
Abstract
Detection of quadruplex structures by visual methods is a major challenge of the quadruplex nucleic acid research area. Consequently, considerable efforts are under way for the discovery of quadruplex specific agents endowed with fluorescence properties. In this review chapter we propose a comprehensive and critical overview of the diverse molecular design and strategies that have been described to identify quadruplex-selective fluorescent probes. Innovative compounds as well as classical DNA dyes are reviewed. The compounds have been divided into three classes: (1) “light-up” probes that display a strong enhancement upon G4 binding, (2) “light-off” probes that display a decreased fluorescence upon binding, and (3) permanent probes (“tagged” G4-binders) that exhibit no variation of fluorescence but display quadruplex binding specificity. The labeling performances of probes in various analytical contexts (in solution, in gel, at the level of chromosomes, and in fixed cells) are also reported and commented on when available. Finally we address the strengths and weaknesses of each probe class and highlight the critical features that must be addressed in developing a practicable quadruplex-specific labeling agent.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Single-stranded regions of nucleic acid containing repeats of bases are known to form alternative secondary structures due to unusual (i.e., non-Watson–Crick) base pairing. These alternative structures are believed to act as roadblocks to proteins processing single-stranded DNA templates transiently formed during replication, homologous recombination, repair, and transcription. In the same manner, transcriptional machineries that use single-stranded messenger RNA as substrate for translation and splicing may also be affected. However, although alternative structures are currently recognized to interfere with the transfer of genetic information, the question as to whether they participate in a natural regulation mechanism or are simply toxic elements is still open [1–7].
Currently the most well-studied and characterized non-B-DNA structures are the four-stranded assemblies called G-quadruplexes (abbreviated G4), which form by fold-over of single-stranded sequences containing guanines repeats and are the subject of this volume. G-Quadruplexes are composed of π-stacks of guanine quartets held together by H-bonding and by monovalent metal cations (K+ in a cellular context) sandwiched between the quartets (for structural details see reviews [8–11]).
Structural recognition of quadruplex nucleic acids by small molecules has been the focus of tremendous developments since the pioneering work of S. Neidle, L. Hurley et al. who paved the way by targeting quadruplex DNA with anthraquinone derivatives [12]. Since then this research area is progressing rapidly in the desire to find small molecules able to probe the formation of quadruplexes in a biological context and to help in deciphering their biological functions. Another objective of this field is to discover new targeted pharmacological agents able to act in a sequence/structure specific manner on DNA or RNA with resulting anticancer properties [13–15].
Although well-studied in vitro, evidence of the formation of quadruplexes in vivo remains mostly indirect but is currently supported by robust biochemical and molecular genetics data [1–7, 16]. Logically, the question of direct visualization of quadruplexes arises. Compelling evidence has been provided by observation of G4-DNA in human DNA by electron and atomic force microscopy [17–20] and by immunochemistry in ciliates [21]. In parallel, the search for fluorescent compounds that could signal the presence of a quadruplex structure is a matter of great interest, especially in view of the increasing use of fluorescent probes in cellular biology and the emergence of new fluorescence-based microscopies [22, 23]. However, currently, direct visualization of quadruplexes in a functional biological system is a challenging task accompanied with technical difficulties and conceptual uncertainties. Indeed, one should bear in mind that only large domains with a high density of quadruplexes able to form fluorescent foci could be imaged by microscopy, since detection of a single monomeric quadruplex unit is largely below the actual resolution; additionally, whether such domains do form and under what conditions is poorly understood. Nonetheless, the first absolute requirement of this quest is the availability of highly specific quadruplex-binding fluorescent dyes. Consequently, the chemistry of fluorescent compounds able to target quadruplexes experienced extensive development during the past decade.
In this review we describe the considerable work in terms of molecular design that has produced a large variety of quadruplex-interactive agents able to achieve G4-DNA labeling in different analytical contexts (e.g., from solution to gel electrophoresis and cellular imaging) and with various efficiencies. The compounds have been divided into three classes: (1) “light-up” probes, i.e., compounds that display a strongly enhanced fluorescence upon binding, (2) “light-off” probes, i.e., compounds that display a strongly decreased fluorescence upon binding, and (3) permanent probes, i.e., compounds the fluorescence of which is not modified by binding, but exhibiting exquisite specificity; these are mainly fluorescently tagged quadruplex binders. Finally, we address the strengths and weaknesses of each probe class and examine the issues and highlight the critical probe design features that must be addressed in developing a practicable G4-DNA probe (especially as a function of the intended application).
In order to help understand the diverse molecular designs of the probes described below, it is necessary to recall that quadruplexes may accommodate small molecules in several ways as depicted in Fig. 1. π-Stacking on external G-quartets on the top or bottom of a quadruplex unit is the most well-known mode characterized both in solid state and in solution [24–26]. In domains containing repeats of quadruplex units, sandwiching of a ligand between two units is likely to occur, as shown by X-ray studies [27, 28] and suggested by indirect evidence [20]. The stacking mode that may eventually involve interactions with surrounding loops is mainly the feature of planar rigid aromatics. Alternatively, insertion in the grooves of quadruplexes, that possess four grooves, is to be considered, especially for nonplanar, flexible molecules as shown by NMR [29] and CD in recent studies [30, 31]. Finally, quadruplexes provide a polyanionic scaffold that is capable of inducing external binding of cationic molecules in more or less ordered aggregates as is known for duplex DNA and certain dyes [32]. We will see that the design of fluorescent G4-DNA ligands relies on these different binding sites that provide various environments differing by their polarity and their capacity to immobilize external ligands and to isolate them from interaction with water – three parameters that strongly affect the fluorescence of dyes.

Binding modes of ligands to quadruplex DNA
2 “Light-Up” Probes
2.1 Classical DNA Probes
2.1.1 Cyanine Dyes
The cyanine (polymethine) dyes, which are among the oldest synthetic dyes, have been widely used as fluorescent probes for nucleic acids as they may display a large fluorescent enhancement upon binding. Their binding modes may be complex depending strongly on the length of the polymethine bridge and on the nucleic acid conformation. Thus, intercalation, dimerization in minor grooves, and template-directed external self-stacking have been characterized [33, 34]. Asymmetric cyanines are built from two distinct heteroaromatic systems connected through a conjugated monomethine or polymethine bridge. Short-bridged cyanines (monomethine and trimethine dyes) are often characterized by a turn-on fluorescence upon binding to DNA. This phenomenon is rationalized by the restriction of the rotation of this conjugated bridge, thus preventing nonradiative relaxation processes from taking place (Fig. 2) [35, 36].

Binding to DNA immobilizes cyanine dyes (here thiazole orange) and hinders the non-radiative deactivation pathways through restriction of the rotation around the conjugated bond, leading to enhanced fluorescence
2.1.1.1 Thiazole Orange
Thiazole orange (TO, Fig. 2) is a well-known monomethine dye that binds to almost all forms of nucleic acids. Therefore TO has been used in a variety of applications such as RNA staining [37], agarose gel DNA staining [38], and capillary electrophoresis [39]. TO represents a paradigm of nucleic acid light-up probes since its fluorescence quantum yield is about 2 × 10−4 when the dye is free in solution but increases to around 0.1 when bound to ds DNA and poly(dA), or 0.4 in the case of poly(dG) [35, 40]. Thus, TO exhibits a fluorescence enhancement upon binding, which varies from 50-fold to 1,000-fold depending on the sequence and conformation of the nucleic acid.
TO was firstly used as a fluorescent indicator for quadruplex DNA in the G4 fluorescent intercalator-displacement (FID) assay [41], by analogy with the previous work of Boger et al. on DNA hairpins [42, 43]. It was found that TO becomes highly fluorescent (500–3,000-fold increase, λ ex = 501 nm, λ max = 539 nm; Fig. 3) when bound to various intramolecular quadruplex structures including those formed from the human telomeric repeats, oncogene promoters, and minisatellite sequences (c-myc, c-kit, CEB1) [44–46].Footnote 1 Significantly lower fluorescent enhancements (around 60–70-fold) were found with tetramolecular parallel quadruplexes (TG3T, TG4T, and TG5T) although these structures also seem to accommodate TO, as estimated by ESI-MS measurements [44, 46]. Apparent affinities of TO to quadruplex and duplex DNA are close, with K d values falling in the micromolar range. Fluorescence titrations, Job plot analysis, and mass spectrometry supported a 1:1 stoichiometry model between TO and several intramolecular quadruplex DNAs and 2:1 with tetramolecular structures TG3T, TG4T, and TG5T. The predominant binding mode is likely to be π-stacking on external G-tetrads, and in this regard the lower fluorescence of TO bound to parallel quadruplexes would be explained by the absence of loops resulting in a poorer immobilization of the dye and/or poorer isolation from water. However, groove binding or external stacking cannot be completely excluded in these cases. We will see below that other light-up probes also show lower fluorescence enhancement in tetramolecular quadruplexes.

Normalized absorbance (dashed lines) and fluorescence spectra (solid lines) of Hoechst 33258 (yellow), thiazole orange (orange), and TO-PRO-3 (blue) in the presence of c-kit2 (concentration of dye =0.5 μM; dye/DNA ratio =2). Reprinted with permission from [44]. Copyright 2011 Springer Science+Business Media
Finally, TO was found to label pretty well RNA quadruplexes and dimeric quadruplex matrices made of longer DNA or RNA sequences ([47] and unpublished data). The displacement of TO from G4-DNA has been exploited to implement a high-throughput screening assay for the discovery of new G4 binders via library screening [48].
In particular conditions (pH 8.0 in the absence of monovalent cations), TO seems to interact and to fluoresce preferentially with G-wires (i.e., long tetramolecular quadruplexes) than with double helical DNA [49]. In these conditions, TO and G-wires form stable complexes in gel experiments whereas this is not the case with ds DNA.
TO has been used in a quadruplex-templated FRET experiment to probe quadruplex conformations through a ternary interaction system [50]. In this system, TO and a quadruplex-specific binder (BOQ1 or MMQ1; cf. Sect. 3.1), displaying appropriate photophysical properties (i.e., emission around 450–500 nm, overlapping the absorption of TO) form a donor–acceptor pair, with TO being the acceptor. Quadruplex structures have two external quartets and can thus accommodate both compounds close enough (about 10 Å) for FRET to occur. When the human telomeric quadruplex structure is added to a mixture of BOQ1 (or MMQ1) and TO, a strong decrease of BOQ1 (MMQ1) fluorescence is observed, while TO emission is greatly increased (see footnote 1). Importantly, the FRET response was found to be specific for the quadruplex structure, indicating that the quadruplex matrix provides a more efficient positioning of the two FRET partners as compared to a duplex matrix.
In vitro fluorescent detection of 5′,3′-cyclic diguanylic acid (c-di-GMP) using TO has recently been achieved [51]. While TO is not fluorescent in the presence of simple nucleotides (GTP, GMP, cGMP), it induces the formation of quadruplex structures composed of c-di-GMP G-tetrads via intercalation or external stacking. Higher cation concentration (up to 1 M), and Na+ preferentially to K+ and NH +4 , were shown to increase the TO fluorescence since it promotes c-di-GMP aggregation. In conclusion, the exceptional fluorescence properties of TO and its strong binding to many quadruplexes make it a very efficient quadruplex stain for in vitro experiments, in particular for fluorescence-based screening assays.
In cells, TO was found to stain preferentially nucleoli (but not nuclear DNA) as well as the cytoplasm [49]. This is consistent with the fact that TO and derivatives (SYTO dyes) are known to stain preferentially RNAs and are commercialized on this purpose [52]. For this reason, and owing to its absence of significant binding selectivity for biological quadruplexes it appears rather irrelevant to use this dye for probing quadruplexes inside cells, although the presence of quadruplex RNA (and also DNA) in the nucleoli is not excluded.
2.1.1.2 TO Derivatives
In order to probe specifically quadruplex DNA, TO has been appended to bisquinolinium compounds, ones of the best and most selective quadruplex binders of the pyridodicarboxamide (PDC) series [53–55]. Two designs were conceived: the first, PDC-L-TO, consists in linking both components by a flexible linker, whereas the second, PDC-M-TO, is built by merging the N-methylquinolinium moiety present in both scaffolds (Fig. 4).

TO derivatives designed for targeting quadruplex DNA
Both designs afforded fluorescent off/on probes which were different in their selectivity. Thus, PDC-L-TO is devoid of selectivity for quadruplex DNA and displays an intense orange–yellow fluorescence (λ em = 530 nm) both with duplex and quadruplex DNA. The absence of selectivity is attributable to the affinity of TO for duplex DNA that counterbalances the quadruplex preference of the PDC motif. On the other hand, PDC-M-TO stabilizes strongly the telomeric quadruplex DNA even in the presence of a strong excess of ds DNA (ΔT 1/2 = 17 °C). The fluorescence enhancement of PDC-M-TO (λ ex = 500 nm, λ em = 550 nm) when bound to the telomeric quadruplex (22AG: d[G3(T2AG3)3]) is decreased by about 36% as compared to TO alone, but displays an eightfold selectivity compared to the complex with ds DNA.Footnote 2 On this basis, selective staining of quadruplex DNA could be observed in gel electrophoresis, but the sensitivity of the merged compound remained weaker than that of the linked PDC-L-TO compound (Fig. 5). This result underlines the difficulties in fluorescence engineering, as the quadruplex selectivity is retained at the expense of the fluorescence quantum yield. In addition, both dyes show some degree of aggregation at high ionic strength. Still, the new merged design described in this study represents an innovative and solid basis for the design of novel G-quadruplex probes.

PAGE of human telomeric quadruplex (22AG) and duplex oligonucleotide (ds26) DNA (0.3–2.0 μM) stained by PDC-M-TO (a, 5 μM) and post-stained with PDC-L-TO (b, 2 μM) (λ ex = 532 nm, λ max = 550 nm). Reprinted with permission from [53]. Copyright 2009 John Wiley and Sons
Based on the same concept, another group designed a benzothiazole-substituted benzofuroquinolinium dye 1 (Fig. 4) [56], in which TO has been merged with benzofuroquinoline, a planar aromatic system known for its quadruplex-interactive properties [57]. Both absorption and fluorescence of this new hybrid derivative are significantly shifted towards the red as compared to TO (λ ex = 527 nm, λ max = 573 nm), thereby indicating a higher degree of conjugation. As expected, the dye is poorly fluorescent in water (Φ F = 0.0012) and exhibits a bright orange fluorescence when bound to quadruplex DNA (λ ex = 527 nm, λ max = 573 nm, Φ F = 0.25 and 0.18 for the telomeric repeat and c-myc, respectively). Conversely, much smaller changes were observed with duplex DNA (26 bp oligonucleotide and calf thymus DNA) and none with the bovine serum albumin protein (Fig. 6). Moreover, quadruplex and duplex DNA could be easily distinguished in gel electrophoresis using this hybrid molecule as a stain.Footnote 3

Pictures of the benzofuroquinolinium dye (5 μM) in the presence of human telomeric quadruplex (telo21), BSA protein, duplex DNA oligonucleotide (ds26) and calf thymus DNA (ct DNA) upon irradiation at 302 nm. Reprinted from [56] with permission of The Royal Society of Chemistry
The dye is supposed to bind by π−π end-stacking in a quasi-planar conformation, the rotation about the methine bridge being efficiently restricted. The absorption spectra in the presence and the absence of DNA show the typical blue-shift observed for cyanine aggregates, thereby suggesting that the free dye also aggregates to a certain extent, although this was not considered. Finally, the dye is slightly fluorescent (by a factor of 3) in the presence of single-stranded and double-stranded RNA as compared to G-quadruplex RNA and DNA. Imaging in fixed cells (MCF7) indicates that the dye is localized in the nuclei with faint staining of genomic chromatin.
2.1.1.3 Other Cyanine Dyes
SYBR Green I is an asymmetric cyanine dye, structurally and photophysically similar to TO (λ ex = 497 nm, λ max = 520 nm upon binding to duplex DNA), and widely used for “in-gel” staining of duplex and, to a lower extent, single-stranded DNA (Fig. 7) [58].

