
Abstract
Mass spectrometry has become a key technique for the structural analysis of carbohydrates. Due to their special properties and requirements carbohydrates and especially chemically modified carbohydrates occupy a position between biopolymers and synthetic polymers. Charged analytes can be obtained by adduct formation with appropriate small ions or by various labeling procedures. Besides molecular mass profiling, tandem mass spectrometry can give more detailed structural information including sugar constituents, sequence and interresidue linkage positions, and some information on stereochemistry. Substitution patterns of polysaccharide derivatives are also studied by ESI IT-MS and MALDI ToF-MS. In this review, ion formation of carbohydrates, their chemical modification, fragmentation pathways of various analyte species, and the applicability of MS for quantitative evaluations are discussed. Mainly ESI applications are presented, but where of general significance MALDI-MS applications are also outlined. Examples of application are given, excluding the well-reviewed area of biologically important O- and N-linked glycans. Molecular mass determination and structural analysis of heteroglycans are followed by examples of cellulose and starch derivatives.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Carbohydrates
- Electrospray-ionization mass spectrometry
- Fragmentation
- Labeling
- Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- Polysaccharides and derivatives
1 Introduction
The development of efficient ionization methods for mass spectrometric analysis of large molecules has triggered tremendous progress in structural analysis of biomacromolecules. The inherent properties of these compounds arising from polarity, non-volatility, and chemical instability, and the often limited availability in pure form cause significant analytical challenges. These were overcome first by fast atom bombardment (FAB), and since the late 1980s more efficiently by electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). In combination with appropriate mass analyzers such as sector field instruments, quadrupoles, ion traps (IT) and time-of-flight (ToF) tubes, these ionization techniques and their advanced development have been widely applied to biomolecules of oligo- and polymeric size. The combination with tandem mass spectrometry (MSn) makes these techniques even more attractive, since in addition to the molecular mass measurement, sequence information can be obtained and substituents can be localized. Although for various reasons nucleic acids and especially proteins and peptides have been preferentially studied, and “proteomics” has even developed into a separate research area, carbohydrates have not been ignored. Unlike proteins, where the nitrogen atoms enable easy and multiple-charged (protonated) ion formation, carbohydrates commonly form sodium adducts at much lower yields. In addition, compared to proteins and peptides, poly- or oligosaccharides show lower surface activity, exhibit higher polarity, are less stable, often show dispersity of molecular mass and chemical structure, and due to many stereochemical centers have isobaric ions, which cannot easily be differentiated by MS.
Nevertheless, carbohydrate analysis has also profited a lot from these methods, and the specific challenges have been tackled. Most of the publications in this field deal with O- and N-linked glycans from the glycosylation sites of proteins, or with other biologically active molecules, e.g., human milk oligosaccharides [1–5] or everninomicins, a class of complex oligosaccharide antibiotics [6]. Many reviews have reported on the progress in the area of O- and N-glycan analysis [7–22]. In this chapter, these compounds will only be considered when more general insights and methods are concerned. On the other hand, ESI-MS and MALDI-MS have also been applied for structural analysis of polysaccharides from plants or microorganisms to gain insights into sequences and branching patterns etc. of these less regular biomacromolecules. In addition to application to native compounds, methods for the analysis of the substitution pattern of starch and cellulose derivatives have been developed [23]. Cyclodextrins (cyclic α-1,4-linked glucooligosaccharides) can form inclusion complexes with a wide range of substrates and have many applications, e.g., for encapsulation of drugs. ESI-MS has turned out to be a useful tool for investigating such molecular recognition phenomena, thus providing a powerful means for the analysis of a wide range of host–guest and other non-covalent complexes present in solution [24, 25]. In the case of defined glycoarchitectures such as glycodendrimers, MS is a valuable method for controlling conversion and uniformity [26, 27].
Quantitative aspects are often not emphasized in applications. On the one hand, structural analysis of glycoconjugates is mainly interesting from the qualitative perspective, but on the other hand relative ion intensities in a mass spectrometer cannot simply be translated into sample portions. However, in the field of polysaccharide derivatives the exact quantification of analytes is of high importance. Fundamental studies dealing with the ionization process have revealed several parameters that influence relative ion intensities, and models for calculations and predictions have also been developed [28–30].
Basic aspects like ion formation, labeling of carbohydrates, fragmentation in collision-induced dissociation (CID) processes, and quantifiability will be addressed, completed by examples of application on plant polysaccharides. The high importance of MS for the analysis of substitution patterns of polysaccharide derivatives will be emphasized and, in this context, methods of sample preparation and quantitative aspects will be discussed. The focus is on ESI-MS, but MALDI-MS has often been applied to the same problems and will be considered where reasonable.
It is assumed that the reader is familiar with the basic principles of the ESI and MALDI processes, and with the instrument setup. With respect to (special) MS techniques [e.g., nanospray, desorption electrospray ionization (DESI), Orbitrap, Fourier transform ion cyclotron (FTIC), ESI-atmospheric pressure ion mobility-ToF MS] and the field of glycoconjugates, the reader is referred to the recent literature [29, 31–34].
2 Carbohydrates as Analytes for Mass Spectrometry
2.1 Ion Formation of Carbohydrates
2.1.1 Positively Charged Analytes
A prerequisite for MS analysis is the existence of charged species. Positive and negative ions can be recorded, but the positive mode is more common and therefore preferentially considered here. Carbohydrates are neutral and, except for amino sugars, usually exhibit no basic groups that are available for protonation. More common is, thus, the formation of adducts with metal cations, mainly alkali ions, and preferentially the ubiquitously present sodium ion. Ion yield and consequently the sensitivity of MS depend on appropriate coordination sites. Coordination is accomplished by the oxygen atoms with their non-bonding electron pairs. However, it is not simply a 1:1 complex that is generated with the most basic oxygen, but a multi-coordinated species in which several oxygen atoms interact in a cooperative way with the cation. As a simplified model of glycans, polyethyleneglycol (PEG) can be considered, which (like carbohydrates) exists in oligomeric and polymeric forms, and in open-chain and cyclic forms. For these, it was found by ion mobility measurements and molecular calculations that the complexation constant for sodium adduct formation increases with the number of (CH2CH2O) units until it reaches an optimum at the favored coordination number. For sodium, energy minima were found for Na+-PEG-9, Na+-PEG-13, and Na+-PEG-17 with seven or eight oxygen coordination sites [35]. Lithium prefers the coordination number 7, and the larger potassium and caesium ions the coordination number 10 or 11 [36]. The PEG chain “wraps” around the cation and adopts the most favored conformation. These studies refer to solvent-free complexes and are not only a model for MALDI, where ionization mainly occurs in the gas phase in vacuum, but also for ESI, where neutral molecules like common carbohydrates only become sodiated after consecutive Coulomb explosions of the primarily formed charged droplets and evaporation of the solvent [28]. Therefore, it should be emphasized here that the relative intensities of ions observed in the mass spectrometer do not simply reflect the equilibrium in solution, but depend on the surface activities of the analytes [37] and (related to these) on the solvation energies (or desorption energies from the droplets), solvent, droplet size, and instrument parameters affecting the ion transfer. Bahr et al. found a huge change in relative intensities even for neutral unmodified oligosaccharides (maltopentaose) when applying nano-ESI QIT-MS (QIT: quadrupole ion trap) (Fig. 1) [38].

ESI-MS analysis of a mixture of maltopentaose and insulin (both 5×10−6 M) with conventional forced-flow (“micro”) (a), and nanospray (b). The averaged absolute intensities for the base peaks are indicated. Reproduced from [38] with kind permission of the publisher
A nanospray capillary with an orifice diameter of 1–2 μm and a flow rate of <1 μL/min displayed sensitivities which for the maltopentaose was comparable to that of the much more surface active peptide applied together with the maltooligomer. Although surface activity can be regarded as a thermodynamic parameter, reaching the surface requires migration from the interior, which is a kind of kinetic control. At the high field strength of ESI (e.g., 106–107 V/m) the electrophoretic mobility is the most relevant property in this regard. The effect of this kinetic parameter is obvious from the dependence of the relative signal intensities of competing ions on the capillary voltage (Fig. 2) [30].

Voltage dependence of relative signal ratios of various positive ions in ESI-MS. Values were normalized to a ratio of 1 at 1 kV. DTMA decyltrimethylammonium iodide, TMA tetramethylammonium bromide, TEA tetraethylammonium bromide. For details see [30]. Reproduced from [30] with kind permission of the publisher
Crown ethers, as the cyclic PEG analogs, have also been studied [39, 40]. Comparing adduct formation of a given crown ether (host) in a certain solvent with various cations (guests), e.g., the alkali ions, the experimentally observed ratio of signal strength is in good agreement with the theoretical data calculated from the complexation constants in solution. In comparison with the deviating behavior of the linear tetraglyme (di-O-methyl-PEG-4), their lower conformational flexibility obviously mimics a behavior more similar to the solvated state.
Against this background, it is not surprising that disaccharides are detected with higher sensitivity than monosaccharides, which do not exhibit a sufficient number of oxygen atoms in appropriate orientation, although the surface activity of monosaccharides is probably comparable to that of disaccharides. In addition, the disaccharide with its glycosidic linkage has a higher flexibility to adopt the appropriate conformation required for efficient complexation. Probably for the same reason, reduction of a disaccharide to an alditol glycoside additionally enhances the response in ESI-MS. Hofmeister et al. [41] have studied the coordination site of Li+ in isomeric disaccharides and open-chain gentiobiose, and showed experimentally and by semi-empirical MNDO (modified neglect of diatomic overlap) molecular orbital calculations that the lithium cation is penta-coordinated between the two sugar rings (Fig. 3).

The location and coordination of the charge-giving cation is also important for the fragmentation of sugar complexes in collision induced dissociation (CID), which will be discussed in Sect. 4.1.
ESI-MS competition experiments with cyclodextrins showed a much larger affinity of the β-cyclodextrin to sodium compared to all other alkali cations, thus demonstrating that it is the size of the cyclic maltooligosaccharide in relation to the cationic radius that drives the formation of the inclusion complex [42], comparable to crown ethers.
Although sodium is ubiquitous, the sample solution is sometimes spiked with sodium salts like sodium acetate [43]. If other adduct types are required, the cations are added as soluble salts, e.g., iodides, chlorides, acetates, trifluoroacetates, perchlorates, or sulfates (less than millimolar concentrations). At high concentrations, slightly acidic protons can be exchanged against Na+ and clusters with the salt are formed, e.g., [M + Na + n NaOAc]+. An increase of sodium adducts with increasing cone (nozzle-skimmer) voltages, but significant in-source fragmentation has been reported by Harvey [44].
Other cations e.g., divalent cations (Mg2+, Ca2+, Mn2+, Co2+, Cu2+) have been studied for their relative complexation ability and cone-voltage-dependent intensities of the different adduct ions formed with maltoheptaose and an high-mannose N-glycan. Beside the [M + X]2+ ions, singly charged fragment ions [M + X2+-(anhydroGlc + H+)] were observed [45]. Silver adducts have also been applied, especially with the aim to find diagnostically valuable fragmentation pathways. [M + Ag]+ and [M + 2Ag]2+ ions have been observed [46].
Variation of counterion is also of interest with respect to stability of the [M + X]+ adduct. In the series of alkali ions, the Li adduct is the most stable, whereas the Cs adduct has the lowest dissociation energy.
A more special application is the coordination with various transition metals under participation of coordinating additives or covalently linked tags, which has been studied by the group of Leary. This approach allows differentiating of diastereoisomers, which is a frequent and important topic in carbohydrate analysis. Established methods in this field, like NMR spectroscopy, are slower and less sensitive, making ESI in combination with tandem MS a promising alternative. Derivatization with diethylenetriamine and complexation with, e.g., Zn2+, differentiates between stereoisomers of hexose (Fig. 4) [48, 49]. Gluco-, galacto- and manno-configuration of the frequently occurring N-acetyl-2-amino-2-deoxy-hexoses have been differentiated by tandem MS of their diaminopropane-cobalt(III) complexes, in which the sugar nitrogen participates in cobalt complexation [47].

Above: Proposed structures of the Co(DAP)(GlcNAc) complexes m/z 426 and 352. Below: MS3 spectra of m/z 352 from top to bottom GlcNAc (A), GalNAc (B) and ManNAc-Co(DAP) (C) complexes in MS2. Reproduced from [47] with kind permission of the publisher
For more basic information on models for ESI, the reader is referred to literature on the charged residue model (CRM) of Dole et al. [50], on the ion evaporation model (IEM) of Iribarne and Thomson [51], to reviews summarizing the progress in this field [28, 29, 52], and to monographs [31–33].
2.1.2 Negatively Charged Analytes
Negative ions, directly formed from native carbohydrates, are of less relevance as long as no additional acidic groups like carboxy, sulfate or phosphate groups are present. Although OH groups in carbohydrates are comparably acidic (pK a 12–14), this is not acidic enough to form [M-H]− ions to sufficient extent under normal conditions. These ions are relatively unstable and tend to fragment within the ion source. But again, with respect to selectivity and fragmentation pathways, it is fruitful to generate negatively charged adducts. Harvey found well-polarizable anions such as halogenides (with exception of fluoride), sulfates, phosphates, and nitrates forming stable anionic products with some N-glycans, with nitrate giving best stability and sensitivity [53–56]. Both, singly and doubly charged ions were observed, and uronic acid as constituent caused even higher charge states. In studies of neutral oligosaccharides obtained from N-glycans, it was found that in the presence of ammonium phosphate (0.5 mM in methanol/water 1:1) more stable [M + H2PO4]− adducts were formed nearly quantitatively. The fragment spectra of these anion adducts resemble that of the [M-H]− ions since the first fragmentation step is the elimination of the corresponding acid, in this case H3PO4, thus finally yielding [M-H]− ions [57]. These negatively charged ions show diagnostically valuable fragmentation behavior without competing mechanisms or rearrangements, thus giving rise to ions presenting unambiguously specific structural features. [1, 2, 58].
2.2 Carbohydrate Derivatives
The physico-chemical properties of carbohydrates are strongly influenced by chemical modification. All parameters affecting ion abundance, such as surface activity, solubility and solvation energy, electron density at O atoms and thus coordination properties, and electrophoretic mobility are either slightly or heavily changed. Furthermore, charged or easily ionizable groups can be introduced by labeling reactions. Surface activity is probably the most decisive factor for the enhanced absolute and higher relative signal intensity of modified carbohydrates in ESI-MS compared to native carbohydrates [37, 38]. In conventional ESI-MS analysis, multiple O-alkylation of (oligo)saccharides affects the stepwise increasing response for [M + Na]+ in MS, which is obvious from mixtures of un-, mono-, di- and trisubstituted glucose, where the higher alkylated constituents are increasingly overestimated. This effect becomes less and less pronounced with increasing degree of polymerization (DP) of the corresponding oligosaccharides. Solvation of the OH-rich native or only modestly modified carbohydrates is probably much stronger in commonly used protic solvents (often methanol), since hydrogen bonds can be formed that are of higher bonding energy than van der Waals interactions. With increasing lipo- or amphiphilic character, the analytes become more and more located at the droplet–air interphase, and thus are preferably transferred into the highly charged progenies in subsequent Coulomb explosions. When higher alkyl or hydroxyalkyl residues are introduced, the sensitivity increases further. In the latter case, additional coordinating oxygen atoms are available in the flexible side groups. Also, polar substituents like carboxymethyl groups, which occur as neutral COOH or COONa, enhance the response in ESI-MS. Chemically modified carbohydrates will be treated in more detail in the section on polysaccharide derivatives (Sect. 6).
2.2.1 Labeling of Carbohydrates
A special type of chemical modification is the selective introduction of a label or tag. The tag can exhibit a permanent charge, often a quaternary ammonium group, or an easily ionizable function, e.g., an amino (positive mode), carboxy or sulfonic acid group (negative mode). Besides, most of the tags are chromophors and some tags are also fluorescent, which can be used for parallel detection in the case of liquid chromatography (LC) or capillary electrophoresis (CE) coupling to the ESI mass spectrometer [17, 18, 58]. For a selective reaction, the dormant terminal carbonyl function of the glycan is used. Formation of hydrazones, oximes, and imines are the most popular reactions, often followed by a reduction to shift reversibly formed intermediates like imines to the stable amines [59–61]. Michael reaction with 1-phenyl-3-methyl-5-pyrazolone has also been applied [62]. Glycan labeling strategies and their applications have been reviewed by several authors, again mainly focusing on glycoconjugate analysis, but of general meaning.
Girard’s T [1-(hydrazinocarbonylmethyl)trimethylammonium chloride, GT] and Girard’s P reagent [1-(hydrazinocarbonylmethyl)pyridinium chloride, GP] were applied to increase the intensity in MS by hydrazone formation [63, 64] and for performing quantitative measurements [65, 66]. Hydrazones are formed by nucleophilic attack of an acid hydrazide such as GT at the anomeric carbon atom of the reducing sugar, followed by elimination of water (Fig. 5). NMR studies have proved that the product is stabilized as its β-N-glycoside [68]. Thus, no reduction step is necessary to shift the equilibrium to the product side. However, products are acid-sensitive and show the highest stability at neutral pH [67, 69–71].

