
Abstract
Although the cellulose crystallinity index (CI) is used widely, its limitations have not been adequately described. In this study, the CI values of a set of reference samples were determined from X-ray diffraction (XRD), nuclear magnetic resonance (NMR), and infrared (IR), Raman, and vibrational sum frequency generation (SFG) spectroscopies. The intensities of certain crystalline peaks in IR, Raman, and SFG spectra positively correlated with the amount of crystalline cellulose in the sample, but the correlation with XRD was nonlinear as a result of fundamental differences in detection sensitivity to crystalline cellulose and improper baseline corrections for amorphous contributions. It is demonstrated that the intensity and shape of the XRD signal is affected by both the amount of crystalline cellulose and crystal size, which makes XRD analysis complicated. It is clear that the methods investigated show the same qualitative trends for samples, but the absolute CI values differ depending on the determination method. This clearly indicates that the CI, as estimated by different methods, is not an absolute value and that for a given set of samples the CI values can be compared only as a qualitative measure.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- X-ray diffraction
- Sum frequency generation spectroscopy
- Infrared spectroscopy
- Raman spectroscopy
- Nuclear magnetic resonance
- Crystallinity index
- Wood pulp
1 Introduction
Cellulose is the most abundant natural polymer on the planet, and constitutes lignocellulosic biomass along with hemicelluloses and lignin [1]. Cellulose is produced from glucose molecules by photosynthesizing plants such as trees, grasses, and algae as well as by bacteria, fungi, and tunicates (i.e., sea squirts) [2]. Cellulose is a linear macromolecule of repeating 1-4-linked β-d-glucopyranose units. In plants, individual cellulose polymer chains are produced by protein complexes in the plasma membrane and aggregate into microfibrils [3–5]. Hydrogen bonding between and within cellulose chains in cellulose microfibrils leads to formation of ordered crystal structures [6–9]. The crystal structure of cellulose plays a key role in the mechanical strength of plant cell walls as well as deconstruction of lignocellulose biomass [10–14].
Naturally occurring cellulose exists as two polymorphs, cellulose Iα and Iβ. Algae and bacteria primarily produce cellulose Iα, whereas land plants tend to produce mostly cellulose Iβ [15, 16]. Cellulose can be irreversibly converted into cellulose Iβ by heat treatment [17]. Cellulose II is produced via mercerization with a strong alkaline solution or precipitation from a dissolved state [18, 19]. Cellulose I and II can be converted to cellulose III1 and III2, respectively, by treatment with dry liquid ammonia [2]. It has been also claimed that cellulose IV1 and IV2 can be produced simply by heating cellulose III1 or III2, but the products could be a form of disordered cellulose Iβ [20]. These polymorphs of cellulose differ in the size of the unit cell, number of chains included in the crystal unit cell, and orientation of the chains in the crystal (parallel or antiparallel).
Several techniques have been used to measure the amount of crystalline structure and the polymorph type in cellulose samples. Although X-ray diffraction (XRD) is the most widely used technique, infrared spectroscopy (IR), Raman spectroscopy, nuclear magnetic resonance (NMR), and vibrational sum frequency generation (SFG) spectroscopy are also used to investigate the crystal structure of cellulose [21–24]. Because XRD measures coherent scattering from crystalline lattices, it is conceptually easy to understand. However, there are several issues concerned with the practical application of XRD to biomass [25]. In fact, XRD analysis of whole biomass can be challenging because of interference from incoherent X-ray scattering of the amorphous phases, which include hemicelluloses and lignin. Several data processing methods have been proposed and used to extract values for the amount of crystalline cellulose from XRD data of biomass [26]; however, these methods have some limitations that are often neglected in data processing or interpretation. These issues are discussed in this study.
There are several peaks in NMR, IR, and Raman spectra that are characteristic of crystalline cellulose. Cellulose and hemicelluloses share the same organic functional groups (C–C, C–H, C–O–C, and C–O–H); the main differences between them are the monomeric constituents and regioselectivity of glycosidic bonds, which eventually govern the packing of these carbohydrate polymer chains. Structural constraints (such as bond distances and bond angles) imposed by specific polymer chain packing can cause certain peaks in NMR, IR, and Raman spectra to be different for crystalline and amorphous structures [24]. Although many studies have shown that crystalline cellulose can be distinguished from other components in biomass using NMR, IR, and Raman spectroscopy, quantitative analysis is still challenging because of subtle differences between the crystalline and amorphous phases.
