon after declared extinct (Wakefield et al. 2012). The almost ~2,000 individuals currently living are the progeny of captive individuals kept in zoos and provide one of the too few success stories of conservation biology, with thriving reintroduction reserves in Mongolia, Russia, and China. However, the pedigree of all living Przewalski’s horses was founded by only 12–16 animals, potentially limiting their evolutionary potential and chances of survival in the future. By using museum specimens to sequence the complete genomes of living Przewalski’s horses representing all founding lineages, as well as those from animals that lived in the late nineteenth and early twentieth century, Der Sarkissian and colleagues found a number of features relevant to the conservation of the population (Der Sarkissian et al. 2015b).
First, for two of the horses analyzed, the genetic data were not compatible with the expected pedigree as described within the conservation studbook. The latter is key to predicting what the expected inbreeding levels would be for any parental pair of males and females. The identification and correction of mistakes in the conservation studbook have, thus, an overall positive impact on the ex situ conservation program by enabling more accurate estimates of individual relatedness and limiting the inbreeding levels in future generations.
Second, the Przewalski’s horses and domestic horses were found to represent the descending populations of two lineages that split some ~45 kyrs ago, in agreement with earlier estimates based on more limited genome-scale data (Orlando et al. 2013). Those separate populations continued to admix with each other even after the horse was domesticated some ~5.5 kyrs ago and virtually up until their discovery by the modern world. The contribution of both populations to gene flow was, however, asymmetrical, with the lineage leading to Przewalski’s horses predominantly contributing to that leading to domesticated horses. Estimates indicate that this contribution into the latter lineage was moderate and altogether corresponded to approximately 2–3% of the genome (Gaunitz et al. 2018).
The situation changed during captivity as Der Sarkissian and colleagues could identify one historical Przewalski’s horse as the F1 hybrid of one Przewalski’s stallion and one Mongolian domestic mare. Additionally, recent introgression from domesticates could be detected in a fraction of the living Przewalski’s horses analyzed. Therefore, while some branches in the Przewalski’s horse pedigree are devoid of such introgression introduced during captivity, some individuals show as much as ~30% of domestic genome ancestry. Explicit modeling based on f4-statistics and including the genome data from Botai horses, the direct ancestors of Przewalski’s horses, revealed that a minimal ~6–7% of the genome of Przewalski’s horses pertained to this recent introgression from domesticates (Gaunitz et al. 2018). Nonetheless, altogether, those studies suggest that the vast majority of the genome of Przewalski’s horses has no equivalent in modern domesticates. All possible efforts must thus be done to preserve this genetically unique population of horses, even if they do not represent the last truly wild horses in the planet.
Improving the chances of success of future conservation plans will, however, not necessarily equate to designing mating schemes aimed at diluting the impact of this domestic ancestry in future generations. Other genomic features, such as the levels of inbreeding and deleterious mutations (i.e., the so-called genomic load), as well as the taxonomic diversity of the gut microbiome, which is now known to be impacted by captivity in horses (McKenzie et al. 2017; Metcalf et al. 2017), will also have to be considered.
4 Domestication
Within the last six millennia, two equine species have been domesticated almost at the same time but in different regions of the world. Archaeological evidence from Egypt suggests that the donkey was already domesticated ~5 kyrs ago (Rossel et al. 2008), probably to help the transportation of people and goods across the Sahara in response to drier climatic conditions. Since then, donkeys have remained important pack animals and facilitated the development of mobile pastoralism and major overland trade routes in both Africa and Eurasia. Similarly, the domestication of the horse also had a far-reaching impact on human history (Kelekna 2009). With horses, humans could travel for the first time well above their own speed and carry their germs, culture, and genes across vast geographic areas. The development of horse-drawn chariots and cavalry also radically changed the history of warfare and was instrumental to the emergence and stability of empires until the mechanization of weaponry during the second half of the twentieth century. Beyond the battlefield, farm horses have massively impacted agricultural productivity, especially in the late Middle Ages (Langdon 2006). Although earlier changes in the human/horse relationship have been suggested (Anthony 2007; Anthony and Brown 2011), the bite wear patterns present on the animal teeth indicate that horses were harnessed during the ~5.5-kyr-old Eneolithic culture of Botai from the North central Kazakh steppes (Outram et al. 2009), where the animal represent >99% of the bone assemblages. Additionally, the isotopic signatures of equine milk found in fatty acid traces preserved on ceramics support horse milking. This suggests that horses were already domesticated in the region some ~5.5 kyrs ago, despite the fact that the important size shifts that are generally taken as a domestication marker in other animals (Vigne et al. 2005) are only observed for horses at least a millennium later (Benecke and von den Driesch 2003). Discoveries of corral structures associated to the Botai culture, including at the eponymous site, confirmed domestication (Gaunitz et al. 2018).
The biological changes that accompanied the domestication processes of horses and donkeys are difficult to reconstruct from current patterns of genetic diversity both due to the development of intensively selected and extremely influential breeds during the last two centuries and the almost extinction and, thus, the massive loss of diversity experienced by their wild relatives (Der Sarkissian et al. 2015b; Kimura et al. 2011). By traveling the past, aDNA offers the possibility to catch evolution red-handed and chart through space and time the important genetic changes underlying horse and donkey domestication. An overview of the main findings related to the existence of one single or multiple domestication center(s), the importance of wild restocking of the livestock, the traits selected from early domestication stages to the emergence of particular breeds, is presented in the following section.