Cyanine dyes evaluated for recognition of quadruplex DNA
SYBR Green I has recently been used in a quadruplex and K+ detection system in cooperation with a water-soluble cationic conjugated polymer [59]. The polymer allows a sixfold fluorescence amplification of the dye bound to quadruplex through a FRET effect (Fig. 8),Footnote 4 but not with single stranded DNA (in the absence of potassium). However, the same effect has been observed previously in an independent study using duplex DNA [60].

(a) Emission spectra and (b) FRET ratio (F 525/F 441) of SYBR Green I (acceptor) and cationic conjugated polymer (donor) with a quadruplex-forming sequence in the presence of increasing concentrations of K+ (0–50 mM). Reprinted with permission from [59]. Copyright 2010 American Chemical Society
TO-PRO-3 is a trimethine analogue of TO, known to bind strongly duplex DNA with a high fluorescence enhancement (Fig. 7) [61]. TO-PRO-3 has recently been used as an alternative to TO in the G4-FID method to evaluate the putative G4-DNA binders absorbing around 500 nm, since its absorbance and fluorescence spectra are red-shifted as compared to TO (λ ex = 642 nm, λ max = 718 nm upon binding to the c-kit2 quadruplex; Fig. 3) [44]. Like TO, it binds nucleic acids with micromolar K d, without strong discrimination between various quadruplex and duplex structures but with high fluorescence enhancements, ranging from 120 to 250 depending on the DNA sequence and structure. The binding modes of TO-PRO-3 to duplex DNA were shown to be multiple, involving both intercalation and groove dimerization/aggregation [34]. Likewise, the binding to quadruplexes might also follow multiple modes, although π-stacking on a G-quartet is most likely.
The monomethine dye Cyan 40 and the trimethine Cyan 2 were examined with respect to their binding to G-wires containing 750 guanine tetrads in the absence of monovalent cations and at pH 8.0 [62]. The fluorescence enhancement of Cyan 2 increases more in the presence of quadruplex (15-fold) and triplex structures than in the presence of duplex DNA. However, it should be noted that both Cyan 2 and Cyan 40 were previously described to act as efficient “light-up” probes for ds DNA [63, 64] and were found to localize in the nucleoli of mammalian cells [25]. Again, this addresses the question of rationalizing cellular localization of G4-DNA stains on the ground of their interaction and selectivity for G-wires that are artificial polymeric quadruplexes without entrapped cations, and thus have no biological relevance.
DODC is a symmetric cyanine that binds to duplex DNA in minor grooves, mainly as a dimer. Its binding to quadruplex DNA was first examined 15 years ago with various dimeric hairpin quadruplex structures (i.e., displaying two diagonal loops) [65]. DODC binds to these structures with micromolar K d and displays an induced circular dichroism (ICD) signal which may result from groove interactions [30]. More importantly, fluorescence intensity of DODC upon binding was dramatically reduced (about 50%) together with a 6 nm red-shift, but increased (18%) with the tetramolecular parallel TG4T quadruplex and not significantly changed with other structures (ss DNA, ds DNA, hairpins).Footnote 5
More recently, DODC was used as a fluorescent reporter for quadruplex DNA in a “mix and measure” fluorescence screening assay [66]. A variety of quadruplex structures were examined and, unlike in previous results, DODC was found to exhibit a high fluorescence enhancement (220–500-fold with various concentrations of DNA; λ ex = 579 nm, λ max = 610 nm).Footnote 6 DTCC, which also binds to minor grooves of ds DNA, was also included in the aforementioned test. Higher fluorescence enhancement were monitored (240–980-fold, λ ex = 554 nm, λ max = 577 nm) but with greater amounts of DNA.
ETC (Fig. 7) is an extended aromatic cyanine dye which was found to recognize human telomeric quadruplexes through end-stacking interactions [67, 68]. In aqueous buffer it readily forms J-aggregates (λ abs = 660 nm) that dissociate in monomer (λ abs = 584 nm) upon binding to quadruplex DNA.Footnote 7 Addition of various quadruplex structures (human telomeric repeat, c-kit1, c-myc, bcl-2) induced a strong fluorescence enhancement of ETC monomer (70-fold, λ max = 600 nm), and allowed specific detection in gel electrophoresis (PAGE) experiments.Footnote 8 Detection at higher (physiological) cation concentrationFootnote 9 was achieved with MTC, a close analog of ETC, which displays a somewhat lower ability to form aggregates [69]. Upon addition of quadruplex structures (human telomeric repeat, c-kit1, c-myc, bcl-2), disappearance of the absorption band of the H-aggregate and appearance of the monomer band (λ abs = 580 nm) were observed, accompanied by a 1,000-fold fluorescence enhancement. At the same time, MTC H-aggregates are almost not dissociated in the presence of ss DNA and ds DNA (Fig. 9).

(A) Apparent color of MTC (4 μM) alone (a) and with 20 μM of human telomeric (b), c-myc 2345 (c), c-kit1 (d), bcl-2 (e) quadruplexes, 22-mer oligonucleotide duplex (f), 17-mer single strand (g), calf-thymus (h), and salmon sperm (i) DNA. (B) Fluorescence microscopy images of MTC-stained oligonucleotides on Au film (same legend). Adapted with permission from [69]. Copyright 2010 American Chemical Society
2.1.2 Quadruplex-Template-Directed Synthesis of Cyanine Dyes
In an elegant approach designed by S. Ladame et al., quadruplex probing was efficiently performed using a biocompatible fluorogenic reaction generating cyanine dyes upon binding of two PNAs, complementary to the quadruplex flanking sequences, and functionalized by nonemissive hemicyanine moieties (Fig. 10) [70]. Upon hybridization of the PNAs with a folded quadruplex structure, the two hemicyanine derivatives are close enough to react, yielding fluorescent trimethine dyes 2 (Fig. 11).

DNA-templated synthesis of cyanine dyes. Reprinted with permission from [70]. Copyright 2010 John Wiley and Sons

Trimethine cyanine dyes generated upon PNA binding to G-quadruplex template
Since the reactants are non-fluorescent when the PNAs are either unbound or bound to unfolded single stranded DNA (no close proximity of reactants), this system offers a high signal-to-noise ratio. Increase in fluorescence intensity in the presence of quadruplex by factors of 45 or 40 were observed for the symmetric cyanine 2a (λ ex = 542 nm, λ max = 563 nm) and the asymmetric cyanine 2b (λ ex = 562 nm, λ max = 606 nm), respectively (Fig. 12).Footnote 10 No fluorescence was observed at low PNA concentration and high calf thymus DNA concentration. Also, the PNA sequence complementarity allows one to target specific quadruplex-forming sequences, and hence potentially to probe single quadruplexes.

Fluorescence emission spectra of a mixture of two functionalized PNA (500 nM each) in absence (circles) and presence of 500 nM of target quadruplex (squares), ss DNA (diamonds), and a randomized flanking sequence quadruplex (triangles). Reprinted with permission from [70]. Copyright 2010 John Wiley and Sons
This system has been applied for the dual sensing of hairpin and quadruplex DNA structures through the templated generation of pentamethine and trimethine cyanines, respectively [71]. Their resulting fluorescent properties were sufficiently different (λ ex = 540 and 625 nm, respectively) to detect these structures simultaneously, but with a significant fluorescence background (about 50%).
Although this strategy needs further improvements and in particular will have to overcome the issue of poor cellular uptake of PNAs, it represents a highly interesting alternative to the use of small fluorescent molecules. In particular, this approach based both on sequence and structure recognition holds the key to the specific detection of a unique type of quadruplex DNA.
2.1.3 Benzimidazole Derivatives
Hoechst 33258 (Fig. 13) is a bis-benzimidazole derivative, binding preferentially to minor grooves of AT-rich domains of ds DNA, routinely used for staining nuclear DNA [72]. The fluorescence enhancement observed upon binding, characterized by a large Stokes shift, is explained by the restriction of the dye mobility and its isolation from water [73]. Interaction of Hoechst 33258 with c-myc quadruplex has been examined by Maiti [74] and Patel [75]. As with duplex DNA, the dye is rotationally restricted and isolated from water and may interact in multiple ways (loop, groove, or quartet binding). Hoechst 33258 was used as an alternative to TO in the G4-FID method to assess the compounds absorbing around 500 nm, since its absorbance and fluorescence spectra are blue-shifted as compared to TO (λ ex = 343 nm, λ max = 473 nm upon binding to the c-kit2 quadruplex; Fig. 3) [44] (see footnote 1). Of note, the fluorescence enhancements observed with a set of quadruplex structures are dramatically lower (7–19-fold) compared to duplex DNA (36-fold), and the same trend is observed with affinity constants (log K a ranging from 5.4 to 6.1 for G4 structures vs 6.4 for duplex DNA) [44, 74], thereby confirming that Hoechst definitely binds better to duplex than to quadruplex DNA.

Bis-benzimidazole quadruplex ligands
Structurally related phenylene-bisbenzimidazole derivatives (Fig. 13) with a more pronounced crescent shape were synthesized to target quadruplex structures via π-stacking on G-quartets. These compounds display dissociation constants in the range (0.7–5.3) × 105 M−1 with the Tetrahymena termophilia quadruplex d[(T2G4)4] [76]. The highest fluorescence enhancement (around 50-fold for a dye-to-DNA ratio of 7.7) was obtained with the EtBzEt derivative, whilst the linear analogue had a poor affinity. These studies demonstrate that duplex minor groove-binding ligands, when suitably modified, may switch their preference from duplex to quadruplex [77].
2.1.4 Ethidium Derivatives
Ethidium bromide (EtBr, Fig. 14) is a classical nucleic acid staining agent, interacting with duplex DNA via intercalation [78]. Fluorescence enhancement upon binding could be explained by protection from the excited-state deprotonation of the solvent [79]. EtBr also binds to triplex DNA and, to a lesser extent, to quadruplex DNA by external stacking [30, 80]. First fluorescence measurements of EtBr in complex with tetramolecular quadruplex DNA T4G4 were published over two decades ago, where it was found slightly less fluorescent than in EtBr–duplex DNA complexes (Fig. 15a; λ ex = 510 nm, λ max = 590 nm) [81]. Further studies confirmed the weaker fluorescence enhancement and hypochromic shift of EtBr in the presence of quadruplex DNA as compared with duplex or triplex DNA [83].

Structures of ethidium and derivatives thereof

(a) Fluorescence intensity of EtBr (10 μM; λ ex = 510 nm, λ max = 597 nm) upon addition of duplex (filled circles) and quadruplex (open circles) DNA. Reprinted with permission from [81]. Copyright 1992 American Chemical Society. (b) Fluorescence titration of compound 4 (400 nM) in the presence of the quadruplex-forming oligonucleotide 28G (40 nM–1.8 μM), λ ex = 500 nm. Circles indicate emission spectrum without 28G. Adapted with permission from [82]. Copyright 2001 Oxford University Press
Ethidium derivatives were prepared with the aim of improving their quadruplex-binding capabilities while retaining interesting fluorescence properties [82, 84]. Two of them (3 and 4, Fig. 14) display strong fluorescence increase upon binding to the human telomeric quadruplex structure (cf. Fig. 15b). The increase of fluorescence quantum yield (from 0.06 and 0.04 to 0.25 and 0.20, respectively) is complemented by the shifts of absorbance and emission wavelengths (λ ex = 477 and 464 nm to 508 and 494 nm and λ max = 597 and 603 nm to 595 and 592 nm, respectively).Footnote 11 It was also observed that binding of 4 to a parallel-stranded quadruplex structure d[(T2G4T)4] leads to a lower fluorescence increase than with an antiparallel structure, probably because the dye is more exposed to the solvent. The two derivatives were successfully used to stain quadruplex DNA in agarose gels, but with a 100-fold lower sensitivity than the cyanine dye DODC.
2.2 Triphenylmethane Dyes
Malachite green (MG), crystal violet (CV), and methyl green (MEG) (Fig. 16) belong to the class of triphenylmethane (TPM) dyes that have been known for more than a century. The characteristic feature of TPM dyes is the existence of several conformers in the fundamental state and the formation of a nonemissive twisted intramolecular charge transfer (TICT) state resulting from diffusional mutual rotation of the phenyl rings [85, 86]. Consequently, the fluorescence quantum yield of the free dyes in organic solvents is usually very low (≈10−5) but increases significantly with the viscosity of the solvent due to restriction of intramolecular rotations [87]. Likewise, the fluorescence of TPM dyes (malachite green and derivatives) has been shown to be enhanced upon binding to RNA aptamer matrices in which the molecule is immobilized [87]. Two TPM dyes have been studied recently for their interaction with quadruplex-forming sequences. MG was shown to bind the G-rich sequence d[(G2T)13G] which resulted in a 100-fold increase in dye fluorescence (Fig. 17) [88]. Again, this phenomenon has been attributed to increased molecular rigidity of the dye when bound to the quadruplex DNA structure. Interestingly, the fluorescence enhancement is much lower (14-fold) with single-stranded and double-stranded oligonucleotides used as controls. However, the interaction appears rather complex and the dye interacts only at low concentration in oligonucleotide, thus suggesting the recognition of an intramolecular quadruplex. The structural information is scarce as the quadruplex that can be formed by this unusual sequence is only characterized by thermal difference spectra (TDS) [89]. Nevertheless, a 70-fold increase in fluorescence is also observed with the more classical (G4T4)4 quadruplex but at high concentration. Follow-up studies by Shen et al. using several TPM dyes (CV, MG, MEG, Fig. 16) and a panel of well-defined quadruplex-forming sequences (intramolecular and tetramolecular G-quadruplexes from the telomeric human sequence [hum21, hum12] or from Oxytricha nova [Oxy28]) have shown that energy transfer spectra of TPM dyes can serve to discriminate intramolecular and tetramolecular quadruplexes from other DNA forms [90, 91]. Energy transfer is a reliable indicator of π-stacking interactions and it is likely that the TPM dyes interact with external G-quartets. CV and MEG bind better than MG with dissociation constants in the low micromolar range (K d = 0.036 μM for CV; 0.026 μM for MEG) [92].Footnote 12 However, a competition dialysis experiment indicated a rather moderate binding selectivity for intramolecular quadruplexes with respect to double-stranded DNA (ct DNA) and no preference for the tetramolecular telomeric quadruplex (hum12). Nonetheless, CV was subsequently proposed for the development of fluorescent switch-on probes for homogeneous detection of potassium ions [93], for i-motif [94], and the development of a G-quadruplex-based turn-on fluorescence assay for the 3′ → 5′ exonuclease activity [95].

Triphenylmethane dyes

Enhancement of the fluorescence intensity of MG (1 μM) in an aqueous solution containing phosphate buffer (5 mM) at pH 7 with d[(G2T)13G] at increasing concentrations: (1) 0 μM, (2) 6 μM, (3) 60 μM, (4) 96 μM, (5) 108 μM, (6) 120 μM. Reprinted with permission from [88]. Copyright 2007 John Wiley and Sons
It is worth noting that, in spite of significant fluorescence enhancements (around 70–100-fold), the fluorescence quantum yield of TPM dyes globally remains low (around 10−3), which is a limiting factor for fluorescence applications using routine equipment (for comparison, fluorescence enhancements of G4-bound TO are in the 1,000–3,000-fold range). This, together with the poor ability of the series to discriminate between different biomacromolecules [87, 96], minimizes the practical interest of TPM dyes for quadruplex sensing applications.
2.3 Carbazoles
In 2003, the group of T.-C. Chang reported the synthesis of a novel carbazole derivative featuring two vinylpyridinium branches in positions 3 and 6 (Fig. 18). This compound (called BMVC) was shown to stabilize significantly the human telomeric quadruplex (ΔT 1/2 = +13 °C measured by CD-melting).