Recently, vibrational SFG spectroscopy has been demonstrated to selectively detect crystalline cellulose dispersed in amorphous phases [27, 28]. SFG is a nonlinear optical process for the study of a noncentrosymmetric optical medium. Crystalline cellulose has noncentrosymmetry over several orders of length scales. At the molecular scale, all carbon centers (C1, C2, C3, C4, C5) in the glucopyranose ring are chiral. The symmetry of the crystal unit cells are also noncentrosymmetric (space groups P1 and P21) [9, 23]. The glucan chains in cellulose Iα and Iβ are arranged in parallel fashion, whereas those of cellulose II are arranged in antiparallel fashion [29]. The noncentrosymmetric crystals can be distributed randomly, centrosymmetrically (for example, antiparallel packing), or noncentrosymmetrically (parallel packing) within the amorphous matrices. Thus, the dispersion pattern of cellulose crystals over the characteristic length of the SFG process (so-called coherence length) can also affect the SFG spectra [30]. Although individual chiral centers could generate weak SFG signals, in principle their signals are generally much weaker or negligible compared with SFG signals from noncentrosymmetric crystals. Thus, SFG can selectively detect multiscale structural aspects (such as crystal structure and mesoscale packing) of crystalline cellulose dispersed in a whole biomass sample without interference from amorphous components [24, 29–33]. This selective detection of crystalline cellulose in its native state is very important for a deep understanding of the role of crystalline cellulose in cell wall properties, both mechanically and chemically. If the packing of crystalline cellulose in biomass remains relatively constant, the SFG signal intensity can be used to estimate crystallinity [28]. However, this becomes challenging if the crystal packing pattern changes drastically between samples [30].
In this paper, we compare the apparent crystallinity index (CI) obtained from several structural characterization techniques. In the literature, the CI values obtained from different techniques and/or samples have been compared in a quantitative manner, and in some cases the results have been taken as “accurate” or “absolute.” We propose that the CI value should be taken or considered only as a qualitative index to demonstrate a trend between samples, and not as an absolute quantity. The main purpose of this paper is to explain how the CI is obtained by different methods and how different the results can be (even for a given technique when the CI is calculated by different methods). We also attempt to draw a correlation between the different methods. The data presented in this paper provide an insight into the usefulness and limitations of CI estimation.
2 Experimental
2.1 Sample Preparation
Several types of cellulose were used in this study. Cotton linter (Justfiber C10CL FCC) was kindly provided by the International Fiber Corporation; Avicel PH-101 and α-cellulose were purchased from Sigma-Aldrich. Fully bleached hardwood and softwood pulps were obtained from a mill in the southeastern United States. These pulps contained 20.0% and 16.3% noncellulosic carbohydrates, respectively.
Decrystallized samples were prepared using a modified version of the procedure described by Schroeder et al. [12, 34]. Whatman filter paper 1 was used in this study as a reference, highly crystalline, and purified sample. It was first soaked in water and the fibers were dispersed under magnetic stirring overnight. The water was then removed by vacuum filtration over a glass filter. The mat of fibers was removed and re-suspended in a beaker filled with acetone. This dispersion was filtered using vacuum filtration. After the third rinsing with acetone, the fiber mat was suspended in dimethyl sulfoxide (DMSO). Following the same procedure, the DMSO was removed via vacuum and re-suspended three times. The purpose of this solvent exchange was to swell the cellulose fibers and make them more accessible to reaction with formaldehyde. The cellulose and DMSO mixture was then heated to 125°C and paraformaldehyde added. As a result of the reaction, methylol-cellulose was formed, which is soluble in DMSO, and the system became clear. The methylol-cellulose and DMSO solution was filtered through a glass crucible. Amorphous cellulose was then precipitated in a stirred bath with 0.2 M sodium methoxide in methanol and propanol (1:1). After precipitation, the fibers were washed thoroughly with methanol, 0.1 M hydrochloric acid, and de-ionized water, and then freeze-dried. Partially de-crystallized samples were obtained in a similar manner but not applying solvent exchange so that the cellulose chains only partially dissolved. In addition, no filtration was used so that these chains were preserved. The same procedure was followed to precipitate and wash the dissolved portion.
2.2 X-Ray Diffraction
Freeze-dried cellulose samples were placed on a low-background quartz holder and measured using a Rigaku SmartLab X-ray diffractometer with a Cu tube (λ = 1.5405 Å). The radiation was generated at 25 mA and 35 kV. A step size of 0.05° and 5 s exposure were used for measuring the scattering angle 2θ in the range 9–41°. The CI was determined by two methods, the peak height (PH) and amorphous subtraction (AS) methods. The PH method, also known as Segal’s method, measures the intensity of the crystalline peak (I 200–I AM) and the total intensity (I 200) after the background spectrum is removed. The ratio of the intensities of these two peaks gives an estimate of the crystallinity of the cellulose sample [35]. The AS method requires the amorphous spectrum to be subtracted from the sample spectrum [36]. This method is challenging because a comparable amorphous sample should be used as a background, and most processes to obtain such a sample (e.g., ball-milling) produce significant modifications. The amorphous background spectrum used here was prepared by utilizing an amorphous cellulose sample from the same given starting material. A scaling factor was used to shift the amorphous spectrum to fit under the sample spectra. The CI was then taken as the area of the crystalline portion (total area minus amorphous background area) divided by the total area.