4.1 Domestication Center(s) and Wild Restocking
Extensive surveys of the mitochondrial diversity present in extant domestic donkeys across the world have indicated the presence of two main clusters, genetically distinct from hemiones (Beja-Pereira et al. 2004). This ruled out hemiones as possible progenitors for the domestic donkey and suggested that two independent domestication processes gave rise to each genetic cluster. The Nubian wild ass, which historically spread across North Africa, from the Red Sea shores of Sudan to the Moroccan Atlantic coast, shows haplotypes typical of the first cluster and thus represents a likely progenitor for this cluster. The critically endangered Somali wild ass, which only survives today in small pockets within Sudan and Erithrea, represented a possible candidate for the progenitor of the second cluster. However, the Somali wild ass was found to form a cluster of its own, distinct and almost equidistant to the other two (Beja-Pereira et al. 2004), leaving the origins of the second cluster unknown. Perhaps the recent demographic collapse of African wild ass populations resulted in the loss of haplotypes in either wild ass population, precluding any identification of direct genetic affinities within extant populations. By extending the genetic analyses to field-collected feces, museum specimens, and subfossil material, Kimura and colleagues have confirmed the limited genetic diversity present in the Somali wild ass (Kimura et al. 2011). This is in line with the signature of a drastic demographic bottleneck found in the sequence data underlying the first diploid genome sequence of the species, which likely took place within the last ~25 kyrs (Jónsson et al. 2014). Despite the large sampling effort in the study from Kimura and colleagues, the Somali wild ass remained genetically distinct. Furthermore, whole genome sequence data confirmed a population time split at ~350 kyrs ago for the lineages of the Somali wild ass and donkey domesticates, which predates the onset of domestication (Rossel et al. 2008). Therefore, the Somali wild ass has not given rise to any of the two main domestic donkey clusters. The sequence data obtained by Kimura and colleagues confirmed instead the relationship between the Nubian wild ass and the first domestic cluster, as a ~3-kyr-old domestic specimen from Central Sahara showed a mitochondrial haplotype identical to museum specimens of Nubian wild asses (Kimura et al. 2011). However, while eight of nine such museum specimens nested with the first mitochondrial cluster, the haplotype of the ninth specimen was typical of the second cluster, suggesting that the Nubian wild ass, or any extinct population also sharing this haplotype – possibly the Atlas wild ass from the Maghreb and the coast of Yemen – was in fact also the progenitor of the second domestication. Testing this hypothesis will require additional sequence data from ancient African wild ass. This might prove difficult but not impossible given the encouraging success in the characterization of the mitogenome or nuclear genome variation present in ancient remains from the Middle East (Almathen et al. 2016; Mohandesan et al. 2017) and Eastern Africa (Gallego Llorente et al. 2015).
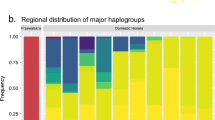
The mitochondrial variation found in present-day and ancient domestic donkeys and African wild asses has revealed something additional, which proved also valid for horses, namely, their domestication process involved substantial restocking of the livestock from wild progenitors (Kimura et al. 2011; Vilà et al. 2001; Jansen et al. 2002; Lippold et al. 2011a; Achilli et al. 2012). In none of these species analyzed does the mitochondrial diversity found in domestic (ancient) animals form a single, monophyletic cluster encompassing only a small fraction of the (ancient) wild genetic diversity. Instead, the haplogroups found in wild (ancient) animals and (ancient) domesticates overlap to a very large extent, suggesting either that lots of such haplogroups were present in the founder group of domesticates or that wild females were mated with male domesticates throughout the domestication process. Analyses based on nuclear data support the latter scenario (Warmuth et al. 2012; Der Sarkissian et al. 2015b). Recent estimates based on whole mitogenome sequences suggest that possibly up to three quarters of the mtDNA diversity once present in the wild has been effectively incorporated into the horse domestic gene pool (Lippold et al. 2011a). Such estimates are not available for donkeys, due to the still relatively limited characterization of the genetic variation in (ancient) wild animals.
Interestingly, in horses, the Y chromosome shows a pattern opposite to mtDNA. There, present-day domesticates show extremely limited variation, with one single haplotype dominating most breeds and populations (Lindgren et al. 2004; Wallner et al. 2013). This has supported the contention that the process of horse domestication involved restocking from the wild mostly through mares, rather than stallions, possibly as the aggressive reproductive behavior of the latter makes them more difficult to manage. However, sequence data from ancient domestic horses, especially from Scythian stallions (Lippold et al. 2011b; Librado et al. 2017), have revealed quite diverse Y-chromosome haplotypes in the Iron Age. Therefore, the number of stallion founders used in early domestication stages was not limited, and the participation of a diversity of stallions has only reduced after the Iron Age. Further studies, charting the Y-chromosome diversity of stallions through time, will reveal when and in which archaeological and/or historical contexts this reduction took place. Although presently unknown in absence of extensive surveys of the Y-chromosome diversity in this species, the wild restocking in the domestic donkey could be expected to show limited sex bias, in contrast to the horse as African pastoralists recruit both jacks and jennets to their herds and trapping of wild animals irrespective of sex is documented historically.
The first complete genome sequences obtained from Late Pleistocene wild horses have revealed that a third population of horses existed some ~16–42 kyrs ago, in addition to the populations underlying present-day domestic and Przewalski’s horses (Schubert et al. 2014). Little is known about this population of wild horses as it is currently known from the genome sequences of three specimens only. It was spread across the Taymir peninsula and Yakutia from ~42 kyrs ago to ~5 kyrs ago and split from the population ancestral to both present-day domestic and Przewalski’s horses by the time of the Eemian interglacial, some ~130 kyrs ago (Schubert et al. 2014; Librado et al. 2015). As none of the genomes from present-day horses clusters together with any of the three known genomes for this population, it is considered to have become extinct within the last ~5 kyrs. This population, however, was proposed to have contributed to the genetic makeup of present-day horses, possibly representing a minimum of ~13% of their genome ancestry using calculations based on D-statistics (Durand et al. 2011) and f4-statistics (Patterson et al. 2012). However, once the earliest domestic horses from Botai were sequenced (Gaunitz et al. 2018), D-statistics showed that Botai and modern domesticates have similar amounts of shared derived polymorphisms with the archaic population (in contrast to Przewalski’s horses, which show a deficit of such variation). This pattern is compatible with two scenarios. Firstly, the admixture from the archaic lineage took place prior to the divergence between the lineages of Botai and modern domesticates but was further eliminated post-Botai in Przewalski’s horses (this component was, however, maintained in modern domesticates). Alternatively, an admixture from a yet unidentified ghost lineage into Przewalski’s horses may be the reason why they look more distant to the archaic lineage identified. Determining which of these scenarios prevailed requires further work. Either way, it nonetheless illustrates that wild, archaic populations of horses have significantly contributed to the genetic makeup of modern horses, be domesticated, or feral.