Hydrazone formation of cellobiose with Girard’s T reagent. Reproduced from [67] with kind permission of the publisher
Another powerful method for labeling of carbohydrates is reductive amination [17, 58]. In this two-step derivatization, an imine is formed by reaction with a primary amine, which is subsequently reduced to the product, usually by NaCNBH3. Some authors [72–74] have studied various parameters that influence the rate and result of the reactions, such as solvent, pH, ratio of reagents, temperature, time, and sample work-up. Selectivity of the reducing agent with respect to the carbonyl function is a crucial point, often not considered [67, 75]. Reductive amination of the carbonyl function is also pH-sensitive [76]. In most cases, large molar excess of amine (up to several 1,000-fold) [77] have been used to avoid twofold reaction of the primary amine and to shift the equilibrium to the imine, which is subsequently reduced to the corresponding aminodeoxyalditol (Fig. 6). Recently, 2-picoline borane has been found to be superior to the cyano compound, since it displays higher reducing selectivity, is nontoxic and does not introduce sodium into the sample [67, 78].

Reductive amination of glucose. Reproduced from [67] with kind permission of the publisher
Aliphatic amines, functionalized amines like aminoethyl crown ethers, but in most cases aromatic amino reagents have been applied for reductive amination, i.e., 2-amino-pyridine (AP), regiosiomeric aminobenzoic acids (AA), their esters (e.g., ethyl: AEE) and amides (AB), but also polycyclic aromatic amines like 2-aminonaphthalene- (ANTS) and 1-amino-pyrene-trisulfonic acids (APTS). Table 1 shows the structures and mass increments of some common labeling reagents [60]. Acetic acid is usually added to catalyze reductive amination reactions by protonation of the carbonyl group and the intermediate imine, which results in a promotion of the hydride transfer to the iminium ion [79]. However, at too-low a pH, the primary amino function is protonated to too-high an extent. Thus, the reactions have to be performed in an optimal pH range, which depends on the basicity of the amine applied. Sun et al. [76] estimated that the pH should be close to, but not lower than, the pK a value of the amine applied. Therefore, aromatic deactivated amines are preferably employed because they are less basic than the aliphatic amines and hence show higher reactivity at low pH values. Electron-withdrawing groups in o- or p-positions further decrease the pK a of the ammonium form, e.g. o-aminobenzoic acid (2-AA) has a pK a of 2.18 [80]. The introduction of acidic groups offers the option of negative ion formation. 2-AA is often used due to its fluorescence and UV activity [72, 81] but also for fragmentation studies in structure analysis of polysaccharides [82–84].
To obtain mass spectra with a high signal-to-noise ratio it is important to remove contaminants such as the excess of reagents, buffer, etc. [58]. MS is therefore often coupled with CE [72, 85, 86] or more commonly with HPLC [87, 88]. Samples are purified by size exclusion chromatography (SEC), precipitation with acetone [89], solid phase extraction [74, 88], or extraction of excess amine with organic solvents [73, 87]. Besides being time-consuming, these steps involve the risk of bias of constituents in a complex mixture.
For the analysis of the substituent distribution of polysaccharide derivatives, the exact quantitative analysis of oligosaccharide mixtures from partial degradation of the polymer plays a key role. Although the strong influence of hydroxy and methoxy substituents in methyl celluloses on pseudomolecular ion yields of oligosaccharides can be overcome simply by permethylation with methyl iodide-d 3 , this approach cannot be applied for hydroxyalkyl derivatives like hydroxyethyl, hydroxypropyl, hydroxyethylmethyl, and hydroxypropylmethyl cellulose (HEC, HPC, HEMC, HPMC). To level off the strong influence of alkoxyalkyl groups on ion formation, amino groups or permanently charged tags have been introduced [90, 91]. These procedures include reductive amination with propyl amine and subsequent quaternization of the nitrogen by methylation (Section 6.2).
3 Molecular Mass Determination
The search for an ionization method that is able to transfer and ionize relatively large polar molecules without decomposition into the gas phase was strongly motivated by the demand for molecular mass information especially of biopolymers. When in the late 1980s MALDI [92, 93] and ESI-MS [94] opened new possibilities compared to FAB-MS, the question arose whether molecular weight distributions could also be determined by these methods. For MALDI ToF-MS, which covers a much wider m/z range, this has been shown to be possible for certain synthetic polymers under appropriate conditions [95]. In the field of biopolymers, polydispersity of molecular mass is only typical for carbohydrates. However, due to the high polarity and limited chemical stability of carbohydrates, there are only a few examples of molecular mass distribution analyses in the higher mass range. In contrast to peptides and proteins, carbohydrates are much less prone to multiply charged ion formation, which could reduce the m/z values.
By means of a SEC-ESI-MS online coupling, the mass spectra of a dextran 5,000 standard with a polydispersity of 1.6 could be successfully obtained by accumulating all spectra recorded during dextran elution [96]. Interestingly, discrete areas of the peak profile showed that dextrans of higher DP were detected as fourfold charged ions (Δm/z = 40.5), followed by [M + 3Na]3+ with a maximum at DP22, [M + 2Na]2+ with a maximum at DP15. In the peak maximum, single-charged [M + Na]+ ions were the dominating species with a maximum for DP7 (M 1,152). Up to DP42 could be detected in the subspectra. Direct infusion of this standard provided up to triply charged ions and the highest DP detected was 26. In contrast, mass spectra obtained by MALDI ToF-MS of the spotted SEC fractions showed only single-sodiated adducts, typical for MALDI, but covered about the same mass range. Up to m/z 7,500 was detectable, but due to the high laser power required for desorption of the larger molecules, a plateau of lower m/z fragment ions was observed with increasing intensity. Similar experiments with dextran 12,000 showed fivefold charged ions in SEC-ESI-MS, and the highest m/z detected corresponded to a mass of approximately 9,500, while MALDI-MS could detect up to DP70 (molecular mass 11,358) as [M + Na]+ (Fig. 7). It was also shown that sensitivity and stability can be improved by permethylation, extending the mass range to about 100,000 Da [96]. Whether the correct molecular weight distribution can be matched by this approach was not proved; however, SEC-MS coupling impressively showed how pre-separation mitigates the competition of analytes in ESI between smaller and larger molecules. On average, seven glucose units complexed one sodium ion under the conditions applied, whereas MALDI leads to single-charged ions.

Above: ESI TIC trace (a) and mass spectra of dextran 5,000 obtained by SEC-MS. Spectra were generated by summing discrete areas beneath the peak in the TIC trace at elution times of (b) 18.94–19.08 min, (c) 19.64–19.72 min, (d) 20.38–20.52 min, and (e) 21.63–21.71 min. Below: MALDI mass spectra obtained from manual fractionation of dextran 12,000, following SEC. Reproduced from [96] with kind permission of the publisher
Since MALDI ToF-MS can cover a wider mass range, most studies in this field have been performed with MALDI. Pre-separation by gel permeation chromatography (GPC) reduces the dispersity of the fractions, which should be no larger than 1.2 to avoid border-discrimination effects [97]. With this restriction, dextran fractions with molecular mass as high as 94,000 (HPSEC-MALLS) could be analyzed (MS: M w = 90,000). Enzymatic digests of hyaluronic acid were analyzed by MALDI-MS up to molecular mass of 15,000, and these data were used for calibration of the GPC system, giving much more realistic results than calibration with chemically and topologically different pullulan and dextran standards [98]. Even better results have been obtained with 2′,4′,6′-trihydroxy-acetophenone (THAP) as a matrix [99]. Pullulans of molecular mass up to 47,000 could at least be detected but were no longer resolved. However, in the presence of insulin mixed with polysaccharides of similar average mass, the latter were detected with only 1/1,500 of the protein signal intensity. Degradation products were detected in all polysaccharide spectra and the molecular mass was lower than that determined by SEC. When the signal intensities were corrected for the detection efficiencies in the microchannel plate detector, which decrease with m/z due to decreasing velocity, the distribution was shifted to higher average molecular mass values, and with 10,618 Da came close to the SEC result of 12,000 Da. In contrast to proteins, a much lower matrix-to-analyte ratio was appropriate. Even at a 100:1 ratio (1 nmol/μL matrix, 0.01 nmol/μL analyte) good mass spectra were obtained. Dextrans, polysialic acid, and glycoproteins were also most successfully measured using THAP as matrix. Addition of CsI allowed recording spectra in positive ([M + Cs]+) and negative ([M + I]−) modes. Permethylation strongly improved the sensitivity and reduced discrimination of higher masses, probably partly due to less fragmentation and partly due to better desorption/ionization properties [98].
In 2008, Schnöll-Bitai et al. reported that a non-crystalline matrix, the ionic liquid 2,5-dihydroxybenzoic acid/butylamine (DHBB), was superior to THAP for the MALDI analysis of the molecular mass distribution of pullulans in the mass range of 5,900 to 112,000 Da [100]. Figure 8 shows the mass spectra of two pullulan standards with DHB and DHBB for comparison. [M + BuNH3]+ ions were detected, and the molecular mass and polydispersity values deviated by 10–25% from the supplier’s data. The authors also developed a theory to explain why the liquid character of the matrix should reduce fragmentation compared to a less flexible solid matrix. However, the low polydispersities of ca. 1.1 of the standard samples are also a prerequisite for the comparably low discrimination effects, since narrow distributions facilitate adjustment of the laser power appropriate for all constituents.

MALDI-ToF mass spectra of the standard samples pullulan-11,800 (left) and pullulan-22,800 (right) measured with the matrices DHB (a, c), and DHBB (2,5-dihydroxybenzoic acid/ butylamine) (b, d, e). e shows an enlarged part of the smoothed distribution given in d. Reproduced from [100] with kind permission of the publisher
Another example is the analysis of fructans, which are extended sucrose-based glycans consisting of 1,2- and 1,6-linked fructofuranosyl residues and various molecular weight distributions and branching patterns [101, 102]. Typical fructan-containing plants are dahlia, chicory, artichoke, agave, and onions. Stahl et al. compared DP distributions obtained by MALDI ToF-MS and high performance anion exchange chromatography–pulsed amperometric detection (HPAEC-PAD), and could detect fructooligosaccharides in the mass range of 2,000–6,000 by both methods, while the quantitative signal profiles were different. Interestingly, cell layers of onion tissue could be placed with the matrix on the sample target, giving the same fructan signals. Molecular masses of hemicelluloses have also been analyzed by means of MALDI ToF-MS [103, 104].
4 Structure Analysis
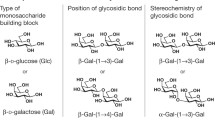
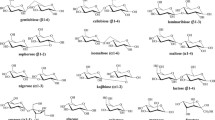
Without any knowledge of the structure of a carbohydrate sample, the molecular weight and molecular weight distribution are of little value. While the constituents of other biopolymers, like the amino acids of proteins, differ in mass, the diversity of glycans is mainly based on isomerism. The existence of five stereocenters – four different positions of alcohol functions and the option of four- or five-membered rings (furanosyl or pyranosyl, respectively) in an aldohexose – means that, at least theoretically, more than one million isobaric disaccharides can be formulated. Only a very small portion of these exist in nature, e.g., nine disaccharides from d-glucose, one of the 16 possible stereoisomers of aldohexoses (Fig. 9).

Structures of nine isomeric disaccharides built from d-glucopyranose
Therefore, besides mass profiling, it is also very important to gain insight into linkage and branching patterns, and into the sequence of oligosaccharides. Although chemical sequencing is well established for peptides and DNA, it is almost non-existent in the case of carbohydrates and has only been successful in special cases and for short sequences, e.g., by Svensson oxidation and subsequent β-elimination [105, 106]. Thus, tandem mass spectrometry is a very important and valuable technique for comprehensive structural analysis of oligosaccharides. Metastable fragment ions have been used in the case of MALDI ToF-MS PSD (post-source decay), but fragmentation is nearly non-controllable. Use of a collision cell (MALDI-CID/PSD-ToF-MS) has improved this analysis [107]. ESI coupled to a triple-quadrupole analyzer allows precursor ion selection in a first MS step, CID in the second quad, and mass analysis in the third quad. Ion traps, often used in ESI-MS instruments, can easily accumulate ions of a distinct m/z, and then be used for CID and mass analysis of fragment ions [29]. Helium is most commonly used as collision gas, and the amplitude for excitation can be controlled for efficient fragmentation. As a further important option of this technique, the process can be repeated by isolating and fragmenting daughter ions in the same manner. Thus, generations of fragment spectra can be obtained, as long as abundance of the ions is sufficient and further fragmentation energetically possible, in practice up to MS4 or MS5. The competing process is the dissociation of the complexed cation, which makes the carbohydrate “invisible.” Nano-ESI-MS is often employed in combination with a Q-ToF (quadrupole and time-of-flight mass analyzer) (CID, as introduced by Jennings [108], or decomposition; some authors use dissociation only for the loss of the charge-giving cation: [M + X]+ → M + X+). For details of instrument set-ups see [29].
Figure 10 summarizes the approach, including optional labeling and/or separation prior to MS and MSn.

Sequence analysis of oligosaccharides by (LC)-ESI-MSn
4.1 Fragmentation of Carbohydrates in Tandem MS for Sequencing and Determination of Linkage Pattern
To gain structural information beyond the molecular mass, tandem mass spectrometry is widely applied in glycan analysis. Fragment ions are assigned according to the nomenclature of Domon and Costello [109], who systematically described the positive and negative ions observed by FAB-CID-MS of protonated oligosaccharides (Fig. 11).

Nomenclature of fragment ions according to Domon and Costello [109]
To deduce the sequence of an oligosaccharide without being misled by fragment ions of only apparently clear origin, it is very important to know the mechanisms of fragmentation and possible artifact formation. Therefore, similar to studies made decades before for electron impact mass spectra of carbohydrate derivatives, basic studies have been performed with defined model substances for understanding the “rules” of decomposition. Although there are some generally observed fragmentation patterns of carbohydrates, which are independent of the instrument, charge state, and individual structure (e.g., cleavage of glycosidic linkages), there are on the other hand significant differences controlled by the charge-giving group or counterion, positive or negative ion mode, chemical modification of OH groups, blocking (methyl glycoside) or labeling at the reducing end, solvent, additives, and pH. These differences are of course valuable for extending the choice of diagnostically valuable fragment ions, but at the same time they complicate matters.
4.1.1 Reducing Oligosaccharides
4.1.1.1 Protonated Oligosaccharides
Protonated oligosaccharides [M + H]+ require the lowest energy for dissociation, and mainly undergo cleavage of the glycosidic linkages. Y and C ions represent protonated shorter oligosaccharides, with the Y-series comprising the reducing side of the starting compound and the C-series the non-reducing side. The complementary B and Z ions differ by 18 Da (H2O). Differentiation of these ions, which in the case of non-derivatized homoglycans are isomeric, has been achieved by isotopic labeling at the reducing end with 18O [110–112]. In accordance with the linkage stabilities, B and Y fragments usually dominate over C/Z-fragments or are even formed exclusively [113]. Usually, one glycosidic linkage is broken to yield two fragments maintaining the original sequence; however, protonated species can also undergo an “internal loss,” where a proton-supported transglycosidation occurs accompanied by loss of one internal anhydroglycose [114–118]. The mechanism is probably similar to a proton-catalyzed transglycosidation, which in the gas phase requires an appropriate sequence and conformation to enable an SN2-like reaction. For a reductively aminated trisaccharide, Harvey et al. have suggested the pathway shown in Fig. 12 [119, 120]. Although this rearrangement can cause erroneous interpretation, it has also been used to obtain additional information from the substitution patterns of internal residues of trisaccharides [121].