2.3 X-Ray Diffraction Simulation
To investigate the effect of cellulose crystal size on the CI value using the AS method, simulated diffraction patterns were created from the .cif file obtained for cellulose Iβ [9] using the Mercury program v3.3 [37], which is similar to that used in a previous report [38]. Several powder diffraction patterns were obtained by varying the full width at half maximum values. The CI values for each simulated spectra were found by applying the AS method in a manner similar to that used for the experimental spectra described earlier.
2.4 Nuclear Magnetic Resonance Spectroscopy
High-resolution solid-state 13C NMR spectra were collected at 9.4 T with cross-polarization and magic angle spinning (CP/MAS) in a Bruker Avance 400 MHz spectrometer. 1H and 13C fields were matched at 53.6 kHz, and a 1 dB ramp was applied to the proton rotating-frame during the matching period. Acquisition time was 0.034 s and sweep-width was 30 kHz. Magic-angle spinning was performed at 7,000 Hz. The average was calculated for 3,000 scans using a 2 ms contact time and a delay time of 4.0 s. Crystalline cellulose peaks can be found in the literature [39]. The CI was determined by dividing the total area of crystalline peaks by the area under C4 carbon peaks.
2.5 Fourier-Transform Infrared and Raman Spectroscopies
Fourier-transform infrared (FT-IR) spectroscopy measurements were performed using a Nicolet 8700 FT-IR Spectrometer (Thermo Scientific) with a deuterated triglycine sulfate (DTGS) detector. Data were collected in the region 650–4,000 cm−1 with a 4 cm−1 resolution, averaged over 100 scans, in reflection mode using a smart iTR diamond plate unit (Thermo Scientific). Each spectrum was baseline-corrected and normalized to the maximum absorbance at 1,030 cm−1. Similar to previous studies, the FT-IR relative crystallinity was calculated using the ratio of the absorption bands at 1,315 and 1,369 cm−1 from the filter paper reference samples [40, 41]. The absorbance of each band was measured relative to the valley value at approximately 1,348 cm−1. These two bands were chosen because the 1,315 cm−1 peak intensity varies substantially in the reference samples and is located close to the 1,369 cm−1 peak, which is used as an internal standard for crystallinity calculations [41]. FT-Raman spectroscopy measurements were taken using a Nicolet NXR Spectrometer (Thermo Scientific) using a 1,064 nm excitation source with a beam power of 1–1.8 W and a germanium detector. Data were collected in the region 250–3,800 cm−1 with an 8 cm−1 resolution, averaged over 1,000 scans. Each spectrum was baseline-corrected and normalized to the maximum absorbance at 2,894 cm−1. Relative crystallinity from the Raman spectra was calculated using the ratio of the absorbance bands at 380 and 1,096 cm−1 [42]. The 380 and 1,096 cm−1 peak intensities were measured relative to the minima at 356 and 942 cm−1, respectively.
2.6 Vibrational Sum Frequency Generation Spectroscopy
SFG spectroscopic measurements were carried out by irradiating the sample with picosecond laser pulses in the IR and visible wavelengths and detecting the emitted SFG signals. The SFG spectrometer (EKSPLA) was pumped by a picosecond Nd:YAG laser (1,064 nm at 10Hz). The infrared light was generated with an optical parameter generator/amplifier (OPG/OPA) pumped with 532 and 1064 nm and tuned to 2.3–10 μm with <6 cm−1 bandwidth. The polarization of incident IR and visible laser pulses were parallel (p-polarized) and perpendicular (s-polarized) to the plane of laser incidence. The emitted SFG signal (s-polarized) was detected in the reflection geometry [27]. The visible (60° to surface normal) and IR (56°) laser pulses were overlapped spatially and temporally on each sample. A beam collimator was used to enhance the signal collection efficiency and the SFG signal was filtered through a monochromator and detected with a photomultiplier tube (Hamamatsu Corp.). The SFG intensity was normalized to incident IR and visible laser intensities. SFG spectra were taken at 4 cm−1 per step in the CH stretching region (2,700–3,050 cm−1) and 8 cm−1 per step in the OH stretching region (3,096–3,800 cm−1). Slight variations in SFG intensity occurred at different locations on the sample pellet, depending on the texture and packing density of the cellulose during sample preparation. Therefore, SFG intensity at the 2,944 and 3,320 cm−1 peaks were monitored at multiple locations (n = 3–8) on the pellet so that each full-scan spectra was representative of the average SFG intensity at the given locations. No baseline corrections or normalization was performed. The relative crystallinity was calculated by measuring the peak intensity at 2,944 cm−1 relative to the minimum at 3,132 cm−1 [28].