Structure of carbazole derivatives
Interestingly, the binding of the dye was accompanied by a strong fluorescence enhancement evaluated to be around two orders of magnitude as compared to the free dye that is poorly fluorescent [97, 98]. BMVC belongs to the family of cationic styryl dyes that are characterized by fast deactivation of their excited state due to rotational vibrations around the vinylic bond. This well-known phenomenon results in a strong quenching of the fluorescence of the unbound compounds whilst their fluorescence is restored upon immobilization in viscous solvents (typically glycerol) or in polymeric biological matrices such as DNA. Subsequently the photophysical properties of this dye were thoroughly investigated in correlation with its properties for quadruplex DNA recognition, focusing mainly on the human telomeric quadruplex. Although BMVC binds the human telomeric quadruplex d[(T2AG3)4] with a nanomolar K d [99], it also seems to bind rather well to duplex DNA structures like calf thymus DNA and linear short duplexes, although the affinity for the latter forms has not been determined. Nonetheless, in certain conditions, the dye appears to have a clear preference for the human telomeric quadruplex-forming sequence d[(T2AG3)4] as shown by a competition assay carried out on gel electrophoresis (Fig. 19) [100]. In this assay the quadruplex is selectively stained when a mixture of duplex and quadruplex is mixed with 0.1 μM dye (called prestaining). Subsequent post-staining, i.e., incubation of the gel after migration in a solution of BMVC at 10 μM, enables labeling of both duplex and quadruplex DNA forms. The dye reveals an extreme sensitivity since as little as 0.2 pmol of quadruplex can be detected in the gel.

Binding preference of BMVC to quadruplex DNA. 20 μM of quadruplex DNA d(T2AG3)4 or linear duplex (d[ATGCGCA2T2GCGCAT])2 were incubated with BMVC (2 or 0.2 μM) and analyzed on a 20% polyacrylamide gel (prestain, lanes 1–6). The same gels were then poststained with 10 μM BMVC to visualize the position and level of DNA loaded (poststain, lanes 7–12). An asterisk indicates position of the free quadruplex DNA. Reprinted with permission from [100]. Copyright 2004 American Chemical Society
Another interesting feature of the BMVC fluorescence is the difference in the maximum emission wavelength when bound to quadruplex or duplex DNA. Indeed, a strong blue shift is observed in the presence of duplex DNA (λ max = 545 nm) as compared to the red-shifted emission in the presence of quadruplex (λ max = 575 nm). This is clearly seen by the different colors (yellow to orange) of the fluorescence emission of the DNA–dye complexes both on gel and in solution (Fig. 20a). Although this is not commented on by the authors, this difference indicates that BMVC is in a less polar environment when bound to duplex than when bound to quadruplex which reflects better shielding from water upon interaction with the double helix structure.

(a) Fluorescence of solutions of BMVC free and in the presence of duplex or quadruplex DNA; from left to right: BMVC alone, in the presence of a linear duplex (d[ATGCGCA2T2GCGCAT])2, and d(T2AG3)4 quadruplex in Na+ and K+ conditions, respectively. Adapted with permission from [101]. Copyright 2006 American Chemical Society. (b, c) Fluorescence collected on chromosomes in metaphases of human CL1-1 (b) and Ca9-22 cells (c). Adapted with permission from [100]. Copyright 2004 American Chemical Society
This ability of BMVC to signal differently duplex and quadruplex structures was tentatively applied for detecting the presence of quadruplex DNA at the level of chromosomes. Chromosomes in metaphases were observed by fluorescence microscopy and the fluorescence was collected from various regions. Telomeric proximal regions showed fluorescence emissions around 565 nm whereas the other chromosomal regions showed fluorescence emission around 545 nm (Fig. 20b). On this basis, the authors concluded on the presence of quadruplex structures at telomeres. However, both the specific conditions required for this experiment (the red-shifted signal is seen only at short staining time, i.e., 5 min and very low dye concentration of 2 nM) and the associated technical difficulties, i.e., the precision in term of spatial resolution and imaging contrast, are not addressed.
In a follow-up study, the lifetime of the excited state of BMVC in the presence of various DNA forms was measured. A longer lifetime was observed in the telomeric quadruplex (1.96 ns with d(T2G3)4 in Na+ conditions), as compared to the linear duplex (1.46 ns with (d[ATGCGCA2T2GCGCAT])2). However, in K+ conditions the lifetime of the dye bound to the quadruplex was also shorter (1.56 ns with d[(T2AG3)4] in K+ conditions). The authors conclude that the long lifetime is characteristic of the antiparallel form whilst the short component is due to the parallel form, assuming that the latter dominates in potassium conditions. Two-photon-excited fluorescence lifetime imaging (2PE-FLIM) was then performed with the aim of detecting the presence of quadruplexes on chromosomes in metaphase. The long-lifetime component was observed in the telomeric regions arguing again for the presence of quadruplex in its antiparallel form although potassium is predominant in the cells (Fig. 21). Consistent with the previous observation on chromosomes using the red-shifted signature [100], the quadruplex-specific long-living component is time and concentration dependant, suggesting that BMVC associates first to the (presumed) very low amount of quadruplexes present in the genome and then redistributes on the massive excess of duplex domains upon equilibration. Kinetic studies would thus be required to fully understand the behavior of BMVC and to conclude firmly on the presence of quadruplex structures at telomeres.

2PE-FLIM micrograph of BMVC-stained metaphase chromosomes of nasopharyngeal carcinoma KJ-1 cells. (a) A typical 2PE-FLIM image of metaphase chromosomes of nasopharyngeal carcinoma KJ-1 cells stained with 0.1 μM BMVC for 90 s. (b) The fluorescence lifetime distribution of the BMVC-stained chromosomes. (c) The color-coded image of the metaphase chromosomes is represented by discrete timemode at 1.85 ns. Red designates mode 1 resulting from the interaction with antiparallel G-quadruplex structures, while white designates mode2 due to the interaction with other DNA structures. The arrows indicate mode 1 for visualization. Adapted with permission from [101]. Copyright 2006 American Chemical Society
In parallel studies, BMVC was used to explore the interconversion between the antiparallel and the parallel forms of the telomeric quadruplex both on short (24 bases) and long (78 bases) sequences in combination with CD spectroscopy. The dye exhibits a strong ICD signal upon interaction with both parallel and antiparallel quadruplexes [99]. Finally, molecular dynamics studies have shown that BMVC likely binds to the basket Na+-form of the telomeric quadruplex by external π-stacking on the two G-quartets combined with interactions with the diagonal and parallel surrounding loops [99]. The strong involvement of loops in the interaction would explain the binding preference for the antiparallel telomeric quadruplex suggested by the initial gel analysis.
More recently, reports by other groups concerning N-alkyl [102] or N-aryl analogues [103] of BMVC (Fig. 18) have been published. The first study based on modeling suggests that the bis(vinylpyridinium)carbazole core could fit nicely in the grooves of the antiparallel form of the telomeric quadruplex. The second study demonstrates that the N-phenyl derivative of BMVC (Cbz-2py) binds both duplexes and various intramolecular quadruplexes (telomeric, c-myc, c-kit). However, this compound exhibits a strong and preferential fluorescence enhancement in AT-rich duplexes likely resulting from a net preference for duplex minor groove binding. This would tend to indicate that BMVC is better in terms of fluorescence selectivity for quadruplex over duplex structures.
The major interest of the studies carried out by T.-C. Chang et al. is to have made use of the particular photophysical characteristics of BMVC to propose diverse fluorescent-based approaches that may allow signaling of quadruplex-containing regions at the level of chromosomes, although the probe has a relatively poor structural preference for quadruplexes. Thus this work sets the stage for future possible design of experiments based on probes with optical features (fluorescence lifetime, emission wavelength) specific for quadruplexes and combined with current advanced technologies (one- and two-photon FLIM mapping).
2.4 Porphyrins
Porphyrins have been thoroughly investigated for their interaction with quadruplex DNA following the pioneering work of L. Hurley et al. on the 5,10,15,20-tetrakis(1-methyl-4-pyridyl)porphine called TMPyP4 (Fig. 22) [104]. Porphyrins are fluorescent compounds but exhibit highly variable quantum yields as a function of the substituents born by the tetrapyrollic scaffold and whether they are in the free-base form or metallated. For instance, most of the pyridinium series developed to interact with quadruplexes [105–107] show either poor fluorescence variations or fluorescence attenuation due to electron transfer from guanines to the porphyrin excited state, likely favored by the strong electron-acceptor character of the pyridinium units (see below Sect. 3, Light-off probes). However, fluorescence increase of TMPyP4 has been mentioned in several cases. Fluorescence quantum yield of free TMPyP4 in water is moderate (Φ F = 0.05) with a typical broad dual-band pattern exhibiting two maxima in the NIR region (660 and 720 nm). Upon association with tetramolecular and bimolecular quadruplexes (d[TG4T])4, (d[G4T4G4])2 and with the intramolecular telomeric sequence d[AG3(T2AG3)3] the emission intensities increase about 1.5–2.5 times [107] in Na+ conditions, displaying a well-structured spectrum (Fig. 23).Footnote 13 In contrast, a strong decrease (>70%) is reported with intramolecular and dimeric hairpin quadruplexes from telomeric sequences d[TAG3(T2AG3)3T] and d[T2AG3T2AG3)2] in K+ conditions (10 mM K+) [106]. Finally, a slightly different pattern was observed upon interaction with the parallel-quadruplex-forming d[G4(T2AG3)4], showing an increase of the band at 658 nm and a decrease of the band at 718 nm, which might be explained by modification of the transition probability of the two vibrational states [108]. On the whole, all these studies suggest that the fluorescence of TMPyP4 is highly dependent on the structure of the bound quadruplex.

Structures of TMPyP4 and TrPyP4

Fluorescence emission spectra of TMPyP4 (4 μM) and its complexes with (TG4T)4, AG3(T2AG3)3, and (G4T4G4)2G-quadruplexes (20 μM). Adapted with permission from [105]. Copyright 2006 American Chemical Society
In the follow-up studies the N-propyl analogue TrPyP4 showed only slight changes of fluorescence intensity upon addition of various G4 sequences; however, as in the case of TMPyP4, binding to G4 DNA induced a better resolution of the emission spectrum with the appearance of clear structured vibrational contributions [109–111].
A series of porphyrin-anthraquinone dyads 5 was prepared [112] with various chain length (from 1 to 10 methylene units; Fig. 24). Compounds with the longer chain length (7 or 10 methylene units) exhibit a higher affinity for (d[TG4T])4 (ΔT 1/2 = 12.8 and 11.2 °C, respectively, as determined by UV-melting) than those with the shorter linkers (ΔT 1/2 = 3.0 and 6.4 °C for derivatives with 1 and 4 methylene units, respectively). Upon addition of increasing concentration of (d[TG4T])4 the derivatives with 4, 7, and 10 methylene units in the chain display similar fluorescence emission spectra, peaking at 665 nm with an enhanced intensity (~30%), whilst such increase is not observed with the shortest-chain analog.

Structures of anthraquinone conjugate 5 and TMe2D4
The metallated forms of the cationic bis(pyridinium)porphyrin TMe2D4 (Fig. 24) exhibit more interesting fluorescence properties. In particular, Cu(TMe2D4) was reported to exhibit a strong fluorescence enhancement (14-fold) with an infrared-shifted emission peaking at 850 nm in the presence of tetramolecular quadruplexes (d[T n G4T n ])4. Interestingly, the emission increases in intensity as the number of thymines increases from 1 to 3, likely due to enhanced shielding from water that is a strong quencher of copper porphyrins. Worth mentioning is the fact that the emission of Cu(TMPyP4) is lower and furthermore is not affected in these conditions.
Anionic mesoporphyrins bearing carboxylate groups (Fig. 25) have also been investigated as fluorescent probes for G4-DNA. The anionic N-methyl-mesoporhyrin IX (NMM) exhibits a marked selectivity for quadruplex structures as compared to duplex with a good affinity, only recently quantified (ΔT 1/2 = 14 °C determined by FRET-melting) [113]. In early studies the fluorescence of NMM was shown to be strongly enhanced in the presence of dimeric and monomeric G4-forming sequences (d[G4T4G4])2 and TBA, whereas no variation was observed in the presence of calf-thymus DNA [114].

Structures of N-methyl-mesoporhyrin IX (NMM) and protoporphyrin IX (PPIX)
Protoporphyrin IX (PPIX) is known as a photodynamic agent in cancer therapy, able to interact with various proteins. On the tracks of NMM, binding of PPIX against various G4-DNA (TBA, human telomeric, or Oxy28) has been investigated [115]. The emission spectrum of PPIX is typical of a porphyrin compound with a dual band in the 600–700 nm range. The fluorescence enhancement efficiencies observed with different G4-DNA are from 16.2 to 17.9-fold, whilst the binding constants are rather different (K d from 0.04–50 μM). In the same work, PPIX has also been used as a “sensor” of parallel quadruplexes formed in K+ conditions as the fluorescence increases rapidly when the K+ concentration rises from 2 to 100 mM. The fluorescence properties of Zn-PPIX have also been used to monitor the lead-driven interconversion between a duplex and a G4 form acting as components of a DNA nanodevice [116].
2.5 Probes Operating via Aggregation-Induced Emission
Aggregation-induced emission (AIE) is a process observed in an unusual fluorogen system, in which aggregation enhances the fluorescence [117]. It has been found that these systems are nonemissive in dilute solutions, but become highly luminescent when the molecules are aggregated in concentrated solutions. 1,1,2,2-Tetrakis[4-(2-triethylammonioethoxy)phenyl]ethene (TTAPE, Fig. 26) can be considered an archetypal AIE luminogen. Structural analysis reveals that unlike conventional luminophores, such as the disk-like planar aromatic molecules (for example perylene derivatives; cf. Sect. 3.3), TTAPE is a propeller-shaped nonplanar molecule. In a dilute solution, four phenyl rotors in TTAPE undergo dynamic intramolecular rotation about its ethene stator, which nonradiatively annihilates the excited state and renders the molecule nonfluorescent. In the aggregates, the molecules cannot efficiently pack through a π–π stacking process due to their propeller shape; at the same time, the intramolecular rotations of the aryl rotors are greatly restricted owing to the physical constraint. This restriction of intramolecular rotation (RIR) blocks the nonradiative pathway and opens up the radiative channel. As a result, the fluorogen becomes emissive in the aggregate state.

