
Abstract
Atomic force spectroscopy (AFM)-based single-molecule force spectroscopy (SMFS) was invented in the 1990s. Since then, SMFS has been developed into a powerful tool to study the inter- and intra-molecular interactions of macromolecules. Using SMFS, a number of problems in the field of supramolecular chemistry and mechanochemistry have been studied at the single-molecule level, which are not accessible by traditional ensemble characterization methods. In this review, the principles of SMFS are introduced, followed by the discussion of several problems of contemporary interest at the interface of supramolecular chemistry and mechanochemistry of macromolecules, including single-chain elasticity of macromolecules, interactions between water and macromolecules, interactions between macromolecules and solid surface, and the interactions in supramolecular polymers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- AFM
- Desorption
- Inherent elasticity
- Macromolecules
- Mechanochemistry
- Molecular motor
- Non-covalent interactions
- Polymer models
- QM calculations
- Single-molecule elasticity
- SMFS
- Supramolecular chemistry
- Supramolecular polymer
- Water rearrangement
1 Introduction
Since 1987, supramolecular chemistry has developed into a major field [1]. New structures with novel properties have been created from existing molecules via non-covalent interactions, including hydrophobic interactions, electrostatic forces, H-bonding, van der Waals forces, etc. At the very beginning, supramolecular chemistry dealt with small molecules such as crown ethers and cryptands. Later, non-covalent interactions were applied to the field of macromolecules, yielding an important concept of a supramolecular polymer [2]. Connected by non-covalent interactions, the dynamic supramolecular polymer fused the two fields of small organic molecules and macromolecules. The principle of supramolecular chemistry is central not only to chemical sciences but also to life and material sciences [3–7].
Mechanochemistry has recently emerged as an important field. Mechanical force has a fundamental role in biological processes and material applications. For a bulk material, the elastic modulus and other mechanical properties can easily be measured with universal testing machines. However, the output force of a molecular motor cannot be obtained by bulk measurements. Biomacromolecules often work in self-assemblies of only one or several molecules. The mechanical properties at the single-molecule level are key parameters for these systems. When using bulk/ensemble methods, only average properties can be deduced from the experimental results. In order to measure the mechanical properties of biomacromolecules directly, several single-molecule manipulation techniques were invented in the 1990s, such as optical tweezers, magnetic tweezers, and atomic force spectroscopy (AFM) based single-molecule force spectroscopy (SMFS) [8–10].
A number of important problems in supramolecular chemistry and mechanochemistry on the single-molecule level, such as the strength of the covalent bond and of intermolecular interactions, and the elasticity of an isolated polymer chain, cannot be addressed by bulk measurements. By solving these problems during the last two decades, AFM-SMFS has become widely accepted as a powerful tool to study inter- and intra-molecular interactions of macromolecules [11–24]. Many reviews on AFM-SMFS have been published, discussing the instrumentation, the applications to the problems of biological and synthetic macromolecules. In this review, we focus on recent advances by AFM-SMFS in the fields of supramolecular chemistry and mechanochemistry. First, the principles of SMFS are briefly introduced. Then, single-chain elasticity of macromolecules and the theoretical models, which form the foundation of our understanding of many aspects of micromechanical behavior of macromolecules, are discussed. Later, the application of SMFS to understand the interactions between macromolecules and surroundings (i.e., water and solid surfaces) is reviewed. Finally, the binding interactions in supramolecular polymers and synthetic molecular machines are discussed.
2 Basic Principles of AFM-SMFS
AFM was invented in 1986 by the Nobel Laureate Gerd Binnig and his colleagues as a tool for the imaging of surfaces with high resolution [25]. Because of the high resolution of force sensing, AFM can also be utilized to detect inter- and intra-molecular interactions in surface-immobilized systems. This feature is the origin of AFM-SMFS. In brief, the principle of SMFS is to manipulate a macromolecule that bridges an AFM tip and a solid support (also called substrate). During the manipulation of the molecule bridge, a cantilever deflection-piezopath curve is recorded and later converted into a force-molecule extension curve (in brief, force curve). A schematic drawing of the SMFS setup and the principles of its operation are shown in Fig. 1 [11, 12, 26, 27].

(a) Schematic of AFM-based SMFS. (b) Schematic of pulling a single polymer chain and the corresponding force curve obtained by SMFS
The molecular bridge can be formed by various methods, which can be classified into two types: physisorption and chemisorption. Physisorption is a simple method, in which molecules are first adsorbed onto the substrate and then the tip captures one or more molecules from the substrate when a compressive force of up to several nanoNewtons (nN) is applied for several seconds [10, 13, 28]. According to many studies, such compressive force is sufficient to facilitate a strong non-specific adsorption between the AFM tip and the target molecule [12, 29–32]. The other strategy to form a macromolecular bridge between the tip and the substrate is to bind the molecule to the surfaces covalently. The terminal groups of the target molecule and the tip (and/or the substrate) are modified with chemically reactive groups, which can form a bond (covalent, coordination, or other intermolecular interactions with similar strength) upon contact at room temperature [33–35]. For both strategies, the interaction between the tip and the target molecule should be stronger than the non-covalent interactions to be measured. In an ideal case, the force resolution of a SMFS is about 10 picoNewtons (pN), which is high enough for detecting the intermolecular interactions involved in the supramolecular self-assembly.
Typically, force-extension curves are measured in a liquid to prevent interference from the capillary force at surfaces. This condition makes force measurements relevant to supramolecular systems, because self-assembly is usually performed in a liquid medium [36]. Aqueous solutions, organic solvents, and mixture of them have been used as the liquid environment for force measurements. The following sections illustrate some typical applications of SMFS on the study of supramolecular chemistry and mechanochemistry of macromolecules.
3 Inherent Single-Chain Elasticity of Macromolecules
In the past 20 years, with the development of single-molecule experimental methods [10, 37–39], the behavior of many macromolecules during stretching has been investigated at the single-chain level [9, 10, 26, 27, 40]. The stretching elasticity of a single polymer chain has been found to depend on several factors, including the structure of the side chains, the solvent and any solutes (such as a denaturant and/or a salt), and the temperature [13, 14, 29, 41]. Molecular understanding of this body of empirical data must start with quantifying the inherent elasticity of the polymer chain, i.e., the micromechanical behavior of an ideal polymer chain without long range interactions and specific interactions with the solvent. A practical condition is a nonpolar organic solvent, in which the interactions between the solvent molecules and the polymer chains are limited to van der Waals forces, which can be ignored in SMFS studies. In recent years, Cui et al. utilized SMFS to investigate the inherent single-chain elasticity of a series of macromolecules [42, 43].
3.1 Three Polymer Models that Combine Elasticity from Quantum Mechanical Calculations
With the development of experimental tools to study single-chain mechanics of polymers, theoretical models have become increasingly important in understanding the measured single-chain stretching behavior of macromolecules. Generally, because of the large number of degrees of freedom, the single-chain stretching behavior has to be treated using the approaches of statistical mechanics, by replacing the atomistic description of the chain structure with simplified models [44]. The three models most commonly used to describe single polymer chains, all based on the seminal work of Flory and others, are the freely jointed chain (FJC) model, the freely rotating chain (FRC) model, and the worm-like chain (WLC) model, mathematically represented by (1–3) below:
In these equations, F is the external stretching force acting across the termini of the chain, R the end-to-end distance of a polymer chain under F, L[F] the F dependent contour length of the polymer chain, k B the Boltzmann constant, T the absolute temperature, and l k , l p , and l b denote the Kuhn length, persistence length, and rotating unit length of the polymer chain, respectively. L[F] and l k (or l p or l b ) are two free parameters for model fitting.
With the advancement of SMFS, the validity of these models has been demonstrated. The WLC model was successfully applied to describe the measured single-molecule elasticity of a double stranded DNA, proteins, and other polymers [45, 46]. The FJC model was exploited to fit the single-chain elasticity of many kinds of polymers, such as amylose and cellulose [26, 27, 29]. However, these models assume that polymer chains have only entropic elasticity, i.e., the contour length of a polymer chain is inextensible, although contour length is extensible by the deformation of bond angles and bond lengths. Enthalpic elasticity is integrated into these models by means of force-dependent contour length, L[F]:
where K 0 is the linear elasticity of the single polymer chain, and L 0 the contour length of free polymer. Together with K 0, there are three free fitting parameters for each of the above-mentioned models. In practice, these free fitting parameters are determined by the trial and error method. In general, the fitted parameter l k (or l p or l b ) has no close relationship with the molecular chain structure of the polymer.
In 2005, Hugel et al. reported the enthalpic elasticity with high-accuracy of a monomer of three kinds of polymers (Fig. 2), which was calculated at the MP2 level of theory [47]. The modulus of the monomer was nonlinear and can be expressed in a polynomial expansion:
where a 0 and a[F] are the length of the monomer at zero force and force F, respectively, and γ n is the nth polynomial coefficient.

Schematic of one unit of three kinds of polymers (propane, DNA, and a di-peptide) used in the MP2 calculations. Figure reproduced with permission from [47]
Because changes of bond angles and bond lengths are already included in the calculations on one repeating unit, (5) can be rewritten for the whole polymer chain (fitting results support this assumption; see Sect. 3.2):
To simplify the calculations involved in the original models, L 0 is introduced into the models as follows:
If L[F]/L 0 is known, the corresponding F can be calculated by (6). Because (7)–(9) incorporate quantum mechanical (QM) results, the respective models are called QM-FJC, QM-FRC, and QM-WLC, each with only one free-fit parameter (l k , l b , and l p , respectively) [48].
3.2 Macromolecules with a Carbon–Carbon (C–C)Backbone
To test these new models, Cui et al. investigated the single-chain mechanics of a series of polymers with the C–C backbone (poly(N,N-diethylacrylamide) (PDEAM), polyacrylamide (PAAm), and polystyrene (PS); Scheme 1) in nonpolar solvents by SMFS [43]. To compare SMFS-behavior of polydisperse polymers with varying contour length, the extension measured in individual experiments is divided by the strain-free contour length to yield normalized extension (see below for detail), which is plotted against applied force. The normalized force curves of these polymers measured in octane were superimposable over the entire force regime; see Fig. 3. The only marked difference is the beginning part of the force curves, which originates from the strong or weak adhesion force between the AFM tip and the sample surface. It can be concluded that the inherent elasticity of a polymer chain with C–C backbone did not depend on the side chains, at least if they were moderately sized. This result is very different from those observed from the bulk measurements; for instance, the sizes of the side chains have a marked influence on the temperature of glass transition, Tg (http://en.wikipedia.org/wiki/Glass_transition).