3 Results and Discussion
3.1 Correlation of “Apparent” Crystallinity Estimated from XRD, IR, Raman, and SFG Analyses of Reference Samples
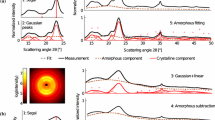
A set of reference samples with three different crystallinities was prepared as described in the “Sample Preparation” section. We used Whatman filter paper because it is readily and widely available and can be taken as a reference for highly crystalline (as-received filter paper), partially crystalline (prepared by adjusting the decrystallization process), and amorphous cellulose (precipitated from solution) [12, 34]. These samples were analyzed using XRD, IR, Raman, and SFG, as shown in Fig. 1. The filter paper samples were not suitable for 13C solid-state NMR analysis, and thus comparison of CI values obtained using this technique was not possible.

Characterization of cellulose reference samples using (a) XRD, (b) Raman, (c) IR, and (d) SFG. Reference samples were prepared from filter paper and are labeled as untreated (black), partially decrystallized (red) and amorphous (blue). All spectra are offset for clarity. Insets in (b) and (c) show the regions used for CI calculations
There are many different methods for calculation of the CI from XRD as shown in Fig. 1a [43, 44]. Of the various methods, the most widely used is the peak height (PH) method originally proposed by Segal et al. as a “time-saving empirical measure of relative crystallinity” [35]. This method assumes the background intensity at 2θ = ~18° to be an amorphous contribution and the peak height at 2θ = 22.8° to result from both crystalline and amorpous contributions. In order to quantify and provide a more realistic value, alternative methods such as curve fitting or amorphous subtraction (AS) have been suggested [26]. Depending on the method used to analyze XRD data, the CI value can vary drastically [28]. Note that most of the methods incorrectly assume that X-ray sensitivity to crystalline and amorphous phases is the same. The Rietveld fitting of XRD data can resolve these uncertainties in the CI calculation [45]. Because these advanced methods are not easily amenable to non-XRD experts, a more user-friendly method has been developed [46].
Figure 1b compares the Raman spectra of the same samples. The 1,480 and 380 cm−1 peaks are known to be characteristic of crystalline cellulose [42, 47]. The former is assigned to the CH2 bending mode of the exocyclic CH2OH side chain and the latter is speculated to originate from one of the torsion or bending modes of the six-membered ring with respect to the glycosidic bonds [24]. In the case of the 1,480 cm−1 peak, the peak deconvolution from the amorphous counterpart (~1,460 cm−1) can be troublesome for calculation of CI. After proper background correction, good correlation between the 380 cm−1 intensity and the XRD CI value has been demonstrated [42]. Hemicelluloses do not show a peak at 380 cm−1, whereas they can show broad peaks in the CH2 bending vibration region. Thus, use of the 380 cm−1 Raman peak intensity is advantageous for CI calculation [48].
Figure 1c displays the IR spectra of the same samples. There are a number of small peaks that vary with the degree of crystallinity. Noticeable peaks are found at 706, 1,056, 1,110, and 1,315 cm−1. The intensities of these peaks are reported to vary in accordance with the XRD CI value [41]. Although the 1,056 and 1,110 cm−1 peaks are sharp, they overap with broad and strong background peaks in the C–C and C–O stretch vibration region. For that reason, the peaks at 706 and 1,315 cm−1 might work better for CI calculation. The peaks at 1,640 cm−1 become larger as the degree of crystallinity decreases. This is a result of the ingress of water into the amorphous cellulose region. For the same reason, the OH stretch peaks (3,000–3,600 cm−1) become broader for the partially and fully decrystallized samples. When applied directly to lignocellulose biomass, the IR method can suffer from peak overlap from noncellulosic components.
Figure 1d exhibits the SFG spectra of the same samples. Although there are many peaks characteristic for crystalline cellulose in the lower wavenumber region, their peak assignment is not straightforward because of substantial coupling between various vibrational modes [31]. The C–H and O–H stretching vibration region alone can provide rich information about the polymorphism (Iα, Iβ, II, III) and mesoscale packing pattern of celluose microfibrils [29, 30]. A strong SFG peak at 2944 cm−1 and a weaker peak at 3,320 cm−1 are characteristic features of antiparallel-packed cellulose Iβ microfibrils [30]. The intensities of these peaks decrease in a nonlinear fashion as the crystalline cellulose portion in the sample decreases [28]. The additional peak at 2,968 cm−1 for the partially decrystallized sample might be a result of some changes in the cellulose crystal packing [30], which could have occurred during the partial decrystallization process involving incomplete dissolution of cellulose crystals. For the fully decrystallized sample (precipitated from fully dissolved cellulose chains), the OH stretch peak is completely missing and the CH peak is shifted to 2,960 cm−1 and is weaker. This implies that a small amount of cellulose II is formed during the precipitation process, because amorphous cellulose chains do not produce measureable SFG signals [29].