Probes operating via aggregation-induced emission
AIE fluorogens are useful analytical tools and have been judiciously utilized as sensitive and selective chemosensors and bioprobes of the “light-up” type. In many of these sensing systems, the aggregation of the free AIE molecules in solution is triggered by electrostatic attraction and coordinative complexation to the analyte [118, 119]. Thus, the emission of the TTAPE that is weak in aqueous buffer (Φ F ≈ 0.5%) is restored in the presence of certain quadruplex-forming sequences [118]. In particular, the largest fluorescence enhancement (45-fold) was obtained for the human telomeric DNA sequence d[G3(T2AG3)3] in the presence of K+ ions, whereas almost no variation was induced by the same sequence in the presence of Na+, indicating selective recognition of the K+ conformers. Subsequently, more modest but still significant fluorescence increase (10–12-fold) was observed with the c-myc derived sequence d[TG(AG3TG3T)2A2] and with the telomeric sequence from Oxytricha nova d[G4(T4G4)3] (Fig. 27). This also enables selective staining of the telomeric quadruplex in gel [119]. Of note, TTAPE also binds to duplex DNA but the emission of TTAPE-G4 and TTAPE-ds DNA complexes display significant differences: the emission in quadruplex is twice as intense and is characterized by a bathochromic shift by 20 nm. This observation is presented as a way to discriminate between the two DNA forms [118]. A molecular modeling experiment indicates that the dye may be stacked on the surface of the quadruplex structure in a loop–groove interaction based on multiple electrostatic interaction with phosphates, which impedes the intramolecular rotations and favors the radiative relaxation [119]. The binding constant determined by fitting the fluorimetric titration curve obtained with the human telomeric G4-DNA in K+ is rather modest (K a = 7.8 × 105 M−1).Footnote 14 A very close value was found by calorimetric titrations (ITC) (K a = 2.4 × 105 M−1), which also enabled to determine a remarkably low affinity constant for the same sequence in Na+ conditions (K a = 4.3 × 102 M−1) [119]. Additional information about the changes in the fluorescence emission of TTAPE in the presence and in the absence of DNA was obtained from the time-resolved fluorescence spectra. This technique offers valuable information about the interactions of the fluorogens in the excited state with the environment, by measuring the fluorescence lifetime. In buffer solution and in absence of DNA, TTAPE decays in a single-exponential way with a very short lifetime of 20 ps, which is consistent with the involvement of a nonradiative process of de-excitation. The fluorescence lifetime is lengthened to a few nanoseconds (3–4 ns) when the human telomeric quadruplex is added to the solution, and the fluorescence decay curve is better fitted by a double-exponential function [119].

(a) Photoluminescence spectra of buffer solutions of TTAPE in the presence of K+ and G-quadruplexes with different conformations. [TTAPE] = 4.5 μM, [DNA] = 9 μM, [K+] = 0.5 M; λ ex = 350 nm. (b) Photographs of TTAPE solutions in the presence of different G-quadruplexes taken under UV illumination (365 nm). Reprinted with permission from [119]. Copyright 2010 John Wiley and Sons
TTAPE was also evaluated in cells: the compound penetrates into living HeLa cells and appears nontoxic. However, the dye does not reach the nucleus as the fluorescence is exclusively observed in the cytoplasmic compartment [119].
Another remarkable AIE molecule is the silole 6, reported for the first time by Tang and co-workers [120]. The fluorescence of silole 6 increases largely upon mixing with DNA, and large fluorescence enhancements were detected for long DNA, indicating that 6 can be used for fluorescence turn-on detection of DNA. More interesting, this behavior enabled following the DNA cleavage process by nucleases [121]. This behavior prompted the authors to exploit silole 6 as a fluorescent light-up probe (λ max ≈ 470 nm) to detect G-quadruplex formation. Compound 6 was employed to study G-quadruplex formation using an exonuclease I hydrolysis assay. The fluorescence of 6, in the presence of a duplex containing the telomeric sequence (21-mer) and in absence of K+, gradually becomes weak by prolonging the enzymatic hydrolysis reaction time. This is due to fragmentation of DNA by exonuclease I, and as a result the aggregation of silole 6 cannot take place efficiently. However, upon addition of KCl, the fluorescence of silole 6 is only slightly affected, which is attributed to the K+-induced formation of quadruplex that inhibits the hydrolysis reaction. Very similar results have been obtained in the presence of various G-quadruplex forming sequences such as c-myc, c-kit, VEGF, and an intermolecular G-quadruplex d(G4T4G4) [122].Footnote 15
2.6 Miscellaneous
2.6.1 Natural Products
Several aromatic natural products are weakly fluorescent when free in aqueous buffer whilst their binding to quadruplex DNA may significantly increase their fluorescence intensity. Among them are the isoquinoline alkaloids, berberine and palmatine (Fig. 28), which were initially identified as inhibitors of topoisomerases I and II, the latter being the targets of several anticancer drugs used in the clinic [123].

Berberine derivatives
Both compounds include an aromatic moiety with a quaternary bridgehead nitrogen atom, and appear suitable for stacking interactions with the G-quartet. Berberine has a very low quantum yield in aqueous solution (Φ F = 4.5 × 10−4) [124], which is increased by a factor of 50 upon addition of quadruplex DNA while the maximum in the fluorescence emission spectrum (λ max = 522 nm) is slightly blue-shifted by 5 nm. The analysis of the binding isotherms revealed a 1:1 binding model with a binding constant of 1.2 × 106 M−1 for berberine binding to human telomeric quadruplex d[AG3(T2AG3)3] (Fig. 29) [125]. Fluorescence polarization anisotropy measurements of berberine and palmatine bound to G-quadruplex structures revealed an increase of fluorescence anisotropy as compared to the free compounds (0.140 and 0.141 in the complex with G4-DNA vs 0.038 and 0.032 in the free state, respectively), indicative of binding. However, the observed values are lower than those found for the planar analogues (e.g., coralyne, cf. Sect. 3.2), indicating that buckling of berberine and palmatine hinders their interaction with quadruplex DNA [126].

Fluorescence emission spectra of 0.5 μM berberine in 50 mM MES buffer (pH 7.4) and 100 mM KCl in the absence and presence of successive addition of quadruplex at 25 °C. The inset is the plot of ΔF vs quadruplex concentration. Reprinted with permission from [125]. Copyright 2008 John Wiley and Sons
By taking advantage of the berberine fluorescence enhancement upon binding to G-quadruplex, a new label-free and homogenous fluorescent assay for K+ has been developed. Upon addition of K+ ions to a mixture containing a G-rich single-stranded DNA (TBA, d[G2T2G2TGTG2T2G2]) and berberine, a remarkable enhancement of fluorescence is induced. Interestingly, the method shows a high selectivity for K+ as detection occurs in the presence of 10,000-fold excess of Na+ [127]. The introduction of an alkylamino group, such as a 3-(1-piperidino)propyl substituent (compound 7) or a positively charged aza-aromatic terminal group (compound 8), into position 9 of berberine improves the binding selectivity for G-quadruplex, while keeping the significant fluorescence “light-up” properties (ΔT 1/2 = 28.2 °C for 7 with the telomeric sequence d[G3(T2AG3)3] vs ΔT 1/2 = 3.9 °C for berberine, calculated from the CD melting curves at 295 nm) [128].Footnote 16 Very similar results were obtained for compound 8 that showed a ΔT 1/2 value of 25 °C (obtained from FRET-melting experiments in the presence of F21T) [129].Footnote 17 , Footnote 18 Similar behavior was observed for the 13-substituted berberine derivative (9) [130].
Isaindigotone (Fig. 30) has a structure including a pyrrolo[2,1-b]quinazoline moiety conjugated with a benzylidene group. It is a naturally occurring alkaloid isolated from the root of Isatis indigotica Fort, a herb commonly used in traditional Chinese medicine. It may be assumed that, like in the case of cyanine dyes (cf. Sect. 2.1), the rotation of the aryl substituent in the isaindigotone scaffold can be hindered upon binding to the quadruplex, leading to enhancement of fluorescence. While isaindigotone itself does not show significant affinity towards quadruplex DNA, derivatives endowed with two aminoalkyl chains (10, 11) were identified as promising G-quadruplex ligands [131]. These two compounds were shown to be potent and selective stabilizers of the telomeric quadruplex (ΔT 1/2 = 21.9 °C for 10 and 17.6 °C for 11 bound to d[(G3(T2AG3)3]; ΔT 1/2 = 0 °C with a 10-bp duplex determined by FRET-melting). Their binding to G4-DNA is accompanied by an increase of fluorescence intensity by a factor of ~2.6 (10: λ max = 448 nm, 11: λ max = 450 nm). The analysis of fluorimetric titration curves revealed an apparent 2:1 ligand–oligonucleotide stoichiometry with an affinity constant per site of around 1.0 × 107 M−1 for both derivatives.Footnote 19

Natural products binding to quadruplex DNA with increase of their fluorescence
Similar behavior has been shown by quercetin (Fig. 30), a flavonoid which has been proved to exhibit antitumor activity. In aqueous solutions quercetin is characterized by two fluorescence bands with maxima at 422 nm (from the locally excited state) and 533 nm (ESIPT band due to the intramolecular proton transfer). In the presence of tetramolecular G-quadruplexes forming either a monomeric quadruplex unit (d[T2AG3T])4 or an end-to-end stacked dimeric quadruplex (d[T2AG3])4 the fluorescence intensity of quercetin increases and the ratio between the two bands changes as a function of the quadruplex structure. By analyzing the fluorescence, the absorption, the CD, and the NMR spectra, it was concluded that quercetin interacts differently with the two structures: external π-stacking mode with a monomeric G-quadruplex and groove-binding mode with a dimeric G-quadruplex [132, 133]. A significant fluorescence enhancement was also observed for rutin, a glycoside derivative of quercetin [133].
The fluorenylium derivative 12, a product of oxidation of papaverine, also exhibits a fluorescence “light-up” behavior. In aqueous solutions 12 is weakly fluorescent due to strong aggregation (H-aggregates in the case of low-salt conditions and J-aggregates at high ionic strength). Upon addition of quadruplex DNA d[(G3T2A)3G3], fluorescence enhancement by a factor of ~8 is induced, whereas interaction with ds DNA resulted only in a twofold increase of fluorescence [134].
Although moderate, the fluorescence increase of those natural products can be used to evidence and characterize the interaction with quadruplexes. However, it is less likely that it is to be of practical use for labeling or detecting quadruplexes due to (1) the poor magnitude of the effect and (2) the rather poor quadruplex vs duplex selectivity of most of these compounds.
2.6.2 Synthetic Compounds
Benzoindoloquinoline derivatives (Fig. 31) were shown to be moderate to good quadruplex binders (ΔT 1/2 = 3–11 °C measured by FRET-melting with F21T) [135]. In this series only SD27 is strongly fluorescent and displays a strong red-shift and a large increase of its emission upon addition of d[(G3T2A)3G3] (Fig. 32).Footnote 20 The structureless emission could originate from formation of excimers and the absence of an isoemissive point indicates the existence of several bound species.

Structures of the benzoindoloquinolone derivatives

Fluorescence titration of SD27 with the G-rich strand at 20 °C. The dashed lines correspond to SD27 with increasing concentrations of 21AG quadruplex (from 0.1 μM to 2.0 μM). Reprinted with permission from [135]. Copyright 2002 Elsevier
2.7 Metal Complexes
Over the past 40 years, metal complexes (mostly cationic) have been extensively studied as DNA binders [136–138]. More recently, this chemical class has been developed for targeting quadruplex nucleic acids [14, 139]. Hereafter, we review the metal-containing species (transition metal complexes, transition metal phthalocyanines, lanthanide cations and their complexes) displaying light-up fluorescence properties upon binding to quadruplex structures.
2.7.1 Diverse Transition Metal Complexes
2.7.1.1 Platinum(II) Complexes
A family of eight platinum(II) complexes, comprising the aromatic ligands dipyridophenazine (dppz) or C-deprotonated 2-phenylpyridine, were synthesized and studied as luminescent quadruplex-DNA probes [140].
One of the dppz complexes, namely [PtII(dppz-COOH)(N^C)]CF3SO3 (N^CH = 2-phenylpyridine, 13, Fig. 33), binds quadruplex structures likely through classical external-quartet stacking with a submicromolar K d and, more importantly, presents a 293-fold fluorescence intensity increase upon addition of quadruplex DNA (λ max = 512 nm, human telomeric sequence) at a rather high concentration of the complex (50 μM).Footnote 21 This emission was assigned to a 3[Pt → π*(dppz)] 3MLCT exited state, which takes place once the complex is isolated from water. Interestingly, the fluorescence enhancement is significantly lower with double-stranded DNA, such as ct DNA, poly(dA-dT)2, and poly(dG-dC)2 (27-, 6-fold, 3-fold, and 6-fold, respectively), which is consistent with the preferential binding to the quadruplex, as shown by competitive dialysis. Thanks to these properties, selective detection of micromolar concentrations of quadruplex DNA has been achieved with 13 in gel electrophoresis (PAGE) using long-wavelength UV irradiation.

Platinum(II) complexes
The same research group (Ma et al.) developed two other families aimed at down-regulating c-myc expression. Hence, nine Schiff base complexes were prepared following a computer-aided structure-based drug design [141]. Emission titrations by a quadruplex structure revealed an eightfold intensity enhancement of the complex 14 (λ max = 652 nm, Fig. 34).

(a) Emission spectra of the complex 14 with increasing concentration of c-myc quadruplex DNA d[TG4AG3TG4AG3TG4A2G2]. (b) Photographs of a solution of this complex (50 mM) in absence and presence of c-myc quadruplex DNA. Reprinted with permission from [141]. Copyright 2009 John Wiley and Sons
The second series is composed of seven tridentate bis(benzimidazole)pyridine (bzimpy) or bis(pyrazole)pyridine (dPzPy) platinum(II) complexes. The bzimpy derivative 15 presented in Fig. 33 displays a broad emission band upon binding to quadruplex DNA, with a 38-fold fluorescence intensity increase (λ max = 622 nm). These complexes bind duplex DNA with at least a tenfold lower affinity than quadruplex DNA, together with a lower fluorescence enhancement (4.4-fold for bzimpy derivative with ct-DNA).
The Pt(II)-terpyridine complex 16 was used to detect G-quadruplex formation via self-assembly of four short guanine-rich strands (Fig. 35) [142]. Thus, short G-rich oligonucleotides were mixed with the terpyridine complex, which resulted in their binding via electrostatic interactions. Upon addition of potassium ions, the oligonucleotides form a quadruplex structure, resulting in a high local concentration of the platinum complex, which in turn leads to emission increase due to the metal–metal-to-ligand charge transfer (3MMLCT) band (λ ex = 550 nm, λ max = 800 nm). However, the binding and fluorescence properties of 16 vis-à-vis prefolded intramolecular quadruplex DNA structures have not been studied, so far.

Schematic cartoon showing the possible self-assembly of 16 via Pt···Pt and π–π interactions induced by G-quadruplex formation upon K+ ion binding
2.7.1.2 Ruthenium(II) Complexes
Although Ru(II) complexes are mostly nonplanar and feature octahedral or pseudo-octahedral geometry (cf. Fig. 40), there is a growing interest in their use as pharmaceuticals, and notably as DNA binders [143]. Barton’s group reported for the first time reversible light-up DNA-binding ruthenium(II) complexes containing dipyrido[3,2-a:2′,3′-c]phenazine (dppz) and bipyridine (bpy) or phenanthroline (phen) ligands [144, 145]. These compounds are almost nonemissive in aqueous buffers but a 3MLCT excited state fluorescence emission occurs upon DNA intercalation.
Although many studies with duplex DNA have been published since then, interaction of [Ru(phen)2(dppz)]2+ and [Ru(bpy)2(dppz)]2+ with quadruplex DNA was only recently examined (Fig. 36) [146, 147]. [Ru(phen)2(dppz)]2+ binds to the human telomeric sequence in potassium conditions more tightly than in sodium conditions, or to the i-motif (one order of magnitude difference in the latter case).Footnote 22 Addition of quadruplex to the complex results in a fluorescence intensity increase (λ max ≈ 625 nm), around five times larger with the quadruplex structure as compared to the i-motif, likely due to a better π-overlapping and a better isolation from the solvent. Analogous results were obtained with [Ru(bpy)2(dppz)]2+. With the human telomeric sequence, both the fluorescence enhancement (fourfold) and the binding affinity (one order of magnitude) of the complex were larger in potassium-rich buffer than in sodium-rich buffer. In either case, the quadruplex-forming sequence was preferred over the i-motif structure.