Primary structures of three polymers with C–C backbone. Figure reproduced with permission from [43]

Comparison of measured normalized force curves of poly(N,N-diethylacrylamide) (PDEAm), polyacrylamide (PAAm), and polystyrene (PS) obtained in octane and the QM-FRC fitting curve. Figure reproduced with permission from [43]
The experimental force curves of these C–C backbone-based polymers were fitted to QM-FRC model using the coefficients of Hugel et al. [47]:
where γ 1 is the linear elasticity of the repeating unit, and the other two coefficients γ 2 and γ 3 are nonlinear corrections. It should be noted that the modulus coefficients have a dimension of force, which is different from the bulk materials (force/area). The area of the cross section of the single-chain is omitted because it is a variable depending on the position of the chain.
During stretching of a single polymer chain, its L[F]/L 0 increases from 1.0 as F increases until the polymer bridge ruptures, i.e., L[F]/L 0 is a monotonically increasing function of F and vice versa. Assuming the upper limit for F is 2,000 pN [8], the upper limit for L[F]/L 0 is about 1.07 according to (10). For any given value of L[F]/L 0, F is calculated by (10). Any pair of values for L[F]/L 0 and F are then converted to the corresponding normalized extension of polymer, R/L 0, according to (7)–(9). In this way, the whole QM-based fitting curve can be generated when L[F]/L 0 is increased from 1.0 to 1.07.
The force curves in Fig. 3 were best reproduced with the QM-FRC model (8) with l b = 0.154 nm [43], which is identical to the C–C bond length. With a single C–C bond as the rotating unit and the chain elasticity deduced from QM calculations, the QM-FRC model has been developed into a parameter free and structure relevant single-chain model, which can quantitatively describe polymers with C–C backbone. Although there are some exceptions when the side chain is large (e.g., dendrimer or long graft chain) [43], this work provides a general result for the inherent elasticity of single neutral polymer chains with C–C backbones. These fitting results confirmed that the QM-FRC model is very successful in describing the micromechanics of these flexible polymers.
3.3 Natural Cellulose
Cellulose is a widely distributed polysaccharide in nature, and is polymerized glucose. It is the most important substance in plants, where it serves as the basic building material of cell walls [49, 50]. Interestingly, the Young's modulus of a perfect cellulose crystal may be as high as that of steel, which is much higher than that of most polymer materials [51, 52]. A single cellulose chain is generally expected to be rigid [49]. Single-molecule measurements of natural cellulose (NC) are complicated by its poor solubility in common solvents. NC is soluble in several ionic liquids (IL) [53], which allowed Cui et al. recently to investigate the single-chain elasticity of NC by SMFS (Fig. 4). To obtain the inherent elasticity of NC, they measured the single-chain force curve of an NC sample prepared from an IL (1-allyl-3-methylimidazolium chloride, AMIMCl) [54] solution [42] and fitted it to the QM-FJC model [47, 48], using the following three parameters derived by QM calculations on a glucose dimer:

(a) Normalized force curves of NC obtained in octane. (b) Comparison of a force curve of NC obtained in octane and the QM-FJC fitting curve. (c) Comparison of force curves of PAAm and NC obtained in octane. (d) Molecular structure of the dimer (cellobiose) used in QM calculations and the QM results. The arrows indicate the atoms defining the constrained distance. Figure reproduced with permission from [42]
Interestingly, the experimental force curve could be fitted well to the QM-FJC model (with l k = 0.514 nm) but not to the QM-WLC alternative. In other words, the NC chain seemed flexible at the single-molecule level, which was also confirmed by the similarity of the normalized force curves of NC and PAAM, with free rotation around the C–C bonds of the backbone. This finding contradicts the traditional view of NC as a rigid polymer [49], which probably reflects the rigidifying effect of extensive H bonding between NC chains in its crystals [55], on which all previous mechanical measurements had been done. The QM-FJC model fitting results revealed that the length of a pyranose ring of NC equaled the length of a Kuhn segment (l k ) in the model [56]. This result means that cellulose on the single chain scale is rather flexible, which is surprising because cellobiose (consisting of two pyranose rings) has been recognized to be the repeat unit of cellulose for a long time. With the fine flexibility, each of the pyranose rings can find its best binding site on the crystal surface, which perfectly lowers the energy of the self-assembled structures. This may explain why NC became the basic building material of plants during plant evolution.
4 Interactions Between Water and Macromolecules
Water can be regarded as the most important substance for life on Earth. It is the most widely used solvent of biomolecules. However, it is also a very complicated solvent. With H-bonding donor and acceptor sites, this polar solvent strongly influences the properties of solutes. It is interesting that water forms supramolecular structures with many kinds of macromolecules, including DNA, proteins, and many synthetic polymers [26, 29, 32, 41, 57, 58]. Several typical water–macromolecule assemblies have been investigated by SMFS.
4.1 PEG
Polyethylene glycol (PEG) is a water soluble polymer with many applications from industrial manufacturing to medicine [59]. PEG is prepared by polymerization of ethylene oxide, and is commercially available. The molecular weight of PEG can be up to several megadaltons (MDa), corresponding to the contour length of up to several microns. Gaub and co-workers studied the single-molecule mechanics of PEG in water and hexadecane [26], and found that the force curves obtained in the two environments were superimposable in the low-force regime, which reflected the entropic elasticity of an ideal polymer; and in the very high force regime, which is dominated by the enthalpic elasticity. However, in the middle force regime, the behavior of PEG in the two solvents was very different; see Fig. 5a. The phenomenon was attributed to the formation of solvent-induced supramolecular structures. In hexadecane, upon stretching the PEG molecule behaved as an ideal polymer chain with pure entropic elasticity, which was confirmed by the subsequent fitting result to the modified FJC (M-FJC) model [44]. In water, however, PEG adopted the trans-trans-gauche state (helical conformation) [60]. Two adjacent oxygen atoms of PEG formed H-bonds to a water molecule, establishing a water-bridged supramolecular structure which stabilized the helical state (Fig. 5b). Above a threshold force, the helical state changed into the all trans state [8, 61], where the adjacent oxygen atoms were separated by a longer distance than is required to maintain the water bridge. Therefore, the difference between force curves obtained in the two solvents was attributed to the free energy cost of breaking the water bridge, which was calculated to be about 3 k B T/monomer.

(a) Force curves of polyethylene glycol (PEG) measured in aqueous solution (red) and hexadecane (blue) along with smoothed curves (black lines). (b) Schematic of conformation transition from the helical state to all trans state. Figure reproduced with permission from [26]
4.2 PAAm and PVA
Besides PEG, other water soluble polymers, such as poly(vinyl alcohol) (PVA) [41] and polyacrylamide (PAAm) [29], can also form water-mediated supramolecular structures in aqueous solution. SMFS results indicated that the force curves of PVA and PAAm measured in water could not be fitted well to the WLC model (for PVA) or the M-FJC model (for PAAm). However, the force curves in aqueous 8 M urea solution (urea is generally considered to break H-bonds) were remarkably distinct from those measured in water and fitted well to the WLC model and M-FJC model for PVA and PAAm, respectively. This fact demonstrated the existence of H-bonded supramolecular structures in PVA and PAAM in water, which were destroyed in 8 M urea.
4.3 DNA
DNA, with its special double helix structure, is a typical supramolecular system, and it plays the central role in the transcription, expression, and conservation of genetic information [62]. The biological function of DNA is closely related to the stability and the integrality of the double strands [63], and the mechanical stability of double-stranded DNA (dsDNA) strongly depends on the ambient conditions. Recently, Cui et al. investigated the important role of water in determining or stabilizing the supramolecular structure of DNA by SMFS [31, 32].
An advantage of SMFS is that the polymer sample can be measured in different environments. Such measurements suggest that the double-stranded DNA is thermodynamically unstable in at least some non-aqueous media. To screen the effects of water on the structure of DNA, Cui et al. performed the SMFS experiments on dsDNA in diethylbenzene (DEB, a poor solvent for DNA) [64]. The force curves obtained in DEB were very different from those obtained in water in that they lacked the characteristic “B-S” transition plateau [38–40, 46] (Fig. 6b). A similar result was observed in another poor solvent for DNA, 1-propanol, which does not form aromatic stacking interactions with DNA. The force curves of dsDNA measured in poor solvents (DBE and 1-propanol) were similar to those of an unstructured polymer chain. These facts implied that dsDNA was already denatured into single-stranded DNA (ssDNA). To confirm this, Cui et al. utilized a theoretical single-chain elasticity (calculated at the MP2 level) of ssDNA to fit the force curve of dsDNA in organic solvents.

(a) Typical “B-S” force curve of dsDNA. (b) Force curves of dsDNA obtained in a poor solvent, DEB. (c) Comparison of forces curve measured on dsDNA and ssDNA in poor solvent and fitting curve to the QM-FRC model. Figure reproduced with permission from [32]
In order to find the most appropriate model to describe the single-chain elasticity of dsDNA in poor solvents, the force curves were fitted to three QM models [48]. Fitting results suggested that l k = 0.59 nm for QM-FJC model and l b = 0.295 nm for QM-FRC model (Fig. 7a, b). Because the length of a repeating unit of ssDNA is 0.59 nm, QM-FJC is a structure relevant model [31, 32]. The inability to fit the experimental data to the QM-WLC model (Fig. 7d) suggested that the rigid dsDNA chain in water became flexible when stretched in a poor solvent. Thus, dsDNA was concluded to have already denatured into two ssDNA strands when it was pulled into the poor solvents. Fitting results also indicated that the QM-FJC and QM-FRC models become identical at force >25 pN (Fig. 7c). Because l k = 2l b , this result can be explained by viewing one repeating unit of ssDNA as composed of two rotating units, i.e., the sugar ring and the phosphate group.