Given the extent of wild introgression and the dispersal capacity of the horse, it is perhaps not surprising that the horse mitochondrial variation shows a lack of phylogeographic structure, with haplotypes spreading over vast regions (Leonard et al. 2005; Cieslak et al. 2010), and no major haplogroups specific for a single breed (Jansen et al. 2002). Haplogroup D1, which is quite frequent in Iberian breeds and North-African barb horses (Jansen et al. 2002), was yet proposed to reflect local domestication in Iberia, but aDNA failed to reveal its presence in the Iberian Neolithic and Bronze Age (Lira et al. 2010). Thus, it likely reflects the descent of horses introduced after the Bronze Age. However, some mtDNA haplotypes that populated the Iberian Bronze Age are still occasionally found in Iberian breeds (Lira et al. 2010; Jansen et al. 2002) and could represent signs of local domestication. This, and the discovery of a hotspot of STR diversity in Iberian breeds (Warmuth et al. 2011), has been proposed to reflect the existence of a second domestication center for horses in Iberia, independent from the Kazakh/Pontic-Caspian steppes (Outram et al. 2009; Warmuth et al. 2012). However, Iberia also represents a well-known glacial refugium (Hewitt 2000), where more diverse populations of indigenous wild horses might have survived until exogenous domestic horses finally arrived in the region. A simple local wild restocking would then equally well explain the hotspot of diversity observed. Therefore, pending additional data testing the genetic continuity of Iberian horse population pre- and post-domestication, the question of a possible Iberian domestication center is open. In Europe, horses, however, represent a rare fraction of bone assemblages in the archaeological record prior to the Chalcolithic and probably survived as fragmented populations islets around open patches in the canopy forest (Sommer et al. 2011). The limited nature of the archaeological record in Europe prior to the Chalcolithic might limit future attempts at addressing whether Iberian populations were fully replaced or assimilated into the domestic pool. The recent sequencing of the genome of Botai horses has revived the debate on the possible multiple origins of the domestic horses since none of the domestic horses sequenced within the last ~4,100 years directly relates to Botai. Instead, they form a second, distinct monophyletic group with no more than ~2–3% of Botai ancestry. Together with the signal of a massive demographic expansion at approximately ~4,500 years ago found in patterns of mitochondrial variation, this suggests that another horse group, not related to Botai horses, could have been domesticated around that time and fueled massive expansion of human groups across Eurasia (Gaunitz et al. 2018). Alternatively, the Botai genetic component could have been diluted to almost disappear during such expansion by means of introgressive capture (Larson and Fuller 2014). Further data, particular from animals that lived in the third millennium BCE, are necessary to tease both scenarios apart and to identify the true temporal and geographic locus of horse domestication.
4.2 Trait Selection and Genetic Load
Given the importance of the horse industry, representing a yearly €100 billion impact for the EU economy alone, genetic investigations of present-day domestic horses have not been limited to neutral markers, such as mtDNA and the Y chromosome. Many studies have successfully identified genes underlying major phenotypic traits and disorders (see Chowdary 2013 and references therein). Besides coat color (e.g., Brooks et al. 2002; Brooks and Bailey 2005; Brunberg et al. 2006; Reissmann et al. 2007; Bellone et al. 2013; Imsland et al. 2016), variants statistically associated with racing performance have been discovered (Hill et al. 2010; McGivney et al. 2010; Tozaki et al. 2010; Petersen et al. 2013). Perhaps the most spectacular example involves a single mutation and a SINE insertion at the MSTN gene, with homozygous mutants showing hypertrophic muscles and top performance at short-distance sprint races (see Rivero and Hill 2016 for a review). One single C ➔ A mutation at the DMRT3 gene is also known to be permissive for alternate gaits complementary to walk, trot, and gallop (Andersson et al. 2012) and is distributed throughout the world (Promerová et al. 2014), mainly in gaited breeds. Additionally, the GYS1 H allele, which can be found at non-negligible frequencies despite causing severe myopathies, has been proposed to represent an example of “thrifty gene” that conferred a selective advantage in early domestication (it increases muscular glycogen storage) but would be maladapted to modern starch-rich diets (McCue et al. 2008). Besides metabolic disorders, the genes underlying size variation have received much attention (Signer-Hasler et al. 2012; Makvandi-Nejad et al. 2012; Metzger et al. 2013), and four loci (LCORL, HMGA2, ZFAT, and LASP1) seem to explain a majority (as much as 83% in Makvandi-Nejad et al. 2012) of the size differences among breeds, which illustrates how humans have successfully manipulated a fraction of the horse genetic potential to create a whole diversity of breed sizes.
This brief overview is certainly not exhaustive as many other genotype/phenotype associations have been unveiled since the horse reference genome, and high-throughput genotyping tools have been developed (Wade et al. 2009; McCue et al. 2012; Petersen et al. 2013). Ancient DNA has offered a unique opportunity to chart these variants through space and time and track the origins and the geographical, cultural, and historical context into which the underlying characters later expanded. For instance, aDNA from museum samples suggested that the “speed” mutation at MSTN probably entered the pedigree of racing Thoroughbred horses from local British mares, as it was absent from all founding Arab stallions (Bower et al. 2012). In addition to such candidate gene approaches, targeting specific loci of known genetic variation, the advent of whole genome sequencing has enabled the first genome scans in horses, providing a first glimpse at the full suite of genetic changes that have been introduced and/or selected in the course of their domestication. The main findings resulting from these two complementary approaches are presented below.
4.3 Candidate Gene Approaches
Most analyses of the genetic variation present at nuclear loci in ancient horses have focused on loci involved in coat coloration (Ludwig et al. 2009; Pruvost et al. 2011; Ludwig et al. 2015; Wutke et al. 2016a). The Dun phenotype is characterized by pigment dilution and typical so-called primitive markings, corresponding to undiluted black stripes, mostly along the spine and the legs. It represents the wild-type coat coloration phenotype in horses and is common to all Przewalski’s horses. The causative variant underlying Dun coloration in horses, a ~1.6 kb-long insertion downstream of the TBX3 gene (Imsland et al. 2016), has only been described recently. The presence/absence of the corresponding 1.6 kb block could not be investigated in earlier studies opting for candidate gene approaches, which can thus not distinguish Dun and non-Dun horses. Deleted alleles have yet been identified in the sequence data underlying two ancient genomes dated to ~43 and ~5 kyrs ago (Schubert et al. 2014; Librado et al. 2015), suggesting that a diversity of color morphs existed prior to domestication (Imsland et al. 2016). The diversity was nonetheless much limited compared to what is seen from the early Bronze Age, as only the black allele at ASIP (Ludwig et al. 2009) and the leopard spotted allele at TRPM1 (Pruvost et al. 2011) could be detected in Late Pleistocene and early Holocene horses. Incidentally, the genetic identification of spotted horses in the Late Pleistocene suggests that the famous cave paintings of the dappled horses of Pech-Merle could well be simple representations of living animals, not symbolic expressions (Pruvost et al. 2011).