Mechanism proposed by Harvey et al. for fucose migration during the fragmentation of 2-AB-derivatized 2′-fucosyl-lactose. Reproduced from [119] with kind permission of the publisher
4.1.1.2 Adducts with Metal Cations
There are marked differences between fragment ions produced from protonated and metal cation-complexed oligosaccharides. In the case of alkali-coordinated oligosaccharides, heterolytic cleavage of the glycosidic bond is always accompanied by proton transfer, so that the part of the molecule that binds the alkali cation is detected, which means usually both parts according to the relative probabilities of retaining the cation. In addition to the B and Y ions, which dominate the positive mode mass spectra of protonated oligosaccharides, A and X fragments from cross-ring cleavages (see Fig. 11) are registered.
The stronger the cation is bound, the higher is the degree of fragmentation, e.g., Li+ adducts form a higher diversity of fragment ions compared to Cs+ adducts, which do not fragment but only dissociate into the carbohydrate and the Cs ion [44]. This is nicely illustrated by comparative MS measurements of a manno-glycan [(GlcNAc)2(Man)5] complexed with Li+, Na+, K+, Rb+, and Cs+ (Fig. 13) by addition of the corresponding iodides (50 ng/μL). Decreasing binding energies of alkali metal ions with increasing size have also been shown by Cancilla et al. [122] and Botek at al. [123].

Comparative ESI-CID-QToF MS measurements of a manno-glycan [(GlcNAc)2(Man)5] complexed with Li+, Na+, K+, Rb+, and Cs+ (from top to bottom). Reproduced from [44] with kind permission of the publisher
Cancilla et al. [124] studied the coordination of chitobiose and chitotriose with various alkali metal cations and found decreasing binding with increasing size of the ions. K+ becomes bound more efficiently when extending the di- to the trisaccharide, a phenomenon that has also been observed for inulin, where potassium adducts become increasingly favored over sodium adducts with growing DP. Molecular modeling indicated preferred coordination of the cation by the oxygens of the glycosidic linkage, O-3, O-5′, and O-6′. (Fig. 14) [124]. The results of this study suggest that glycosidic bond cleavages are charge-induced whereas cross-ring cleavages are charge-remote processes. Adams et al. reported fundamental studies of charge-remote fragmentations [125].

Coordination of K+ to chitobiose and chitotriose. Average K+–O distance is 5.779 Å for chitobiose and 6.220 Å for chitotriose.Reproduced from [124] with kind permission of the publisher
Thus, the most commonly observed [M + Na]+ adducts of reducing oligosaccharides undergo additional cross-ring cleavages, requiring the rupture of two linkages. Depending on which side retains the charge, additional A or X fragments are observed. Since most of these fragmentations start from the reducing end, eliminating small neutral molecules, the A-type are usually favored over X-type ions. The larger fragment has (at comparable chemical structure) the greater affinity to maintain the alkali cation. However, 1,5X ions, which correspond to Yi fragments bearing C1, and ring oxygen of the next sugar residue (m/z = Y + 28) were observed by CID with air as collision gas at 1–2 kV for malto-, manno- and dextranoligosaccharides (MALDI-ToF/ToF-MS) [126]. Primary fragments of sufficient energy undergo further fragmentation. Therefore, many ions are the result of further successive losses. This can be proved by MSn experiments and has to be considered in the interpretation, e.g., Ai ions formed from Yj fragment ions only give redundant information, since they all represent the same original reducing end.
The mechanisms of fragmentation have been studied by Hofmeister et al. using isotope-labeled (18O, 2H) model compounds, representing all α- and β-linked positional isomers of disaccharides (see Fig. 9) [41], and were found to follow a retro-aldol cleavage of the ring-opened aldehyde form (Fig. 15). Since a new carbonyl group is formed in this 0,2A-fragmentation (including linkage 0 and 2 of the sugar ring, starting with numbering of linkages with 0 for O-5–C-1, proceeding clockwise), a second retro-aldol reaction can occur in an aldohexose. This reaction splits off C2H4O2 (M-60-60) again, yielding 2,4An. 0,3A-cleavages (M-90) can be rationalized by a preceding tautomerization to the 2-keto form. Further tautomerization to the 3-keto isomer could then lead to a precursor for direct formation of 2,4An. Another plausible retro-ene mechanism for the formation of 0,2An [127] has not been confirmed by isotopic labeling studies [41].

Fragmentation pathways for a 1,6-linked disaccharide (isomaltose) by subsequent retro-aldol cleavages (0,2A, 2,4A) or after tautomerization to the ketosugar (0,3A); according to Hofmeister [41]
Whether these cleavages are observed in the tandem mass spectra depends on the availability of the involved OH group. If this is blocked by other sugars or by substituents, then cleavage is inhibited. Thus, the pattern of fragment ions is of high diagnostic value for the elucidation of linkage positions: M-120 is observed for 1,2-linked aldohexose disaccharides (0,2X2), M-90 for 1,3-linked (0,3X2), a loss of 60 (0,2A2) and 120 u (2,4A2) for 1,4-linked, and of 60, 90 and 120 u for 1,6-linked positional isomers. For pentoses the corresponding fragmentations can be deduced.
Figure 16 shows the ESI-CID mass spectra of the isomeric disaccharides sophorose (glc-β-1,2-glc) and gentiobiose (glc-β-1,6-glc).

ESI-CID-MS of sophorose (a) and gentiobiose (b)
Although all mentioned fragmentations can be observed for the 1,6-linked disaccharides, substitution of 2-OH in sophorose only allows the 0,2-cleavage. However, since the second glucosyl residue is linked to the C1-C2-fragment, formally a 0,2X- instead of the 0,2A-fragment is observed. Apart from B and Y ions, elimination of water (M-18) and probably formaldehyde (M-30) are observed.
In a similar way, the position of non-sugar substituents can be deduced from shifts in the daughter mass spectra, as will be outlined later (Sects. 5.1 and 6.3) [128–132].
If the molecule is branched, it is usually difficult to differentiate between sugar residue losses of the different branches. The branches are assigned Greek letters α, β, and γ in order of decreasing molecular weight [109], whereas ions resulting from cleavage of the core unit are not designated a Greek letter. The numbering continues in parallel into the branches (“antennae” in the case of glycoconjugates).
In 2004, Garozzo et al. reported on three new fragment ions in the MALDI-ToF/ToF-tandem mass spectra of sodiated ions of well-known human milk oligosaccharides, and suggested a pathway for their formation involving a six-member-ring rearrangement [5]. These fragments were also of high diagnostic value since they allowed the discrimination of linkage positions. These unexpected ions, assigned E, F, and G, are illustrated in Fig. 17.

Formation of E, F and G fragment ions as proposed by Spina et al. F ions are indicative of a 1,3-linkage. Reproduced from [5] with kind permission of the publisher
Each of the ions represents disproportionations of the mother ion since a lactone and a deoxysugar, or an oxo-sugar and an anhydroalditol, are formed from the two aldehydes constituting the disaccharides. All fragmentations are accompanied by additional elimination of HX (HX = ROH or H2O or NH2Ac) from position 2 or 4 of the later observed fragment ion. Surprisingly, only the reduced products, but never the oxidized counterparts are observed in the mass spectra. There is no information on whether these structures and mechanisms have been made likely by isotopic labeling studies. But, independent of the pathway, F and G ions are indicative of a glycosyl unit β-linked to the neighboring 3-position, while E was observed for β-1,4-linkages to glucose. It should be kept in mind that these fragments were observed in higher energy MALDI-ToF/ToF-MS/MS experiments, and not in MALDI-PSD mass spectra. Usually, there is no fundamental difference in the fragmentation routes observed by the various tandem MS methods, but only an influence on the extent of fragmentation due to the different energies.
4.1.1.3 Negative Ions
As described above, negative ions of carbohydrates are obtained with high abundance if appropriate counterions like nitrate or dihydrogenphosphate are added. Since elimination of the corresponding acid HY is the first fragmentation step, tandem mass spectra of these [M + Y]− adducts do not differ significantly from those of the deprotonated oligosaccharides. Principally, negative ions can form the same fragments as positive ions (A, B, C, X, Y, Z), but due to the negative charge as a driving force, other pathways are favored. The negative charge is assumed to be located at the most acidic position, which often is the hemiacetal function. Thus, the anion formed corresponds to a Cn ion (n = number glycosyl units), which induces consecutive C-type fragmentation probably by electron pair shifts. From the anomeric anion, alcoholates (RO−) can be pushed out from the 3- and 6-position, while for 4-linked glycosides the negative charge must be located at O-2. Due to this charge-controlled fragmentation, MSn spectra of deprotonated oligosaccharides can be read “from right to left” and are therefore of high interpretive value, which has been explicitly outlined by Harvey [53–56]. Pfenninger et al. have successfully studied and applied this procedure to human milk oligosaccharides, which have essential biological functions [1, 2]. An example is given in Fig. 18.

Consecutive C-type fragmentation of deprotonated oligosaccharides (negative mode) as proposed by Pfenninger et al. [1]; two alternative mechanisms (black and striped arrows) are shown. Cleavage for 1,3- (a), 1,6- (b), 1,4-linked hexopyranosides (c), and a 4-linked 2-deoxy-N-acetylhexosamine (d) are shown. Reproduced from [1] with kind permission of the publisher
4.1.2 Non-Reducing Oligosaccharides
4.1.2.1 Methyl Glycosides
If the carbohydrate is permethylated or obtained by partial methanolysis from a polysaccharide, the reducing end is blocked. For these methyl glycosides, no retro-aldol cleavages occur, which is also obvious from the tandem mass spectrum of the non-reducing disaccharide trehalose (structure shown in Fig. 9) [41].
Depending on the collision energy and the gas used, different cross-ring cleavage ions are observed: 1,3A, 3,5A, and 1,5X (Fig. 19, left) [133]. These are formed by charge-remote fragmentation processes at high collision energies and are much more favored if the heavier argon is used rather than helium. At low collision energies, only the more labile glycosidic linkages are cleaved. At 4 kV, pronounced cross-ring cleavages are observed. Depending on the linkage and branching positions, in addition to the mentioned 1,3A, 3,5A, and 1,5X ions, 0,4A, 2,4A, and 0,2X fragments could be observed (Fig. 19, right). 1,5X ions allow the differentiation of Y and C ions, since they are always related to Y ions with m/z (1,5Xi) = m/z (Yi + 28) [107, 134]. Permethylated oligosaccharides fragmented with argon at high collision energy also showed W ions from C5–C6-cleavages, maintaining the charge on the “right” end of the oligosaccharide and being diagnostic for 1,6-glyclosidic linkages.

Proposed pathways for cross-ring cleavages in permethylated oligosaccharides, according to Lemoine et al. [133]
4.1.2.2 Sequencing of Permethylated Carbohydrates
Reinhold and coworkers have thoroughly studied the fragmentation behavior of per-O-methylated di- and higher oligosaccharides comprising various linkage positions and stereochemistry of the hexapyranosyl monomer units (Glc, Gal, Man) [135, 136]. Although Y-type ions are more abundant, they focused on the B-type ions from glycosidic linkage cleavage, which are more informative. The consequence of substitution of hydroxyl group in these permethylated compounds is a proton transfer from C-2 instead of 2-OH to the cleaved part of the glycan. The C = C double bond formed induces a retro-Diels–Alder (RDA) reaction, resulting in a 3,5A fragment ion (Fig. 20). Abundance of these ions strongly depends on the stereochemistry of the pyranose involved. Since, with respect to the dienophile, the Diels–Alder reaction is stereospecifically syn, reaction is favored for galactose, which yields cis-1,3-dimethoxypropene whereas glucose and mannose derivatives give the trans-isomer. The differences in MSn spectra are reproducible and the relative intensities of various fragments relate to the stereochemistry [136]. Figure 21 illustrates how the different free and permethylated B2 ions obtained from reduced maltotriose undergo further disassembly. Depending on the location of the linked sugar residue, further cross-ring cleavages were observed, probably also the result of RDA reaction after isomerization of the double bond. Comprehensive studies of permethylated oligosaccharide standard compounds resulted in a spectral library, which allows the facile evaluation of structural details including interresidue linkage, monomer identification, anomeric configuration, and branching [137]. An algorithm was developed for this congruent strategy for carbohydrate sequencing [138], requiring up to MS5 measurements, thus in principle achieving a gas-phase separation of isomer-derived fragments. Based on the knowledge of precursor–product relationships, the individual structures can be deduced [139]. Recently this new tool resulted in a patent [140].

(a) Retro-Diels–Alder CID fragmentation of B-type ions from permethylated oligosaccharides. (b) Further generic cross-ring cleavages observed for B ions of regioisomeric oligosaccharides (R mono- or oligosaccharide substituent). Reproduced from [136] with kind permission of the publisher

ESI-MS3 spectra of B2 ions obtained by MS2 from (a) native and (b) O-methylated maltotriositol. Reproduced from [136] with kind permission of the publisher
A similar approach has been applied by Mischnick and coworkers for the analysis of isomeric mixtures of partially methylated disaccharides. Also without chromatographic preseparation, the qualitative and quantitative composition of these isomeric mixtures were evaluated from the MS2 and MS3 spectra as outlined in Sect. 5.1 [130]. Elucidation was based on the knowledge of the fragmentation pathways of isotope-labeled O-methylated but reducing di- and trisaccharides [131].
4.1.2.3 Labeled Compounds
Labeling of the oligosaccharide at the reducing end as described above (Sect. 2.2) can influence the fragmentation behavior differently. It causes a mass shift of Y and Z fragments and thus allows them to be distinguished from the corresponding C and B ions comprising the non-reducing end. In the positive ion mode, these analytes can be protonated due to the basic amino group introduced by reductive amination, but are often sodiated. The label can contain further nitrogen groups, which strongly favors the location of the charge at the tag. Labeling reagents with quaternary ammonium groups like Girard’s T or acidic labels like the sulfonic acid reagents ANTS and APTS or AA fix the charge side. This can be advantageous for a straightforward interpretation of the tandem mass spectra with respect to sequence.
However, the observed fragment pattern again strongly depends on whether the molecule is protonated or sodiated. In [M + H]+ of cellooligosaccharides reductively aminated with dimethylamine, the proton is located at the amino group and, consequently, a ladder of Yi ions (i = 1, 2, 3… n) are detected. In contrast, tandem MS of [M + Na]+ of the same analyte shows daughter ions from cross-ring cleavages, thus indicating that the sodium is (de)located at the carbohydrate chain, and that the larger aglycon (amino-deoxy-alditol compared to methyl) can be cleaved off (Fig. 22).

Above: cellotetraose, reductively aminated with dimethylamine (DMA). Below: ESI CID-MS showing comparison of [M + H]+ and [M + Na]+ fragmentation. Reproduced from [128] with kind permission of the publisher
These fragments can be separated by one of the various methods mentioned. Although MALDI-MS is very appropriate for profiling the molecular masses of the oligosaccharides in the digest, ESI-MSn was superior to MALDI-ToF-MS-PSD for unambiguous sequence analysis due to the applicability of MS3 and MS4 where MS2 did not give sufficient structure information. An example of the ESI-MS2 of a xyloglucan oligomer that contains additional fucosyl-O-acetyl-galactose in the side chain is given in Sect. 4.2.1.
Consequently, a free carbonyl side chain is generated, enabling retro-aldol cleavages as outlined above (Fig. 15), for example C3 at m/z 527, giving m/z 467 and 407, or C2 m/z 365 yielding m/z 305 and 245, due to the 1,4-linkage. The formation of C fragments has been proved for 13C-cellotriose labeled with dimethylamine [128]. In addition to Y fragments, Y* ions with a Δm/z of 45 are observed at even higher abundance, corresponding to a loss of dimethylamine.
In a comprehensive study, Harvey introduced various tags by reductive amination in a high mannose glycan, and compared the relative ion intensities in ESI-Q-ToF and MALDI ToF-MS (positive ion mode), as well as the fragmentation behavior of the products [60]. There was nearly no difference in the MS2 spectra for aminobenzoic acid, aminopyridine, and aminoquinoline derivatives, since the label was lost in the primary fragmentation step, indicating that the sodium ion is more probably coordinated to the carbohydrate part (Fig. 23) [60]. Fragmentation differed slightly for aminoacridone and p-amino-N-(2-diethylaminoethyl)benzamide.

4.1.2.4 Negative Ions
Gennaro et al. detected ANTS-derivatized maltooligosaccharides after ion pair reversed phase (RP)-HPLC (NEt3H+ as counter ion) as doubly charged ions (two of three SO3H were dissociated). Under CID conditions, SO3 is first eliminated, followed by nearly exclusive formation of a “ladder” of Y 2−i fragment ion. (Fig. 24) [88]. Intensity is reported to be enhanced by a factor of 20 for Glc7-ANTS compared to Glc7.