Experimental force curves fitted to the QM-FRC (a) and the QM-FJC (b) models. Low-force regime of the two fits is compared in (c). QM-WLC fitting results (d) of ssDNA with various l b values from 0.3 nm (left) to 10 nm (right). Figure reproduced with permission from [48]
As further corroboration of the dsDNA denaturation in poor solvents, force measurements were performed in a denaturing environment, an aqueous solution of guanidine chloride (Gua), which is a typical denaturant of dsDNA. The force curves obtained in this case were identical to those measured in DEB. The perfect superposition of normalized force curves measured on dsDNA and ssDNA in DEB undoubtedly demonstrates that dsDNA is denatured into ssDNA upon pulling it in a poor solvent.
Molecular dynamics (MD) simulations were carried out to observe the details of the denaturation of dsDNA [65]. It was observed dsDNA unwind spontaneously when put into the apolar solvent directly, which may explain why dsDNA are denatured in both DEB (water immiscible) and propanol (water miscible). The main reason should be the absence of hydrophobic force in non-aqueous media [66], which contributes greatly to the stability of dsDNA in aqueous media. The thermal disturbance at room temperature could be strong enough to destroy the supramolecular structure of dsDNA when hydrophobic force is absent.
The single-chain mechanics of ssDNA in an aqueous environment was also measured, and then the force curves obtained in solution of phosphate buffered saline (PBS) and DEB were normalized and compared (Fig. 8a). It was found that there was a clear difference in the middle force regime, but no deviation in the low and high force regimes [31].

(a) Comparison of the force curve of ssDNA obtained in DEB and the QM-FRC fitting curve. (b) Comparison between the smoothed force curve of ssDNA obtained in aqueous solution and in DEB. (c) Comparison between the force curves of ssDNA obtained in various aqueous media and QM-FRC fitting curve. (d) Comparison between the force curves obtained in aqueous guanidine chloride and DEB. Figure reproduced with permission from [31]
By excluding the effects of salt and poor solvent types, the deviation was attributed to the water rearrangement upon elongation [26]. At the free state (F = 0), there is more bound water around ssDNA. Upon elongation, the distances between the H-bond donors and acceptors of ssDNA are increased, which caused a water rearrangement. Thus, an addition energy (0.58 k B T/unit) is needed for the water rearrangement, which is reflected by the deviation between the force curves obtained in water and organic solvent. Comparing with other water soluble polymers [26, 30], one can find that the ssDNA system consumes the lowest energy for the water rearrangement. It was speculated that the weak competition influence of water may be an important factor for DNA to form a stable double helix in water.
In fact, the water rearrangement around the ssDNA chain can be expressed in another form, i.e., it can be regarded as a partial dehydration process:
The self-organization from ssDNA to dsDNA is an important process for life. This process can be formularized below, where ssDNA′ denotes the complimentary chain of ssDNA:
However, the formula is incomplete, because the bound water around dsDNA and ssDNA chains is not considered [67]. It is known that both ssDNA and dsDNA chains are hydrated in the aqueous solution. Because the bases are hidden in the duplex, one dsDNA chain has less binding sites with water than that of the sum of the two corresponding ssDNA chains [68]. Therefore, before the self-organization from ssDNA to dsDNA, a partial dehydration process of ssDNA should occur. Thus, (13) should be modified into a more rigorous form as follows:
For the reaction, a typical value for the free energy change (ΔG 14) measured by differential scanning calorimetry (DSC) is −4.3 kJ/(mol · residue) [69]. In this process, the formation of dsDNA can be separated into two steps, including the partial dehydration and the supramolecular self-assembly, as described by the following two equations:
In the two steps, all the water rearrangement is completed in (15), and all the assembly occurs in (16), which are a non-spontaneous (ΔG 15 > 0) and a spontaneous process (ΔG 16 < 0), respectively. However, for the total process, which is a favorable reaction with the physiological conditions, the free energy change (ΔG 14 = ΔG 15 + ΔG 16) is negative. Thus, the free energy necessary to remove partially the hydration shell of ssDNA must be compensated by the free energy of the formation of the dsDNA (that is, ΔG 15 < ΔG 16). However, neither ΔG 15 or ΔG 16 is readily available in references or a value which can be easily measured.
It can be seen that the process of partial dehydration in (15) is very similar to the process of the loss of bound water in (12). Then, it is expected that the free energy changes of (12) and (15) are very close; ΔG 12 ≈ ΔG 15 ≈ 1.4 kJ/(mol · residue). Thus, one can calculate that ΔG 15 is about −7.1 kJ/(mol · base), which is not far from zero. Based on these calculations, it is found that the value of ΔG 15 is vital to the process of (14). If ΔG 15 is a much larger value (e.g., >3.6), the whole process of DNA organization is not be favorable, because ΔG 14 would be a positive value. Although the water rearrangement upon stretching also occurs in other macromolecule/water systems, such as the poly(N-vinyl-2-pyrrolidone)/water system (13.0 kJ mol−1 unit)[30] or the PEG/water system (7.2 kJ mol−1 unit) [26], it is found that the dsDNA system involves the lowest energy among them.
As discussed above, water plays a key role in the self-assembly process of dsDNA (see Scheme 2). On the one hand, it is the weak disturbance of water molecules that ensures the stability of the dsDNA in aqueous solution. On the other, by shaving off the water molecules, dsDNA can be destabilized and tends to unwind. Another factor is that there are many kinds of water soluble macromolecules, but few can form a stable supramolecular structure in water. This may imply that DNA is somewhat special in the molecular structure.

Hydration/dehydration processes regulate the supramolecular structures of DNA. Figure reproduced with permission from [57]
In fact, the specific structure of DNA is not occasional, it is more likely to be the final result of natural selection. Cui conceived a possible route for the prebiotic evolution as follows.
Because of the complexity involved, the whole prebiotic chemical evolution is fractionized into six levels; see Scheme 3. First, the primitive Earth is an inorganic world, in which there are gas mixtures, water, and so on (Level 1). The inorganic compounds may be converted into organic monomers by the Miller experiment [70] and other possible routes (Level 2). After that, many macromolecules can be generated by polymerization reactions (Level 3). For the three levels, these reactions are in a dynamic equilibrium because of the reversibility. To prevent the degradation into monomers, these macromolecules must form a stable supramolecular structure in the aqueous solution (Level 4). However, water is a strong solvent, which usually has remarkable impact on the solute molecules. With the interference from water, only a few macromolecules can form stable supramolecular structures in water. As a result of long-term chemical evolution [71], the primary structure of dsDNA has been selected in a Darwinian fashion to adapt to the water environment. In further evolution, combined with the function of self-replication (Level 5), it is expected that the primitive life would emerge (Level 6).

Possible evolution route from inorganic molecules to the primitive living systems. Figure reproduced with permission from [57]
During the whole chemical evolution, water is the most important substance. As a reactant in the Miller experiment and the only solvent in Level 2, water directly participated in the early prebiotic chemical evolution. In addition, as a “selector” from Level 3 to Level 4, most of the macromolecules in Level 3 were screened out by the water environment. Water is the primary environment of terrestrial life, which supports life and, at the same time, restricts the form of life. It is therefore water that defines and shapes life. It is believed that this notion casts a new light on the origin of life
4.4 Hyaluronan
Hyaluronan (also called hyaluronic acid, HA) is a linear polysaccharide widely present in the extracellular matrix [72]. Because of direct communication with proteins and cells present in tissues, HA is an important molecule in the regulation of many cellular and biological processes. Hydration is believed to be one of the key factors influencing its functions. In 2007, Vancso et al. reported the single chain behavior of HA in different conditions. In aqueous media, HA showed marked deviation between the single-chain force curves obtained at different temperatures. The force curves obtained at 46°C can be described well by the m-FJC model. For those obtained at 29°C, only the high force regime (>700 pN) can be fitted by the model. This deviation was at various temperatures. After normalization, these two kinds of force curves can be superposed well in the high force regime. The deviation between the two kinds of force curves was attributed to the superstructure of HA in aqueous media at lower temperature, i.e., a local structure involving a H-bonded network along the polymeric chain, with H-bonds between the polar groups of HA and possibly water and water-mediated intramolecular bonds. This hypothesis is reasonable, because it was reported that the superstructure becomes increasingly destabilized when the temperature is raised to 46°C [73]. This conclusion was further supported by the result obtained in DMSO (an effective breaker of H-bonds), in which the force curve was similar to that obtained in aqueous media at 46°C.
4.5 PNIPAM
Since Pelton and Chibante reported the synthesis of the first temperature-sensitive microgel from N-isopropylacrylamide (NIPAM) and crosslinker in 1986 [74], responsive microgels have attracted numerous attempts to explore their potential application in many fields, such as sensing and drug delivery [75]. Now the most extensively studied responsive microgels are prepared with poly(N-isopropylacrylamide) (PNIPAM) [2]. It is well known that the phase transition temperature of PNIPAM is about 32°C [76]. When T < LCST, the PNIPAM chain is soluble in water as a random coil. When T ~ LCST, the subtle balance between PNIPAM and water is broken and phase transition occurs [77]. However, the single-molecule level mechanism of the phase transition of PNIPAM had not been proposed until very recently.
Since 2012, Cui et al. have carried out a series of studies to try to understand further the mechanism by means of SMFS at the single-molecule level [78–80]. One of the key features of PNIPAM is that the single-chain conformation in aqueous solution can be tuned easily by changing the temperature around its LCST [81–84]. When the temperature is below its LCST, it is a random coil state. However, the conformation of a single PNIPAM chain is changed from coil to globule state spontaneously if the temperature rises above its LCST. The single-chain elasticity of PNIPAM at different temperatures in water has been measured; see Fig. 9. When the temperature was below LCST, there was no difference in the whole force regime among the normalized force curves obtained at different temperatures. Above LCST, an obvious deviation in the middle force range can be found with increase of temperature. Interestingly, T = 35°C was a turning point of the whole variation; when T ranged from 31 to 35°C, the middle parts of the force curves dropped gradually, whereas from 35 to 40°C, the middle parts rose gradually.