Figure 2 presents the correlations between CI values calculated from the data shown in Fig. 1. The XRD data were processed using the PH and AS methods. For the AS method, the XRD data of the fully depolymerized sample (blue curve in Fig. 1a) was used as a reference for CI = 0%. Note that the PH method always gives higher CI values than the AS method. This is an artifact and limitation of the PH method caused by simply reading the height at 2θ = 22° as total diffraction, even though the amorphous phase alters the baseline. For the fully depolymerized sample, the PH method gives 34% CI whereas the AS method gives 0% CI (simply because it is the reference for CI = 0%). The difference between the two methods becomes smaller for samples of higher crystallinity (CIPH = 94% versus CIAS = 78% for the as-received filter paper). It should be noted that both methods ignore the fact that XRD sensitivity to the amorphous background and crystalline peaks are different.

Correlation between crystalline peak intensities in (a) Raman, (b) IR, and (c) SFG vibration spectroscopy of reference samples with respect to XRD crystallinities calculated using the peak height method (circles) and amorphous subtraction method (triangles)
Because there are peaks characteristic for crystalline cellulose in IR, Raman, and SFG spectra, the intensities of these peaks can be correlated with the absolute amount of crystalline cellulose in the sample. However, it is difficult to know the absolute value unless the sample is prepared by mixing known amounts of fully crystalline and fully amorphous cellulose [28]. Thus, we simply compared their intensities with the XRD CI values. Figure 2 shows nonlinear but reasonably good correlation between the CI values obtained using the different techniques. In the case of IR and Raman data, the crystalline peaks appear above the nonzero background or sometimes overlap with the amorphous peaks. Thus, proper background subtraction and intensity normalization using an internal reference (the peak that is not sensitive to crystallinity) is necessary [41, 42]. The nonlinearity of the correlations in Fig. 2 could be a result of improper baseline correction or nonlinearity of the relationship between XRD CI and the true crystallinity of the sample [28]. In the case of SFG, the background from amorphous cellulose is typically zero; thus, the absolute intensity can be used for CI calculations. However, because of the nonlinear nature of the SFG process and scattering of the incident and emitted light from rough samples, as well as birefringence of crystalline cellulose, it is difficult to obtain theoretical predictions of SFG intensity [28].
3.2 Simulation of X-Ray Diffraction Data for Cellulose Crystals of Different Sizes
In plant cell walls, cellulose microfibrils are synthesized by cellulose synthase complexes (CSCs). The CSC consists of a rosette of six subunits, and each subunit contains putatively six cellulose synthesis proteins. If all units are active in cellulose synthesis, then cellulose microfibrils would contain 36 chains [49]. However, some models assumed microfibrils with 18 chains [6]. The cross-sectional shape of a cellulose microfibril and the exact number of cellulose chains it contains are difficult to determine [50]. Based on the average cross-section of a glucose chain, the diameter of a microfibril containing 36 chains is estimated to be ~3.8 nm (Fig. 3a). The exact diameter is dependent on an assumption of the shape of the microfibril cross-section. As the number of chains in the crystalline microfibrils varies, the size of the coherent lattice that produce XRD peaks also varies. In plant cell walls, several individual microfibrils have been observed to bundle into larger aggregates called macrofibrils [51]. Some algae and tunicates produce much thicker cellulose crystals (up to 15–20 nm in diameter).

(a) Estimation of the diameter (d) of a cellulose microfibril with circular cross-section (A) as a function of the number of chains in the microfibril. (b) Simulated XRD diffractograms using the Mercury program at different peak input values of full width at half maximum (FWHM). Inset in (b) shows the calculated CI value from the amorphous subraction (AS) method versus FWHM value
In XRD, the crystal size affects the width of the diffraction peak. The mathematical relationship between the peak width (full width at half maximum, FWHM) of the diffraction peaks and the crystal size (d) is known as the Scherrer equation [52]:
where k is the proportionality constant, λ is the X-ray wavelength, and θ is the Bragg diffraction angle. Note that the proportionality constant k has been derived to be about 0.9, assuming Gaussian line profiles of XRD and small perfect cubic crystals of uniform size without any distortion of the lattice [53]. This value is widely used for estimation of cellulose crystal size because it is not sensitive to crystallite shape and symmetry.
In the literature, it is often assumed that the CI and crystal size are independent of each other and are calculated as separate quantities. However, these two terms may not be totally independent but are coupled. In order to demonstrate their dependency, XRD diffractograms were simulated using the Mercury software for the unit cell of cellulose Iβ crystal with different FWHM settings (Fig. 3b) [38]. Thus, this data could be considered as 100% crystalline samples with different crystal sizes. In Fig. 3b, the background region close to the assumed amorphous phase rises as FWHM increases. The inset of Fig. 3b shows the CI values calculated for the data in Fig. 3b using the AS method. The CI value decreases in proportion to the FWHM value. This simulation clearly shows that, as cellulose microfibril diameter decreases, the calculated CI values also decrease although the true crystallinity does not change. For a 36-chain cellulose microfibril, the Scherrer equation predicts the FWHM to be around 2.3°, which sets the maximum CI value calculated from the AS method at ~78%. Should 78% CI from AS analysis be taken to correspond to a 100% crystalline sample? To consider the aggregation of individual cellulose microfibrils in plant cell walls, pristine samples of high uniformity are needed [54]. More in-depth analysis with independent experimental design is needed to address this question.