ANTS-derivatized (8-aminonaphthalene-1,3,6-tri-sulfonic acid) maltooligosaccharides analyzed by LC-ESI-MS. Above: Selective ion current chromatogram. Below: CID-MS (negative mode) of DP7 (m/z 759.4) giving a ladder of Y2− ions after elimination of one sulfonic acid. Reproduced from [88] with kind permission of the publisher
Amination without reduction has also been recommended, using p-amino-benzoic acid ethyl ester [141]. Instead of the aminodeoxyalditol, an aminosugar is formed because the imine formed from the aldehyde and the amine is “trapped” by aminal formation (see Girard’s T labeling in Fig. 5), which is also performed without reduction and yields the β-N-glycoside (“closed-ring labeling”). However, this approach requires the reagent in high excess and subsequent purification. About 0.01–10 nmol of substrate was applied for syringe pump injection. Negative ions were studied and behaved similarly to [M-H]− of unlabeled oligosaccharides, “pushing out” a series of subsequent C ions as described by Pfenninger et al. for negative ion ESI QIT-MS of milk oligosaccharides [1]. Some additional fragments of diagnostic value with respect to the linkage position were detected for these labeled non-reduced compounds. Fragments comprising the labeled end of an oligosaccharide resembled the behavior of corresponding disaccharide derivatives, while fragment ions involving the other end of the molecule were similar to those of the unlabeled analog. The presence of 2-amino-2-deoxy sugars favored C/Z fragmentation and cross-ring cleavage. Elimination of ROH from position 3 is also favored by 2-amino-2-deoxy sugars [141].
4.2 Applications in Structural Analysis
Literature on structural analysis of carbohydrates by mass spectrometry mainly deals with O- and N-linked glycans after release from biologically active glycoproteins, and also, but less often, with bacterial lipopolysaccharides and polysaccharides derived from microbes or plants. Human milk oligosaccharides have been extensively studied [1, 2, 5]. The ratio of the comprised sugars, their linkage positions and branching pattern, sequence, and stereochemistry need to be elucidated. Molecular mass and molecular mass distribution, and non-sugar substituents and their location or pattern within the carbohydrate backbone are additional structural features that have to be studied.
What can mass spectrometry contribute to this field? With the “soft” ionization methods ESI and MALDI, information on molecular mass can be obtained at least in a qualitative manner and, as outlined in Sect. 3, limited to a certain mass range and with higher sensitivity and accuracy if coupled with SEC separation.
Substituents can be recognized by the mass shifts they produce. Sequence and linkage positions can be deduced from tandem mass spectra, although it is not generally possible to deduce them unambiguously, especially in complex branched structures. Isotopic labeling, periodate oxidation, or other chemical modifications have been applied in sample preparation for MS analysis to increase the specificity of structure information [43]. Stereochemistry of glycosidic linkages as well as ring size is still mainly deduced from NMR spectra or enzymatic digestibility. Sugar constituents are determined by various chromatographic or electrophoretic methods after hydrolysis of glycosidic linkages, but Reinhold et al. and Leary et al. have demonstrated that even the stereochemistry of isobaric sugar units can be differentiated from their tandem mass spectra if permethylated oligosaccharides are fragmented (Fig. 21) [136–140], or if certain complex-forming additives are applied that are sensitive to the stereochemistry and cause distinct intensity differences in the daughter mass spectra (Fig. 3) [47–49, 142, 143].
Progress in the field of glycobiology has been widely reviewed [7–16] and shall therefore not be outlined in this article. Structural analysis of cell wall polysaccharides or exopolysaccharides from microbes involves special demands, which are different from the popular O- and N-glycan area. The decisive difference is their dispersity with respect to molecular mass, composition, and branching pattern. Separation into molecularly uniform fractions is no longer possible. Mixtures have to be dealt with and averaged data like relative ratios of sugar constituents, average degree of branching, or average length of certain sequences of side chains have to be determined. Qualitatively, the existence of certain structural features can be deduced by MSn methods. Naturally, pretreatment by enzymatic, chemical, and/or separation methods specifies structural information and thus improves the resolution of the final image. This will be demonstrated by a few examples, emphasizing the contribution of mass spectrometry to this area of structure elucidation.
4.2.1 Plant Polysaccharides
4.2.1.1 Arabinoxylans
Arabinoxylans and arabinogalactans are widespread in plants. Arbinoxylans represent the main so-called hemicelluloses of cereals. They consist of β-1,4-linked xylopyranosyl residues (Xylp, X) with arabinofuranosyl residues (Araf, A) attached to O-2 or O-3 of the xylan backbone (Fig. 25). Other xylans also contain some glucuronic acid and galactose in the side chains. Fractions from acid- and enzyme-hydrolyzed arabinoxylans have been analyzed by SEC-ESI-MS. Thus, fractions eluting in the same mass range, but of different hydrodynamic volume due to different chemical composition (acidic or neutral), could be distinguished. Subsequent fragmentation by CID up to MS3 gave additional sequence information [95].

Structural features of an arabinoxylan
For the analysis of the microheterogeneity, Roepstorff et al. [144–146] studied arabinoxylooligosaccharides (AX) up to DP5 derived by enzymatic digestion with endoxylanase prior to and after permethylation by ESI QToF-MSn. Fragment ions at M-60 (0,2An) and M-90 (2,4An) indicated the 1,4-linkage of the pentosan backbone; however, due to the isobaric character of Xyl and Ara, branching patterns could not be deduced directly. After permethylation, mainly B and Y fragments were obtained, but 1,5X fragments (corresponding to 4-O-formyl derivatives; see Fig. 19) and 2,4A and 3,5A ions were also formed (Fig. 26) [83, 145]. Further fragmentation (MS3) of isobaric mixtures of B and Y fragments allowed differentiating branching patterns since the detectable number of methyl groups of a pentosyl unit corresponds to the linkage or branching pattern. However, CID measurements up to MS4 were required to distinguish positions of Ara linkages unequivocally, while also taking into account information from methylation analysis and NMR spectroscopy [146].

ESI IT-MS2 of m/z 709.6 obtained from enzymatic hydrolysis of arabinoxylan and subsequent permethylation. I and II represent possible structures of the tetrasaccharides comprising four pentosyl residues (I Xyl4, II AraXyl3). Reproduced from [146] with kind permission of the publisher
For more complex mixtures with less restricted options of linkage patterns, labeling and/or chromatographic separation prior to ESI-MS will be necessary to avoid misinterpretation of spectra of isomeric compounds. This has been performed by Maslen et al. [83] who labeled arabinoxylooligomers with o-amino-benzoic acid. In addition to ions from glycosidic and cross-ring cleavages, the D, E, F, G, H, and W ions mentioned above were detected (for the nomenclature, see Fig. 17) [5, 126]. These fragments, resulting from elimination and secondary oxidative eliminations from B-ions, indicated the position of arabinosyl residues in the pre-separated isomeric oligosaccharides.
4.2.1.2 Xyloglucans
Xyloglucans are the main portion of the so-called hemicelluloses of dicotyledons, although their composition depends on the taxonomic family. The xylose residues attached to the β-1,4-linked glucan chain can be capped by galactosyl or additional fucosyl residues. Voragen et al. have analyzed xyloglucan structure in blackcurrants by using different approaches, including online CE- and RP-HPLC-ESI-MSn and off-line HPAEC-MALDI ToF-MS [147]. The general principle in the analysis of heteroglycans with a certain diversity of size and chemical structure uses a prefractionation and a chemical or enzymatic partial hydrolysis to oligosaccharides. If enzymes with a known specificity are available, these are very valuable tools for retracing the puzzle pieces obtained to the polymer structure. In the case of xyloglucans, xylan-specific endoglucanases can be applied, which cleave linkages between an unsubstituted and a xylose-substituted glucose in the glucan backbone, thus producing a limited number of specific oligosaccharide building blocks. These can be separated by one of the various methods mentioned. Although MALDI-MS is very appropriate for profiling the molecular masses of the oligosaccharides in the digest, ESI-MSn was superior to MALDI ToF-MS-PSD for unambiguous sequence analysis due to the applicability of MS3 and MS4 where MS2 did not give sufficient structure information. Figure 27 shows an example of ESI-MS2 of a xyloglucan oligomer that contains additional fucosyl-O-acetyl-galactose in the side chain.

ESI-MS2 of a xyloglucan oligosaccharide with fucose and O-acetyl-galactose in the side chain (XXFG) obtained from enzymatic degradation of xyloglucan of blackcurrants. Reproduced from [147] with kind permission of the publisher
Coupling with capillary electrophoresis (CE), although not often employed, was applied in this case and shown to be superior to LC-ESI-MS. Labeling with a charged tag, APTS, which also allowed laser-induced fluorescence detection (LIFD), was necessary for CE and could solve some problems observed with RP-HPLC.
However, previous knowledge of the structural features of such glycans was considered for interpretation. This is helpful but bears the risk of misinterpretation if unexpected new structural features occur. Data and observations from various methods, their power and limitations, as well as the accumulated knowledge from biology (e.g., specificity of enzymes, relationships of taxonomy and structural features), chemistry, and instrumental analysis have to be combined in a plausible manner to generate a representative picture of such complex structures of biopolymers. HPEAC-ESI-MS has also been applied to enzymatic digests of legumes [148].
4.2.1.3 Pectins
Pectins are also ubiquitously occurring, very complex, and important heteropolysaccharides. They are found in the primary cell wall of all plants and form the middle lamella of higher plants. The main chain consists of α-1,4-linked galacturonic acid, which is partially methyl esterified and interrupted by rhamnose residues. Attached to this backbone, arabinans and other oligomeric heteroglycosidic side chains are found (“hairy regions”) (Fig. 28).

Schematic structure of pectin. The main chain is constituted of α-1,4-galcturonic acid, which is partially methyl esterified. Gelation mechanism and gel strength depend on the degree of methyl esterification and the distribution of methyl ester groups in the smooth regions
Therefore, it is not surprising that structural analysis of pectin has profited much from the recent developments in the field of mass spectrometry and other instrumental techniques [149]. As described before, a combination of isolation and fractionation steps, partial degradation by enzymes or chemical methods, labeling, various chromatographic and electrophoretic separations, and finally off-line or online MS and tandem-MS give a more and more detailed insight into the structural features. The methyl esterification pattern could be analyzed after enzymatic digestion by MALDI ToF-MS [149]. Although sequencing is established in the field of proteins/peptides and nucleotides, it is still a challenge in the field of carbohydrates, although the work of Reinhold et al. is an impressing milestone [135–140]. Jensen et al. have reported the initial steps of a solid phase-supported sequencing approach for pectins [150].
5 Quantitative Analysis by Mass Spectrometry
The question of whether data obtained by ESI or MALDI mass spectrometry can be used for quantitative evaluations has already been addressed above with regard to the molecular weight distribution of polysaccharides. Quantitative analysis of known and available compounds using internal standards, often performed by online-LC-ESI-MS methods, will not be addressed here. However, we will now discuss the conditions under which signal strength can be used to determine the relative composition of a carbohydrate mixture.
The MALDI process is basically more suitable for quantitative measurements than the ESI methodology. Linear relationships of concentration (in a certain matrix) and signal height or area have been found for various compounds [151, 152]. Linearity of the individual response nevertheless requires calibration with the authentic compound or an appropriate internal standard. However, the relative response values in mixtures is of greater relevance for the determination of molar compositions e.g., of oligosaccharides released from O- or N-linked glycans. Therefore, Naven and Harvey studied the relative signal strength of equimolar mixtures of such oligosaccharides (28 pmol of each per target spot) of roughly similar type and covering the mass range from 420 to 2,400 m/z [151]. Although data are usually averaged in MALDI to level the shot-to-shot variations caused by the heterogeneity of the spot (here from 240 shots), standard deviation of a triple determination was in most cases between 7 and 18% when a sector field instrument was coupled to the ion source as mass analyzer (the matrix was 2,5-dihydroxy benzoic acid, DHB). Choice of the matrix had little influence, although for connection with a ToF analyzer, the response of oligosaccharides increased until m/z 1,000 then remained stable but with poor precision. Bias towards the low molecular mass analytes is probably caused by detector saturation through matrix molecules in the lower m/z area.
By labeling with a charge-providing tag, higher intensity and (important for quantitative MS) independency of sodium adduct and avoidance of multiple ion adduct formation is achieved. Powerful reagents are the already mentioned positively charged GT as well as o-aminobenzoic acid (2-AA) for negative mode MS. Kim et al. applied this procedure to oligosaccharides released from neutral N-linked glycans, and proved the method with an equimolar mixture of gluco- and mannooligosaccharides [65, 66]. Evaluation of signal areas but not of heights from MALDI ToF-mass spectra of GT-labeled oligosaccharides agreed well with data obtained by normal phase (NP)-HPLC of the 2-AA labeled mixture (using fluorescence detection) as reference method. Although 100 pmol of substance was required for HPLC, only 20 pmol was necessary for MS analysis [65].
In a similar approach, quantification of a roughly equimolar mixture of glucose up to maltohexaose was carried out [152a]. The exact composition of the maltooligosaccharides was determined by HPTLC (high performance thin layer chromatography) of the 2-AA-labeled compounds. Sample spots for MALDI-ToF-MS contained about 10 pmol of each constituent of the GT-labeled mixture. Matrix and the laser power were varied, with HABA [2-(4-hydroxyphenylazo)-benzoic acid] turning out to be the most appropriate matrix, although the laser power was adjusted significantly above the usually recommended threshold of ion formation (Fig. 29, left). For ESI IT-MS, a mixture containing ca. 160 pmol/μL of each compound was applied using a syringe pump (200 μL/h), and instrument parameters as target mass and, related to this, RF amplitude and capillary exit voltage were varied. Best results were obtained at highest target mass, i.e., m/z of DP 6, for the GT-labeled maltooligosaccharides (Fig. 29, right). Average deviation compared to the reference method (HPTLC) was 1–2% under these conditions. Only slight differences were observed for area and height evaluations.

Quantitative mass spectrometric analysis (positive mode) of a mixture of maltooligosaccharides (DP1–DP6), labeled with Girard’s T (see Fig. 5). Left: MALDI ToF mass spectra with HABA [2-(4-hydroxyphenylazo)-benzoic acid] as matrix under variation of the laser power (45, 50, 55, and 60%, top to bottom); M matrix signals. Right: ESI IT mass spectra under variation of the target mass from m/z of DP1 to DP6 (top to bottom)
5.1 Tandem Mass Spectrometry for Quantification
Tandem mass spectrometry opens a chance for quantification of isobaric mixtures, which are not separated in the mother spectrum. This is a very typical problem in carbohydrate analysis, since many constituents only differ in stereochemistry or are regioisomers with different patterns of the same functional groups or substituents, as in polysaccharide derivatives. With the exception of bacterial lipopolysaccharides, which beside the core region consist of repeating units of oligomeric size, polysaccharides show a high diversity with respect to the distribution of their building bricks, branching pattern, side chain length, and substituents like acetyl or sulfate groups. On the other hand, structural diversity is not simply random, since enzymes involved in the biosynthesis act with certain selectivity, and often various topological patterns exist. Since various enzymes are available, nature produces different types of basically the same polysaccharides, e.g., alginates, galactomannans, pectins, or carrageenans, in random, regular, or block-like patterns.
Analysis of such patterns usually involves partial degradation to oligosaccharides, which can be performed in a more or a less selective manner, using various enzymes or chemical methods, most commonly acid hydrolysis. Chromatographic separation of oligosaccharides obtained by such procedures is limited due to the high complexity, which rapidly increases with DP. A nice example of how tandem MS can help to elucidate the composition of isobars in such mixtures has been reported by Haebel et al. [153]. They analyzed the qualitative and quantitative composition of oligosaccharides derived from partially deacetylated chitin by enzymatic digestion (Fig. 30). These hetero-chitooligosaccharides DnAm (D = GlcNH 2, A = GlcNAc) were chromatographically fractionated and the isomeric mixtures analyzed by MALDI linear ion trap-MS, using CID up to MS3. As usually applied in the analysis of polysaccharide derivatives [23], chemical uniformity of the chitooligomers was achieved by N-acetylation with Ac2O-d 6 . Thus, the originally present and later introduced acetyl groups can be differentiated by Δm/z of 3. By reductive amination with 3-acetylamino-6-aminoacridine, derivatives were generated that exclusively produced Y fragments, always bearing the protonated tag. (Fig. 31) [153]. From their pattern and from additional MS3 experiments, the contribution of individual sequences to isobaric mixtures could be determined up to DP 8 and were shown to consist of an equal number of GlcNAc and GlcNH 2 (D4A4) sequences. However, due to selectivity of the enzyme applied, 96% of this mixture comprised only eight of the theoretically possible 70 different sequences with individual contributions of between 2 and 34%.