Comparisons of force curves of PNIPAM obtained in water at different temperatures and in octane at RT. Figure reproduced with permission from [78]
When PNIPAM was dissolved in water, both the hydrophilic group and the hydrophobic group of the PNIPAM chain were hydrated in water [85–87]. It was expected that there were many fewer bound water molecules in the fully stretched state than that in the free coil state because of the limitation in chain conformation [8]. Thus, the hydrated PNIPAM chain would lose bound water gradually during the force extension process. Then the bound water molecules around the PNIPAM chain were forced to undergo rearrangement upon stretching. During the elongation, the water rearrangement would consume additional energy over and above that needed for the inherent elasticity of the chain. This energy cost for the water rearrangement upon stretching was reflected by the deviation between the force curves obtained in organic solvents and in water at room temperature (RT). The deviation was calculated to be about 2.1 k B T/unit.
However, it could be seen that the middle part of the force curve in water at 35°C was even lower than that in organic solvents, which could be explained by two factors. First, water was a poor solvent for PNIPAM at 35°C, and the amount of bound water around the PNIPAM chain was considerably reduced in this condition compared to the case at RT [85–87]. Thus, compared to the case in water at RT, the energy needed for the water rearrangement upon stretching PNIPAM chain was greatly reduced in water at 35°C. This factor may roughly correspond to the energy difference between water at RT and organic solvents.
According to Boltzmann’s entropy formula (S = k B ln Ω), the entropy of a polymer chain approaches zero when the chain is highly stretched by an external stretching force. That is, the entropies of the final state are the same (S ≈ 0) in the two conditions. However, it is known that the conformation of PNIPAM in water at 35°C is much more compact than that in water at RT, i.e., the initial entropy of PNIPAM at 35°C is much less than that at RT. Thus, the energy cost of a compact conformation would be less than that of a coil conformation. This factor may roughly correspond to the energy difference between water at 35°C and organic solvents.
When the temperature was increased from 35 to 40°C, the middle parts of the force curves began to rise gradually, which may be attributed to the formation of intrachain H-bonds [81]. This assumption could be well supported by the work of Peiyi Wu's group [87], who proposed that the formation of H-bonds between the amide groups was the final step in the multistep conformational change. In the free state, the globule conformation of the PNIPAM chain (T > LCST) could be stabilized by the formation of intrachain H-bonds. However, under tension it was expected that the elongation of the chain would lead to breakage of the intrachain H-bonds. Because the H-bonds between PNIPAM and water were greatly depleted at T > LCST, it was anticipated that new H-bonds would be unlikely to be formed between PNIPAM and water upon the breakage of the intrachain H-bonds. Therefore, there was almost no energy compensation from the formation of H-bonds with water. Thus, the deviation between the force curves obtained at T > 35 and 35°C was mainly contributed by the intrachain H-bonds of PNIPAM. This change of single-chain mechanics from 35°C to 40°C can be used to realize the thermal-mechanical energy conversion at the single-molecule level. That is, the single PNIPAM chain can convert the thermal energy to mechanical work in the range of 35–40°C (see Fig. 10). Thus, this study provided a theoretical basis for the design of nano-devices, namely molecular heat engines, for thermal-mechanical energy conversion [88].

(a) Force curves of PNIPAM obtained at 35°C and 40°C in water. (b) Novel synthetic molecular heat engine can be proposed according to the variation of the single PNIPAM chain mechanics between 35°C and 40°C. Figure reproduced with permission from [78]
Besides temperature, the variation of the solvent composition can also induce the phase transition of PNIPAM, which is often called cononsolvency [89]. PNIPAM in the mixed solvent of water and methanol is the most studied cononsolvency system. The PNIPAM chain acts as a random coil in pure water or pure methanol, whereas it exists as a globule in a range of concentrations in the water/methanol mixed solvents. As the methanol molar fraction (χmethanol) increases from 0% to 35%, the LCST of PNIPAM in the water/methanol mixed solvent is decreased from 32°C to 7.5°C Then from 35% to 45%, a sharp increase follows [90]. Therefore, an interesting coil-to-globule-to-coil transition of PNIPAM takes place in the water/methanol mixed solvent with gradual increase of χmethanol at RT [84, 89, 91, 92]. To gain a better understanding, Cui et al. used the SMFS method to investigate the single-chain mechanics of PNIPAM in the water/methanol mixed solvent at RT [79] (Fig. 11). It was found that the mechanical properties of the PNIPAM chain were greatly dependent on χmethanol of the mixed solvents. From 0% to 10% there was no obvious difference between the obtained force curve. The complexes of methanol and water were formed and dispersed well in the mixed solvents. Therefore, the complexes had almost no influence on the PNIPAM chains because of the low density. Further increased from 10% to 51%, all the force curves obtained in different χmethanol were superposed very well after normalization in the high and low force regimes, but, in the middle regime, remarkable deviation can be observed with the increase of χmethanol. As with the temperature-dependent transition, there was also a turning point at χmethanol 16 %, which was found by Swanson et al. in another study [93]. When χmethanol gradually increased from 10% to 16%, more and more water–methanol complexes were formed and, at the same time, the bound water molecules of the PNIPAM chain became fewer and fewer. Then the dehydrated PNIPAM chain is unstable and the conformation of the polymer chain changes from the coil state to the collapsed state. Because the rearrangement of bound water consumes additional energy upon stretching, the fewer water molecule bound collapsed state was expected to cost less energy. That is why the middle part of the force curve obtained at χmethanol = 16% was lower than that at 10%. However, as χmethanol further increased up to 18%, the water–methanol complexes continued to be formed. Because of the continuous dehydration of PNIPAM, intrachain C=O…H–N H-bonds were formed [81]. Thus, during chain elongation, the additional energy would be consumed to break the intrachain H-bonds, which was reflected by the deviation of force curves. It should be noted that the energy cost to stretch the PNIPAM chain at χmethanol = 18% was lower than that in water at 40°C. This is because the qualities of the intrachain H-bonds formed at χmethanol = 18% were lower than in water at T = 40°C [89]. That is, the methanol-induced collapsed state of PNIPAM chain was less compact than that induced by thermal at T = 40°C. With an increase of methanol from 18% to <50%, the solvent quality was still worse than that of water at RT. At χmethanol = 51%, the force curves superposed very well with those obtained in water at RT, which indicated that the conformations of the PNIPAM chain in the mixed solvent of χmethanol = 51% were similar to those in water at RT. Therefore, the solvent quality of the mixed solvent of 51% < χmethanol <100% was better than that of water for PNIPAM. In addition, based on the results obtained at χmethanol = 16% and pure water, it was possible to convert the chemical energy to the mechanical energy at RT; see Fig. 12. Therefore, the current work cast new light on the design of nano-devices with a function of chemical–mechanical energy conversion.

Comparisons of force curves of PNIPAM obtained in different water/methanol mixed solvents. Figure reproduced with permission from [79]

(a) Force curves of PNIPAM obtained in 0% and 16% of methanol/water mixed solvent. (b) Potential design of a molecular motor for chemical–mechanical energy conversion. Figure reproduced with permission from [79]
PDEAM is one of the polymers most similar to PNIPAM, which also has a coil-to-globule transition in aqueous solution above its LCST (~30°C) [94] (Scheme 4). To understand PNIPAM better, Cui et al. measured the single-molecule mechanical properties of PDEAM, and investigated the coil-to-globule transition and the hydration of a single PDEAM chain [80]. The normalized force curves of the two polymers obtained in nonpolar organic solvents, in which the inherent elasticity of individual polymer chain was reflected, superposed very well in the entire force curve. It was expected that, in most cases, the inherent elasticity of a polymer chain was only related to the backbone but was independent of the side groups (Fig. 13a). However, it was shown that the behaviors of the two polymers in water at RT were very different in the SMFS results; see Fig. 13b–d. During the chain elongation at RT, the additional energy consumed for the water rearrangement showed a large difference (~5.19 ± 0.10 kJ/mol for PNIPAM and ~1.13 ± 0.10 kJ/mol for PDEAM), which was attributed to the different polymer side groups [94]. The key point was that, on the N atom of the side groups of the two polymers, there are an H atom and a hydrophobic isopropyl group for PNIPAM, but for PDEAM there are only two hydrophobic ethyl groups. Because the PNIPAM chain contains both H-bond donor and acceptor, it was more hydrated in water than the PDEAM chain, which only contains acceptor. Therefore, more energy would be consumed for PNIPAM upon elongation.