3.3 Comparison of “Apparent” Crystallinity Estimated from XRD, NMR, IR, Raman, and SFG of Isolated and Natural Cellulose Samples
Five different samples containing cellulose were analyzed using XRD, NMR, IR, Raman, and SFG. Cotton linter is one of the purest native (biologically intact) sources of cellulose and contains the highest amount of cellulose (>95% by dry mass). Avicel and α-cellulose are commercially available purified celluloses. Avicel is produced via acid hydrolysis of pulp or cotton linter; thus, the degree of polymerization of cellulose in Avicel is low. α-cellulose is produced via base-catalyzed hydrolysis of wood pulp. Thus, commercial α-cellulose contains mostly cellulose Iβ, which should not be confused with cellulose Iα. Bleached hardwood (HW) and softwood (SW) pulps were included in this analysis for comparison with commercial cellulose samples.
Figure 4 and Table 1 compare the XRD, NMR, IR, Raman, and SFG intensity data of the five samples tested. The CI values from XRD were calculated using both PH and AS methods. For the IR, Raman, and SFG data, the signal intensities were converted to values equivalent to the XRD CI values using the calibration curves shown in Fig. 2. In 13C ss-NMR, the C4 peak chemical shifts of amorphous and crystalline cellulose phases were 80–85 ppm and 85–89 ppm, respectively. Thus, their relative intensity can be taken as the CI. The processed CI values are plotted in Table 1. It is clear that all methods show the same qualitative trends for samples (e.g., CI is the highest for cotton linter and lowest for α-cellulose) but that the absolute values are different, depending on the method. This indicates that the CI values estimated using different methods are not “absolute” and should not be compared directly. The CI values obtained for different samples using one method can be compared only qualitatively.

Characterization of isolated and natural cellulose samples using (a) XRD, (b) 13C NMR, (c) Raman, (d) IR, and (e) SFG. Samples are cotton linter (black), Avicel (red), α-cellulose (green), hardwood (HW) pulp (blue), and softwood (SW) pulp (orange). In (a) the amorphous standard is plotted as a dotted line. Insets in (c) and (d) show the spectral regions used for CI calculations. All spectra are offset for clarity
There are a few details that deserve further discussion. First, NMR CI values are much lower than the values calculated from XRD data, even those determined by the AS method. As pointed out earlier, XRD is sensitive to the crystalline phase, but less sensitive to the amorphous phase. Thus, the XRD analysis inevitably discriminates between amorphous and crystalline phases. In contrast, NMR is equally sensitive to both crystalline and amorphous phases. This fundamental difference in detection sensitivity of these two methods might be responsible for the discrepancy between the CI values determined using these methods.
Second, XRD CI values calculated for hardwood and softwood fiber samples with the AS method are high, close to those calculated using the PH method. The source for this high value can be seen in Fig. 4a, which shows the amorphous background portion determined using the AS method. The reference diffractogram used as the amorphous background (blue curve in Fig. 1a) has low intensities at 2θ above 25°, but the hardwood and softwood fiber samples show high background in this region (Fig. 4a). Thus, the AS method cannot properly subtract the background portion in this high 2θ region, adding this difference into the crystalline portion. Figure 3b shows that the background intensities in the 2θ > 25° and 2θ = 18° regions rise as the FWHM increases. However, the reference sample for the amorphous phase demonstrates negligible intensities at 2θ > 25°. This makes the CI values calculated using the AS method for the fiber samples much higher than the true mass fraction of crystalline cellulose over the total mass.
4 Conclusions
The apparent CI values of cellulose and biomass samples were evaluated using XRD, solid-state NMR, IR, Raman, and SFG. For a set of filter paper samples, the CI calculated from IR, Raman, and SFG spectra were compared with values from XRD and a nonlinear, positive correlation was observed. The size of cellulose crystals is a factor that can affect the XRD CI value. When different types of cellulose were used, it was clear that all characterization methods show similar trends; but the absolute values are quantitatively different. This indicates that the CI value determined using a given method can be used only as a qualitative and relative measurement.