MALDI LIT-CID-MS spectrum of [M + H]+of isobaric mixture of N-perdeuteroacetylated D3A4-T chitooligosaccharides at m/z 1,684. D GlcNAc-d 3 from GlcNH 2 , A GlcNAc, T tag = 3-N-acetyl-aminoacridine. Quantitative evaluation is outlined in Fig. 31. Reproduced from [153] with kind permission of the publisher

Quantitative sequencing procedure for a mixture of D3A4 isomers. The structure of the main constituent (DADADAA-T) after N-acetylation-d 3 of D units is shown above. Sequences that may be excluded due to the absence of a peak at the corresponding mass are printed in gray. Results were obtained from MS2 spectra (shown in Fig. 30) and from the MS3 spectra of the two Y6 fragments at m/z 1,481 and 1,478 (not shown). Reproduced in modified form from [153] with kind permission of the publisher
Another example deals with the elucidation of the complete monomer composition of methyl amylose or cellulose (Fig. 32) by ESI IT-MS2 and by MS3 of maltose or cellobiose derivatives obtained after permethylation with MeI-d 3 and subsequent partial hydrolysis [130].

Eight monomers with different methylation patterns present in a 1,4-glucan after partial O-methylation
Based on ESI IT-MSn studies with regioselectively O-methyl/O-deuteromethyl maltoses, ions could be assigned to certain glucose fragments [131]. By combining the data obtained for the isotopic patterns of the various fragment ions, all molar ratios of the eight different glucosyl units present in methyl cellulose could be calculated. Since no total hydrolysis is required, as it is for chromatographic or electrophoretic separation and determination of these eight constituents, this approach can be regarded as an independent reference method.
Figure 33 illustrates the evaluation of the mass spectra for a monosubstituted dimer bearing one CH3 and six CD3 groups. The m/z of [M + Na]+ is 464. The Y1 fragment ion of the CID-MS presents the reducing part of the disaccharide. The 1:1 ratio of the intensities at m/z 251 and 254 indicates that the methyl group is located with the same probability at both units of the dimer. The ratio of the abundances of the 0,2A2 ions reflects the probabilities of a CH3-group at O-2 and O-3, or O-6. Elimination of methanol from Y1 is known to involve RO-3. Thus, the ratio of O-3- to (O-2 + O-6)-substitution can be calculated from these fragments. Finally, each signal of the MS1 spectrum (Fig. 34a) is distributed to the contributing isomeric methyl patterns. Since information obtained by MS2 (Fig. 34c) is not sufficient for all isobaric mixtures, MS3 is performed (Fig. 34e, f). For this fragmentation, Li+ adducts are required (Fig. 34b, d) because Na+ adducts dissociate at the collision energy required, as outlined above (Fig. 35). Finally, all data are summed and the complete monomer composition of methylcelluloses, methylamylose or methylcyclodextrin is obtained. For details see [130].

Evaluation of the monomer composition of the monosubstituted fraction of OMe/OMe-d 3 disaccharides obtained from methyl amylose. As an example, the step-by-step-distributions of the signal intensity of the penta-O-methyl-d 3 -mono-O-methyl-dimer ([M + Na]+ m/z 464) on non- (50%), 2-, 3- and 6-O-monosubstitution are shown above. U unsubstituted. Reproduced in modified form from [130] with kind permission of the publisher

ESI-MSn of OMe/OMe-d 3 disaccharides obtained from methyl amylose. ESI-MS of [M + Na]+ and [M + Li]+ (a, c), ESI-MS2 of dimers with 3 Me and 3 Me-d 3 of Na+ and Li+ adducts (b, d), and ESI-MS3 of Y1 at m/z 232 (e), and ESI-MS3 of Y1 at m/z 235 (f). For fragment evaluation see Fig. 35. Reproduced from [130] with kind permission of the publisher

6 Polysaccharide Derivatives
Polysaccharide derivatives, i.e., glycans with non-sugar substituents, are produced both naturally and chemically. Sulfates are common functional groups, e.g., in glycosaminoglycans (e.g., heparin, chondroitin sulfate) and algae polysaccharides (e.g., carrageenan, agar); phosphate esters occur in bacterial lipopolysaccharides or in potato starch; acetate groups are known from hemicelluloses (galactoglucomannan, xylan), acemannan from Aloe vera, or xanthan produced by Xanthomonas campestris. Methyl ethers are found in the terminal glucuronic acid of xylans, in cell wall polysaccharides of Chlorella vulgaris [154, 155], or in various bacterial and fungal polysaccharides, e.g., 2-O-methyl-mannose in mucoralean extracellular polysaccharides [156, 157]. On the other hand, chemical modification of widely available polysaccharides, especially cellulose and starch, is performed on an industrial scale [23, 158–160]. Less abundant glycans such as galactomannans (guar, locust bean gum) or dextrans have also been functionalized by chemical transformation [161].
Chemically modified polysaccharides can be considered as semisynthetic copolymers of high complexity. In addition to the dispersity of the starting material (e.g., concerning their molecular weight), chemical dispersity is established in the modification process. Distribution of substituents has to be considered on various structural levels, which will be outlined here using cellulose as an example. In the glucosyl unit, three different OH groups are available in positions 2, 3, and 6 (see Fig. 32). The distribution in the next dimension, the cellulose chain, depends on the content and arrangement of non-, mono-, di- and trisubstituted monomer residues. If all units of the chain are equally accessible during the reaction, a random pattern is obtained. Depending on the reaction system (homogeneous, heterogeneous, protic, aprotic etc.) and the interactions between primary substituents and remaining OH-groups or reagent, the pattern can deviate from the random model in various ways. Related to this heterogeneity of second order, a heterogeneity of first order also exists, i.e., along the polymer chains within the material (third dimension). Although the material can be fractionated with respect to the heterogeneity of first order, the second order heterogeneity is located on single macromolecules and therefore only analyzable after partial depolymerization [23, 162–167]. The latter can be achieved either by chemical or enzymatic degradation [121, 128, 168, 169].
Method development to gain detailed knowledge of the substituent distributions in all these dimensions is mainly motivated by the fact that the properties of such compounds, e.g., solubility, degradability, or flocculation points in thermoreversible gelation (and the dynamics of these processes), are affected by the chemical structure of the material. Within this very challenging field of structure analysis, MS has always been a very important instrumental method and is still of high potential. Due to the lack of standard compounds, GLC/electron impact (EI) MS is usually applied in monomer analysis because it allows elucidation of the substitution pattern in glucose-based derivatives due to characteristic shifts of fragment ions [23, 170].
The development of soft ionization methods has enabled the analysis of less volatile oligomeric analytes, giving information about the probabilities of various domains in the chains, i.e., the distribution in the chain. Since basic aspects of the ionization processes, labeling, quantification, tandem MS, and fragmentation mechanisms have already been addressed above, this section will focus on the application of these methods to cellulose and starch derivatives to illustrate the high value of MS for this field of carbohydrate analysis [171].
6.1 Methyl Ethers
Methyl ethers are not only important commercial products derived from cellulose and are used as adhesives and thickeners, but are also appropriate model compounds for analytical method development because the methyl group is chemically stable, small, neutral, and available in isotope-labeled version. Therefore, several basic studies, including MS, have been performed on methyl ethers of cellulose [128, 130, 166, 167, 169, 171, 172], amylose [164], starch [121], and dextrans [173].
Analysis of the substitution pattern in the polymer chain of such methyl glucans has been tackled by the following four-step approach:
-
1.
Perdeuteromethylation, which makes the compound chemically uniform, but allows differentiation of original and introduced methyl groups. This is also a prerequisite for the next step.
-
2.
Partial random degradation, which can be performed by aqueous hydrolysis or methanolysis.
-
3.
Mass spectrometric analysis by FAB- [162, 174], ESI- [167, 169, 173] or MALDI ToF-MS (3) [128, 129, 162].
-
4.
Quantitative evaluation of MS data and comparison of the substituent distributions in a certain oligosaccharide fraction with the theoretical distribution calculated from the monomer data, determined independently (Fig. 36).
Fig. 36 
Sample preparation (above) and ESI-MS (below) of methyl cellulose (DS 1.32) for analysis of the substituent distribution along the polymer chain. (DP2 is shown in extended scale in Figs. 33 and 34). Quantifiability of signal abundances is proved by perdeuteromethylation, giving chemically uniform analytes in a narrow m/z range. Methyl pattern of each DP is calculated and compared with the random distribution for the glucose constituents present (compare Fig. 32) [23, 162, 174]

With respect to MS, the most important point in this analytical approach is the quantifiability of the constituents of a certain DP on the basis of signal strength. It is not essential to quantify the relative portions of oligomers, although it would be of additional value to control the randomness and degree of partial depolymerization. In contrast, this is of higher importance for enzymatically digested cellulose or starch ethers.
In the case of methyl ethers, the required quantifiability is achieved by permethylation with MeI-d 3 , to get a chemically uniform, isotope-labeled product. The m/z-range for the oligomers obtained by partial hydrolysis of these compounds is 449–467 for DP2, 653–680 for DP3, and 857–893 for DP4, which means an appropriate mass range for ESI IT-MS. The signals within these oligomer fractions differ by m/z 3, and thus are of sufficient similarity that they are not discriminated by the ionization process or the mass analyzer, but just differing enough to avoid significant overlapping of 13C-related isotopic signals of compounds with various degrees of methylation. At DP4, the relative intensity of the [M + Na + 3]+ ion, which is isobaric with the next higher deuterated homolog, is 2.77% of the main peak or 1.77% of the total intensity (Fig. 37). Since this overlap extends throughout the whole pattern, the final distortion is not significant at this DP and not even very pronounced at DP 9 as long as the distribution is not very narrow or bimodal.

Isotope composition of tetrakis[2,3,6-tri-O-methyl]-cellotetraose
Although in the pioneer work in this field (still employing FAB-MS), correction for the abundant noise was an important factor in evaluation [162, 174], this is often not necessary for the much higher quality ESI and MALDI mass spectra. However, for ions of low intensity at the profile borders, this can be critical, depending on the S/N ratio. One has also to decide whether to summarize the 13C-isotope peaks at [M + Na]+, [M + Na + 1]+, and [M + Na + 2]+ for each signal, or to use only the base peak for evaluation. To sum up all these intensities could average the contribution of noise, but at the same time could impair the quality of small signals, since their isotopic sister ions often strongly deviate from theory due to decreasing S/N ratio. Therefore, as long as all compounds within a set for quantitative evaluation comprise the same C number, as is the case for O-methyl-O-methyl-d 3 -oligoglycosides, and the [M + Na]+ signal is the most abundant, then quantitative evaluation is best based on these main peaks. The situation is different when the number of C atoms, and thus the relative ratios of isotope peaks, differs within a certain DP, as is the case for hydroxyalkylmethyl ethers (see Sect. 6.2). By this approach, various types and extents of deviation from calculated patterns have been determined for methyl polysaccharides [23, 162].
Higher oligomers are only present in lower amounts in a partially degraded polysaccharide derivative and are discriminated in ESI-MS by direct infusion with a syringe pump due to the facts outlined above. To record mass spectra of good quality for these higher homologs, LC-ESI-MS can be applied. The coupling with the chromatographic system allows ionization of each set of analytes of the same DP without competition by compounds with higher surface activity or electrophoretic mobility, and at the same time enables automation. The continuously recorded mass spectra are accumulated for the DP-corresponding intervals. Separation should not be optimized with respect to fast elution and narrow peaks, since broad peaks eluting over a longer time period allow recording and summing of more mass spectra, and thus enhance the sensitivity and quality of the data. The scan range should be programmed for each DP according to the m/z range of interest. To detect the analytes independently by UV detector, a chromophor was introduced by reductive amination with m-amino benzoic acid (3-AA) [172].
6.2 Hydroxyalkyl Methyl Ethers
Cellulose derivatives other than methyl ethers have also been transformed to O-methyl-O-methyl-d 3 -celluloses. This approach was applicable to cellulose acetates [175] and sulfates [176]. In the case of hydroxyalkyl methyl ethers such as hydroxyethyl methyl cellulose (HEMC), and the corresponding hydroxypropyl derivative (HPMC), which are widely applied in the construction, pharmacy, and food areas, this is impossible because the substituent also bears a free OH group. These hydroxyl groups will undergo the same transformations as (or like?) the remaining OH of the cellulose backbone and, as a consequence, the chemical and mass differences will be maintained. In addition, some of the OH groups are already methylated. Therefore, permethylation or perdeuteromethylation are the methods of choice to increase, at least chemical uniformity of the compounds. However, since hydroxyalkyl and methoxyalkyl groups are flexible, and alter polarity and sodium complexation ability, oligosaccharides bearing these groups are extremely overrepresented in ESI mass spectra. Therefore, a permanent cationic group has been introduced by reductive amination with n-propylamine and subsequent permethylation. In ESI-MS, a decreasing trend in degree of substitution (DS)/DP was still observed. DS should be constant over all DPs and resemble the average DS of the sample if all steps of the analytical procedure (partial degradation, derivatization, work-up, MS analysis) are free of discrimination. MALDI-MS gave representative results for HEMCs, HPMS, and HECs [90, 91, 177].
In Fig. 38, the ESI-IT-mass spectrum of an O-deuteriomethylated partially hydrolyzed HEMC is compared with those recorded after reductive amination and subsequent quaternization. Total abundance and S/N ratio is stepwise enhanced. Correct quantitative evaluation of the hydroxyethyl pattern was only possible using the MALDI ToF mass spectra.

ESI-IT-MS of hydroxyethylmethylcellulose (HEMC). The sample was perdeuteromethylated and partially hydrolyzed (a), reductively aminated (b), and quaternized with MeI (c). The number of hydroxyethyl (HE) and methyl (Me) groups are shown. For explanation see text or for more details [91]. Reproduced in modified form from [91] with kind permission of the publisher
The methyl pattern and hydroxyalkyl pattern are analyzed independently. Although for conservation of the methyl pattern permethylation with MeI-d 3 is necessary, the sensitivity for the analysis of the hydroxyalkyl pattern can be enhanced by using permethylation, since all oligosaccharides representing a certain number of hydroxyalkyl groups are then concentrated in one peak rather than being split into several peaks with various ratios of Me and Me-d 3 . Other labels, described above, can also be introduced instead of labeling with an aliphatic amine and additional quaternization.
6.2.1 Quantitative Evaluation
Since the isotopic pattern slightly shifts with increasing number of C atoms, the question of whether quantitative evaluation should be based on the main signal only, or whether 13C isotope peaks should be included, is of higher relevance for these mixed ethers. In the case of O-methylated hydroxyethyl ethers, the number of C atoms range from nine [n(HE) = 0] to 15 [n(HE) = 3 per glucosyl unit; n is theoretically unlimited because the substituent can undergo tandem reactions]. As a consequence for DP 2, the first ion with n(HE) = 0 represents 73.87% of the total ions, and for n(HE) = 6, it represents only 63.83% of all isotope peaks. All other signals [n(HE) = 1…5] lie in between (see Fig. 39). Therefore, it is important to sum up the isotope peaks at least at M + 1 and M + 2 for these derivatives [corresponding to 99.23% (Me6) of the total ion abundance, and to 98.12% (HEme6), respectively as shown in Fig. 39]. This can be achieved by taking the values from the mass spectrum, but, if smaller peaks are not accurately measured, by calculating the entire intensity from the base peak using the known isotopic composition. If necessary, overlapping of [M + 3]+ isotope peaks with the next higher [M]+ must be corrected by subtraction of the calculated amount to avoid a distortion in favor of higher deuteromethylated isomers of a certain DP profile.

6.3 Application of Enzymes
Enzymes have also been applied in combination with MS to analyze the substituent pattern in cellulose and starch derivatives [23, 112, 132, 178–186]. Degradability decreases with increasing number of substituents, since they interfere with the formation of the active complex. However, at the same DS and for a given enzyme, digestibility strongly depends on the location of the substituents on the various hierarchical structure levels. MS is again a very valuable tool for the study of oligosaccharides released by enzymes. Patterns can be analyzed in the same manner as described above for chemically degraded glycans. In contrast to the latter, not only the DS, but also the DP pattern is of interest here. As is obvious from what has been outlined above, MALDI-MS is superior to ESI-MS in this respect, but derivatization can improve ESI-MS, enabling quantitative evaluation under appropriate conditions.
To elucidate the enzyme specificity, i.e., in which positions substituents are accepted at the cleavage site, tandem MS is of high value. Fragmentation provides information about the number and position at the reducing end. For example, unsubstituted residues of the n-mer derived from a 1,4-glucan like starch or cellulose will give 0,2An (M-60) and subsequent 2,4An (M-120). One methyl group at the new reducing end (position −1 in the active complex) causes a loss of 194 instead of 180u to form Bn−1. Location of this methyl group at O-6 can be recognized from a loss of 60 and, subsequently, a shift to −74. Location at O-2 will shift the first cross-ring cleavage to M-74, followed by a loss of 60, while blocking of 3-OH suppresses A-fragment formation (see Figs. 15 and 16). To differentiate overlapping patterns and avoid erroneous interpretation, separation is required. However, due to the enzymes’ specificity, substitution at the cleavage site is usually very restricted. Enebro et al. applied a different approach for this type of position analysis. The oligosaccharides obtained by enzymatic digestion from carboxymethylcellulose (CMC) were permethylated, after the end groups had been reduced to alditols, and after total methanolysis analyzed by LC/ESI-MS2. CID mass spectra of the non-carboxymethyl-substituted glucitol (Fig. 40a) showed subsequent losses of 3 × MeOH. Diagnostically more valuable fragmentations were induced by the free OH at C4 between C3–C4 and C4–C5 of the alditol. Carboxymethylation at O-6 induced a shift for the C4–C6 fragment from m/z 125 t to m/z 183 (Fig. 40b). For the 2- and 3-O-carboxymethyl isomers, the C1–C4 fragment was shifted from m/z 196 to m/z 254 (Fig. 40c, d). These two secondary ethers could not be unambiguously differentiated from the tandem mass spectra, but the independent assignment agreed with an assumed higher probability of ROH elimination from position 3 (m/z 196 and m/z 228 are higher for 3-O-CM than for 2-O-CM). Thus it was found that one of the enzymes (endoglucanases) tolerated carboxymethyl groups at O-6, and another only at O-3, while position 2 was not substituted at all in the released reducing units [132].