Mononuclear ruthenium(II) complexes. Counter-ions are hexafluorophosphates
Two analogous complexes, but containing two dppz ligands, were prepared [148]. The third ligand is a bpy derivative, functionalized by either trimethylammonium (17) or triethylammonium terminal groups (18) (Fig. 36). Stabilization of human telomeric sequence (determined by UV-melting at a 1:1 ratio) induced by both complexes was higher than with [Ru(bpy)2(dppz)]2+ [146]. The derivative 17 binds more tightly than 18 (9.4°C vs 7.0 °C), with an apparent 1:1 stoichiometry. Both complexes exhibit a marked fluorescence intensity increase upon addition of the human telomeric quadruplex, but to a larger extent for 18 than for 19 (7.6-fold and 4.8-fold, respectively; λ max = 630 nm; Fig. 37).Footnote 23

Emission spectra of complexes [Ru(dppz)2(R2-bpy)]2+ (a: 17, b: 18; 2 μM) in the presence of telomeric quadruplex DNA (AG3(T2AG3)3, 0–5 μM). Reprinted with permission from [148]. Copyright 2011 Elsevier
The Thomas’ group investigated the spectroscopic properties of two dinuclear Ru(II) complexes 19 and 20 containing a tppz (tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2″,3″-j]phenazine) and bipyridine or phenanthroline ligands, upon duplex or quadruplex DNA binding (Fig. 38) [149]. Interaction of these two compounds with double-stranded ct DNA results in a large fluorescence enhancement (>60-fold, λ max = 658 and 637 nm for the phen and bpy complexes, respectively).Footnote 24 The light-up effect has been rationalized by analogy with the aforementioned dppz complexes via a computational study [150]. Addition of human telomeric quadruplex DNA in high ionic strength conditionsFootnote 25 induced a fluorescence intensity enhancement of the metal complexes, ca. 2.5 times higher than with duplex DNA (150-fold), together with a substantial hypsochromic shift as compared to ds DNA (λ max = 631 and 605 nm for the phen and bpy complexes, respectively) and longer emission lifetimes (129 and 123 ns for G4-DNA vs 84 and 92 ns for ds DNA). These data were explained by a better isolation of the complexes from the aqueous medium and a greater aromatic overlap with the DNA bases through a 1:1 end-stacking mode. It was also proposed, on the basis of calorimetric studies, that stacking of 19 to a quadruplex structure is more favorable than intercalation into duplex DNA (5 kJ mol−1 difference). However, the proposed end-stacking mode is still questionable considering the large size and complex 3D topology of these dinuclear octahedral Ru(II) complexes.

Dinuclear ruthenium(II) complexes
Other quadruplex-forming sequences (TBA, Pu27 from c-myc, the Oxytricha telomeric sequence) were studied with the phenanthroline complex 19 [206]. The intensity of the 3MLCT emission band and maximum wavelength are both strongly affected by the quadruplex structure. Indeed, fluorescence enhancement ranges from threefold (with TBA) to 138-fold (with the human telomeric sequence), with the maximum emission wavelength varying accordingly (the higher the enhancement, the larger the hypsochromic shift) from 635 nm (human telomere) to 670 nm (TBA). However, the affinities (associated with the 1:1 end-stacking) and fluorescence enhancements are not directly correlated, since larger emission enhancement are also dependent on isolation from the solvent [145]. The authors have explained these differences by the nature and the length of the quadruplex loops, since external loops containing at least three bases result in higher fluorescence intensities of the bound ligand than small lateral loops. Results obtained with complex 20 are analogous.
Finally, it should be noted that 19 was used as a multifunctional biological imaging agent for staining nuclear DNA of living eukaryotic and prokaryotic cells. However, exceptionally large concentrations of compound are required (i.e., 500 μM) which is attributed to an inefficient nonendocytotic cellular uptake [151]. More interestingly, a dual emission peaking at 630 and 680 nm has been observed from the nuclei stained by the Ru complex, which was interpreted as concomitant labeling of duplex DNA and alternative DNA structures (potentially quadruplexes). The blue-shifted band (630 nm) is observed only when the probe is incubated with the cells before fixation by formaldehyde, thereby suggesting that these alternative DNA structures may be induced by probe binding (Fig. 39).

(a) Multiple-emission profile of 19 in live MCF-7 cells, in two separate cellular regions (red and blue); (b) Confocal microscopy images with separate detection channels: 670–700 nm (red) and 630–640 nm (yellow). Reprinted with permission from [151]. Copyright 2009 Nature Publishing Group
Another rigid, dinuclear but unsymmetrical ruthenium(II) complex 21, containing the obip [2-(2-pyridyl)imidazo[4,5-f][1,10]phenanthroline] and bpy ligands, has been studied (Fig. 40) [152]. Addition of the human telomeric quadruplex in sodium conditions resulted in a moderate fluorescence enhancement (1.5-fold, λ ex = 460 nm, λ max ≈ 620 nm), although higher than with calf-thymus DNA.Footnote 26 Concurrently a tenfold preference for the quadruplex over duplex structure was found by fluorimetric titration (1:1 electrostatic external and/or end-stacking model).

Structure of Ru2(obip)(bpy)4. Right: crystal structure by X-ray diffraction (C: gray, H: white, N: blue, Ru: turquoise)
A dinuclear ruthenium(II) complex 22 was designed using a partially flexible acyclic chain instead of a rigid ditopic ligand [153]. Both Ru2+ cations are complexed by two bpy and one hpip (2-phenyl-1H-imidazo[4,5-f][1,10]phenanthroline) ligands (Fig. 41). It is supposed that upon addition of potassium cations, the podand linker adopts a pseudocyclic conformation in order to bind potassium, resulting in a fluorescence intensity decrease, likely due to ruthenium self-quenching when both metal centers come into close proximity. When various quadruplex structures (human telomere, Bcl2, Pu18, c-kit, Vegf) were added, a fivefold increase of fluorescence intensity was observed, presumably due to the opening of the pseudocyclic system (λ ex = 468 nm, λ max ≈ 600 nm).Footnote 27 Conversely, only a slight increase was observed with ds DNA and even a decrease with ss DNA.

Structure of the flexible dinuclear ruthenium complex. The podand linker has been depicted in a pseudocyclic conformation, but remains flexible in the absence of potassium cations
However, strong background fluorescence remains caused by the unbound ruthenium species. This was eliminated using the heavy atom effect by the addition of 50 mM iodide ions. The authors assumed that iodide anions cannot access the bound complexes due to electronic repulsion of DNA phosphates, and fluorescence was indeed recovered upon addition of quadruplex structures, with a 15-fold fluorescence selectivity for c-kit against ds DNA. Using this system, a high sensitivity was obtained, enabling the distinction between quadruplex and duplex DNA at submicromolar concentrations of both 22 and DNA. Thus, as little as 50 nM of quadruplex DNA could be detected in the presence of 1 μM of ds DNA. As found with various other probes described previously, fluorescence enhancement is not only influenced by the binding affinity but also by the binding mode as this complex binds quadruplex structures with only a slight preference over duplex DNA as shown by SPR measurements. A double external stacking model was proposed for this compound, where each ruthenium unit binds one of the external G-quartets.
2.7.2 Transition-Metal Phthalocyanines
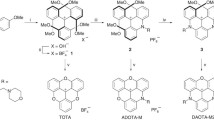
Since the discovery of phthalocyanines more than 80 years ago, this class of macrocyclic compounds, and the corresponding coordination complexes, have been used extensively as fluorescent dyes. First studies of the quadruplex-binding properties of pyridinium-porphyrazine and ammonium-porphyrazine derivatives revealed an enhanced selectivity compared to the well-known porphyrin TMPyP4 (see Sect. 2.4) [154, 155].
Luedtke’s group developed zinc complexes of guanidinium derivatives of phthalocyanine to improve both the cellular uptake and the nucleic acid affinity [156]. Zn-DIGP (Fig. 42) displays a large fluorescence enhancement (200-fold, λ ex = 620 nm, λ max = 705 nm) upon addition of saturating amounts of nucleic acids.Footnote 28 The resulting fluorescence quantum yields are rather low (Φ F = 0.06) but counterbalanced by very large molar extinction coefficients (ε = 30,000–130,000cm−1 M−1) that result in a strong brightness (Φ F × ε), the relevant figure of merit for imaging. Remarkably high binding constants were extracted from fluorimetric titration with various quadruplex structures. In particular, with the c-myc sequence a nanomolar K d was found (K d = 2 × 10−9 M per site with a 2:1 stoichiometry), with a preference of one order of magnitude over unfolded G-rich ss DNA, and 1,000-fold, 100-fold, and 5,000-fold selectivity over C-rich unfolded ss DNA, tRNA, and calf thymus DNA, respectively.

Structure of the guanidinium-modified and amido-modified phthalocyanines
Fixed and living cell microscopy (wide-field or confocal) revealed a successful internalization of Zn-DIGP, mostly probing trafficking vesicles and perinuclear organelles, with no staining of duplex DNA as seen by Hoechst 33342 co-staining experiments (Fig. 43).

Fixed SK-Mel-28 cells stained with 3 μM Zn-DIGP (a; λ ex = 620 nm, λ max = 700 nm) and 8 μM Hoechst 33342 (b; λ ex = 360 nm, λ max = 470 nm) and overlay (c). Adapted with permission from [156]. Copyright 2009 John Wiley and Sons
A K+ detection system has been designed, using Zn-DIGP and the parallel quadruplex forming sequence c-myc [157]. This system relies on the promotion of the c-myc folding by K+, followed by an easily observable fluorescence intensity increase of Zn-DIGP, once bound to the quadruplex structure. A 0.8 μM detection limit of K+ was determined. The absence of promotion of c-myc quadruplex structure by various other cations (NH +4 , Na+, Ca2+, Mg2+, Zn2+, Fe3+, and Cu2+) allows a specific detection of potassium. Hence, 40 μM of K+ could be detected in the presence of a 3,500-fold excess of Na+ ions.
Recently, Zn-DIGP was used jointly with the N-methylmesoporphyrin IX (NMM) in a nucleic acid detection assay [158]. Briefly, the c-myc sequence was separated into two fragments, and flanking segments complementary to a target DNA sequence were added (Fig. 44). In the presence of potassium cations, the two sequences readily form a dimeric quadruplex structure, which is further stabilized by the bound fluorescent probes. Upon addition of the target sequence, base-pairing interactions with both flanking sequences induce the formation of a quadruplex-duplex three-way junction, which modifies the environment and consequently the fluorescence of the probe. This fluorescence modification (increase for NMM, decrease for Zn-DIGP) allows the detection of the target sequence.

Schematic illustration of DNA sensors based on turn-off (pathway A) and turn-on (pathway B) fluorescence changes, utilizing split G-quadruplex probes. In the presence of K+, a split c-myc (blue) forms an associated G-quadruplex-fluorescent dye complex. Sequence-specific DNA hybridization results in reduced fluorescence from Zn-DIGP (pathway A) or increased fluorescence from NMM (pathway B). Reprinted with permission from [158]. Copyright 2011 Springer
A second class of acylamino-substituted phthalocyanine derivatives, in which systematic variations of the length of the alkyl chain and the nature of the terminal function were performed (APc, Fig. 42), was studied by the same group [159]. The influence of these variations on the fluorescence light-up and quadruplex binding properties was subsequently examined. Large enhancements were observed with APc 2–5 (100–400-fold), but not with APc 1 and 6, in the presence of the human telomeric quadruplex (λ ex = 630 nm, λ max = 705 nm) (see footnote 28). APc 2 displayed the best performances (400-fold increase, Φ F = 0.05, ε = 30 000–150,000 cm−1 M−1). Tenfold lower binding constants were determined for APc 2–5 as compared to Zn-DIGP, but the high selectivity against tRNA and calf thymus DNA is preserved (50-fold and 500-fold, respectively). Large fluorescence enhancements were also observed with c-myc and c-kit2 structures (200–800-fold), and the following relative binding affinity sorting was suggested: c-myc > c-kit2 > human telomeric quadruplex. It was hypothesized that APc 1 forms J-aggregates unable to bind quadruplex DNA whereas APc 2–5, which contain more methylene units and hence increased pK a values, form nonfluorescent H-aggregates. Fluorescence enhancement is thus the result of dissociation of the APc aggregates into quadruplex-binding monomers.
2.7.3 Lanthanides
Lanthanide complexes are extensively used as emitters for luminescent resonance energy transfer and as contrast agents in magnetic resonance imaging, but have also already been used to detect duplex DNA via fluorescence enhancement [160–163]. Terbium(III) and europium(III) ions have well-suited luminescence properties for nucleic acid probing, such as narrow emission band widths and large Stokes shifts (see Fig. 47 for an example), but Tb3+ emitting level fits better the triplet state of nucleic acid bases [163, 165] (for a review on the “antenna effect” see [166]).
Terbium(III) emission in aqueous medium is very weak because of the solvent quenching and a low intrinsic molar absorptivity [165]. Upon binding of TbCl3 to quadruplex DNA, energy is transferred via a FRET mechanism from the bases (donor, λ ex = 290 nm) to the terbium ion (acceptor) inducing a 5D4 long-lived excited state emission (λ max = 545 nm), which results in a ~25-fold luminescence increase of Tb3+ in the presence of human telomeric DNA [167]. Interaction of Tb3+ ions with quadruplex DNA is believed to occur through coordination of nitrogen and oxygen atoms of bases and phosphate groups, with a micromolar affinity and a 6:1 stoichiometry.Footnote 29 Putative binding sites are the quadruplex central channel and TTA loops. Cation-displacement experiments were performed with potassium, sodium, and lithium (in that ranking of efficiency), resulting in quenching of terbium fluorescence, which may evidence the presence of terbium in the quadruplex central channel (but also increase the ionic strength that may affect the fluorescence).
Förster energy transfer was used with reverse conditions, i.e., terbium(III) ions (chelated by DTPA or TTHA, Fig. 45) as donors and quadruplex DNA as acceptor [168]. The change in fluorescence lifetime (λ ex = 488 nm) was monitored in the presence of small amounts of quadruplex DNA (20–800 nM), allowing to calculate the amount of transferred energy. Hence, this amount increased with quadruplex DNA concentration (single-stranded 22-mer and double-stranded 12-mer).