References
Pauly M, Keegstra K (2008) Cell-wall carbohydrates and their modification as a resource for biofuels. Plant J 54(4):559–568
Zugenmaier P (2008) Crystalline cellulose and cellulose derivatives: characterization and structures. Springer, Berlin
Cosgrove DJ (2005) Growth of the plant cell wall. Nat Rev Mol Cell Biol 6(11):850–861
Paredez AR, Somerville CR, Ehrhardt DW (2006) Visualization of cellulose synthase demonstrates functional association with microtubules. Science 312(5779):1491–1495
Somerville C, Bauer S, Brininstool G, Facette M, Hamann T, Milne J, Osborne E, Paredez A, Persson S, Raab T, Vorwerk S, Youngs H (2004) Toward a systems approach to understanding plant cell walls. Science 306(5705):2206–2211
Thomas LH, Forsyth VT, Sturcova A, Kennedy CJ, May RP, Altaner CM, Apperley DC, Wess TJ, Jarvis MC (2013) Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Physiol 161(1):465–476
Doblin MS, Kurek I, Jacob-Wilk D, Delmer DP (2002) Cellulose biosynthesis in plants: from genes to rosettes. Plant Cell Physiol 43(12):1407–1420
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci U S A 108(47):E1195–E1203
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose 1 beta from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124(31):9074–9082
Himmel ME, Ding S-Y, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315(5813):804–807
Pérez S, Samain D (2010) Structure and engineering of celluloses. In: Derek H (ed) Advances in carbohydrate chemistry and biochemistry, vol 64. Academic, New York, pp 25–116
Park S, Johnson DK, Ishizawa CI, Parilla PA, Davis MF (2009) Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose 16(4):641–647
Carroll A, Somerville C (2009) Cellulosic biofuels. Annu Rev Plant Biol 60:165–182
Mittal A, Katahira R, Himmel ME, Johnson DK (2011) Effects of alkaline or liquid-ammonia treatment on crystalline cellulose: changes in crystalline structure and effects on enzymatic digestibility. Biotechnol Biofuels 4:41
Atalla RH, Vanderhart DL (1984) Native cellulose – a composite of 2 distinct crystalline forms. Science 223(4633):283–285
Jarvis M (2003) Cellulose stacks up. Nature 426(6967):611–612
Horii F, Yamamoto H, Kitamaru R, Tanahashi M, Higuchi T (1987) Transformation of native cellulose crystals induced by saturated steam at high-temperatures. Macromolecules 20(11):2946–2949
Langan P, Nishiyama Y, Chanzy H (2001) X-ray structure of mercerized cellulose II at 1 angstrom resolution. Biomacromolecules 2(2):410–416
Ruan D, Zhang LN, Zhou JP, Jin HM, Chen H (2004) Structure and properties of novel fibers spun from cellulose in NaOH/thiourea aqueous solution. Macromol Biosci 4(12):1105–1112
Wada M, Heux L, Sugiyama J (2004) Polymorphism of cellulose I family: reinvestigation of cellulose IVI. Biomacromolecules 5(4):1385–1391
Atalla RH, VanderHart DL (1999) The role of solid state C-13 NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl Magn Reson 15(1):1–19
Sugiyama J, Persson J, Chanzy H (1991) Combined infrared and electron diffraction study of the polymorphism of native celluloses. Macromolecules 24(9):2461–2466
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose Iα from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125(47):14300–14306
Kim SH, Lee CM, Kafle K (2013) Characterization of crystalline cellulose in biomass: basic principles, applications, and limitations of XRD, NMR, IR, Raman, and SFG. Korean J Chem Eng 30(12):2127–2141
Ruland W (1961) X-ray determination of crystallinity and diffuse disorder scattering. Acta Crystallogr 14(11):1180–1185
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10
Barnette AL, Bradley LC, Veres BD, Schreiner EP, Park YB, Park J, Park S, Kim SH (2011) Selective detection of crystalline cellulose in plant cell walls with sum-frequency-generation (SFG) vibration spectroscopy. Biomacromolecules 12(7):2434–2439
Barnette AL, Lee C, Bradley LC, Schreiner EP, Park YB, Shin H, Cosgrove DJ, Park S, Kim SH (2012) Quantification of crystalline cellulose in lignocellulosic biomass using sum frequency generation (SFG) vibration spectroscopy and comparison with other analytical methods. Carbohydr Polym 89(3):802–809
Lee CM, Mittal A, Barnette AL, Kafle K, Park YB, Shin H, Johnson DK, Park S, Kim SH (2013) Cellulose polymorphism study with sum-frequency-generation (SFG) vibration spectroscopy: identification of exocyclic CH2OH conformation and chain orientation. Cellulose 20(3):991–1000
Lee CM, Kafle K, Park YB, Kim SH (2014) Probing crystal structure and mesoscale assembly of cellulose microfibrils in plant cell walls, tunicate tests, and bacterial films using vibrational sum frequency generation (SFG) spectroscopy. Phys Chem Chem Phys 16(22):10844–10853
Lee CM, Mohamed NM, Watts HD, Kubicki JD, Kim SH (2013) Sum-frequency-generation vibration spectroscopy and density functional theory calculations with dispersion corrections (DFT-D2) for cellulose Iα and Iβ. J Phys Chem B 117(22):6681–6692