ESI CID-MS ([M + Li]+) of 1,2,3,5,6-penta-O-methyl- d-glucitols-1-d (a) and isomeric 2-, 3- and 6-mono-O-methoxycarbonyl-tetra-O-methyl-d-glucitols-1-d (b, c, d, respectively) obtained from CMC by partial degradation (here acid hydrolysis), reduction with NaBD4, permethylation, and methanolysis. The position of the carboxymethyl group as shown in the formulae can be deduced from certain fragment shifts of b, c, and d compared to a. For details see text. Reproduced from [132] with kind permission of the publisher
Cationic starches [O-(2-hydroxy)propyl-3-trimethylammonium] have also been studied by ESI-MS-CID after exhaustive enzymatic degradation [112]. Due to multiple charged state of the oligosaccharides released, up to fourfold substituted dodecamers (DP12) were detected (Fig. 41). Tandem MS and evaluation of the fragmentation pathway by 2H and 18O labeling allowed differentiation of more homogeneous and more heterogeneous modification of the starch granules under various cationization conditions. H–D exchange of OH indicated that in the first loss of 60 u, two exchangeable H, i.e. from OH, are involved (shift of Δm/z to 62), The second loss is only shifted from 60 to 61 u, which means that one H is abstracted from a C–H (Fig. 42). ESI-MS2 of the isolated monosubstituted cationic maltotrioses, the main products from enzymic digestion (see Fig. 41), proved that no substitution is tolerated at the new formed reducing glucosyl unit (i.e., at the −1 cleavage site) by the α-amylase (from B. licheniformis) as has been found for methyl starches [179]. From the fragment ratios of the shifted B and Y ions it is evident that the cationic substituent is mainly located at the inner glucosyl unit and to a less extent at the new non-reducing end.

ESI-MS and ESI-MS2 of O-(2-hydroxypropyl-3-trimethylammonium) maltooligosaccharides obtained from cationic starch (DS 0.24) by enzymatic hydrolysis with α-amylase and amyloglucosidase. Signals are assigned with the DP (first number), and the number of cationic substituents (n, second number). The main product (3/1) is a monosubstituted maltotriose, for which a structure example is given. Fragmentation of 3/1 is shown in Fig. 42

ESI-MS2 of (a) the mono-O-(2-hydroxy-3-trimethylammonium)propyl-maltotriose (3/1 in Fig. 40) obtained from cationic starch (DS 0.24) by enzymic degradation in D2O and (b) of maltotriose
One has to keep in mind that in contrast to charge-remote fragmentations of sodiated molecules, only those fragments bearing the covalently linked charged substituent – and thus only shifted ions – can be observed. While B2 comprises the maltotriose isomers functionalized at the terminal and the inner 1,4-linked glucosyl residue, Y2 is only observable for the latter.
Therefore, the enhanced B2/Y2 ratio and the occurrence of B1 in the daughter spectrum of the monosubstituted maltotriose undoubtedly indicate the acceptance of substitution at the +1 and +2 cleavage sites of the exo-enzyme amyloglucosidase. With respect to the endo-enzyme α-amylase, tolerance of substitution at the −2 and −3 cleavage sites can be revealed from the ESI-CID-MS data. Cationic starches have also been studied by HPAEC-MS after enzymic hydrolysis [178].
6.4 Carbohydrate-Based Block Copolymers: Determination of Block Length
Another semisynthetic type of oligo- or polysaccharide is presented by copolymers prepared by cationic ring-opening polymerization (CROP) from cyclodextrin derivatives as macromonomers. In nature, block structures can be produced by certain enzymes, e.g., in alginates. These polyuronic acids are formed by postmodification of a β-1,4-d-mannuronan by inversion at C-5, thus forming α-1,4-guluronic acids which, depending on the type of enzyme, can be distributed in random, regular, or block-like patterns [23, 162].
No real multiblock polysaccharide structures can be established using chemical modification, and the number of monomer patterns from the eight possible ones (see Fig. 32) can only be reduced by temporary protection of one or two OH positions. However, since block structures are of great interest for model studies of e.g. gelation properties, cyclodextrin derivatives have been applied as macromonomers. Heptakis[2,3,6-tri-O-methyl]-cyclomaltoheptaose (A7) and heptakis[2,3,6-tri-O-methyl-d 3 ]-cyclomaltoheptaose (B7) were polymerized using various catalysts [186–188]. In addition to NMR spectroscopy, MS is also a very powerful method for analyzing the block length of such products. The polymer obtained was partially degraded and the oligomeric products measured with ESI-MS. From the ratio of AA, AB/BA, and BB dimers the average block lengths were calculated. In an early stage of the reaction the block length was 14 (2 × 7), as expected from theory. However, block length rapidly decreased because transglycosidation randomized the original block structure in a competing process, which dominated as soon as the substrate had been consumed. Figure 43 shows the corresponding reaction, MS, and evaluation scheme.