Structures of PNIPAM and PDEAM. Figure reproduced with permission from [80]

(a) Comparison among the QM-FRC fitting curves and force curves of PNIPAM and PDEAM obtained in octane. (b) Comparison among the force curves of PNIPAM and PDEAM obtained in water and that obtained in octane. (c) Comparison of force curves of PDEAM at RT and 35°C. (d) Comparison of force curves of PNIPAM at RT and 35°C. Figure reproduced with permission from [80]
When the temperature was above its LCST, the single-chain elasticity of PDEAM was not dependent on the temperature, remarkably different to PNIPAM. This may indicate that the variation of the mechanical properties of PDEAM chain during the phase transition is too small to be distinguished (beyond the force resolution of AFM). The significant different temperature-dependence of the two polymers can be explained by two factors: conformation and hydration. First, the globule conformation of PNIPAM was more compact than that of PDEAM at T > LCST. Second, the formation of the intrachain H-bonds of PDEAM was not possible because of the absence of the H-bonds donors in the chain. As a result, with increase of temperature the amount of water molecules dehydrated from the polymer chain of PNIPAM was more than that of PDEAM. That is, the PNIPAM consumes more energy upon stretching [95]. By SMFS, the minor change in structure of the polymer chain was clearly distinguished.
5 Interactions Between Macromolecules and Solid Surface
Great progress has been made recently in the preparation and characterization of organic and polymeric ultra-thin multilayer films. Particularly, the layer-by-layer (LbL) assembly technique, which can be traced back to the pioneering work of Iler in 1966, has been developed very rapidly and has produced promising results [96–99]. The growing interest in LbL assembly is because of the unusual properties of the resulting nanostructured materials and their anticipated applications in the fields of advanced devices and sensors. To date, many delicate methods have been well established to fabricate layered assemblies with tailored architectures. These methods are mainly based on either one or in most cases several combined intermolecular interactions, such as electrostatic forces, H-bonding, and van der Waals interactions. Although the assembly methods are well established, there are still many problems to be addressed, above all the strength of the driving force in LbL assembly. Unfortunately, the strength at the single-molecule level cannot be directly measured by traditional methods. Since the early 2000s, Zhang and coworkers have combined the LbL construction technique and SMFS to investigate directly the strength of the driving force in LbL assembly by detaching the target polymer chain from a substrate [100–103].
5.1 Desorption of Polyelectrolytes from Solid Surface
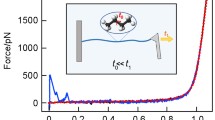
Utilizing the electrostatic interactions as the driving force, various polyelectrolytes are often used as building blocks in LbL assembly. To initiate the target first layer assembly, the substrate is usually treated to bear charge. For instance, the unmodified clean quartz substrate is nearly neutral in charge. However, a chemical modification by amino groups results in a positively charged surface when immersed into neutral or acidic aqueous solutions [101, 103]. A widely used polyanion, poly(2-acrylamido-2-methyl propane sulfonic acid) (PAMPS), can be adsorbed onto the oppositely charged amino-modified quartz. The strength of the driving force for this case has been measured by SMFS in aqueous environment [103]. The typical force curve of the desorption of single chain of PAMPS from the amino-modified quartz is shown in Fig. 14. The sharp peak in the initial stage of each force curve corresponds to the strong adhesion force between the bare tip and the uncovered regions of the substrate. For the case that one polymer chain is captured by the tip, only a little of the apex surface area is affected and the strong interaction between the tip and substrate is retained. Subsequently followed by a long plateau, the force drops to zero, indicating a rupture of the polymer bridge. The distance from the initial stage to the end of plateau is about 230 nm, giving an apparent contour length of the polymer chain being stretched (see Fig. 14). The applied force remains constant over the whole range of the plateau, which indicates that no remarkable elastic elongation occurs on the polymer chain being stretched. Because of the repulsion between monomers, the strongly charged polyelectrolyte chain assumes an extended conformation in solution [104], which facilitates an adsorption of single chains in a train-like conformation on the surface with opposite charge. When the long adsorbed train is attached, the AFM tip retraction progressively unzips the sequence of its binding sites with the surface and the force should remain constant, resulting in a long plateau in the force curve [101, 105]. The desorption force of the single PAMPS chain from the substrate, indicated by the height of the long plateau, remains ~120 pN along the desorption (unzipping) process (see more clearly from the inset of Fig. 14). The loading rate-independent result implies that the lifetime of the bond between the PAMPS chain and the substrate is much shorter than the time scale in our experiments (0.1–1 ms), and the desorption process is carried out in an quasi-equilibrium state. Thus, the adhesion force between PAMPS chain and the substrate is equal to the desorption force obtained.

Typical force curve of PAMPS that shows a long plateau with height of ~120 pN. Figure reproduced with permission from [103]
5.2 Desorption of Neutral Polymers from Solid Surface
In many studies, poly(4-vinyl pyridine) (P4VP) was deposited as the first layer on the substrate, such as the amino group modified quartz. It was assumed that the main driving force is H-bonding [106–108]. By utilizing the SMFS method, Zhang et al. exploited the same system to investigate the strength of the driving force of the first P4VP layer in LbL assembly [100]. Each peak in the force curve after the initial large peak corresponds to the detachment of a polymer segment from the substrate. Several peaks appear in each curve, producing an overall sawtooth pattern. The sawtooth pattern in the force curves has been attributed to the detachment of polymer loops from the substrate in series.
Statistical analysis revealed that the most probable desorption force is ~180 pN. Besides, the desorption force distribution showed that the integer multiples of 180 pN are also frequently observed. This result suggests that the value of 180 pN is an elementary force quantum, which can be attributed to the desorption of a single anchor point of P4VP from the substrate. When the substrate is changed to hydroxyl group-modified quartz, the most probable desorption force shifts to higher values. This result confirms that the signals observed in the force curve correspond to the desorption of P4VP from the substrate, not from the AFM tip. Further experiments shows that the desorption force of a single anchor point is independent of the loading rate, implying that the measurement is carried out in a quasi-equilibrium condition. In other words, the adsorption force between P4VP and the substrate is equal to the measured desorption force. It should be noted that the adsorption force of a single anchor point of P4VP is much stronger than a single H-bond [14, 109], which may suggest that, besides the H-bonding, other interactions (such as the solvophobic interactions) contribute to the driving force of the LbL assembly [110].
It is generally accepted that hydrophobic forces play a central role in the self-assembly carried out in aqueous medium, especially when large building blocks are involved. Yet, a quantitative understanding of this role has been elusive [111]. Again, SMFS is more powerful than traditional methods in this case, which was demonstrated by the work of Zhang et al. in 2003 [102]. To measure the hydrophobic forces per monomer unit, they prepared a segmented copolymer called PNIPAM-seg-PS, in which short hydrophobic PS segments were more uniformly inserted into a linear PNIPAM hydrophilic backbone [112]. The structure and composition of such a copolymer chain is schematically shown in Fig. 15a. It is reasonable to expect that the adsorption of insoluble short hydrophobic PS segments onto the hydrophobic PS substrate result in many PNIPAM “loops”. Figure 15c shows that the force curves exhibit a similar characteristic, namely, a sawtooth pattern. To find why such a pattern exists and how it is related to the chain structure, the distance between each two adjacent peaks in the force curves was analyzed statistically. The Gaussian fitting of the histogram led to an average distance of ~114 nm, which is very close to the average length of the “repeat unit” (one long PNIPAM segment plus one short PS segment, ~100–130 nm in length) in the copolymer chain. These results suggest that the copolymer chain forms loops with a similar size on the PS substrate. Therefore, the sawtooth pattern corresponds to the detachment of the adsorbed PS segments in a single chain from the PS substrate. It was found that for a given stretching velocity (V stretch = 4,600 nm/s) the desorption force essentially follows a Gaussian distribution and the most probable desorption force (F MPD) is ~41 pN. An interesting observation is that FMPD increases with increasing V stretch. The linear dependence of FMPD on log(V stretch) indicates that the adsorption and desorption of the PS segments on the PS substrate is a dynamic process. Because each PS segment contains 20 monomer units on average, it is estimated that the desorption force per PS monomer unit from the PS substrate in water is in the range of 1.3–2.1 pN, depending on the imposed stretching velocity. Compared with previous achievements using different methods [66, 113], this study provides, for the first time, a more direct determination because of its single chain manipulation.

(a) Molecular structure of PNIPAM-seg-PS. (b) Schematic of the adsorbed conformation of PNIPAM-seg-PS on PS substrate. (c) Typical force curves obtained in DI water. Figure reproduced with permission from [102]
6 SMFS Studies on the Interactions Involved in Supramolecular Polymers
In recent years, the development of supramolecular chemistry is usually related to the finding of new acceptors, including cyclodextrins [114], calixarenes [115], cucurbiturils [116], and pillararenes [117], which have frequently been used in the supramolecular systems. Since the concept of “supramolecular polymer” was proposed [2], the interest of supramolecular chemistry has been extended from the host–guest recognition interactions to the discovery of polymerization methods and functional materials with different applications, which include stimuli responsiveness, self-healing, and environmental adaptation [3, 118–120]. With the development of a series of supramolecular polymerization methods, a variety of novel supramolecular polymers are prepared with various non-covalently interactions [121], including host–guest, metal–ligand, H-bonding, π–π, and charge-transfer interactions.
6.1 Host–Guest Interactions
Molecular recognition is the specific binding of a guest molecule to a complementary host molecule to form a host–guest complex [36]. This concept was proposed in the very early stages during the developing of supramolecular chemistry. There are many examples utilizing host–guest interactions to construct supramolecular systems. Among others, the β-cyclodextrin (β-CD), a cyclic oligosaccharide consisting of seven glucose units linked via α-1-4 glycosidic bonds, is a commonly used host molecule, and various molecules (e.g., ferrocene) can be bound in the cavity as guest, mainly via hydrophobic forces [122, 123]. SMFS has been utilized to investigate this important supramolecular system at the single-molecule level [124–128].
The strategy of the experiment involves two steps. The first step is to chemically modify the AFM tip and substrate with host and guest molecules, respectively. The second step is to enable the formation of host–guest complex by bringing together the two surfaces, and rupturing the complex by separating them (see Fig. 16). During the latter step, the force curve is recorded, which provides extensive information on the system. One useful piece of information which can be extracted from the force curve is the rupture force of the host–guest complex. In an ideal case, an individual rupture event is observed in one force curve. However, multiple rupture events can occur simultaneously, which is actually the usual case. Therefore, a statistical analysis on plenty of similar force curves is necessary. Such analysis has shown that the observed rupture forces are integer multiples of one fundamental force quantum of 55 ± 10 pN, which is attributed to the rupture of an individual ferrocene–CD complexes in aqueous medium [124]. Further investigation of ferrocene–CD complexes showed that this force quantum is independent of the spacer length, and independent of the unloading rate [125]. These results indicate that the host–guest complex rupture forces were probed under conditions of thermodynamic equilibrium. This is different from the previously studied biological systems, which are dependent of the unloading rate [129–131].