Structure of DTPA and TTHA ligands
Nonchelated europium(III) was used in the same study to detect low quadruplex DNA concentrations (70% and 25% fluorescence enhancements with 800 nM of quadruplex or duplex calf thymus DNA, respectively; λ max = 453 nm).
Finally, interactions between two terbium-amino acid (dl-cysteine or dl-valine) complexes (23; Fig. 46) and quadruplex or i-motif DNA were studied in 2006 [169]. Both complexes destabilize slightly the human telomeric quadruplex (ΔT m = −3.0 to −4.0 °C, UV-melting), presumably through coordination binding of terbium to N7 of purines and exocyclic C2 of pyrimidines [164, 170]. Binding constants with quadruplex and i-motif DNA were analogous and rather low (ca. 4 × 104 M−1). A 253-fold and 88-fold fluorescence increase (for cysteine and valine complexes, respectively) were observed in the presence of quadruplex DNA, as compared with a 32-fold increase for the control complex TbCl3 (λ ex = 260 nm, λ max = 488, 534, 584 nm; Fig. 47).Footnote 30

Terbium-amino acid complex structure and solid-state structure (from X-ray diffraction; C: gray, H: white, N: blue, Tb: green)

Fluorescence spectra of [Tb2(dl-HVal)4(H2O)8]Cl6 in absence (black) or presence of quadruplex (red) and i-motif (green). Reprinted with permission from [164]. Copyright 2006 Elsevier
Interestingly, the fluorescence increase in the presence of i-motif was significantly lower (around threefold), despite a comparable affinity of the complex for both structures. The authors suggested that the more efficient energy transfer between guanine bases and terbium may account for these results [163, 171].
3 “Light-Off” Probes
Beyond the ligands whose binding to quadruplex DNA is accompanied by an increase or shift of the fluorescence signal, a number of small molecules – usually heterocyclic derivatives – have been described which are intrinsically fluorescent in the absence of DNA, but whose fluorescence intensity decreases to a smaller or larger extent upon binding to the quadruplex target. This fluorescence quenching phenomenon is in most cases due to an electron transfer reaction between the excited fluorophore and DNA, which is exergonic when the reduction potential of the excited dye (E *red ) is larger than the oxidation potential of the nucleic bases. As guanine has the lowest oxidation potential among the DNA bases (1.02 V vs NHE for guanosine and GMP at pH 7.4) [172], it represents the base which is most easily oxidized, and the high density of guanine residues in a quadruplex DNA makes the electron-transfer reaction with a fluorophore even more favorable. Moreover, it was reported that the oxidative damage upon photooxidation of quadruplex DNA is localized on the guanines of the external G-tetrad [173], which gives some evidence that their oxidation potential is lower than the one of guanine bases in duplex DNA. Since the external G-tetrad represents the binding site for a vast majority of quadruplex ligands, it is not unexpected that binding of many fluorescent ligands is accompanied by an efficient quenching of their fluorescence. Nonetheless, it should be mentioned that quenching of fluorescence is not as desired in analytical or biomedical applications as fluorescence shift or fluorescence enhancement, since quenching might be also due to other species present in samples, such as molecules favoring the intersystem crossing (amines, carbonyl derivatives), inorganic quenchers (I−, [Fe(CN)6]3−), or other processes such as ligand aggregation. Still, the fluorescence quenching phenomenon is very useful when additional information about the binding event has to be obtained (binding constant, stoichiometry) and, if the ligand would be selectively quenched by quadruplex DNA and not by the duplex form, sensitive detection of the quadruplex form may be envisaged.
3.1 Heterocyclic Ligands with a Neutral Core
Most planar aromatic heterocyclic compounds exhibit moderate to strong fluorescence if they are not aggregated in aqueous solutions. Some representative examples of quadruplex DNA ligands whose fluorescence quenching behavior upon binding to the quadruplex target was well-documented are given in Fig. 48. Among them the quinacridine derivatives, such as MOQ2, were ones of the first identified quadruplex-stabilizing telomerase inhibitors. These heterocycles bearing polyammonium side chains show a bright blue fluorescence in water (e.g., λ max = 462 nm, Φ F = 0.10 for MOQ2), which is quenched nearly completely upon binding to the telomeric G-quadruplex 22AG (Fig. 49). The analysis of fluorimetric titration curves performed at increasing concentration of 22AG revealed a binding constant of 3.4 × 106 M−1 and a 2:1 stoichiometry [175].Footnote 31 The bis-quinacridine macrocycle BOQ1 was shown to bind 22AG with a higher affinity than the monomeric quinacridine and a tenfold selectivity with regard to duplex DNA (K a = 1 × 107 M−1 with 22AG; K a = 1.2 × 106 M−1 with a 20-bp duplex, 2:1 stoichiometry in both cases, as determined by SPR) (see footnote 31). The fluorescence of BOQ1 in the free state is reduced with respect to the monomeric quinacridine due to the intramolecular stacking effect (λ max = 466 nm, Φ F = 0.03), and binding to the quadruplex leads to further quenching of the fluorescence down to ≈50% of the original value. In contrast, addition of duplex DNA leads to only a slight decrease of the fluorescence consistent with the selectivity inferred from SPR data [174].

Heterocyclic ligands with fluorescence-quenching behavior upon binding to quadruplex DNA

(a) Fluorescence spectra of MOQ 2 (0.1 μM; λ ex = 360 nm) recorded at increasing concentration of oligonucleotide 22AG. (b) Experimental (diamonds) and calculated (solid line) titration curves obtained for the 1:2 stoichiometry. R = DNA/ligand molar ratio. Reprinted with permission from [174]. Copyright 2003 American Chemical Society
The trisubstituted naphthalimides 24 and 25 were selected as potent G-quadruplex ligands from a series of similar derivatives by means of fluorimetric titrations. Their interaction with 22AG leads to quenching of naphthalimide fluorescence, which was useful for determination of the binding stoichiometry and the binding constants (2:1 complexes, K = 3.0 × 106 and 2.4 × 106 M−1 for 24 and 25, respectively) [176].Footnote 32
AZATRUX is a heterocycle with a large C 3h-symmetric aromatic core, designed to stack over the G4 tetrad, and three aminoalkyl chains [177, 178]. In solution AZATRUX shows a maximum of fluorescence at 394 nm (λ ex = 320 nm) that is strongly decreased upon interaction with the telomeric sequences forming dimeric and monomeric quadruplexes d[(TTAG3)8TT], d[(AG3TT)4]. At saturation the fluorescence is completely quenched in the presence of the dimeric quadruplex, whereas a residual fluorescence is detected in the presence of the monomeric quadruplex. Binding curves extrapolated from the titrations show binding constants in the (0.5–1) × 106 M−1 range (Fig. 50).

Fluorescence emission spectra of AZATRUX (8 μM) in the presence of successive additions of telomeric G4-DNA (inset) and plot of the fraction of bound AZATRUX vs DNA concentration. Reprinted with permission from [179]. Copyright 2011 Elsevier
The neutral chiral cyclic helicene M1 was designed as a ligand for higher-order quadruplexes, such as quadruplex dimers. It is a strongly fluorescent compound (λ em ≈ 475 nm) whose fluorescence is efficiently quenched (by ≈80%) upon binding to a quadruplex dimer in which the two quadruplex units are separated by a short TTA loop. Interestingly, the fluorescence of the corresponding P-isomer is not affected in the same conditions, which indicates that only the M-isomer M1 binds to the quadruplex dimer. This unique binding mode was confirmed by molecular modeling calculations [180, 181].
Finally, the acyclic polyheteroaromatic pyridyloxazole ligand TOxaPy represents a neutral quadruplex-DNA ligand with an exceptionally high selectivity for human telomeric quadruplex in Na+-rich conditions, presumably due to its quadruplex groove-binding mode. TOxaPy is characterized by an intense blue fluorescence in water (λ em = 390 nm, Φ F = 0.50) which is strongly quenched upon addition of 22AG in Na+ buffer but much more moderately in the K+-rich one, which is consistent with the melting data showing unprecedented selectivity for the Na+ form of the telomeric quadruplex (ΔT m = 10.8 and <1.0 °C with F21T in Na+ and K+, respectively). Analysis of fluorimetric titration data gave a binding constant value of K = 5 × 106 M−1 with a 1:1 binding stoichiometry [31].Footnote 33
3.2 Heterocyclic Ligands with a Cationic Core
Among other reported G-quadruplex ligands, the azacyanine aza3 (Fig. 51) was developed as a readily accessible, efficient and selective ligand for telomeric quadruplex DNA (association constant of 1.3 × 106 M−1 with sequence d[T2G3(T2AG3)3A] vs 9.2 × 103 M−1 for calf thymus DNA from fluorimetric titrations, i.e., more than a 100-fold selectivity, confirmed also by SPR) [182].Footnote 34 Binding of aza3 to G-quadruplex leads to about twofold quenching of its blue fluorescence (λ max ≈ 375 nm). It may be speculated that this high selectivity with respect to duplex DNA could lead to development of a fluorescence quenching assay for detection of quadruplex DNA.

Cationic ligands whose fluorescence quenching upon binding to quadruplex DNA was documented
The cryptolepine derivative 26 is a member of a series of quadruplex-selective quindoline derivatives [56]. Its interaction with the human telomeric G-quadruplex leads to fluorescence quenching accompanied by a hypsochromic shift of the emission maximum (from 472 nm to 461 nm) [183]. Similar quenching behavior was observed for isoquinoline alkaloids coralyne (λ max = 472 nm), which is also known for its interaction with triplex DNA, and sanguinarine [126]. Synthetic diazoniadibenzochryzene 27 and tetraazoniapentaphenopentaphene 28 belong, like coralyne, to the family of annelated quinolizinium derivatives and possess high fluorescence quantum yields in the absence of DNA (27: λ em = 456 and 481 nm, Φ F = 0.47; 28: λ em = 511 nm, Φ F = 0.14). Interaction of these highly charged organic cations with human telomeric quadruplex results essentially in complete quenching of their fluorescence, which was used for the calculation of binding constants (K a = 1.6 × 106 and 9.0 × 105 M−1 for 27 and 28, respectively).Footnote 35 Interestingly, both 27 and 28 have relatively high quadruplex-vs-duplex selectivity, as demonstrated by thermal denaturation and FID studies [184].
Finally, the N-quaternized quino[4,3,2-kl]acridinium salts 29 were developed as quadruplex binders with associated potential antitumor activity. Of note, the most well studied member of this series is the well-known quadruplex binder RHPS4 (R1 = R3 = F, R2 = H), whose fluorescence properties have not been studied in detail. The fluorimetric titrations with 22AG evidenced moderate quenching effect and were used for calculating the quadruplex-binding constants, which were in the range from 0.9 × 105 M−1 (29a, R1 = R2 = R3 = H) to 1.1 × 106 M−1 (29b, R1 = 2-NHCOtBu, R2 = R3 = H) (see footnote 35). In some cases, high selectivity with respect to duplex DNA (with a binding constant ratio of up to 15) was observed in the fluorimetric titrations. However, the quenching was not complete even for the most quadruplex-affine member of the series (F/F 0 = 0.85) [185]. These results demonstrate that the efficiency of the fluorescence quenching (represented by the F/F 0 value in the presence of excess quadruplex DNA) and the quadruplex-binding affinity are, in general, not interconnected values, and data analysis should always be performed carefully in order to obtain correct values of binding constants.
3.3 Perylene Diimide Derivatives
Water-soluble perylene diimide derivatives, such as PIPER and analogues (Fig. 52a), represent a widely studied class of quadruplex-DNA ligands. Since the fluorescence of PIPER is quenched in aqueous solutions at neutral pH even in the absence of DNA due to strong self-aggregation, it is not particularly useful as a fluorescent probe [186]. However, an alternative study demonstrated significant quenching of fluorescence of PIPER (λ max = 545–500 nm) in very diluted solutions (33.8 nM) upon binding to quadruplex-forming DNA sequences NHE-27Footnote 36 or 2HTR,Footnote 37 and the values of binding constants of 1.8 × 108 and 2.9 × 108 M−1 upon binding of seven or six molecules of ligand to these structures were obtained from the fluorimetric titrations [187].Footnote 38 Less aggregating analogues such as Tel11 are characterized by an intense fluorescence in more concentrated aqueous solutions (up to 1 μM), which is efficiently quenched upon interaction with quadruplex DNA. A nearly-complete quenching of Tel11 was obtained upon addition of substoichiometric amounts of 22AG, which indicates a positive cooperativity of binding, with apparent saturation upon binding of six molecules of the ligand per quadruplex [188]. However, it was shown that PIPER and Tel11 have rather similar affinity for duplex and quadruplex DNA [187, 189].

(a) Representative perylene diimide derivatives. (b) Quadruplex-binding anthracycline antibiotics
3.4 Anthracyclines
The anticancer antibiotics of the anthracycline series, doxorubicin and sabarubicin (Fig. 52b), are characterized by a fluorescence emission with maxima at 590 nm (Φ F = 0.039) and 565 nm (Φ F = 0.063), respectively. Their interaction with quadruplex DNA (human telomeric: d[G3(T2AG3)3]) results in efficient quenching of the fluorescence. In both cases, 1:1 drug–G4-DNA complexes are not fluorescent; instead the 2:1 complexes are fluorescent in Na+-rich conditions (Φ F = 0.009 and 0.017 for doxorubicin and sabarubicin, respectively) but not in the K+-rich buffer. The quadruplex-binding constants, calculated from the fluorimetric data, are log K 11 = 6.0, log K 21 = 10.9 for doxorubicin, and log K 11 = 5.8, log K 21 = 11.5 for sabarubicin in K+ conditionsFootnote 39; similar values were obtained in Na+-rich conditions. Notably, these values are slightly lower than those measured with duplex DNA (log K of 6.5 for doxorubicin and 6.1 for sabarubicin, respectively), thereby indicating the absence of quadruplex-vs-duplex selectivity of these drugs [190, 191].
3.5 Classical Organic Dyes
Several studies focused on the changes of fluorescent properties of well-established organic dyes, commonly used as stains in biochemistry and histology, upon their binding to quadruplex DNA. Thus, it was shown that methylene blue binds to intermolecular G-quadruplex DNA (d[TTAAGG])4) with formation of 1:1 and 1:2 complexes (i.e., one cation of methylene blue stacked between two quadruplexes) with binding constants of 1.0 × 105 and 8.8 × 104 M−1, respectively,Footnote 40 which leads to quenching of its fluorescence [192]. However, interaction with single-stranded and double-stranded DNA structures also leads to fluorescence quenching of methylene blue [193]. Binding of acridine dyes, such as proflavine and acriflavine, to cyclic diguanylic acid (c-di-GMP) was shown to induce formation of G-quadruplex structure in the absence of monovalent metal cations, which was accompanied by fluorescence quenching of these dyes (e.g., at λ em ≈ 510 nm for proflavine) [194]. Interestingly, mononucleotides such as cGMP or rGTP did not induce fluorescence quenching of these acridine dyes; nonetheless, fluorescence quenching of proflavine upon binding to guanine-containing double-stranded DNA has been documented decades ago [195].
A four-branched pyrazine-styryl derivative TASPI (Fig. 53) was initially developed as a two-photon-absorbing dye, strongly fluorescent in aqueous solutions. However, the fluorescence is almost completely quenched upon addition of a stoichiometric amount of the thrombin-binding aptamer (TBA) quadruplex. Interestingly, addition of thrombin to the TASPI-TBA complex resulted in restoration of the fluorescence due to displacement of TASPI. This behavior has been applied to the selective fluorimetric detection of thrombin in the sub-micromolar concentration range [196].

Fluorescent dyes binding to quadruplex DNA with a decrease of fluorescence intensity
3.6 Metal Complexes
In addition to the larger number of Ru(II) complexes whose luminescence increases upon interaction with quadruplex DNA (cf. Sect. 2.7), several complexes which act instead as “light-off” probes have been described. Like in the case of organic fluorophores, the quenching of the luminescent excited state of Ru(II) complexes is due to the photoinduced electron transfer from the guanine bases of quadruplex DNA to the 3MLCT of the complex. Nice examples are provided by the trinuclear Ru(II) polypyridyl complexes 30 and 31 (Fig. 54), whose luminescence (λ max ≈ 600 nm) decreases by ~40% upon binding to 22AG (1:1 binding mode) [197], as well as the dinuclear [(tap)2Ru(tpac)Ru(tap)2]4+ (32). In the latter case it was demonstrated that quadruplex DNA (d(T2AG3)4) induces luminescence quenching much more efficiently than duplex DNA. Interestingly, this quenching behavior is in sharp contrast with the luminescence “light-up” effect of [(phen)2Ru(tppz)Ru(phen)2], differing from 32 only by nitrogen substituents in the ligands (cf. Sect. 2.7.1). It was also shown that 32 is able to produce cross-links of guanine residues of the quadruplex upon photoillumination [198] (Fig. 54).