From the Bronze Age onward, a diversity of variants including chestnut horses, cream and silver dilutions, and sabino and tobiano spotting were present at detectable frequencies and were found to increase in frequency much faster than expected by chance (Ludwig et al. 2009). They were thus likely selection targets to horse herders. This work demonstrated that changes in coat coloration patterns have represented one of the early targets in the process of horse domestication, with different color morphs perhaps providing early herders with a means to differentiate their herds (Wutke et al. 2016a). Importantly, selection patterns were not homogeneous through space and time as the genetic variant underlying leopard spotting was quite common in Western Europe during the early Bronze Age but remained undetectable until it reappeared in Siberia during the Iron Age (Ludwig et al. 2015). Horse herders from different regions and cultures had thus fluctuating selection targets through time.
Importantly, leopard spotting is associated with congenital night blindness in horses (Bellone et al. 2013). Some herders might have had different opinions as to whether the spotted phenotype typical of present-day Appaloosa horses outweighed a diminished vision capacity at night and the related increased risks in terms of predation and thievery. For instance, no spotted (and dilution) alleles were found within Celtic Swiss horses (Elsner et al. 2016), but spotted horses were not uncommon among Iron Age Scythian horses (Ludwig et al. 2015; Librado et al. 2017). Leopard spotting alleles, and more generally other spotting alleles, however dropped in frequency following 400 AD in Europe (Wutke et al. 2016a). While these alleles were selected against, chestnut and black alleles were positively selected and rose to high frequencies, suggesting that strong changes in coat color phenotypes took place in the course of the Middle Ages. Surprisingly, no spotted alleles were detected in early Norse (Viking) horses from Iceland. As these traits are relatively frequent in present-day Icelandic horses, it is likely that the prohibition of horse import in the island was not as strict as commonly believed. Early Norse Icelandic horses were, however, frequent carriers of the DMRT3 allele associated with alternate gaits, such as ambling, which are more comfortable to riders than classical canters and gallops (Wutke et al. 2016b). The mutation was left undetected in mainland Europe, including Scandinavia (although Norway, an important Norse region, was not formally tested), prior to 850–900 AD, where it appeared in Britain. This suggested that Norse first acquired ambling horses on the British Isles before they transported them to Iceland, and possibly further, throughout their whole distribution range.
4.4 Genome Scans
Recent methodological developments in aDNA research have opened access to genome-scale information from past populations. Target enrichment offers the most economical approach for characterizing up to several millions of loci (Haak et al. 2015; Mathieson et al. 2015), whole chromosomes (Fu et al. 2013), and even the non-repeated fraction of nuclear genomes (Carpenter et al. 2013). For DNA extracts showing relatively high levels of endogenous DNA, shotgun sequencing also provides a fast- and cost-effective approach to whole genome sequencing (Orlando et al. 2015). So far, target enrichment has been limited to a handful of loci in ancient equids, mostly the whole mitogenome (Vilstrup et al. 2013), but also several thousands of nuclear loci showing functional and neutral variation (Cruz-Dávalos et al. 2016). Shotgun sequencing has, however, delivered no less than 22 ancient horse genomes (Orlando et al. 2013; Schubert et al. 2014; Der Sarkissian et al. 2015b; Librado et al. 2015, 2017), which, compared to present-day genomes, provided important insights into the selection process underlying horse domestication.
First, by comparing the genomes of two Late Pleistocene horses to the genomes representing a broad range of present-day domestic breeds, Schubert and colleagues could identify 50 kb-long regions showing selection signatures in modern horses (Schubert et al. 2014). The authors implemented four independent tests of positive selection and considered as good candidates those regions supported by a minimum of two independent tests. This provided a total number of 125 regions, which carried genes enriched for four main functional categories of genes. A number of genes were involved in horse locomotion, with genes participating to the organization of skeletal muscles, myotendons, and articular junctions but also to balance and motor coordination (Schubert et al. 2014). Additionally, the candidates included many genes associated with the cardiovascular system and represented possible selection targets to adapt the equine physiology to the energetic demands related to sustained efforts. A third category of candidates included genes associated with skeletal and facial development, possibly echoing the diversity of sizes and faces seen in modern horses. Finally, the last category of selection candidates was indicative of changes potentially associated with behavior and learning capacity, two traits that are central to the development of the human/horse relationship.
As it contrasted to the genomic variation in present-day horses and horses that lived prior to domestication, this work could only detect signatures associated to the domestication process. It could, however, not determine when the detected changes were exactly introduced in the course of horse domestication. The genome sequences of 11 Scythian horses that lived ~2.3 kyrs ago provided a first attempt toward this objective, enabling to look at the changes introduced prior to 2.3 kyrs ago during early domestication stages and those introduced during the last 2.3 kyrs (Librado et al. 2017). Interestingly, the list of candidate genes showing selection signatures in early domestication stages was enriched in genes involved in the neural crest development. Yet, the “neural crest” hypothesis (Wilkins et al. 2014) posits that the full suite of traits that are common to many domestic animals (floppy ears, depigmentation, juvenile behavior, docility, etc., all grouped into the so-called domestication syndrome) was first selected by imposing a selective pressure on central pathways affecting the development of the underlying organs and structures. Genes involved in the formation, migration, and differentiation of neural crest cells were natural good candidates for such pathways, given the large number of cell types and tissues deriving from neural crest cells and their general overlap with traits associated with the domestication syndrome. The variation present in the genomes of Scythian horses lends supports to this hypothesis. Interestingly, the authors could also detect selection signatures within the Scythian horses themselves, mostly of genes involved in the development of forelimbs, which confirmed morphometric measurements of metacarpals showing that Scythian horses were more robust than present-day horses living in the same region of Kazakhstan and Mongolia (Librado et al. 2017). Additionally, Scythian horse riders seem to have selected for genes involved in the posterior pituitary, a key production center of vasopressin and oxytocin. The latter neurohypophysal hormone is essential to both lactation and is known to be involved in the development of strong bonds between humans and dogs (Nagasawa et al. 2015). This suggested that Scythian herders might have selected variants facilitating both the milking of horses and their managing.