Schematic representation (not to scale) of SMFS of ferrocene guest immobilized in a hydroxyl terminated SAM on an AFM tip and a SAM of CD on Au(111). Figure reproduced with permission from [124]
Besides ferrocene, other molecules with similar shape, such as adamantane and benzene, can be guest molecules in the β-CD host. Systematic studies of SMFS found similar results. The analysis of the histograms revealed periodic distributions of forces with loading-rate independent maxima at integer multiples of a certain force quantum characteristic of each guest. For instance, the observed force quanta were 39 ± 15 pN for benzene and 102 ± 15 pN for adamantane, respectively. The force values followed the same trend as the free binding energy ΔG measured for model guest compounds in solution or on β-CD monolayers, as determined by microcalorimetry and surface plasmon resonance measurements, respectively [126].
6.2 Metal–Ligand Interactions
Metal–ligand interactions are often used to fabricate supramolecular systems with regular shapes, e.g., double helicates, racks, grids, and linear rods [36]. These systems are well understood with respect to their behaviors in the solid state as well as on surfaces. However, little was known about the binding force of the supramolecular complexes. Several systems on the topic of metallo-supramolecules have been studied by SMFS [33, 132]. For such purpose, the researchers chemically modified the substrate with a layer of terpyridine Ru(II) mono-complexes, as shown in Fig. 17. The tip was chemically modified with a layer of non-complexed terpyridine ligand. By bringing together the tip and the substrate, the direct proximity of the free ligand and the mono-complex allow the formation of bisterpyridine complexes. By retracting the tip from the substrate, the complex is stretched till rupture, where the rupture force corresponds to the binding force of the bisterpyridine Ru(II) complexes. Statistical analysis of the rupture force shows that, at a given velocity of 118 nm/s, the histogram exhibits one pronounced peak at 95 pN followed by weaker peaks at 171 and 253 pN. The weaker peaks were attributed to the simultaneous rupture of two or three parallel complexes, respectively. This hypothesis was supported by elasticity model fitting and Monte Carlo simulations [33].

Schematic of the SMFS experiment. Figure reproduced with permission from [33]
6.3 H-Bonding
Supramolecular polymers [2, 133] (also called reversible polymers) are comprised of bifunctional monomeric units that are reversibly aggregated through relatively strong non-covalent interactions. For instance, the self-complementary and recognition of the quadruple H-bonded bis(2-ureido-4[1H]-pyrimidinone) (bisUPy) can form rather stable supramolecular polymers. By utilizing SMFS, Zou et al. measured the strength of the driving force of the self-assembly between the monomers (Fig. 18) [134, 135].

Schematic of the SMFS experiment and typical force curve obtained in a hexadecane. Figure reproduced with permission from [134]
As shown in Fig. 18, the apex of the peak of the force curve corresponds to the rupture of the supramolecular polymer upon stretching, from which the “degree of polymerization” (DP) can be estimated to be 2–15. At 301 K and a loading rate of 35 nN/s, the rupture force of the supramolecular polymer upon stretching, which is indicated by the apex of the peak, is 172 ± 23 pN. For the pure dimmers, the rupture force is 180 ± 21 pN [134]. The rupture forces decrease with increasing lengths of the stretched supramolecular polymer chains. This observation is in agreement with the theory developed by Evans et al. [136, 137], who predicted a decrease in the magnitude of the single-complex rupture force with increasing spacer length [138].
By contrast, for measurements carried out in situ at 330 K, no loading-rate dependence was observed for rates between 5 and 200 nN/s. This observation indicates that the corresponding experiment was carried out under quasi-equilibrium conditions. These data obtained at 330 K are equivalent to those obtained at 301 K, but at a lower loading rate [135, 139].
To obtain the thermal force and dimer equilibrium constant K eq using the Evans model, the most probable rupture force between the binding motifs is usually measured for various loading rate. By utilizing the theory of Evans and Williams, Vancso et al. found that those parameters can be obtained even with one data set at a single loading rate. The value of the dimer equilibrium constant obtained for UPy–UPy dimers in hexadecane agrees very well with the value of K eq ~ 1 × 109 M−1 predicted by previous studies with bulk measurements [140].
More recently, the binding force of another self-complementary quadruple H-bonding motif, urea-aminotriazine (UAT), was investigated by SMFS [141, 142]. By measuring the rupture force between UAT at various loading rates, the bond lifetime at zero force, t off(0), can be estimated to be 100 ± 80 ms. This SMFS result is nearly three times higher than that estimated from the data of bulk measurements. This marked discrepancy shows that the complex is much more stable than predicted on the basis of the simple model, in which the stabilization effect of the planarized molecular geometry is not covered. These studies demonstrated that SMFS is an indispensable supplement to the characterization methods.
6.4 π–π Interactions
The π–π interactions (aromatic interactions) are ubiquitous and important in diverse phenomena, including stabilizing DNA and protein structures, packing aromatic molecules in crystals, and binding guest moieties to host systems. It has been suggested that π-π interactions consist of electrostatic, hydrophobic, and van der Waals forces [143]. By modifying the AFM tip with a pyrene tailored polymer chain, Zhang et al. successfully detected the desorption force of a single pyrene unit from a graphite surface (see Fig. 19) [144]. Interestingly, the measured force, 55 ± 16 pN, is independent of the loading rate, implying that the measurement is carried out under quasi-equilibrium conditions. In other words, the adsorption force between the pyrene unit and the graphite surface is equal to the desorption force obtained.