Metal complexes displaying luminescence quenching upon binding to quadruplex DNA
4 Permanent (“Tagged”) Quadruplex-DNA Probes
Most of the above-mentioned studies rely on the intrinsic fluorescence of quadruplex binders or are based on rational design for adapting already known fluorophores to quadruplex recognition. Another approach consists of the covalent linking of a fluorescent moiety to a quadruplex-recognizing motif, resulting in a “tagged” quadruplex binder. This type of hybrid compound can also be called permanent probes as the fluorescence is generally not affected by the binding event. The main issue of this approach is the choice of the fluorescent tag that should not affect the binding features (affinity and selectivity) of the quadruplex-interactive moiety. In that sense, small-sized, neutral or poorly charged tags are recommended. Amongst the panel of dyes that fulfil these criteria and furthermore are chemically accessible and easily derivatizable we can find the boron-dipyrromethene (BODIPY) dye family.
Recently Nagasawa et al. developed a fluorescent derivative of telomestatin, the latter representing for many years the gold standard for quadruplex recognition. The macrocycle 7OTD (Fig. 55) is a synthetic analogue of telomestatin, which is more easily prepared and as efficient in terms of affinity and selectivity for quadruplexes in particular telomeric sequences. 7OTD was therefore a good candidate for functionalization by a fluorescent tag. L1BOD-7OTD exhibits a green fluorescence (λ em = 501 nm) remaining unchanged in the absence or in the presence of various G4-forming sequences [199]. This stable fluorescence allowed the use of the labeled macrocycle to stain specifically G4-forming sequences in gel electrophoresis experiments (Fig. 56). Selective green staining of the telomeric quadruplex d[T2AG3]4 was observed, whilst no fluorescence was detected with non-G4-forming sequences (duplex DNA or single-stranded sequence). Selective staining was also observed with other G4 forming sequences (c-myc, c-kit22, bcl2) and significant retardation of the band corresponding to the ligand–G4 complex is seen with increasing concentration of L1BOD-7OTD. Apparent dissociation constants of L1BOD-7OTD with various G4 forming sequences have been estimated by fluorescence polarization titration and shown to lie in the nanomolar range (0.2–80 nM depending on the G4 sequence). Finally, fixed HeLa cells were stained with L1BOD-7OTD that colocalizes with DAPI in the nuclei, indicating that the labeled compound can be used for imaging in cells.

Structures of telomestatin, L1H1-7OTD and the fluorescent labeled L1BOD-7OTD (LB)

Visualization of the telo24 G4 by L1BOD-7OTD by gel electrophoresis (12% polyacrylamide, 1X TBE buffer, 4 °C) in the presence of 10 μM oligonucleotides (a–c: telo 24, d–f: ds-telo 24) and various concentrations of L1BOD-7OTD. (a, d) All bands were detected using the 526 nm short pass filter. The gel was stained with (b) Stains-All and (e) ethidium bromide then all bands were detected using the 580–640 nm band pass filter. (c, f) Merged images of (a) and (b) or (d) and (e). Reprinted with permission from [199] with permission of The Royal Society of Chemistry
Recently BODIPY has been used to label the bisquinolinum pyrido-dicarboxamide motif (PDC) affording the PDC-BODIPY compound (Fig. 57). In contrast to the PDC-conjugate built with TO as fluorescent moiety (PDC-TO see Sect. 2.1) the selectivity of PDC-BODIPY is retained as demonstrated by means of a streptavidin-paramagnetic-particle based assay recently set up to evaluate optical selectivity of fluorescent G4-binders [200].