Additional work applying similar methodology focused on reconstructing the genomic history of one of the most iconic horse breeds on the planet, namely, the Yakutian horses (Librado et al. 2015). Yakutia represents the coldest country in the Northern hemisphere, with winter temperature records dropping below −70°C. Sequencing the genome of both ancient and present-day Yakutian horses has helped reveal the genetic basis for the suite of morphological and physiological adaptations that Yakutian horses develop to survive in this extreme environment. Interestingly, in addition to identifying candidate genes that also show adaptive signatures among other cold mammals, such as the woolly mammoth and Siberian humans, the study revealed that the population of Yakutian horses developed within an extremely short timeframe, probably within the last thousand years, following the migration of the first Yakut settlers in the region (Keyser et al. 2015). The authors proposed that selection at regulatory regions, as supported by the genomic evidence, is key to the success of such fast episodes of adaptation (Librado et al. 2015).
Finally, the whole genome data available for horses have revealed that the domestication process was accompanied with an increase in the genetic load, as present-day domesticates show a higher fraction of potentially deleterious mutations in protein-coding genes (Schubert et al. 2014). The demographic collapse associated with horse domestication, especially within the last 2.3 kyrs (Librado et al. 2017), is proposed to have reduced the effect of negative selection in purging out (slightly) deleterious mutations from the domestic horse gene pool. Therefore, the domestication of the horse did not only come with an improvement of functions of key interest to humans but also resulted in an overall inflation in deleterious mutations, potentially enhancing the odds that horses develop important genetic disorders.
5 Conclusions and Future Perspectives
Members of the horse family have accompanied more than three decades of aDNA research. Even though a wealth of taxonomic groups has been studied the horse has focused most of the attention, especially since the advent of high-throughput DNA sequencing, most logically given the impact that horses have had on human history. The genome-scale methodology currently applied to horses will likely be used for other members of the family in the near future, particularly the donkey, where de novo reference genomes and genome-scale data have now become available (Orlando et al. 2013; Huang et al. 2015; Bertolini et al. 2015; Renaud et al. 2018), but also for endangered close relatives. There is also a lot to be learnt from the extinct members of the family, especially for helping implement sound conservation programs. This chapter strictly focused on how the changes in the genetic variation observed through space and time could help better understand the biology and evolutionary history of equine species. However, with recent developments in aDNA research, it is now possible to recover information of the host itself but also of its pathogens and the whole community of microbes living in the gut and/or in the mouth (see Warinner et al. 2015 for a review). Additionally, epigenetic information can be gathered from aDNA extracts, potentially revealing how environmental cues are integrated at the genomic level to regulate gene expression (Orlando et al. 2015; Gokhman et al. 2016). The application of such methodology to the horse, as well as all other members of its family, will undoubtedly open new exciting avenues for equine research.
References
Achilli A, Olivieri A, Soares P, et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc Natl Acad Sci U S A. 2012;109:2449–54.
Alberdi MT, Prado JL. Review of the genus Hippidion Owen, 1869 (Mammalia: Perissofactyla) from the Pleistocene of South America. Zool J Linn Soc. 1993;108:1–22.
Alberdi MT, Prado JL. Comments on Pleistocene horses from Tarija, Bolivia, and the validity of the genus Onohippidium (Mammalia: Equidae), by B.J. MacFadden. J Vert Paleontol. 1998;18:669–72.
Alberdi MT, Prado JL, Prieto A. Considerations on the paper “morphological convergence in Hippidion and Equus (Amerhippus) South American equids elucidated by ancient DNA analysis”, by Ludovic Orlando, Véra Eisenmann, Frédéric Reynier, Paul Sondaar, Catherine Hänni. J Mol Evol. 2005;61:145–7.
Almathen F, Charruau P, Mohandesan E, et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc Natl Acad Sci U S A. 2016;113:6707–12.
Andersson LS, Larhammar M, Memic F, et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature. 2012;488:642–6.
Anthony DW. The horse, the wheel and language. Oxford: Princeton University Press; 2007.
Anthony DW, Brown DE. The secondary products revolution, horse-riding, and mounted warfare. J World Prehist. 2011;24:131.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Beja-Pereira A, England PR, Ferrand N, et al. African origins of the domestic donkey. Science. 2004;304:1781.
Benecke N, von den Driesch A. Horse exploitation in the Kazakh steppes during the Eneolithic and Bronze Age. In: Levine M, Renfrew C, Boyle K, editors. Prehistoric steppe adaptation and the horse. Cambridge: McDonald Institute for Archaeological Research; 2003. p. 69–82.
Bennett EA, Champlot S, Peters J, et al. Taming the late Quaternary phylogeography of the Eurasiatic wild ass through ancient and modern DNA. BioArXiv. 2017. https://doi.org/10.1101/090928.
Bertolini F, Scimone C, Geraci C, Schiavo G, Utzeri VJ, Chiofalo V, Fontanesi L. Next generation semiconductor based sequencing of the donkey (Equus asinus) genome provided comparative sequence data against the horse genome and a few millions of single nucleotide polymorphisms. PLoS One. 2015;10:e0131925.
Bellone RR, Holl H, Setaluri V, et al. Evidence for a retroviral insertion in TRPM1 as the cause of congenital stationary night blindness and leopard complex spotting in the horse. PLoS One. 2013;8:e78280.
Bower MA, McGivney BA, Campana MG, et al. The genetic origin and history of speed in the Thoroughbred racehorse. Nat Commun. 2012;3:643.
Boyd L, Houpt KA. Przewalski’s horse: the history and biology of an endangered species. Albany, New York: State University of New York Press; 1994. isbn:10-ISBN 0-791-41889-8; 13-ISBN 978-0-791-41889-5; OCLC 28256312.
Braud M, Magee DA, Park SD, et al. Genome-wide microRNA binding site variation between extinct wild aurochs and modern cattle identifies candidate microRNA-regulated domestication genes. Front Genet. 2017;8:3.
Brooks SA, Bailey E. Exon skipping in the KIT gene causes a Sabino spotting pattern in horses. Mamm Genome. 2005;16:893–902.