Schematic setup for measuring the π–π interaction between pyrene and graphite in aqueous medium by SMFS. Figure reproduced with permission from [144]
6.5 Host-Stabilized Charge Transfer Interactions
Host-stabilized charge transfer interaction (HSCT) is a concept that was proposed by Kim et al. in which donor and acceptor are both encapsulated in the cavity of the host cucurbit[8]uril (CB[8]) [145, 146]. Because of the short distance between the donor and the acceptor, the CT interaction is remarkably enhanced. However, attempts to obtain supramolecular polymers with a high degree of polymerization utilizing only single HSCT interactions have failed for the following reasons: (1) the strength of single HSCT interactions was not sufficient and (2) single HSCT interactions do not have high orientation selectivity, thus leading to the formation of cyclic species and suppressing the formation of polymeric species.
Recently, Zhang et al. have developed a new idea of utilizing multiple HSCT interactions for fabricating supramolecular polymers [121, 128]. By using a carefully designed multifunctional monomer, the cyclization is successfully suppressed, leading to the formation of supramolecular polymers. SMFS results revealed that the rupture force of the supramolecular polymer can be nearly 200 pN, indicating that the binding force within the supramolecular polymer is rather strong. The contour length measured by SMFS can reach as high as 600 nm, implying that the supramolecular polymer has a high degree of polymerization.
7 SMFS Studies on Synthetic Molecular Machines and Analog Systems
To mimic the fantastic biomolecules that can generate directional forces, people have attempted to design and synthesize a number of molecular machines during the last two decades. Because molecular machines usually work at the single-molecule level, SMFS is an ideal method to investigate those systems. We would like to provide four examples of SMFS studies of the synthetic molecular machines in this section.
Azobenzene is a light-sensitive molecule. Under irradiation by UV light of 365 nm, the azobenzene in trans configuration is converted to cis isomer, and under irradiation by light of 420 nm, an inverse process takes place. A change of end-to-end distance is expected with these transitions, i.e., the molecule is shortened upon transition from trans to cis and vice versa. By utilizing a polymer containing azobenzene groups in the backbone, Gaub et al. first realized the conversion from light energy to mechanical work at the single-molecule level [147, 148]. To minimize the negative effects to the AFM cantilever, the evanescent wave from total internal reflection was used. A shortening of ~3 % of the polymer chain was observed from the saturated trans-azo state to the saturated cis-azo state. The efficiency of converting optical energy to mechanical work was up to 10 %.
In 2007, Vancso and coworkers realized the closed-cycle conversion from electric energy to mechanical work at the single-molecule level [149]. The electrochemically responsive polymer, poly(ferrocenylsilane) (PFS), was covalently attached to a gold electrode. Using SMFS it was found that the PFS chain shrank when the oxidized state was converted into the neutral state. The shrinkage against an external load can be used to design a molecular device. A maximum conversion efficiency of 26% was obtained in the single molecule experiments.
Rotaxanes are paradigm synthetic molecular machine systems which enable controlled large-amplitude movement and positioning of one mechanically interlocked component with respect to another. Differing from the previously studied biological/synthetic molecular machines, a rotaxane is a small molecule (less than 5 nm long). Recently, Leigh and his coworkers investigated an elegant system with rotaxanes by SMFS [150, 151]. In a rotaxane, the molecular ring is threaded onto a molecular axle. There are two H-bond binding site in the axle, but with different affinities to the motif in the ring. A peak of ~30 pN can be found upon elongation of the PEO chain, which is linked to the ring of rotaxane. This peak can be observed upon elongation or relaxation, which indicates that the process is reversible. Confirmed by control experiments, the peak is attributed to the process of breaking/forming of the H-bonding between the two motifs in the molecule ring and axle of the rotaxane, respectively. In the relaxation process, the macrocycle travels back from the weak binding site to the strong one and it is able to generate a force against the external load of 30 pN, similar to those generated by biological machines. The mechanical work produced by this sub-molecular motion is ~6 kcal mol−1. This work demonstrates that AFM-SMFS can be used to investigate the mechanochemical behavior of a molecule less than 5 nm in size.
During the past 20 years, proteins and synthetic polymers have been investigated by SMFS, which has aided in the understanding of their nanomechanical properties. However, it remains a challenge to correlate directly the bulk mechanical performance to the nanomechanical properties of individual constituent macromolecules. Guan et al. recently made major progress towards this goal [152–154]. They synthesized a biomimetic modular polymer with two UPy (see also Sect. 6.3) motifs in each module. By stretching/relaxing, the UPy motifs in single modular polymer chain can be unfolded/refolded, resulting in a sawtooth pattern in the force curve, which resembles that of a modular protein, titin. By increasing the stretching velocity, an increase of the rupture force of the UPy motifs can be observed, through which the parameters of the single-molecule energy landscape can be derived. When the mechanical properties of bulk material from the same polymer were compared with those at the single-molecule level, Guan et al. found direct correlations between these two block of data: the high rupture force and quantitative passive re-folding observed at the single-molecule level can be associated with the large energy dissipation during plastic deformation, as well as the slow but complete recovery of strength, strain, and toughness observed during the course of the load relaxation recovery cycle in the bulk experiment. These results illustrated the potential for SMFS to serve as a guide for future rational design of advanced multifunctional materials.
8 Summary and Perspective
During the last two decades, the supramolecular chemistry and mechanochemistry of macromolecules have been developed greatly. The SMFS method provided an effective way to access the macromolecules at the single-chain level. With the single-molecule mechanics results, the classic polymer chain theory of Flory has been verified for the first time. The bound water of macromolecules and the rearrangement upon elongation have been investigated, showing that the energies associated with the bound water is very important to the self-assembly of biomacromolecules. For supramolecular polymers, the binding force between the moieties has been measured directly. With the great effort of researchers, a clearer image of macromolecules system has been revealed at the molecular level.
However, there are still some challenges in this field. Here are two examples. (1) The database of single-chain elasticity of macromolecules is not yet complete. As with the periodic table of elements, a complete database of macromolecules would certainly be helpful for the development of science and engineering. (2) The noise level of SMFS is still too high. A typical noise of 5–10 pN conceals some important data. However, it is greatly anticipated that, in the future, SMFS can contribute further to the development of supramolecular chemistry and mechanochemistry of macromolecules.
Abbreviations
- AFM:
-
Atomic force spectroscopy
- AMIMCl:
-
Allyl-3-methylimidazolium chloride
- bisUPy:
-
Bis(2-ureido-4[1H]-pyrimidinone)
- CB[8]:
-
Cucurbit[8]uril
- DEBenzene:
-
Diethylbenzene
- DP:
-
Degree of polymerization
- dsDNA:
-
Double-stranded DNA
- FJC:
-
Freely jointed chain
- Force curve:
-
Force-extension curve
- FRC:
-
Freely rotating chain
- Gua:
-
Guanidine chloride
- HSCT:
-
Host-stabilized charge transfer
- IL:
-
Ionic liquid
- LbL:
-
Layer-by-layer
- MD:
-
Molecular dynamics
- M-FJC:
-
Modified FJC
- NIPAM:
-
N-Isopropylacrylamide
- nN:
-
NanoNewtons
- P4VP:
-
Poly(4-vinyl pyridine)
- PAAM:
-
Polyacrylamide
- PAMPS:
-
Poly(2-acrylamido-2-methyl propane sulfonic acid)
- PBS:
-
Phosphate buffered saline
- PDEAM:
-
Poly(N,N-diethylacrylamide)
- PEG:
-
Polyethylene glycol
- PFS:
-
Poly(ferrocenylsilane)
- pN:
-
PicoNewtons
- PNIPAM:
-
Poly(N-isopropylacrylamide)
- PS:
-
Polystyrene
- PVA:
-
Poly(vinyl alcohol)
- RT:
-
Room temperature
- SMFS:
-
Single-molecule force spectroscopy
- ssDNA:
-
Single-stranded DNA
- UAT:
-
Urea-aminotriazine
- WLC:
-
Worm-like chain
- β-CD:
-
β-Cyclodextrin
- χmethanol :
-
Methanol molar fraction
References
Lehn J-M (1985) Science 227:849
Brunsveld L, Folmer BJB, Meijer EW, Sijbesma RP (2001) Chem Rev 101:4071
Lehn J-M (2013) Angew Chem Int Ed 52:2836
Lehn J-M (2007) Chem Soc Rev 36:151
Lehn JM (2002) Proc Natl Acad Sci U S A 99:4763
Lehn JM (2002) Polym Int 51:825
Gulik-Krzywicki T, Fouquey C, Lehn J (1993) Proc Natl Acad Sci U S A 90:163
Grandbois M, Beyer M, Rief M, Clausen-Schaumann H, Gaub HE (1999) Science 283:1727
Rief M, Oesterhelt F, Heymann B, Gaub HE (1997) Science (Washington D.C.) 275:1295
Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE (1997) Science 276:1109
Hugel T, Seitz M (2001) Macromol. Rapid Commun 22:989
Janshoff A, Neitzert M, Oberdorfer Y, Fuchs H (2000) Angew Chem Int Ed 39:3212
Clausen-Schaumann H, Rief M, Tolksdorf C, Gaub HE (2000) Biophys J 78:1997
Rief M, Clausen-Schaumann H, Gaub HE (1999) Nat Struct Mol Biol 6:346
Marszalek PE, Lu H, Li HB, Carrion-Vazquez M, Oberhauser AF, Schulten K, Fernandez JM (1999) Nature 402:100
Li YR, Qin M, Li Y, Cao Y, Wang W (2014) Langmuir 30:4358