With respect to the accuracy of this method, two sources of error must be emphasized. First of all, in the case of incomplete conversion, residual substrate must be carefully removed, since its partial hydrolysis will produce only AA and BB sequences and thus cause too-high apparent block lengths. Second, the new glucosidic linkages formed in the polymerization process preferably show β-configuration, while the starting material is α-linked. However, hydrolysis rates for α- and β-glycosides are different [189]. Faster cleavage of β-linkages will cause enrichment of homogeneous AA and BB α-rich dimers at a high degree of degradation; consequently the block lengths are observed too high. Therefore, short and harsh hydrolysis conditions should be applied to suppress selectivity. Furthermore, it should be mentioned that end groups are neglected in the calculation of block length. Therefore, the DP must be high enough to justify this. Otherwise, terminal residues should be labeled for differentiation from the inner chain residues.
7 Conclusion and Outlook
Impressive progress has been achieved in mass spectrometric analysis of complex carbohydrates during the last decades. On the instrumental side, speed, sensitivity, and resolution have increased at the same time as detailed knowledge on fragmentation mechanisms has been gained. How fragmentation pathways can be influenced by, e.g., appropriate labeling procedures or complexation has been established. In addition, in the popular and important field of glycoconjugate analysis, significant progress has been achieved for plant and bacterial polysaccharides. The analysis of substituent distribution in polysaccharide derivatives has also immensely profited from these new possibilities. It is expected that this trend will continue during the coming years, generating algorithms for automated data evaluation, improving quantifiability, and refining techniques for separation and differentiation of isobars and isomers, e.g., by making use of the topological effects on ion mobility. Such progress will allow more differentiated and higher throughput analysis, which could produce sufficient data for the statistical evaluation of substitution patterns in complex mixtures of polysaccharide derivatives. Further development of complexing agents and labeling procedures for specific fragmentation pathways in CID will probably improve and extend the applicability of sequence analysis, still a tedious and challenging endeavor.
Abbreviations
- AA:
-
Aminobenzoic acid
- AB:
-
Aminobenzamide
- AEE:
-
Aminobenzoic acid ethyl ester
- ANTS:
-
2-Aminonaphthalene-trisulfonic acid
- AP:
-
Aminopyridine
- APTS:
-
1-Amino-pyrene-trisulfonic acid
- CE:
-
Capillary electrophoresis
- CID:
-
Collision induced dissociation
- CMC:
-
Carboxymethylcellulose
- CROP:
-
Cationic ring-opening polymerization
- DESI:
-
Desorption electrospray ionization
- DHB:
-
Dihydroxybenzoic acid
- DHBB:
-
2,5-Dihydroxybenzoic acid/butylamine
- DNA:
-
Deoxy ribonucleic acid
- DP:
-
Degree of polymerization
- ESI:
-
Electrospray ionization
- FAB:
-
Fast atom bombardment
- FTIC:
-
Fourier transform ion cyclotron
- GLC:
-
Gas liquid chromatography
- GP:
-
Girard’s P [1-(hydrazinocarbonylmethyl)pyridinium chloride]
- GPC:
-
Gel permeation chromatography
- GT:
-
Girard’s T [1-(hydrazinocarbonylmethyl)trimethylammonium chloride]
- HABA:
-
2-(4-Hydroxyphenylazo)-benzoic acid
- HEC:
-
Hydroxyethyl cellulose
- HEMC:
-
Hydroxyethylmethyl cellulose
- HPAEC-PAD:
-
High performance anion exchange chromatography-pulsed amperometric detection
- HPC:
-
Hydroxypropyl cellulose
- HPMC:
-
Hydroxypropylmethyl cellulose
- HPSEC-MALLS:
-
High performance size-exclusion chromatography–multi-angle laser light scattering
- HPTLC:
-
High performance thin layer chromatography
- IEM:
-
Ion evaporation model
- IT:
-
Ion trap
- LIFD:
-
Laser-induced fluorescence detection
- MALDI:
-
Matrix-assisted laser desorption/ionization
- MS:
-
Mass spectrometry
- NMR:
-
Nuclear magnetic resonance
- NP:
-
Normal phase high performance liquid chromatography
- PEG:
-
Polyethyleneglycol
- PSD:
-
Post-source decay
- QIT:
-
Quadrupole ion trap
- Q-ToF:
-
Quadrupole time-of-flight mass analyzer
- RDA:
-
Retro-Diels–Alder
- RP-HPLC:
-
Reversed phase high performance liquid chromatography
- SEC:
-
Size-exclusion chromatography
- THAP:
-
2′,4′,6′-Trihydroxy-acetophenone
- ToF:
-
Time-of-flight
- UV:
-
Ultraviolet
References
Pfenninger A, Karas M, Finke B, Stahl B (2002) Structural analysis of underivatized neutral human milk oligosaccharides in the negative ion mode by nano-electrospray MSn (part 1: methodology). J Am Soc Mass Spectrom 13:1331–1340
Pfenninger A, Karas M, Finke B, Stahl B (2002) Structural analysis of underivatized neutral human milk oligosaccharides in the negative ion mode by nano-electrospray MSn (part 2: application to isomeric mixtures). J Am Soc Mass Spectrom 13:1341–1348
Schmid D, Behnke B, Metzger J, Kuhn R (2002) Nano-HPLC-mass spectrometry and MEKC for the analysis of oligosaccharides from human milk. Biomed Chromatrogr 16:151–156
Dreisewerd K, Kölbl S, Peter-Katalinic J, Berkenkamp S, Pohlentz G (2006) Analysis of native milk oligosaccharides directly from thin-layer chromatography plates by matrix –assisted laser desorption/ionization orthogonal time-of-flight mass spectrometry with a glycerol matrix. J Am Soc Mass Spectrom 17:139–150
Spina E, Sturiale L, Romeo D, Impallomeni G, Garozzo D, Waidelich D, Glueckmann M (2004) New fragmentation mechanisms in matrix-assisted laser desorption/ionization time-of-flight/time-of-flight tandem mass spectrometry of carbohydrates. Rapid Commun Mass Spectrom 18:392–398
Chen G, Pramanik BN, Bartner PK, Saksena AK, Gross ML (2002) Multiple-stage mass spectrometric analysis of complex oligosaccharide antibiotics (everninomicins) in a quadrupole ion trap. J Am Soc Mass Spectrom 13:1313–1321
Harvey DJ (2011) Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update for the period 2005–2006. Mass Spectrom Rev 30:1–100
Harvey DJ (2009) Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update for 2003–2004. Mass Spectrom Rev 28:273–361
Harvey DJ (2008) Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update covering the period 2001–2002. Mass Spectrom Rev 27:125–201
Harvey DJ (2006) Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update covering the period 1999–2000. Mass Spectrom Rev 25:595–662
Kirsch S, Bindila L (2009) Nano-LC and HPLC-chip-ESI-MS: an emerging technique for glycobioanalysis. Bioanalysis 1:1307–1327
Huhn C, Selman MHJ, Ruhaak LR, Deelder AM, Wuhrer M (2009) IgG glycosylation analysis. Proteomics 9:882–913
Ruhaak LR, Deelder AM, Wuhrer M (2009) Oligosaccharide analysis by graphitized carbon liquid chromatography-mass spectrometry. Anal Bioanal Chem 394:163–174
Grice ID, Wilson JC (2009) Analytical approaches towards the structural characterization of microbial wall glycopolymers. In: Moran AP, Holst O, Brennan PJ, von Itzstein M (eds) Microbial glycobiology: structures, relevance and applications. Academic, Oxford, pp 233–252
Harvey DJ (2005) Structural determination of N-linked glycans by matrix-assisted laser desorption/ionization and electrospray ionization mass spectrometry. Proteomics 5:1774–1786
Harvey DJ (2005) Proteomic analysis of glycosylation: structural determination of N- and O-linked glycans by mass spectrometry. Proteomics 2:87–101
Harvey DJ (1999) Matrix-assisted laser desorption/ionization mass spectrometry of carbohydrates. Mass Spectrom Rev 18:349–450
Harvey DJ (2003) Matrix-assisted laser desorption/ionization mass spectrometry of carbohydrates and glycoconjugates. Int J Mass Spectrom 226:1–35
Zaia J (2004) Mass spectrometry of oligosaccharides. Mass Spectrom Rev 23:161–227
Jang-Lee J, North SJ, Sutton-Smith M, Goldberg D, Panico M, Morris H, Haslam S, Dell A (2006) Glycomic profiling of cells and tissues by mass spectrometry: fingerprinting and sequencing methodologies. Methods Enzymol 415:59–86
Haslam SM, North SJ, Dell A (2006) Mass spectrometric analysis of N- and O-glycosylation of tissues and cells. Curr Opin Struct Biol 16:584–591
Campa C, Coslovi A, Flamigni A, Rossi M (2006) Overview on advances in capillary electrophoresis-mass spectrometry of carbohydrates: a tabulated review. Electrophoresis 27:2027–2050
Mischnick P, Momcilovic D (2010) Structure analysis of starch and cellulose derivatives. Adv Carbohydr Chem Biochem 64:117–210
Short LC, Syage JA (2008) Electrospray photoionization (ESPI) liquid chromatography/ mass spectrometry for the simultaneous analysis of cyclodextrin and pharmaceuticals and their binding interactions. Rapid Commun Mass Spectrom 22:541–548
Zhang H, Zhang H, Qu C, Bai L, Ding L (2007) Fluorimetric and mass spectrometric study of the interaction of β-cyclodextrin and osthole. Spectrochim Acta A Mol Biomol Spectrosc 68A:474–477
Kensinger RD, Yowler BC, Benesi AJ, Schengrund C-L (2004) Synthesis of novel, multivalent glycodendrimers as ligands for HIV-1 gp120. Bioconjug Chem 15:349–358
Han S, Yoshida T, Uryu T (2007) Synthesis of a new polylysine-dendritic oligosaccharide with alkyl spacer having peptide linkage. Carbohydr Polym 69:436–444
Kebarle P, Verkerk UH (2009) Electrospray: from ions in solution to ions in the gas phase: what we know now. Mass Spectrom Rev 28:898–917, and references cited herein
Rodrigues JA, Taylor AM, Sumpton DP, Reynolds JC, Pickford R, Thomas-Oates J (2007) Mass spectrometry of carbohydrates: newer aspects. Adv Carbohydr Chem Biochem 62:58–141
Zhou S, Cook KC (2001) A mechanistic study of electrospray mass spectrometry: charge gradients within electrospray droplets and their influence on ion response. J Am Soc Mass Spectrom 12:206–214
Cole RB (2010) Electrospray and MALDI mass spectrometry, 2nd edn. Wiley-VCH, Weinheim
De Hoffmann E, Stroobant V (2007) Mass spectrometry, 3rd edn. Wiley-VCH, Weinheim
Hillenkamp F, Peter-Katalinic J (eds) (2007) MALDI MS. Wiley-VCH, Weinheim
Clowers BH, Dwivedi P, Steiner WE, Hill HH Jr (2005) Separation of sodiated isobaric disaccharides and trisaccharides using electrospray ionization atmospheric pressure ion mobility-time of flight mass spectrometry. J Am Soc Mass Spectrom 16:660–669
Von Helden G, Wyttenbach T, Bowers MT (1995) Inclusion of a MALDI ion source in the ion chromatography technique: conformational information on polymer and biomolecular ions. Int J Mass Spectrom Ion Proc 146(147):349–364
Wyttenbach T, Von Helden G, Bowers MT (1997) Conformations of alkali ion cationized polyethers in the gas phase: polyethylene glycol and bis[(benzo-15-crown-5)-15-ylmethyl] pimelate. Int J Mass Spectrom Ion Proc 165(166):377–390
Schmidt A, Karas M, Dülcks T (2003) Effect of different solution flow rates on analyte ion signals in nano-ESI MS, or: when does ESI turn into nano-ESI? J Am Chem Soc Mass Spectrom 14:492–500
Bahr U, Pfenninger A, Karas M, Stahl B (1997) High-sensitivity analysis of neutral underivatized oligosaccharides by nanoelectrospray mass spectrometry. Anal Chem 69:4530–4535
Blair SM, Kempen EC, Brodbelt JS (1998) Determination of binding selectivities in host-guest complexation by electrospray/quadrupole ion trap mass spectrometry. J Am Soc Mass Spectrom 9:1049–1059, and references cited herein
Lee S, Wyttenbach T, Von Helden G (1995) Gas phase conformations of Li+, Na+, K+, and Cs + complexes with 18-crown-6. J Am Chem Soc 117:10159–101560
Hofmeister GE, Zhou Z, Leary JA (1991) Linkage position determination in lithium-cationized disaccharides: tandem mass spectrometry and semiempirical calculations. J Am Chem Soc 113:5964–5970
Reale S, Teixido E, De Angelis F (2005) Study of alkali metal cations binding selectivity of β-cyclodextrin by ESI-MS. Ann Chim 95:375–381
Reinhold VN, Reinhold BB, Costello CE (1995) Carbohydrate molecular weight profiling; sequence, linkages, and branching data: ES-MS and CID. Anal Chem 67:1772–1784
Harvey DJ (2000) Collison-induced fragmentation of underivatized N-linked carbohydrates ionized by electrospray. J Mass Spectrom 35:1178–1190
Harvey DJ (2001) Ionization and collision-induced fragmentation of N-linked and related carbohydrates using divalent cations. J Am Soc Mass Spectrom 12:926–937
Harvey DJ (2005) Ionization and fragmentation of N-linked glycans as silver adducts by electrospray mass spectrometry. Rapid Commun Mass Spectrom 19:484–492
Desaire H, Leary JA (1999) Differentiation of diastereomeric N-acetylhexosamine monosaccharides using ion trap tandem mass spectrometry. Anal Chem 71:1997–2002
Gaucher SP, Leary JA (1998) Stereochemical differentiation of mannose, glucose, galactose, and talose using zinc(II) diethylenetriamine and ESI-ion trap mass spectrometry. Anal Chem 70:3009–3014
Desaire H, Laevell MD, Leary JA (2002) Solvent effects in tandem mass spectrometry: mechanistic studies indicating how a change in solvent conditions an pH can dramatically alter CID spectra. J Org Chem 67:3693–3699
Dole M, Mack LL, Hines RL, Mobley RC, Ferguson LS, Alice MB (1968) Molecular beams of macroions. J Chem Phys 49:2240–2249
Iribarne JV, Thomson BA (1976) On the evaporation of small ions from charged droplets. J Chem Phys 64:2287–2294
Kebarle P, Tang L (1993) From ions in solution to ions in the gas phase. Anal Chem 65:A972–A986
Harvey DJ (2005) Fragmentation of negative ions from carbohydrates: part 1. use of nitrate and other anionic aducts for the productions of negative ion electrospray spectra from N-linked carbohydrates. J Am Soc Mass Spectrom 16:622–630
Harvey DJ (2005) Fragmentation of negative ions from carbohydrates: part 2. Fragmentation of high-mannose N-linked glycans. J Am Soc Mass Spectrom 16:631–646
Harvey DJ (2005) Fragmentation of negative ions from carbohydrates: part 3. Fragmentation of hybrid and complex N-linked. J Am Soc Mass Spectrom 16:647–659
Harvey DJ, Jaeken J, Butler M, Armitage AJ, Rudd PM, Dwek RA (2010) Fragmentation of negative ions from N-linked carbohydrates: Part 4. Fragmentation of complex glycans lacking substitution on the 6-antenna. J Am Soc Mass Spectrom 45:528–535
Harvey DJ, Royle L, Radcliffe CM, Rudd PM, Dwek RA (2008) Structural and quantitative analysis of N-linked glycans by matrix-assisted laser desorption ionization and negative ion nanospray mass spectrometry. Anal Biochem 376:44–60
Lamari FN, Kuhn R, Karamanos NK (2003) Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometric structure analysis. J Chromatogr B 793:15–36
Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M (2010) Glycan labeling strategies and their use in identification and quantification. Anal Bioanal Chem 397:3457–3481
Harvey DJ (2000) Electrospray mass spectrometry and fragmentation of N-linked carbohydrates derivatized at the reducing terminus. J Am Soc Mass Spectrom 11:900–915
Nicotra F, Cipolla L, Peri F, Ferla BL, Redaelli C, Derek H (2007) Chemoselective neoglycosylation. Adv Carbohydr Chem Biochem 61:353–398
You J, Sheng X, Ding D, Sun Z, Suo Y, Wang H, Li Y (2008) Detection of carbohydrates using new labeling reagent 1-(2-naphthyl)-3-methyl-5-pyrazolone by capillary zone electrophoresis with absorbance (UV). Anal Chim Acta 609:66–75
Gouw JW, Burgers PC, Trikoupis MA, Terlouw JK (2002) Derivatization of small oligosaccharides prior to analysis by matrix-assisted laser desorption/ionization using glycidyltrimethylammonium chloride and Girard's reagent T. Rapid Commun Mass Spectrom 16:905–912
Naven TJP, Harvey DJ (1996) Cationic derivatization of oligosaccharides with Girard's T reagent for improved performance in matrix-assisted laser desorption/ionization and electrospray mass spectrometry. Rapid Commun Mass Spectrom 10:829–834
Gil G-C, Kim Y-G, Kim B-G (2008) A relative and absolute quantification of neutral N-linked oligosaccharides using modification with carboxymethyl trimethylammonium hydrazide and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem 379:45–59
Jang K-S, Kim Y-G, Gil G-C, Park S-H, Kim B-G (2009) Mass spectrometric quantification of neutral and sialylated N-glycans from a recombinant therapeutic glycoprotein produced in the two Chinese hamster ovary cell lines. Anal Biochem 386:228–236
Unterieser I, Mischnick P (2011) Labeling of oligosaccharides for quantitative mass spectrometry. Carbohydr Res 346:68–75
Takeda Y (1979) Determination of sugar hydrazide and hydrazone structures in solution. Carbohydr Res 77:9–23
Girard A, Sandulesco G (1936) Sur une nouvelle série de réactifs du groupe carbonyle, leur utilisation à l'extraction des substances cétoniques et à la caractérisation microchimique des aldéhydes et cétones. Helv Chim Acta 19:1095–1107
Gudmundsdottir AV, Nitz M (2007) Hydrolysis rates of 1-glucosyl-2-benzoylhydrazines in aqueous solution. Carbohydr Res 342:749–752
Gudmundsdottir AV, Paul CE, Nitz M (2009) Stability studies of hydrazide and hydroxylamine-based glycoconjugates in aqueous solution. Carbohydr Res 344:278–284
Sato K, Sato K, Okubo A, Yamazaki S (1998) Optimization of derivatization with 2-aminobenzoic acid for determination of monosaccharide composition by capillary electrophoresis. Anal Biochem 262:195–197
Zhang Y, Huang L-J, Wang Z-F (2007) A sensitive derivatization method for the determination of the sugar composition after pre-column reductive amination with 3-amino-9-ethylcarbazole (AEC) by high-performance liquid chromatography. Chin J Chem 25:1522–1528
Bigge JC, Patel TP, Bruce JA, Goulding PN, Charles SM, Parekh RB (1995) Nonselective and efficient fluorescent labeling of glycans using 2-amino benzamide and anthranilic acid. Anal Biochem 230:229–238
Borch RF, Bernstein MD, Durst HD (1971) Cyanohydridoborate anion as a selective reducing agent. J Am Chem Soc 93:2897–2904
Sun Z, Wei Z, Wei K (2009) A model for predicting the optimal conditions for labeling the carbohydrates with the amine derivatives by reductive amination. Lett Org Chem 6:549–551
Xia B, Feasley CL, Sachdev GP, Smith DF, Cummings RD (2009) Glycan reductive isotope labeling for quantitative glycomics. Anal Biochem 387:162–170
Ruhaak LR, Steenvoorden E, Koeleman CAM, Deelden AM, Wuhrer M (2010) 2-Picoline-borane: a non-toxic reducing agent for oligosaccharide labeling by reductive. Proteomics 10:2330–2336
Schellenberg KA (1963) The synthesis of secondary and tertiary amines by borohydride reduction1. J Org Chem 28:3259–3261
Zapala L, Kalembkiewicz J, Sitarz-Palczak E (2009) Studies on equilibrium of anthranilic acid in aqueous solutions and in two-phase systems: aromatic solvent-water. Biophys Chem 140:91–98
Anumula KR, Dhume ST (1998) High resolution and high sensitivity methods for oligosaccharide mapping and characterization by normal phase high performance liquid chromatography following derivatization with highly fluorescent anthranilic acid. Glycobiology 8:685–694
Harvey DJ (2005) Halogeno-substituted 2-aminobenzoic acid derivatives for negative ion fragmentation studies of N-linked carbohydrates. Rapid Commun Mass Spectrom 19:397–400
Maslen SL, Goubet F, Adam A, Dupree P, Stephens E (2007) Structure elucidation of arabinoxylan isomers by normal phase HPLC-MALDI-TOF/TOF-MS/MS. Carbohydr Res 342:724–735
Harvey DJ (2005) Collision-induced fragmentation of negative ions from N-linked glycans derivatized with 2-aminobenzoic acid. J Mass Spectrom 40:642–653
Gennaro LA, Delaney J, Vouros P, Harvey DJ, Domon B (2002) Capillary electrophoresis/electrospray ion trap mass spectrometry for the analysis of negatively charged derivatized and underivatized glycans. Rapid Commun Mass Spectrom 16:192–200
Doliška A, Strnad S, Ribitsch V, Kleinschek K, Willför S, Saake B (2009) Analysis of galactoglucomannans from spruce wood by capillary electrophoresis. Cellulose 16:1089–1097
Yuen CT, Gee CK, Jones C (2002) High-performance liquid chromatographic profiling of fluorescent labelled N-glycans on glycoproteins. Biomed Chromatogr 16:247–254
Gennaro LA, Harvey DJ, Vouros P (2003) Reversed-phase ion-pairing liquid chromatography/ion trap mass spectrometry for the analysis of negatively charged, derivatized glycans. Rapid Commun Mass Spectrom 17:1528–1534