Brooks SA, Terry RB, Bailey E. A PCR-RFLP for KIT associated with tobiano spotting pattern in horses. Anim Genet. 2002;33:301–3.
Brunberg E, Andersson L, Cothran G, et al. A missense mutation in PMEL17 is associated with the silver coat color in the horse. BMC Genet. 2006;7:46.
Cardoso JL, Vilstrup JT, Eisenman V, et al. First evidence of Equus asinus L. in the chalcolithic disputes the Phoenicians as the first to introduce donkeys into the Iberian Peninsula. J Archaeol Sci. 2013;40:4483–90.
Carpenter ML, Buenrostro JD, Valdiosera C, et al. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am J Hum Genet. 2013;93:852–64.
Chowdary BP. Equine genomics. Oxford: Wiley-Blackwell; 2013.
Cieslak M, Pruvost M, Benecke N, et al. Origin and history of mitochondrial DNA lineages in domestic horses. PLoS One. 2010;5:e15311.
Cruz-Dávalos DI, Llamas B, Gaunitz C, et al. Experimental conditions improving in-solution target enrichment for ancient DNA. Mol Ecol Resour. 2016. https://doi.org/10.1111/1755-0998.
Cucchi T, Mohaseb A, Debue K, et al. Detecting taxonomic and phylogenetic signals in equids cheek teeth with geometric morphometrics: towards new paleontological and archaeological proxies. R Soc Open Sci. 2017;4:160997. https://doi.org/10.1098/rsos.160997.
Da Fonseca RA, Smith BD, Wales N, et al. The origin and evolution of maize in the Southwestern United States. Nat Plants. 2015;1:14003.
Der Sarkissian C, Vilstrup JT, Schubert M, et al. Mitochondrial genomes reveal the extinct Hippidion as an outgroup to all living equids. Biol Lett. 2015a;11.
Der Sarkissian C, Ermini L, Schubert M, et al. Evolutionary genomics and conservation of the endangered Przewalski’s horse. Curr Biol. 2015b;25:2577–83.
Durand EY, Patterson N, Reich D, et al. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–52.
Eisenman V. Folivores et tondeurs d’herbe: forme de la symphyse mandibulaire des Equidés et des Tapiridés (Perissodactyla, Mammalia). Geobios. 1998;31:113–23.
Eisenman V. Pliocene and Pleistocene Equids: palaeontology versus molecular biology. Cour Forsch Inst Senckenberg. 2006;256:71–89.
Eisenman V. Sussemionus, a new subgenus of Equus (Perissodactyla, Mammalia). C R Biol. 2010;333:235–40.
Eisenmann V, Baylac M. Extant and fossil Equus (Mammalia, Perissodactyla) skulls: a morphometric definition of the subgenus Equus. Zool Scr. 2000;29:89–100.
Elsner J, Deschler-Erb S, Stopp B, et al. Mitochondrial d-loop variation, coat colour and sex identification of Late Iron Age horses in Switzerland. J Archaeol Sci. 2016;6:386–96.
Ermini L, Der Sarkissian C, Willerslev E, et al. Major transitions in human evolution revisited: a tribute to ancient DNA. J Hum Evol. 2015;79:4–20.
Frantz LA, Mullin VE, Pionnier-Capitan M, et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science. 2016;352:1228–31.
Franzen JL. The rise of the horse family. Baltimore, MD: Johns Hopkins University Press; 2010.
Froese DG, Westgate JA, Reyes AV, et al. Ancient permafrost and a future, warmer Arctic. Science. 2008;321:1648.
Fu Q, Meyer M, Gao X, et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci U S A. 2013;110:2223–7.
Gaunitz C, Fages A, Hanghøj K, et al. Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science. 2018. https://doi.org/10.1126/science.aao3297.
Geigl EM, Grange T. Eurasian wild asses in time and space: morphological versus genetic diversity. Ann Anat. 2012;194:88–102.
Gallego Llorente M, Jones ER, Eriksson A, et al. Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science. 2015;350:820–2.
Gokhman D, Meshorer E, Carmel L. Epigenetics: it’s getting old. Past meets future in Paleoepigenetics. Trends Ecol Evol. 2016;31:290–300.
Green RE, Krause J, Briggs AW, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–22.
Groves CP, Willoughby DP. Studies on the taxonomy and phylogeny of the genus Equus-1. Subgeneric classification of the recent species. Mammalia. 1981;45:321–54.
Guthrie RD. Rapid body size decline in Alaskan Pleistocene horses before extinction. Nature. 2003;426:169–71.
Guthrie RD. New carbon dates link climatic change with human colonization and Pleistocene extinctions. Nature. 2006;441:207–9.
Haak W, Lazaridis I, Patterson N, et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–11.
Haile J, Froese DG, Macphee RD, et al. Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc Natl Acad Sci U S A. 2009;106:22352–7.
Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–13.
Higuchi R, Bowman B, Freiberger M, et al. DNA sequences from the quagga, an extinct member of the horse family. Nature. 1984;312:282–4.
Hill EW, Gu J, Eivers SS, et al. A sequence polymorphism in MSTN predicts sprinting ability and racing stamina in Thoroughbred horses. PLoS One. 2010;5:e8645.
Hofreiter M, Paijmans JL, Goodchild H, et al. The future of ancient DNA: technical advances and conceptual shifts. BioEssays. 2015;37:284–93.
Huang J, Zhao Y, Bai D, et al. Donkey genome and insight into the imprinting of fast karyotype evolution. Sci Rep. 2015;5:14106.
Imsland F, McGowan K, Rubin CJ, et al. Regulatory mutations in TBX3 disrupt asymmetric hair pigmentation that underlies Dun camouflage color in horses. Nat Genet. 2016;48:152–8.
Jansen T, Forster P, Levine MA, et al. Mitochondrial DNA and the origins of the domestic horse. Proc Natl Acad Sci U S A. 2002;99:10905–10.
Johnstone C (2004) A biometric study of equids in the Roman world. PhD. Department of Archaeology, University of York.
Jónsson H, Schubert M, Seguin-Orlando A, et al. Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc Natl Acad Sci U S A. 2014;111:18655–60.
Kelekna P. The horse in human history. Cambridge: Cambridge University Press; 2009.
Keyser C, Hollard C, Gonzalez A, et al. The ancient Yakuts: a population genetic enigma. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370:20130385.