Park YB, Lee CM, Koo B-W, Park S, Cosgrove DJ, Kim SH (2013) Monitoring meso-scale ordering of cellulose in intact plant cell walls using sum frequency generation spectroscopy. Plant Physiol 163(2):907–913
Kafle K, Xi X, Lee CM, Tittmann BR, Cosgrove DJ, Park YB, Kim SH (2014) Cellulose microfibril orientation in onion (Allium cepa L.) epidermis studied by atomic force microscopy (AFM) and vibrational sum frequency generation (SFG) spectroscopy. Cellulose 21:1075–1086
Schroeder LR, Gentile VM, Atalla RH (1986) Nondegradative preparation of amorphous cellulose. J Wood Chem Technol 6(1):1–14
Segal L, Creely J, Martin A, Conrad C (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29(10):786–794
Thygesen A, Oddershede J, Lilholt H, Thomsen AB, Stahl K (2005) On the determination of crystallinity and cellulose content in plant fibers. Cellulose 12:563–576
Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, Streek JV, Wood PA (2008) Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J Appl Crystallogr 41(2):466–470
French AD, Cintrón MS (2013) Cellulose polymorphy, crystallite size, and the segal crystallinity index. Cellulose 20(1):583–588
Atalla RH (1999) Individual structures of native celluloses. In: Proceedings 10th international symposium on wood and pulping chemistry: main symposium, 7–10 June 1999, Yokohama, Japan. Tappi Press, Atlanta, pp 608–614
Nelson ML, O'Connor RT (1964) Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part II. A new infrared ratio for estimation of crystallinity in celluloses I and II. J Appl Polym Sci 8(3):1325–1341
Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, Park WH, Youk JH (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res 340(15):2376–2391
Agarwal UP, Reiner RS, Ralph SA (2010) Cellulose I crystallinity determination using FT–Raman spectroscopy: univariate and multivariate methods. Cellulose 17(4):721–733
Bansal P, Hall M, Realff MJ, Lee JH, Bommarius AS (2010) Multivariate statistical analysis of X-ray data from cellulose: a new method to determine degree of crystallinity and predict hydrolysis rates. Bioresour Technol 101(12):4461–4471
Terinte N, Ibbett R, Schuster KC (2011) Overview on native cellulose and microcrystalline cellulose I structure studied by X-ray diffraction (WAXD): comparison between measurement techniques. Lenzinger Ber 89:118–131
Driemeier C, Calligaris GA (2010) Theoretical and experimental developments for accurate determination of crystallinity of cellulose I materials. J Appl Crystallogr 44(1):184–192
Driemeier C (2014) Two-dimensional Rietveld analysis of celluloses from higher plants. Cellulose 21(2):1065–1073
Schenzel K, Fischer S, Brendler E (2005) New method for determining the degree of cellulose I crystallinity by means of FT Raman spectroscopy. Cellulose 12(3):223–231
Agarwal UP, Reiner RR, Ralph SA (2012) Estimation of cellulose crystallinity of lignocelluloses using near-IR FT-Raman spectroscopy and comparison of the Raman and Segal-WAXS methods. J Agric Food Chem 61(1):103–113
Ding SY, Himmel ME (2006) The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem 54(3):597–606
Newman RH, Hill SJ, Harris PJ (2013) Wide-angle X-ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol 163(4):1558–1567
Zhang T, Mahgsoudy-Louyeh S, Tittmann B, Cosgrove DJ (2014) Visualization of the nanoscale pattern of recently-deposited cellulose microfibrils and matrix materials in never-dried primary walls of the onion epidermis. Cellulose 21:853–862
Patterson A (1939) The Scherrer formula for X-ray particle size determination. Phys Rev 56(10):978
Oliveira RP, Driemeier C (2013) CRAFS: a model to analyze two-dimensional X-ray diffraction patterns of plant cellulose. J Appl Crystallogr 46(4):1196–1210
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci 108(47):E1195–E1203
Acknowledgements
This work was supported by the Center for Lignocellulose Structure and Formation (CLSF), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DE-SC0001090. This work was also supported by Subcontract No. XGB-3-23024-01 with the National Renewable Energy Laboratory (NREL), under Contract No. DE-AC36-08-GO28308 with the U.S. Department of Energy. Sample preparation and XRD and NMR data collection were carried out with funding from the NREL. IR, Raman, and SFG analyses as well as XRD simulation and data analyses were carried out with CLSF support.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Lee, C. et al. (2015). Correlations of Apparent Cellulose Crystallinity Determined by XRD, NMR, IR, Raman, and SFG Methods. In: Rojas, O. (eds) Cellulose Chemistry and Properties: Fibers, Nanocelluloses and Advanced Materials. Advances in Polymer Science, vol 271. Springer, Cham. https://doi.org/10.1007/12_2015_320
Download citation
DOI: https://doi.org/10.1007/12_2015_320
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26013-6
Online ISBN: 978-3-319-26015-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)