Pabst M, Kolarich D, Pöltl G, Dalik T, Lubec G, Hofinger A, Altmann F (2009) Comparison of fluorescent labels for oligosaccharides and introduction of a new postlabeling purification method. Anal Biochem 384:263–273
Adden R, Müller R, Mischnick P (2006) Analysis of the substituent distribution in the glucosyl units and along the polymer chain of hydroxypropylmethylcelluloses and statistical evaluation. Cellulose 13:459–476
Adden R, Niedner W, Müller R, Mischnick P (2006) Comprehensive analysis of the substituent distribution in the glucosyl units and along the polymer chain of hydroxyl-ethylmethylcelluloses and statistical evaluation. Anal Chem 78:1146–1157
Karas M, Hillenkamp F (1988) Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem 60:2299–2301
Tanaka K, Ido Y, Akita S, Yoshida Y, Yoshida T, Matsuo T (1988) Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2:151–153
Fenn JB, Mann M, Meng CK, Wong SF (1989) Electrospray ionization for mass spectrometry of large biomolecules. Science 246(4926):64–71
Nielen MWF (1999) MALDI time-of-flight mass spectrometry of synthetic polymers. Mass Spectrom Rev 18:309–344
Deery MJ, Stimson E, Chappell CG (2001) Size exclusion chromatography/mass spectrometry applied to the analysis of polysaccharides. Rapid Commun Mass Spectrom 15:2273–2283
Garozzo D, Impallomeni G, Spina E, Sturiale L, Zanetti F (1995) Matrix-assisted laser desorption/ionization mass spectrometry of polysaccharides. Rapid Commun Mass Spectrom 9:937–941
Yeung B, Marecak D (1999) Molecular weight determination of hyaluronic acid by gel filtration chromatrography coupled to matrix-assisted laser desorption ionizaton mass spectrometry. J Chromatogr A 852:573–581
Hsu N-Y, Yang W-B, Wong C-H, Lee Y-C, Lee RT, Wang Y-S, Chen C-H (2007) Matrix-assisted laser desorption/ionization mass spectrometry of polysaccharides with 2’,4’,6’-trihydroxyacetophenone as matrix. Rapid Commun Mass Spectrom 21:2137–2146
Schnöll-Bitai I, Ullmer R, Hrebicek T, Rizzi A, Lacik I (2008) Characterization of the molecualr mass distribution of pullulans by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using 2,5-dihydroxy-benzoic acid butylamine (DHBB) as liquid matrix. Rapid Commun Mass Spectrom 22:2961–2970
Stahl B, Linos A, Karas M, Hillenkamp F, Streup M (1997) Analysis of fructans from higher plants by matrix-assisted laser desorption/ioniszation mass spectrometry. Anal Biochem 246:195–204
Haská L, Nyman M, Andersson R (2008) Distribution and characterisation of fructan in wheat milling fractions. J Cereal Sci 48:768–774
Jacobs A, Dahlman O (2001) Characterization of the molar masses of hemicelluloses from wood and pulps employing size exclusion chromatography and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Biomacromolecules 2:894–905
Jacobs A, Lundqvist J, Stålbrand H, Tjerneld F, Dahlman O (2002) Characterization of water-soluble hemicelluloses from spruce and aspen employing SEC/MALDI mass spectroscopy. Carbohydr Res 337:711–717
Kenne L, Lönngren J, Svensson S (1973) A new method for specific degradation of polysaccharides. Acta Chem Scan 27:3692–3698
Jansson PE, Kenne L, Lindberg B, Ljunggren H, Lonngren J, Ruden U, Svensson S (1977) Demonstration of an octasaccharide repeating unit in the extracellular polysaccharide of Rhizobium meliloti by sequential degradation. J Am Chem Soc 99:3812–3815
Spina E, Cozzolino R, Ryan E, Garozzo D (2000) Sequencing of oligosaccharides by collision-induced dissociation matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 35:1042–1048
Jennings K (1968) Collision-induced decompositions of aromatic molecular ions. Int J Mass Spectrom Ion Phys 1:227–235
Domon B, Costello CE (1988) A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J 5:397–409
Garozzo G, Impallomeni E, Spina E, Green BN, Hutton T (1991) Linkage analysis in disaccharides by electrospray mass spectrometry. Carbohydr Res 221:253–257
Friedl CH, Lochnit G, Geyer R, Karas M, Bahr U (2000) Elucidation of zwitterionic sugar cores from glycosphingolipids by nanoelectrospray ionization-ion-trap mass spectrometry. Anal Biochem 284:279–287
Tüting W, Wegemann K, Mischnick P (2004) Enzymatic degradation and electrospray tandem mass spectrometry as tools for determining the structure of cationic starches prepared by wet and dry methods. Carbohydr Res 339:637–648
Viseux N, de Hoffmann E, Domon B (1998) Structural assignment of permethylated oligosaccharide subunits using sequential tandem mass spectrometry. Anal Chem 70:4951–4959
McNeil M (1983) Elimination of internal glycosyl residues during chemical ionization-mass spectrometry of per-O-alkylated oligosaccharide-alditols. Carbohydr Res 123:31–40
Kovácik V, Hirsch J, Kovác P, Heerma W, Thomas-Oates J, Haverkamp J (1995) Oligosaccharide characterization using collision-induced dissociation fast atom bombardment mass spectrometry. J Mass Spectrom 30:949–958
Brüll LP, Heerma W, Thomas-Oates J, Haverkamp J, Kovácik V, Kovác P (1997) Loss of internal 1 → 6 substituted monosaccharide residues from underivatized and per-O-methylated trisaccharides. J Am Soc Mass Spectrom 8:43–49
Brüll LP, Kovácik V, Thomas-Oates J, Heerma W, Haverkamp J (1998) Sodium-cationized oligosaccharides do not appear to undergo ‘internal residue loss’ rearrangement processes on tandem mass spectrometry. Rapid Commun Mass Spectrom 12:1520–1532
Ernst B, Müller DR, Richter WJ (1997) False sugar sequence ions in electrospray tandem mass spectrometry of underivatized sialyl-Lewis-type oligosaccharides. Int J Mass Spectrom Ion Proc 160:283–290
Harvey DJ, Mattu TS, Wormald MR, Royle L, Dwek A, Rudd PM (2002) “Internal residue loss”: rearrangements occurring during the fragmentation of carbohydrates derivatized at the reducing terminus. Anal Chem 74:734–774
Ma Y-L, Vedernikova I, Van den Heuvel H, Claeys M (2000) Internal glucose residue loss in protonated O-diglycosyl flavonoids upon low-energy collision-induced dissociation. J Am Soc Mass Spectrom 11:136–144
Van der Burgt YEM, Bergsma J, Bleeker IP, Mijland PJHC, Van der Kerk-van HA, Kamerling JP, Vliegenthart JFG (2000) FAB CIDMS:MS analysis of partially methylated maltotrioses derived from methylated amylose: a study of the substituent distribution. Carbohydr Res 329:341–349
Cancilla MT, Penn SG, Caroll JA, Lebrilla CB (1996) Coordination of alkali metals to oligosaccharides dictates fragmentation behavior in matrix assisted laser desorption ionization/Fourier transform mass spectrometry. J Am Chem Soc 118:6736–6745
Botek E, Debrun JL, Hakim B, Morin-Allory L (2001) Attachment of alkali cations on β-D-glucopyranose: matrix-assisted laser desorption/ionization time-of-flight studies and ab initio calculations. Rapid Commun Mass Spectrom 15:273–276
Cancilla MT, Wong AW, Voss LR, Lebrilla CB (1999) Fragmentation reactions in the mass spectrometry analysis of neutral oligosaccharides. Anal Chem 71:3206–3218
Adams J (1990) Charge-remote fragmentations: analytical applications and fundamental studies. Mass Spectrom Rev 9:141–186
Mechref Y, Novotny V, Krishnan C (2003) Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal Chem 75:4895–4903
Spengler B, Dolce JW, Cotter RJ (1990) Infrared laser desorption mass spectrometry of oligosaccharides: fragmentation mechanisms and isomer analysis. Anal Chem 62:1731–1737
Momcilovic D, Schagerlöf H, Wittgren B, Wahlund K-G, Brinkmalm G (2005) Improved chemical analysis of cellulsoe ethers using dialkylamine derivatization and mass spectrometry. Anal Chem 77:2948–2959
Momcilovic D, Schagerlöf H, Röme D, Jörntén-Karlsoon M, Karlsson K-E, Wittgren B, Tjerneld F, Wahlund K-G, Brinkmalm G (2005) Novel derivatisation using dimethylamine for tandem mass spectrometric structure analysis of enzymatically and acidically depolymerised methyl cellulose. Biomacromolecules 6:2793–2799
Adden R, Mischnick P (2005) A novel method for the analysis of the substitution pattern of O-methyl- α- and β-1,4-glucans by means of electrospray ionisation-mass spectrometry/ collision induced dissociation. Int J Mass Spectrom 242:63–73
Tüting W, Adden R, Mischnick P (2004) Fragmentation pattern of regioselectively O-methylated maltooligosaccharides in electrospray ionisation-mass spectrometry/collision induced dissociation. Int J Mass Spectrom 232:107–115
Enebro J, Momcilovic D, Siika-aho M, Karlsson S (2009) Liquid chromatography combined with mass spectrometry for investigation of endoglucanase selectivity on carboxymethyl cellulose. Carbohydr Res 344:2173–2181
Lemoine J, Fournet B, Despeyroux D, Jennings KR, Rosenberg R, de Hoffmann E (1993) Collision-induced dissociation of alkali metal cationized and permethylated oligosaccharides: influence of the collision energy and of the collision gas for the assignment of linkage position. J Am Soc Mass Spectrom 4:197–203
Muzikar J, Mechref Y, Huang Y, Novotny MV (2004) Enhanced post-source decay and cross-ring fragmentation of oligosaccharides facilitated by conversion to amino derivatives. Rapid Commun Mass Spectrom 18:1513–1518
Sheeley DM, Reinhold VN (1998) Structural characterization of carbohydrate sequence, linkage, and branching in quadrupole ion trap mass spectrometer: neutral oligosaccahrides and N-linkes glycans. Anal Chem 70:3053–3059
Ashline D, Singh S, Hanneman A, Reinhold V (2005) Congruent strategies for carbohydrate sequencing. 1. Mining structural details by MSn. Anal Chem 77:6250–6262
Zhang H, Singh S, Reinhold VN (2005) Congruent strategies for carbohydrate sequencing. 2. FragLib: an MSn spectral library. Anal Chem 77:6263–6270
Lapadula AJ, Hatcher PJ, Hanneman AJ, Ashline DJ, Zhang H, Reinhold VN (2005) Congruent strategies for carbohydrate sequencing. 3. OSCAR: an algorithm for assigning oligosaccharide topology from MSn data. Anal Chem 77:6271–6279
Ashline DJ, Lapadula AJ, Liu Y-H, Lin M, Grace M, Pramanik B, Reinhold VN (2007) Carbohydrae structural isomers analyzed by sequential mass spectrometry. Anal Chem 79:3830–3842
Reinhold VN, Lapadula AJ, Ashline DJ, Zhang H (2008) Systems and methods for sequencing carbohydrates by mass spectrometry. PCT International Patent Application WO 2008027599 A2
Cheng HL, Her GR (2002) Determination of linkages of linear and branched oligosaccharides using closed-ring-chromophore labeling and negative ion trap mass spectrometry. J Am Soc Mass Spectrom 13:1322–1330
Sible EM, Brimmer SP, Leary JA (1997) Interaction of first row transition metals with a 1–3, a 1–6 mannotriose, and conserved trimannosyl core oligosaccharides: a comparative electrospray ionization study of doubly and singly charged complexes. J Am Chem Soc 8:32–42
König SR, Leary JA (1998) Evidence for linkage position determination in cobalt coordinated pentasaccharides using ion trap mass spectrometry. J Am Chem Soc Mass Spectrom 9:1125–1134
Matamoros Fernandéz LE, Obel N, Scheller HV, Roepstorff P (2004) Differentiation of isomeric oligosaccharide structures by ESI tandem MS and GC-MS. Carbohydr Res 339:655–664
Matamoros Fernandéz LE, Obel N, Scheller HV, Roepstorff P (2003) Characterization of plant oligosaccharides by matrix-assisted laser desorption/ionization and electrospray mass spectrometry. J Mass Spectrom 38:427–437
Matamoros Fernandéz LE, Sorensen HR, Jorgensen C, Pedersen S, Meyer AS, Roepstorff P (2007) Characterization of oligosaccharides from industrial fermentation residues by matrix-assisted laser desorption/ionization, electrospray mass spectrometry, and gas chromatography mass spectrometry. Mol Biotechnol 35:149–160
Hilz H, De Jong LE, Mabel MA, Schols HA, Voragen AGJ (2006) A comparison of liquid chromatography, capillary electrophoresis, and mass spectrometry methods to determine xyloglucan structures in black currants. J Chromatogr A 1133:275–286
Okatch H, Torto N, Armateifio J (2003) Characterisation of legumes by enzymatic hydrolysis, microdialysis sampling, and micro-high-performance anion-exchange chromatrography with electrospray ionization mass spectrometry. J Chromatogr A 992:67–74
Körner R, Limberg G, Mikkelsen JD, Roepstorff P (1998) Characterization of enzymatic pectin digests by matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 33:836–842
Guillaumie F, Justesen SFL, Mutenda KE, Roepstorff P, Jensen KJ, Thomas ORT (2006) Fractionation, solid-phase immobilization and chemical degradation of long pectin oligogalacturonides. Initial steps towards sequencing of oligosaccharides. Carbohydr Res 341:118–129
Naven TJP, Harvey DJ (1996) Effect of structure on the signal strength of oligosaccharides in matrix-assisted laser desorption/ionization mass spectrometry on time-of-flight and magnetic sector instruments. Rapid Commun Mass Spectrom 10:1361–1366
Harvey DJ (1993) Quantitative aspects of the matrix-assisted laser desorption mass spectrometry of complex oligosaccharides. Rapid Commun Mass Spectrom 7:614–619
Unterieser I, Cuers J, Voiges K, Enebro J, Mischnick P (2011) Quantitative aspects in electrospray ionization ion trap and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of maltooligosaccharides. Rap Commun Mass Spectrom 25:2201–2208. DOI:10.1002/rcm.5105
Haebel S, Bahrke S, Peter MG (2007) Quantitative sequencing of complex mixtures of heterochitooligosaccharides by vMALDI linear ion trap mass spectrometry. Anal Chem 79:5557–5566
Ogawa K, Yamaura M, Maruyama I (1994) Isolation and identification of 3-O-methyl-D-galactose as a constituent of a neutral polysaccharides of Chlorella vulgaris. Biosci Biotech Biochem 58:942–944
Ogawa K, Yamaura M, Maruyama I (1997) Isolation and identification of 2-O-methyl-L-rhamnose and 3-O-methyl-L-rhamnose as constituents of an acidic polysaccharide of Chlorella vulgaris. Biosci Biotech Biochem 61:539–540
Aspinall GO (1982) The polysaccharides. Academic, New York
De Ruiter GA, Bruggen-van V, der Lugt AW, Mischnick P, Smid P, Van Boom JH, Notermans SH, Rombouts FM (1994) 2-O-methyl-D-mannose residues are immunodominant in extracellular polysaccharides of Mucor racemosus and related molds. J Biol Chem 269:4299–4306
Dönges R (1990) Non-ionic cellulose ethers. Brit Polym J 23:315–326
Klemm D, Philipp B, Heinze T, Heinze U, Wagenknecht W (1998) Comprehensive cellulose chemistry: vol 1 fundamentals and analytical methods, vol 2 functionalization of cellulose. Wiley VCH, Weinheim
Wurzburg OB (ed) (1986) Modified starches: properties and uses. CRC, Boca Raton
Heinze T, Liebert T, Heublein B, Hornig S (2006) Functional polymers based on dextrans. Adv Polym Sci 205:199–291
Mischnick P, Kühn G (1996) Correlation between reaction conditions and primary structure: model studies on methyl amylose. Carbohydr Res 290:199–207
Mischnick P, Hennig C (2001) A new model for the substitution patterns in the polymer chain of polysaccharide derivatives. Biomacromolecules 2:180–184
Adden R, Müller R, Mischnick P (2006) Fractionation of methyl cellulose according to polarity - a tool to differentiate first and second order heterogeneity of the substituent distribution. Macromol Chem Phys 207:954–965
Mischnick P, Adden R (2008) Fractionation of polysaccharide derivatives and subsequent analysis to differentiate heterogeneities on various hierarchical levels. Macromol Symp 262:1–7
Mischnick P (2001) Challenges in structure analysis of polysaccharide derivatives. Cellulose 8:245–257
Mischnick P, Heinrich J, Gohdes M, Wilke O, Rogmann N (2000) Structure analysis of 1,4-glucan derivatives macromol. Chem Phys 201:1985–1995
Adden R, Melander C, Brinkmalm G, Gorton L, Mischnick P (2006) New approaches to the analysis of enzymatically hydrolyzed methyl cellulose. part 1. Investigation of structural parameters on the extent of degradation. Biomacromolecules 7:1399–1409
Melander C, Adden R, Brinkmalm G, Gorton L, Mischnick P (2006) New approaches to the analysis of enzymatically hydrolyzed methyl cellulose. Part 2. Comparison of various enzyme preparations. Biomacromolecules 7:1410–1421
Kochetkov NK, Chizov OS (1971) Mass spectrometry of carbohydrate derivatives. Adv Carbohydr Chem 21:29–38
Mischnick P, Niedner W, Adden R (2005) Possibilities of mass spectrometry and tandem-mass spectrometry in the analysis of cellulose ethers. Macromol Symp 223:67–77
Cuers J, Unterieser I, Adden R, Rinken M, Mischnick P (2011) Analysis of the substituent pattern in cellulose ethers by quantitative HPLC-ESI-MS. Abstracts SCM5, Amsterdam
Vollmer A, Voiges K, Bork C, Fiege K, Cuber K, Mischnick P (2009) Comprehensive analysis of the substitution pattern in dextran ethers with respect to the reaction conditions. Anal Bioanal Chem 395:1749–1768
Arisz P, Kauw HJJ, Boon JJ (1995) Substituent distribution along the cellulose backbone in O-methylcelluloses using GC and FAB MS for monomer and oligomer analysis. Carbohydr Res 271:1–14
Heinrich J, Mischnick P (1999) Determination of the substitution pattern in the polymer chain of cellulose acetates. J Polym Sci A Polym Chem 37:3011–3016
Gohdes M, Mischnick P (1998) Determination of the substitution pattern in the polymer chain of cellulose sulfates. Carbohydr Res 309:109–115
Adden R, Müller R, Brinkmalm G, Ehrler R, Mischnick P (2006) Comprehensive analysis of the substituent distribution in hydroxyethyl celluloses by quantitative MALDI-TOF-MS. Macromol Biosci 6:435–444
Richardson S, Nilsson G, Cohen A, Momcilovic D, Brinkmalm G, Gorton L (2003) Enzyme-aided investigation of the substituent distribution in cationic potato amylopectin starch. Anal Chem 75:6499–6508
Mischnick P (2001) Selectivity in enzymic degradation of methyl amylose. Starch/Stärke 53:110–120
Momcilovic D, Wittgren B, Wahlund K-G, Karlsson J, Brinkmalm G (2003) Sample preparation effects in matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry of partially depolymerized carboxymethyl cellulose. Rapid Commun Mass Spectrom 17:1107–1115
Enebro J, Karlsson S (2006) Improved matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry of carboxymethylcellulose. Rapid Commun Mass Spectrom 20:3693–3698
Schagerlöf U, Schagerlöf H, Momcilovic D, Brinkmalm G, Tjerneld F (2007) Endoglucanase sensitivity for substituents in methyl cellulose hydrolysis studied using MALDI-TOFMS for oligosaccharide analysis and structural analysis of enzyme active sites. Biomacromolecules 8:2358–2365
Enebro J, Momcilovic D, Siika-aho M, Karlsson S (2007) A new approach for studying correlations between the chemical structure and the rheological properties in carboxymethyl cellulose. Biomacromolecules 8:3253–3257
Enebro J, Momcilovic D, Siika-aho M, Karlsson S (2009) Investigation of endoglucanase selectivity on carboxymethyl cellulose by mass spectrometric techniques. Cellulose 16:271–280
Momcilovic D, Wittgren B, Wahlund K-G, Karlsson J, Brinkmalm G (2003) Sample preparation effects in matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry of partially depolymerised methyl cellulose. Rapid Commun Mass Spectrom 17:1116–1124
Adden R, Bösch A, Mischnick P (2004) Novel possibilities by cationic ring-opening polymerisation of cyclodextrin derivatives: preparation of a copolymer bearing block-like sequences of Tri-O-methyl-glucosyl units. Macromol Chem Phys 205:2072–2079
Bösch A, Nimtz M, Mischnick P (2006) Mechanistic studies on cationic ring-opening polymerization of cyclodextrin derivatives using various Lewis acids. Cellulose 13:493–507
Bösch A, Mischnick P (2007) Bifunctional building blocks for glyco-architectures by TiCl4-promoted ring opening of cyclodextrin derivatives. Biomacromolecules 8:2311–2320
Capon B (1969) Mechanism in carbohydrate chemistry. Chem Rev 69:407–498
Acknowledgments
I thank Christian Bork, Inga Unterieser, and Julia Cuers for the preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Mischnick, P. (2011). Mass Spectrometric Characterization of Oligo- and Polysaccharides and Their Derivatives. In: Hakkarainen, M. (eds) Mass Spectrometry of Polymers – New Techniques. Advances in Polymer Science, vol 248. Springer, Berlin, Heidelberg. https://doi.org/10.1007/12_2011_134
Download citation
DOI: https://doi.org/10.1007/12_2011_134
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-28040-5
Online ISBN: 978-3-642-28041-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)