Kimura B, Marshall FB, Chen S, et al. Ancient DNA from Nubian and Somali wild ass provides insights into donkey ancestry and domestication. Proc Biol Sci. 2011;278:50–7.
Langdon J. Horses, oxen and technological innovation: the use of draught animals in English farming from 1066-1500. Cambridge: Cambridge University Press; 2006.
Larson G, Fuller DQ. The evolution of animal domestication. Annu Rev Ecol Evol Syst. 2014;45:115–36.
Lawling AM, Polly PD. Geometric morphometrics: recent applications to the study of evolution and development. J Zool. 2010;280:1–7.
Lindgren G, Backström N, Swinburne J, et al. Limited number of patrilines in horse domestication. Nat Genet. 2004;36:335–6.
Leonard JA, Rohland N, Glaberman S, et al. A rapid loss of stripes: the evolutionary history of the extinct quagga. Biol Lett. 2005;1:291–5.
Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–6.
Librado P, Der Sarkissian C, Ermini L, et al. Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc Natl Acad Sci U S A. 2015;112:E6889–97.
Librado P, Fages A, Gaunitz C, Leonardi M, Wagner S, Khan N, Hanghøj K, Alquraishi SA, Alfarhan AH, Al-Rasheid KA, Der Sarkissian C, Schubert M, Orlando L. The evolutionary origin and genetic makeup of domestic horses. Genetics. 2016;204:423–34.
Librado P, Gamba C, Gaunitz C, et al. Ancient genomic changes associated with domestication of the horse. Science. 2017;356:442–5.
Lippold S, Matzke NJ, Reissmann M, et al. Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication. BMC Evol Biol. 2011a;11:328.
Lippold S, Knapp M, Kuznetsova T, et al. Discovery of lost diversity of paternal horse lineages using ancient DNA. Nat Commun. 2011b;2:450.
Lira J, Linderholm A, Olaria C, et al. Ancient DNA reveals traces of Iberian Neolithic and Bronze Age lineages in modern Iberian horses. Mol Ecol. 2010;19:64–78.
Llamas B, Willerslev E, Orlando L. Human evolution: a tale from ancient genomes. Philos Trans R Soc Lond Ser B Biol Sci. 2017;372.
Lorenzen ED, Nogués-Bravo D, Orlando L, et al. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature. 2011;479:359–64.
Ludwig A, Pruvost M, Reissmann M, et al. Coat color variation at the beginning of horse domestication. Science. 2009;324:485.
Ludwig A, Reissmann M, Benecke N, et al. Twenty-five thousand years of fluctuating selection on leopard complex spotting and congenital night blindness in horses. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370:20130386.
MacFadden BJ. Pleistocene horses from Tarija, Bolivia, and the validity of the genus Onohippidium (Mammalia: Equidae). J Vert Paleontol. 1997;17:199–218.
MacFadden BJ, Carranza-Castaneda O. Cranium of Dinohippus mexicanus (Mammalia Equidae) from the early Pliocene (latest Hemphillian) of central Mexico and the origin of Equus. Bull Florida Mus Nat Hist. 2002;43:163–85.
MacHugh DE, Larson G, Orlando L, et al. Taming the past: ancient DNA and the study of animal domestication. Annu Rev Anim Biosci. 2017;5:329–51.
Mailund T, Halager AE, Westergaard M, et al. A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species. PLoS Genet. 2012;8:e1003125.
Makvandi-Nejad S, Hoffman GE, Allen JJ, et al. Four loci explain 83% of size variation in the horse. PLoS One. 2012;7:e39929.
Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One. 2010;5:e14004.
Mathieson I, Lazaridis I, Rohland N, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503.
McCue ME, Valberg SJ, Miller MB, et al. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91:458–66.
McCue ME, Bannasch DL, Petersen JL, et al. A high density SNP array for the domestic horse and extant Perissodactyla: utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012;8:e1002451.
McGivney BA, McGettigan PA, Browne JA, et al. Characterization of the equine skeletal muscle transcriptome identifies novel functional responses to exercise training. BMC Genomics. 2010;11:398.
McKenzie VJ, Song SJ, Delsuc F, et al. The effects of captivity on the mammalian gut microbiome. Integr Comp Biol. 2017;57:690–704.
Metcalf JL, Song SJ, Morton JT, et al. Evaluating the impact of domestication and captivity on the horse gut microbiome. Sci Rep. 2017;7:15497.
Metzger J, Philipp U, Lopes MS, et al. Analysis of copy number variants by three detection algorithms and their association with body size in horses. BMC Genomics. 2013;14:487.
Meyer M, Kircher M, Gansauge MT, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–6.
Miller W, Drautz DI, Ratan A, et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature. 2008;456:387–90.
Mohandesan E, Speller CF, Peters J, et al. Combined hybridization capture and shotgun sequencing for ancient DNA analysis of extinct wild and domestic dromedary camel. Mol Ecol Resour. 2017;17:300–13.
Nagasawa M, Mitsui S, En S, et al. Oxytocin-gaze positive loop and the coevolution of human-dog bonds. Science. 2015;348:333–6.
Orlando L. Equids. Curr Biol. 2015;25:R973–8.
Orlando L, Eisenmann V, Reynier F, et al. Morphological convergence in Hippidion and Equus (Amerhippus) South American equids elucidated by ancient DNA analysis. J Mol Evol. 2003;57(suppl 1):S29–40.
Orlando L, Mashkour M, Burke A, et al. Geographic distribution of an extinct equid (Equus hydruntinus: Mammalia, Equidae) revealed by morphological and genetical analyses of fossils. Mol Ecol. 2006;15:2083–93.
Orlando L, Metcalf JL, Alberdi MT, et al. Revising the recent evolutionary history of equids using ancient DNA. Proc Natl Acad Sci U S A. 2009;106:21754–9.
Orlando L, Ginolhac A, Raghavan M, et al. True single-molecule DNA sequencing of a pleistocene horse bone. Genome Res. 2011;21:1705–19.
Orlando L, Ginolhac A, Zhang G, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499:74–8.
Orlando L, Gilbert MT, Willerslev E. Reconstructing ancient genomes and epigenomes. Nat Rev Genet. 2015;16:395–408.
Outram AK, Stear NA, Bendrey R, et al. The earliest horse harnessing and milking. Science. 2009;323:1332–5.
Park SD, Magee DA, McGettigan PA, et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015;16:234.