Fuhrmann A, Getfert S, Fu Q, Reimann P, Lindsay S, Ros R (2012) Biophys J 102:2381
Friddle RW, Noy A, De Yoreo JJ (2012) Proc Natl Acad Sci U S A 109:13573
Jiang ZH, Zhang YH, Yu Y, Wang ZQ, Zhang X, Duan XR, Wang S (2010) Langmuir 26:13773
Song B, Schönherr H (2012) Supramolecular chemistry. Wiley, Ltd
Schroeder T, Walhorn V, Mattay J, Anselmetti D (2012) Analytical Methods in Supramolecular Chemistry. Volume 1&2, Second Edition: 559 Wiley Online Library
Neuman KC, Nagy A (2008) Nat Methods 5:491
Liu K, Song Y, Feng W, Liu N, Zhang W, Zhang X (2011) J Am Chem Soc 133:3226
Liu N, Peng B, Lin Y, Su Z, Niu Z, Wang Q, Zhang W, Li H, Shen J (2010) J Am Chem Soc 132:11036
Binnig G, Quate CF, Gerber C (1986) Phys Rev Lett 56:930
Oesterhelt F, Rief M, Gaub HE (1999) New J Phys 1:6.1
Zhang W, Zhang X (2003) Prog Polym Sci 28:1271
Marszalek PE, Oberhauser AF, Pang Y, Fernandez JM (1998) Nature 396:661
Zhang WK, Zou S, Wang C, Zhang X (2000) J Phys Chem B 104:10258
Liu C, Cui S, Wang Z, Zhang X (2005) J Phys Chem B 109:14807
Cui S, Albrecht C, Kühner F, Gaub HE (2006) J Am Chem Soc 128:6636
Cui S, Yu J, Kuehner F, Schulten K, Gaub HE (2007) J Am Chem Soc 129:14710
Kudera M, Eschbaumer C, Gaub HE, Schubert US (2003) Adv Func Mater 13:615
Kuehner F, Erdmann M, Sonnenberg L, Serr A, Morfill J, Gaub HE (2006) Langmuir 22:11180
Sonnenberg L, Luo Y, Schlaad H, Seitz M, Coelfen H, Gaub HE (2007) J Am Chem Soc 129:15364
Lehn J-M (1995) Supramolecular chemistry: concepts and perspectives. VCH, Weinheim
Kellermayer MSZ, Smith SB, Granzier HL, Bustamante C (1997) Science 276:1112
Bustamante C, Marko JF, Siggia ED, Smith S (1994) Science (Washington, D.C.) 265:1599
Smith SB, Finzi L, Bustamante C (1992) Science 258:1122
Smith SB, Cui Y, Bustamante C (1996) Science 271:795
Li HB, Zhang WK, Xu WQ, Zhang X (2000) Macromolecules 33:465
Bao Y, Qian HJ, Lu ZY, Cui SX (2014) Nanoscale 6:13421
Wang K, Pang X, Cui S (2013) Langmuir 29:4315
Flory P, Volkenstein M (1969) Statistical mechanics of chain molecules. Wiley Online Library
Tskhovrebova L, Trinick J, Sleep JA, Simmons RM (1997) Nature 387:308
Marko JF, Siggia ED (1995) Macromolecules 28:8759
Hugel T, Rief M, Seitz M, Gaub HE, Netz RR (2005) Phys Rev Lett 94:048301
Cui S, Yu Y, Lin Z (2009) Polymer 50:930
Klemm D, Heublein B, Fink HP, Bohn A (2005) Angew Chem Int Ed 44:3358
Wan Z, Li L, Cui S (2008) Biopolymers 89:1170
Azizi Samir MAS, Alloin F, Dufresne A (2005) Biomacromolecules 6:612
Sakurada I, Nukushina Y, Ito T (1962) J Polym Sci Pol Chem 57:651
Wang H, Gurau G, Rogers RD (2012) Chem Soc Rev 41:1519
Zhang H, Wu J, Zhang J, He J (2005) Macromolecules 38:8272
Eichhorn S, Young R (2001) Cellulose 8:197
French AD, Johnson GP (2009) Cellulose 16:959
Cui S (2010) Phys Chem Chem Phys 12:10147
Tanford C (1970) Adv Prot Chem 24:195
Bergstrom K, Holmberg K, Safranj A, Hoffman AS, Edgell MJ, Kozlowski A, Hovanes BA, Harris JM (1992) J Biomed Mater Res 26:779
Mandelkern L (1990) Acc Chem Res 23:380
Begum R, Matsuura H (1997) J Am Chem Soc 93:3839
Watson JD, Crick FH (1953) Nature 171:737
Kool ET, Morales JC, Guckian KM (2000) Angew Chem Int Ed 39:990
Tanaka K, Okahata Y (1996) J Am Chem Soc 118:10679
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) J Comput Chem 4:187
Pashley RM, McGuiggan PM, Ninham BW, Evans DF (1985) Science 229:1088
Ball P (2008) Nature 452:291
Brovchenko I, Krukau A, Oleinikova A, Mazur AK (2008) J Am Chem Soc 130:121
Breslauer KJ, Frank R, Blöcker H, Marky LA (1986) Proc Natl Acad Sci U S A 83:3746
Miller SL (1953) Science 117:528
Knight RD, Landweber LF (2000) Cell 101:569
Giannotti MI, Rinaudo M, Vancso GJ (2007) Biomacromolecules 8:2648
Haxaire K, Buhler E, Milas M, Perez S, Rinaudo M (2002) Hyaluronan, vol 1. Woodhead Publishing Ltd., Cambridge, p. 37
Pelton R, Chibante P (1986) Colloids Surf 20:247
Meyer DE, Shin B, Kong G, Dewhirst M, Chilkoti A (2001) J Control Release 74:213
Schild H (1992) Prog Polym Sci 17:163
Lessard D, Ousalem M, Zhu X, Eisenberg A, Carreau P (2003) J Polym Sci Pol Phys 41:1627
Cui S, Pang X, Zhang S, Yu Y, Ma H, Zhang X (2012) Langmuir 28:5151
Pang X, Wang K, Cui S (2013) Polymer 54:3737
Pang X, Cui S (2013) Langmuir 29:12176
Wang X, Qiu X, Wu C (1998) Macromolecules 31:2972
Okada Y, Tanaka F (2005) Macromolecules 38:4465
Cheng H, Shen L, Wu C (2006) Macromolecules 39:2325
Tanaka F, Koga T, Winnik FM (2008) Phys Rev Lett 101:028302
Ono Y, Shikata T (2006) J Am Chem Soc 128:10030
Cho EC, Lee J, Cho K (2003) Macromolecules 36:9929
Sun B, Lin Y, Wu P, Siesler HW (2008) Macromolecules 41:1512
Kay ER, Leigh DA, Zerbetto F (2007) Angew Chem Int Ed 46:72
Zhang G, Wu C (2001) Phys Rev Lett 86:822
Winnik FM, Ringsdorf H, Venzmer J (1990) Macromolecules 23:2415
Tanaka F, Koga T, Kojima H, Winnik FM (2009) Macromolecules 42:1321
Katsumoto Y, Tanaka T, Ihara K, Koyama M, Ozaki Y (2007) J Phys Chem B 111:12730
Chee CK, Hunt BJ, Rimmer S, Soutar I, Swanson L (2011) Soft Matter 7:1176
Idziak I, Avoce D, Lessard D, Gravel D, Zhu X (1999) Macromolecules 32:1260
Maeda Y, Nakamura T, Ikeda I (2001) Macromolecules 34:1391
Iler RK (1966) J Colloid Interface Sci 21:569
Decher G, Hong JD (1991) Makromol Chem Macromol Symp 46:321
Decher G (1997) Science 277:1232
Zhang X, Chen H, Zhang H (2007) Chem Commun 1395
Zhang W, Cui S, Fu Y, Zhang X (2002) J Phys Chem B 106:12705
Cui S, Liu C, Zhang X (2003) Nano Lett 3:245
Cui SX, Liu CJ, Zhang WK, Zhang X, Wu C (2003) Macromolecules 36:3779
Cui S, Liu C, Wang Z, Zhang X, Strandman S, Tenhu H (2004) Macromolecules 37:946
Netz R, Joanny J (1999) Macromolecules 32:9013
Conti M, Bustanji Y, Falini G, Ferruti P, Stefoni S, Samori B (2001) ChemPhysChem 10:610
Wang LY, Wang ZQ, Zhang X, Shen JC (1997) Macromol. Rapid Commun 18:509
Wang L, Cui S, Zhang X, Jiang M, Chi L, Fuchs H (2000) Langmuir 16:10490
Fu Y, Bai SL, Cui SX, Qiu DL, Wang ZQ, Zhang X (2002) Macromolecules 35:9451
Han T, Williams JM, Beebe TP (1995) Anal Chim Acta 307:365
Ray A (1971) Nature 231:313
Lum K, Chandler D, Weeks JD (1999) J Phys Chem B 103:4570
Zhang G, Wu C (2003) Phys Rev Lett 90:035506
Zhao X, Zhao W, Zheng X, Rafailovich M, Sokolov J, Schwarz S, Pudensi M, Russell T, Kumar S, Fetters L (1992) Phys Rev Lett 69:776
Harada A, Hashidzume A, Yamaguchi H, Takashima Y (2009) Chem Rev 109:5974
Botana E, Da Silva E, Benet-Buchholz J, Ballester P, de Mendoza J (2007) Angew Chem Int Ed 46:198
Bhasikuttan AC, Pal H, Mohanty J (2011) Chem Commun (Camb) 47:9959
Xue M, Yang Y, Chi X, Zhang Z, Huang F (2012) Acc Chem Res 45:1294
Dong S, Zheng B, Wang F, Huang F (2014) Acc Chem Res 47:1982
Huang Z, Yang L, Liu Y, Wang Z, Scherman OA, Zhang X (2014) Angew Chem Int Ed 53:5351
Yan X, Wang F, Zheng B, Huang F (2012) Chem Soc Rev 41:6042
Liu YL, Liu K, Wang ZQ, Zhang X (2011) Chem Eur J 17:9930
Wenz G (1994) Angew Chem Int Ed 33:803
Rekharsky MV, Inoue Y (1998) Chem Rev 98:1875
Schoenherr H, Beulen MWJ, Buegler J, Huskens J, van Veggel FC, Reinhoudt DN, Vancso GJ (2000) J Am Chem Soc 122:4963
Zapotoczny S, Auletta T, Jong MRD, Schoenherr H, Huskens J, van Veggel FC, Reinhoudt DN, Vancso GJ (2002) Langmuir 18:6988
Auletta T, de Jong MR, Mulder A, van Veggel FC, Huskens J, Reinhoudt DN, Zou S, Zapotoczny S, Schoenherr H, Vancso GJ, Kuipers L (2004) J Am Chem Soc 126:1577
Kado S, Kimura K (2003) J Am Chem Soc 125:4560
Liu Y, Wang Z, Zhang X (2012) Chem Soc Rev 41:5922
Kienberger F, Kada G, Gruber HJ, Pastushenko VP, Riener C, Trieb M, Knaus H-G, Schindler H, Hinterdorfer P (2000) Single Mol 1:59
Merkel R, Nassoy P, Leung A, Ritchie K, Evans E (1999) Nature 397:50
Evans EA, Calderwood DA (2007) Science 316:1148
Kersey FR, Yount WC, Craig SL (2006) J Am Chem Soc 128:3886
Lehn JM (1993) Makromol. Chem. Macromol Symp 69:1
Zou S, Schoenherr H, Vancso GJ (2005) Angew Chem Int Ed 44:956
Zou S, Schoenherr H, Vancso GJ (2005) J Am Chem Soc 127:11230
Evans E, Ritchie K, Merkel R (1995) Biophys J 68:2580
Evans E, Ritchie K (1997) Biophys J 72:1541
Evans E (2001) Annu Rev Biophys Biomol Struct 30:105
Vancso GJ (2007) Angew Chem Int Ed 46:3794
Embrechts A, Schonherr H, Vancso GJ (2008) J Phys Chem B 112:7359
Embrechts A, Schonherr H, Vancso GJ (2012) J Phys Chem B 116:565
Embrechts A, Velders AH, Schonherr H, Vancso GJ (2011) Langmuir 27:14272
Hoeben FJM, Jonkheijm P, Meijer EW, Schenning AP (2005) Chem Rev 105:1491
Zhang Y, Liu C, Shi W, Wang Z, Dai L, Zhang X (2007) Langmuir 23:7911
Ko YH, Kim E, Hwang I, Kim K (2007) Chem Commun 1305
Ko YH, Kim K, Kang JK, Chun H, Lee JW, Sakamoto S, Yamaguchi K, Fettinger JC, Kim K (2004) J Am Chem Soc 126:1932
Holland NB, Hugel T, Neuert G, Cattani-Scholz A, Renner C, Oesterhelt D, Moroder L, Seitz M, Gaub HE (2003) Macromolecules 36:2015
Hugel T, Holland NB, Cattani A, Moroder L, Seitz M, Gaub HE (2002) Science 296:1103
Shi WQ, Giannotti MI, Zhang X, Hempenius MA, Sconherr H, Vancso GJ (2007) Angew Chem Int Ed 46:8400
Van Quaethem A, Lussis P, Leigh DA, Duwez AS, Fustin CA (2014) Chem Sci 5:1449
Lussis P, Svaldo-Lanero T, Bertocco A, Fustin CA, Leigh DA, Duwez AS (2011) Nat Nanotechnol 6:553
Roland JT, Guan Z (2004) J Am Chem Soc 126:14328
Chung J, Kushner AM, Weisman AC, Guan ZB (2014) Nat Mater 13:1055
Kushner AM, Gabuchian V, Johnson EG, Guan ZB (2007) J Am Chem Soc 129:14110
Acknowledgements
This work was supported by the Natural Science Foundation of China (21222401, 21074102), the program for New Century Excellent Talents in University (NCET-11-0708), and the Fundamental Research Funds for the Central Universities (SWJTU11ZT05, SWJTU12CX001).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cheng, B., Cui, S. (2015). Supramolecular Chemistry and Mechanochemistry of Macromolecules: Recent Advances by Single-Molecule Force Spectroscopy. In: Boulatov, R. (eds) Polymer Mechanochemistry. Topics in Current Chemistry, vol 369. Springer, Cham. https://doi.org/10.1007/128_2015_628
Download citation
DOI: https://doi.org/10.1007/128_2015_628
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22824-2
Online ISBN: 978-3-319-22825-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)