Patterson N, Moorjani P, Luo Y, et al. Ancient admixture in human history. Genetics. 2012;192:1065–93.
Petersen JL, Mickelson JR, Rendahl AK, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9:e1003211.
Pedersen MW, Overballe-Petersen S, Ermini L, et al. Ancient and modern environmental DNA. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370:20130383.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967.
Promerová M, Andersson LS, Juras R, et al. Worldwide frequency distribution of the ‘Gait keeper’ mutation in the DMRT3 gene. Anim Genet. 2014;45:274–82.
Pruvost M, Bellone R, Benecke N, et al. Genotypes of predomestic horses match phenotypes painted in Paleolithic works of cave art. Proc Natl Acad Sci U S A. 2011;108:18626–30.
Ramos-Madrigal J, Smith BD, Moreno-Mayar JV, et al. Genome sequence of a 5,310-year-old maize cob provides insights into the early stages of maize domestication. Curr Biol. 2016;26:3195–201.
Rasmussen M, Li Y, Lindgreen S, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–62.
Rasmussen M, Guo X, Wang Y, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–8.
Rasmussen M, Anzick SL, Waters MR, et al. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature. 2014;506:225–9.
Reich D, Green RE, Kircher M, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–60.
Reissmann M, Bierwolf J, Brockmann GA. Two SNPs in the SILV gene are associated with silver coat colour in ponies. Anim Genet. 2007;38:1–6.
Renaud G, Petersen B, Seguin-Orlando A, Bertelsen MF, Waller A, Newton R, Paillot R, Bryant N, Vaudin M, Librado P, Orlando L. Improved de novo genomic assembly for the domestic donkey. Sci Adv. 2018;4:eaaq0392. https://doi.org/10.1126/sciadv.aaq0392.
Rivero JL, Hill EW. Skeletal muscle adaptations and muscle genomics of performance horses. Vet J. 2016;209:5–13.
Rossel S, Marshall F, Peters J, et al. Domestication of the donkey: timing, processes, and indicators. Proc Natl Acad Sci U S A. 2008;105:3715–20.
Scheu A (2017) Neolithic animal domestication as seen from ancient DNA. Quat Int. https://doi.org/10.1016/j.quaint.2017.02.009.
Schubert M, Jónsson H, Chang D, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci U S A. 2014;111:E5661–9.
Schubert M, Mashkour M, Gaunitz C, et al. Fast, accurate and sensitive pipeline to genetically identify equine F1-hybrids in archaeological assemblages. J Archaeol Sci. 2017;78:147–57.
Steiner CC, Mitelberg A, Tursi R, et al. Molecular phylogeny of extant equids and effects of ancestral polymorphism in resolving species-level phylogenies. Mol Phylogenet Evol. 2012;65:573–81.
Signer-Hasler H, Flury C, Haase B, et al. A genome-wide association study reveals loci influencing height and other conformation traits in horses. PLoS One. 2012;7:e37282.
Skoglund P, Ersmark E, Palkopoulou E. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr Biol. 2015;25:1515–9.
Sommer RS, Benecke L, Lougas O, et al. Holocene survival of the wild horse in Europe: a matter of open landscape? J Quat Sci. 2011;26:1099–417.
Stoneking M, Krause J. Learning about human population history from ancient and modern genomes. Nat Rev Genet. 2011;12:603–14.
Stuart AJ. Late quaternary megafaunal extinctions on the continents. Geol J. 2015;50:338–63.
Tozaki T, Miyake T, Kakoi H, et al. A genome-wide association study for racing performances in Thoroughbreds clarifies a candidate region near the MSTN gene. Anim Genet. 2010;41(suppl 2):28–35.
Vilà C, Leonard JA, Gotherstrom A, et al. Widespread origins of domestic horse lineages. Science. 2001;291:474–7.
Vigne FD, Helmer D, Peters J. First steps of animal domestication: new archaeozoological approaches. Oxford: Oxbow Books; 2005.
Vilstrup JT, Seguin-Orlando A, Stiller M, et al. Mitochondrial phylogenomics of modern and ancient equids. PLoS One. 2013;8:e55950.
Wade CM, Giulotto E, Sigurdsson S, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–7.
Wakefield S, Knowles J, Zimmermann W, et al. Status and action plan for the Przewalski’s horse (Equus ferus przewalskii). In: Moehlman P, editor. Equids: zebras, asses and horses, vol. 2002. Cambridge: IUNC/SSC Equid Specialist Group, IUCN Publications Services Unit; 2012. p. 82–92.
Warmuth V, Eriksson A, Bower MA, et al. European domestic horses originated in two holocene refugia. PLoS One. 2011;6:e18194.
Warmuth V, Eriksson A, Bower MA, et al. Reconstructing the origin and spread of horse domestication in the Eurasian steppe. Proc Natl Acad Sci U S A. 2012;109:8202–6.
Warinner C, Speller C, Collins M. A new era in palaeomicrobiology: prospects for ancient dental calculus as a long-term record of the human oral microbiome. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370:20130376.
Weinstock J, Willerslev E, Sher A, et al. Evolution, systematics, and phylogeography of pleistocene horses in the new world: a molecular perspective. PLoS Biol. 2005;3:e241.
Wilkins AS, Wrangham RW, Fitch WT. The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics. 2014;197:795–808.
Willerslev E, Davison J, Moora M, et al. Fifty thousand years of Arctic vegetation and megafaunal diet. Nature. 2014;506:47–51.
Wallner B, Vogl C, Shukla P, et al. Identification of genetic variation on the horse y chromosome and the tracing of male founder lineages in modern breeds. PLoS One. 2013;8:e60015.
Wutke S, Benecke N, Sandoval-Castellanos E, et al. Spotted phenotypes in horses lost attractiveness in the Middle Ages. Sci Rep. 2016a;6:38548.
Wutke S, Andersson L, Benecke N, et al. The origin of ambling horses. Curr Biol. 2016b;26:R697–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Orlando, L. (2018). An Ancient DNA Perspective on Horse Evolution. In: Lindqvist, C., Rajora, O. (eds) Paleogenomics. Population Genomics. Springer, Cham. https://doi.org/10.1007/13836_2018_23
Download citation
DOI: https://doi.org/10.1007/13836_2018_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-04752-8
Online ISBN: 978-3-030-04753-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)