Structure of PDC-BODIPY derivative
Finally, during the period of preparation of this manuscript, Rodriguez et al published an “in-situ” labeling strategy using click chemistry [201]. This result will not be described in detail but is nonetheless important with respect to this review. Click chemistry (i.e., the biocompatible 1,3-dipolar cycloaddition between azide-derivatized and alkyne-derivatized partners) has been successfully applied for fluorescent detection of replication and transcription in living cells using alkynyl uracil nucleotides and an azido derivative of Alexa Fluor 549 [202, 203]. The authors prepared an alkynyl derivative of pyridostatin [204]. The cells were first incubated with this compound and subsequently with an azido derivatized fluorophore (Alexa Fluor 549). The red fluorescence characteristic of Alexa Fluor 549 was found localized in the nucleoli with small foci in the nucleus. It was concluded that the compound accumulation in the nucleolar subcompartment results from preferential binding of alternative DNA and RNA structures. The latter may correspond to quadruplexes in consistency with the high GC rich content of ribosomal DNA and its transcriptional status in the nucleoli [205]. However, the rules that govern nucleolar targeting by small molecules are not well-established. For instance, several DNA dyes (propidium iodide, thiazole orange) that intercalate in ds DNA are also known to label RNAs and to stain nucleoli; as well certain anticancer drugs (doxorubicin) with affinity for ds DNA have been shown to act in the nucleoli at the level of rRNA transcription. Thus the real significance of the nucleolar accumulation of G4 binders remains an open question. Finally, technical difficulties are not particularly addressed in the report, although “in-cell” click chemistry is notoriously known to require enormous amounts of copper salt (CuSO4, 1 mM) that is strongly deleterious to cells. Nonetheless, from a more general point of view, this “in situ” labeling strategy opens highly interesting perspectives and may allow one to circumvent the issues frequently associated with the labeling of active principles (i.e., modification of target binding and off target effects).
5 Conclusions and Perspectives
The selected examples above illustrate the tremendous growth of quadruplex-selective fluorescent ligands and new luminescence-based strategies in recent years. Considering the amount of literature devoted to the design and properties of quadruplex fluorescent probes one can easily appreciate the promising future for this class of agents.
A large number of quadruplex-selective probes are now available and many molecular designs have been explored. The choice of a particular probe design will be guided by the specific application of the study and instrumentation thereof, which in turn will determine the stringency of the chemical, photophysical and biocompatibility criteria required.
For instance, light-off probes are a priori less suitable for imaging in cells, whereas they may be of great interest for monitoring interactions and evaluating binding strength and selectivity for quadruplexes; they may also find application for various cation sensing in solution. In the case of light-up probes, the degree of signal enhancement for G4 vs duplex should be sufficient to allow this approach to be used in various situations. We have seen that this criterium is not always satisfied, although it is somewhat difficult to evaluate as it is often not clearly addressed or even obscured.
Although some dyes described in this review are only of academic interest, others seem to hold real promise in terms of practical applications. For instance, several scaffolds with exquisite quadruplex selectivity have been clearly identified, being already usable in various in vitro labeling experiments. Although in certain cases improvements are still needed in terms of solubility and self-aggregating aspects, we are confident that optimized second generations will appear in the near future.
We have seen that a number of probes active in vitro do not penetrate in living cells and thus have not been studied further although application in fixed cells could have been envisaged. A limited number have been used for imaging in cells or at the level of chromosomes. For those that are internalized in cells, most have been found in the nucleus and some more specifically sequestered in nucleoli. For the probes having a low in vitro affinity for duplex DNA, this might be associated with binding to quadruplex DNA or RNA structures. However it should be overstressed that whether the bound structures correspond to quadruplexes or to the many existing complex 3D RNA topologies remains to be determined.
Finally, particular attention should be paid to probes displaying a quadruplex-specific variation of their photophysical properties such as variable emission wavelength or distinct fluorescence lifetime. These agents can be useful tools for mapping quadruplex-containing domains, especially in view of the rapid developments of ultrasensitive imaging instrumentation. In this regard it is worth mentioning that the performances of probes in term of brightness, photostability, and lifetime, represent currently the limit of super resolution and single molecule imaging, thereby opening a bright future for fluorescence engineering of quadruplex-responsive small molecules.
We have deliberately focused on reports addressing the potentiality of quadruplex-interactive small molecules for biological applications and have limited those concerning technological and nanomaterials applications. In addition, although fluorescently labeled oligonucleotides represent major players in the field and are widely used tools in cell biology, they are outside the scope of this review. Finally, every attempt has been made to ensure the comprehensiveness of this review but, due to the overwhelming number of publications in the field, omissions may have been made. The authors would like to apologize in advance for any incompleteness which might, therefore, result.
To conclude, the proposals mentioned here by no means represent the limits of the possible, but merely the most obvious next directions for the future development of the field.
Notes
- 1.
Experiments performed in lithium cacodylate buffer, at pH 7.2 with 100 mM KCl or NaCl.
- 2.
Experiments performed at pH 7.2 with 1 mM KCl.
- 3.
Experiments performed in 10 mM Tris–HCl buffer with 60 mM KCl.
- 4.
Experiments performed in ultra-pure water with 50 mM KCl.
- 5.
Experiments performed in 10 mM Tris buffer with 150 mM NaCl.
- 6.
Experiments performed in 2.5 mM phosphate buffer, 5 mM Na+, and 100 mM K+.
- 7.
Experiments performed in 10 mM PBS K+ or Na+.
- 8.
Experiments performed with 17.2 mM K+ and no Na+.
- 9.
Experiments performed with 140 mM K+, 10 mM Na+, and 2 mM Mg2+.
- 10.
Experiments performed in phosphate buffer pH 7.4 with 100 mM K+.
- 11.
Experiments performed in 10 mM cacodylate buffer pH 7.3 with 100 mM K+.
- 12.
[CV] fixed at 15 μM in 25 mM Tris–HCl (pH = 7.0), the DNA concentrations (strand concentration) at intersection points on the titration curves is 8 ± 1 μM for Hum21.
- 13.
All solutions contain 10 mM Tris–HCl, 1 mM Na2EDTA, and 100 mM NaCl at pH 7.5.
- 14.
[TTAPE] = 4.5 μM, [K+] = 0.15 M in 5 mM Tris–HCl buffer; λ ex = 350 nm.
- 15.
μM 6 in 20 mM pH 7.4 Tris buffer, containing 2.5 μM quadruplex in absence or in the presence of 100 mM KCl.
- 16.
Measured by CD spectra in 10 mM Tris–HCl buffer (pH 7.4) without metal ions.
- 17.
ΔT 1/2 values of 0.2 μM F21T (21-mer human telomeric sequence Fluo-GGG(TTAGGG)3]-TAMRA) incubated in the presence of KCl 60 mM, compound 2 2.0 μM.
- 18.
mM Tris–HCl buffer (pH 7.4) and 100 mM KCl.
- 19.
[ligand] 2 μM in Tris–HCl buffer (10 mM, pH 7.2) with 100 mM KCl.
- 20.
Buffer conditions: 10 mM sodium cacodylate, 100 mM KCl, pH 7.5. Excitation at 274 nm.
- 21.
Experiments performed in 10 mM Tris HCl buffer pH 7.5 with 100 mM K+.
- 22.
Experiments performed at pH 5.5.
- 23.
Experiments performed at pH 7.0, in 10 mM potassium phosphate buffer, with 100 mM KCl and 1 mM K2EDTA.
- 24.
Experiments performed at pH 7.0, in 5 mM Tris buffer, 25 mM NaCl.
- 25.
Experiments performed at pH 7.0 in 10 mM potassium phosphate buffer with 200 mM KCl and 1 mM K2EDTA.
- 26.
Experiments performed in 100 mM NaCl, 10 mM NaH2PO4/Na2HPO4, 1 mM Na2EDTA, pH 7.0.
- 27.
Experiments performed in 10 mM Tris–HCl buffer, pH 7.4, with 100 mM KCl.
- 28.
Experiments performed in 50 mM Tris–HCl, pH 7.4, with 150 mM KCl and 0.5 mM EDTA.
- 29.
Experiments performed in 10 mM Tris–HCl buffer, pH 7.0, without other cations.
- 30.
Experiments performed in 50 mM Tris-HCl buffer, pH 7.1, with 100 mM NaCl.
- 31.
In 10 mM HEPES, 200 mM KCl, 150 mM NaCl, pH 7.4.
- 32.
In 20 mM potassium phosphate, 70 mM KCl, 0.1 mM EDTA, pH 7.0.
- 33.
In 10 mM LiAsMe2O2, 100 mM NaCl, pH 7.2.
- 34.
In 25 mM K2HPO4/KH2PO4, 70 mM KCl, pH 7.
- 35.
In 6 mM Na2HPO4, 2 mM NaH2PO4, 185 mM NaCl, 0.1 mM EDTA, pH 7.0.
- 36.
′-TG3GA G3 TG4A G3 TG4A 2 G2-3′ (c-MYC oncogene).
- 37.
′-A2TC2GTCGAGCAGAGT2(AG3T2)2 AG-3′ (human telomeric repeat).
- 38.
In 10 mM Tris, 100 mM KCl, 1 mM EDTA.
- 39.
In 10 mM Tris, 1 mM EDTA, 50 mM KCl, pH 7.4.
- 40.
In 10 mM K2HPO4/KH2PO4, 1 mM EDTA, pH 7.04.
Abbreviations
- AIE:
-
Aggregation-induced emission
- CD:
-
Circular dichroism
- ct DNA:
-
Calf thymus DNA
- ds DNA:
-
Double-stranded DNA
- ESIPT:
-
Excited-state intramolecular proton transfer
- FID:
-
Fluorescent intercalator displacement
- FRET:
-
Förster resonance energy transfer
- G4-DNA:
-
Quadruplex DNA
- ITC:
-
Isothermal titration calorimetry
- PAGE:
-
Polyacrylamide gel electrophoresis
- PNA:
-
Peptide nucleic acid
- ss DNA:
-
Single-stranded DNA
- SPR:
-
Surface plasmon resonance
- TBA:
-
Thrombin-binding aptamer
- TO:
-
Thiazole orange
- TPM:
-
Triphenylmethane
References
Bacolla A, Wells RD (2009) Mol Carcinog 48:273
Cahoon LA, Seifert HS (2009) Science 325:764
Maizels N (2006) Nat Struct Mol Biol 13:1055
Paeschke K, Capra JA, Zakian VA (2011) Cell 145:678
Lopes J, Piazza A, Bermejo R, Kriegsman B, Colosio A, Teulade-Fichou M-P, Foiani M, Nicolas A (2011) EMBO J 30:4033
Broxson C, Beckett J, Tornaletti S (2011) Biochemistry 50:4162
Beaudoin J-D, Perreault J-P (2010) Nucleic Acids Res 38:7022
Davis JT (2004) Angew Chem Int Ed 43:668
Collie GW, Parkinson GN (2011) Chem Soc Rev 40:5867
Phan AT (2010) FEBS J 277:1107
Neidle S, Balasubramanian S (2006) RSC biomolecular sciences. The Royal Society of Chemistry, Cambridge, UK, p 316
Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH (1997) J Med Chem 40:2113
Monchaud D, Teulade-Fichou M-P (2008) Org Biomol Chem 6:627
Georgiades SN, Abd Karim NH, Suntharalingam K, Vilar R (2010) Angew Chem Int Ed 49:4020
Balasubramanian S, Neidle S (2009) Curr Opin Chem Biol 13:345
Lipps HJ, Rhodes D (2009) Trends Cell Biol 19:414
Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N (2004) Genes Dev 18:1618
Neaves KJ, Huppert JL, Henderson RM, Edwardson JM (2009) Nucleic Acids Res 37:6269
Mela I, Kranaster R, Henderson RM, Balasubramanian S, Edwardson JM (2011) Biochemistry 51:578
Xu Y, Ishizuka T, Kurabayashi K, Komiyama M (2009) Angew Chem Int Ed 48:7833
Schaffitzel C, Berger I, Postberg J, Hanes J, Lipps HJ, Plückthun A (2001) Proc Natl Acad Sci USA 98:8572
Fernandez-Suarez M, Ting AY (2008) Nat Rev Mol Cell Biol 9:929
Kobayashi H, Ogawa M, Alford R, Choyke PL, Urano Y (2009) Chem Rev 110:2620
Neidle S (2009) Curr Opin Struct Biol 19:239
Patel DJ, Phan AT, Kuryavyi V (2007) Nucleic Acids Res 35:7429
Yang D, Okamoto K (2010) Future Med Chem 2:619
Collie GW, Sparapani S, Parkinson GN, Neidle S (2011) J Am Chem Soc 133:2721
Campbell NH, Parkinson GN, Reszka AP, Neidle S (2008) J Am Chem Soc 130:6722
Cosconati S, Marinelli L, Trotta R, Virno A, Mayol L, Novellino E, Olson AJ, Randazzo A (2009) J Am Chem Soc 131:16336
White EW, Tanious F, Ismail MA, Reszka AP, Neidle S, Boykin DW, Wilson WD (2007) Biophys Chem 126:140
Hamon F, Largy E, Guédin A, Rouchon-Dagois M, Sidibe A, Monchaud D, Mergny J-L, Riou J-F, Nguyen C-H, Teulade-Fichou M-P (2011) Angew Chem Int Ed 50:8745
Lusvarghi S, Murphy CT, Roy S, Tanious FA, Sacui I, Wilson WD, Ly DH, Armitage BA (2009) J Am Chem Soc 131:18415
Armitage BA (2005) In: Waring MJ, Chaires JB (eds) DNA binders and related subjects. Topics in Current Chemistry, vol 253. Springer, Berlin/Heidelberg, p 55
Sovenyhazy KM, Bordelon JA, Petty JT (2003) Nucleic Acids Res 31:2561
Nygren J, Svanvik N, Kubista M (1998) Biopolymers 46:39
Karunakaran V, Pérez Lustres JL, Zhao L, Ernsting NP, Seitz O (2006) J Am Chem Soc 128:2954
Lee LG, Chen C-H, Chiu LA (1986) Cytometry 7:508
Rye HS, Quesada MA, Peck K, Mathies RA, GIazer AN (1991) Nucleic Acids Res 19:327
Zhu H, Clark SM, Benson SC, Rye HS, Glazer AN, Mathies RA (1994) Anal Chem 66:1941
Netzel TL, Nafisi K, Zhao M, Lenhard JR, Johnson I (1995) J Phys Chem 99:17936
Monchaud D, Allain C, Teulade-Fichou M-P (2006) Bioorg Med Chem Lett 16:4842
Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP (2001) J Am Chem Soc 123:5878
Boger DL, Tse WC (2001) Bioorg Med Chem 9:2511
Largy E, Hamon F, Teulade-Fichou M-P (2011) Anal Bioanal Chem 400:3419
Monchaud D, Allain C, Teulade-Fichou MP (2007) Nucleosides Nucleotides Nucleic Acids 26:1585
Monchaud D, Allain C, Bertrand H, Smargiasso N, Rosu F, Gabelica V, De Cian A, Mergny JL, Teulade-Fichou MP (2008) Biochimie 90:1207
Halder K, Largy E, Benzler M, Teulade-Fichou M-P, Hartig JS (2011) ChemBioChem 12:1663
Largy E, Saettel N, Hamon F, Dubruille S, Teulade-Fichou M-P (2012) Curr Pharm Des 18:1992
Lubitz I, Zikich D, Kotlyar A (2010) Biochemistry 49:3567
Allain C, Monchaud D, Teulade-Fichou M-P (2006) J Am Chem Soc 128:11890
Nakayama S, Kelsey I, Wang J, Roelofs K, Stefane B, Luo Y, Lee VT, Sintim HO (2011) J Am Chem Soc 133:4856
Haugland RP, The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies (2002), 9th edn. Molecular Probes, Eugene, OR (USA)
Yang P, De Cian A, Teulade-Fichou MP, Mergny JL, Monchaud D (2009) Angew Chem Int Ed 48:2188
Lemarteleur T, Gomez D, Paterski R, Mandine E, Mailliet P, Riou JF (2004) Biochem Biophys Res Commun 323:802
Granotier C, Pennarun G, Riou L, Hoffschir F, Gauthier LR, De Cian A, Gomez D, Mandine E, Riou J-F, Mergny J-L, Mailliet P, Dutrillaux B, Boussin FD (2005) Nucleic Acids Res 33:4182
Lu Y-J, Ou T-M, Tan J-H, Hou J-Q, Shao W-Y, Peng D, Sun N, Wang X-D, Wu W-B, Bu X-Z, Huang Z-S, Ma D-L, Wong K-Y, Gu L-Q (2008) J Med Chem 51:6381
Lu Y-J, Yan S-C, Chan F-Y, Zou L, Chung W-H, Wong W-L, Qiu B, Sun N, Chan P-H, Huang Z-S, Gu L-Q, Wong K-Y (2011) Chem Commun 47:4971
Zipper H, Brunner H, Bernhagen J, Vitzthum F (2004) Nucleic Acids Res 32:e103
Xu H, Gao S, Yang Q, Pan D, Wang L, Fan C (2010) ACS Appl Mater Interfaces 2:3211
Xu Q-H, Wang S, Korystov D, Mikhailovsky A, Bazan GC, Moses D, Heeger AJ (2005) Proc Natl Acad Sci USA 102:530
Haugland RP (1996) Handbook of fluorescent probes and research chemicals. Molecular Probes, Eugene, OR
Kovalska V, Losytskyy M, Yarmoluk S, Lubitz I, Kotlyar A (2011) J Fluoresc 21:223
Yarmoluk S, Lukashova SS, Losytskyya MY, Akermanb B, Kornyushynac OS (2002) Spectrochim Acta A 58:3223
Ohulchanskyy TY, Pudavar HE, Yarmoluk SM, Yashchuk VM, Bergey EJ, Prasad PN (2007) Photochem Photobiol 77:138
Chen Q, Kuntz ID, Shafer RH (1996) Proc Natl Acad Sci USA 93:2635
Paramasivan S, Bolton PH (2008) Nucleic Acids Res 36:e106
Yang Q, Xiang J, Yang S, Zhou Q, Li Q, Tang Y, Xu G (2009) Chem Commun 2009:1103
Yang Q, Xiang J, Yang S, Li Q, Zhou Q, Guan A, Zhang X, Zhang H, Tang Y, Xu G (2010) Nucleic Acids Res 38:1022
Yang Q, Xiang J-F, Yang S, Li Q, Zhou Q, Guan A, Li L, Zhang Y, Zhang X, Zhang H, Tang Y, Xu G (2010) Anal Chem 82:9135
Meguellati K, Koripelly G, Ladame S (2010) Angew Chem Int Ed 49:2738
Koripelly G, Meguellati K, Ladame S (2010) Bioconjug Chem 21:2103
Latt SA, Stetten G (1976) J Histochem Cytochem 24:24
Demchenko AP (2009) In: Demchenko AP (ed) Introduction to fluorescence sensing. Springer, Netherlands, p 407
Maiti S, Chaudhury NK, Chowdhury S (2003) Biochem Biophys Res Commun 310:505
Phan AT, Kuryavyi V, Gaw HY, Patel DJ (2005) Nat Chem Biol 1:167
Jain AK, Reddy VV, Paul A, Muniyappa K, Bhattacharya S (2009) Biochemistry 48:10693
Jain AK, Bhattacharya S (2011) Bioconjug Chem 22:2355
Lepecq JB, Paoletti C (1967) J Mol Biol 27:87
Olmsted J, Kearns DR (1977) Biochemistry 16:3647
Ren J, Chaires JB (1999) Biochemistry 38:16067
Guo Q, Lu M, Marky LA, Kallenbach NR (1992) Biochemistry 31:2451
Koeppel F, Riou J-F, Laoui A, Mailliet P, Arimondo PB, Labit D, Petitgenet O, Hélène C, Mergny J-L (2001) Nucleic Acids Res 29:1087
Sun X, Cao E, He Y, Qin J (1999) Sci China Ser B Chem 42:62
Rosu F, De Pauw E, Guittat L, Alberti P, Lacroix L, Mailliet P, Riou J-F, Mergny J-L (2003) Biochemistry 42:10361
Martin MM, Plaza P, Meyer YH (1991) Chem Phys 153:297
Jurczok M, Plaza P, Martin MM, Rettig W (1999) J Phys Chem A 103:3372
Babendure JR, Adams SR, Tsien RY (2003) J Am Chem Soc 125:14716
Bhasikuttan AC, Mohanty J, Pal H (2007) Angew Chem Int Ed 46:9305
Mergny JL, Li J, Lacroix L, Amrane S, Chaires JB (2005) Nucleic Acids Res 33:e138
Kong DM, Ma YE, Wu J, Shen HX (2009) Chem Eur J 15:901
Guo J-H, Zhu L-N, Kong D-M, Shen H-X (2009) Talanta 80:607
Kong D-M, Ma Y-E, Guo J-H, Yang W, Shen H-X (2009) Anal Chem 81:2678
Kong D-M, Guo J-H, Yang W, Ma Y-E, Shen H-X (2009) Biosens Bioelectron 25:88
Ma DL, Kwan MHT, Chan DSH, Lee P, Yang H, Ma VPY, Bai LP, Jiang ZH, Leung CH (2011) Analyst 136:2692
Leung CH, Chan DS, Man BY, Wang CJ, Lam W, Cheng YC, Fong WF, Hsiao WL, Ma DL (2011) Anal Chem 83:463
Baptista MS, Indig GL (1998) J Phys Chem B 102:4678
Chang C-C, Wu JY, Chang TC (2003) J Chin Chem Soc 50:185
Chang C-C, Wu J-Y, Chien C-W, Wu W-S, Liu H, Kang C-C, Yu L-J, Chang T-C (2003) Anal Chem 75:6177
Chang C-C, Chien C-W, Lin Y-H, Kang C-C, Chang T-C (2007) Nucleic Acids Res 35:2846
Chang C-C, Kuo IC, Ling IF, Chen C-T, Chen H-C, Lou P-J, Lin J-J, Chang T-C (2004) Anal Chem 76:4490
Chang C-C, Chu J-F, Kao F-J, Chiu Y-C, Lou P-J, Chen H-C, Chang T-C (2006) Anal Chem 78:2810
Zhang X-F, Zhang H-J, Xiang J-F, Li Q, Yang Q-f, Shang Q, Zhang Y-X, Tang Y-L (2010) J Mol Struct 982:133
Dumat B, Bordeau G, Faurel-Paul E, Mahuteau-Betzer F, Saettel N, Bombled M, Metgé G, Charra F, Fiorini-Debuisschert C, Teulade-Fichou MP (2011) Biochimie 93:1209
Han FX, Wheelhouse RT, Hurley LH (1999) J Am Chem Soc 121:3561
Wei C, Jia G, Yuan J, Feng Z, Li C (2006) Biochemistry 45:6681
Zhang H-J, Wang X-F, Wang P, Ai X-C, Zhang J-P (2008) Photochem Photobiol Sci 7:948
Anantha NV, Azam M, Sheardy RD (1998) Biochemistry 37:2709
Zhang HJ, Wang XF, Wang P, Pang SP, Ai XC, Zhang JP (2008) Sci China Ser B Chem 51:452
Wei C, Han G, Jia G, Zhou J, Li C (2008) Biophys Chem 137:19
Wei C, Wang L, Jia G, Zhou J, Han G, Li C (2009) Biophys Chem 143:79
Wei C, Jia G, Zhou J, Han G, Li C (2009) Phys Chem Chem Phys 11:4025
Zhao P, Xu L-C, Huang J-W, Fu B, Yu H-C, Ji L-N (2009) Dyes Pigm 83:81
Nicoludis JM, Barrett SP, Mergny J-L, Yatsunyk LA (2012) Nucleic Acids Res 10.1093/nar/gks152
Arthanari H, Basu S, Kawano TL, Bolton PH (1998) Nucleic Acids Res 26:3724
Li T, Wang E, Dong S (2010) Anal Chem 82:7576
Li T, Dong S, Wang E (2010) J Am Chem Soc 132:13156
Hong Y, Lam JWY, Tang BZ (2011) Chem Soc Rev 40:5361
Hong Y, Häuβler M, Lam JWY, Li Z, Sin KK, Dong Y, Tong H, Liu J, Qin A, Renneberg R, Tang BZ (2008) Chem Eur J 14:6428
Hong Y, Xiong H, Lam JW, Haussler M, Liu J, Yu Y, Zhong Y, Sung HH, Williams ID, Wong KS, Tang BZ (2010) Chem Eur J 16:1232
Luo J, Xie Z, Lam JWY, Cheng L, Chen H, Qiu C, Kwok HS, Zhan X, Liu Y, Zhu D, Tang BZ (2001) Chem Commun 1740
Wang M, Zhang D, Zhang G, Tang Y, Wang S, Zhu D (2008) Anal Chem 80:6443
Huang J, Wang M, Zhou Y, Weng X, Shuai L, Zhou X, Zhang D (2009) Bioorg Med Chem 17:7743
Sanders MM (1998) Biochem Pharmacol 56:1157
Diaz MS, Freile ML, Gutierrez MI (2009) Photochem Photobiol Sci 8:970
Arora A, Balasubramanian C, Kumar N, Agrawal S, Ojha RP, Maiti S (2008) FEBS J 275:3971
Bhadra K, Kumar GS (2011) Biochim Biophys Acta 1810:485
Liu Y, Li B, Cheng D, Duan X (2011) Microchem J 99:503
Zhang W-J, Ou T-M, Lu Y-J, Huang Y-Y, Wu W-B, Huang Z-S, Zhou J-L, Wong K-Y, Gu L-Q (2007) Bioorg Med Chem 15:5493
Ma Y, Ou TM, Tan JH, Hou JQ, Huang SL, Gu LQ, Huang ZS (2009) Bioorg Med Chem Lett 19:3414
Franceschin M, Rossetti L, D’Ambrosio A, Schirripa S, Bianco A, Ortaggi G, Savino M, Schultes C, Neidle S (2006) Bioorg Med Chem Lett 16:1707
Tan J-H, Ou T-M, Hou J-Q, Lu Y-J, Huang S-L, Luo H-B, Wu J-Y, Huang Z-S, Wong K-Y, Gu L-Q (2009) J Med Chem 52:2825
Sun H, Tang Y, Xiang J, Xu G, Zhang Y, Zhang H, Xu L (2006) Bioorg Med Chem Lett 16:3586
Sun H, Xiang J, Tang Y, Xu G (2007) Biochem Biophys Res Commun 352:942
Juskowiak B, Galezowska E, Koczorowska N, Hermann TW (2004) Bioorg Med Chem Lett 14:3627
Alberti P, Schmitt P, Nguyen C-H, Rivalle C, Hoarau MS, Grierson D, Mergny J-L (2002) Bioorg Med Chem Lett 12:1071
Erkkila KE, Odom DT, Barton JK (1999) Chem Rev 99:2777
Keene FR, Smith JA, Collins JG (2009) Coord Chem Rev 253:2021
Kieltyka R, Englebienne P, Moitessier N, Sleiman H (2010) Methods Mol Biol (Clifton, NJ) 608:223
Jiang YL, Liu ZP (2010) Mini Rev Med Chem 10:726
Ma D-L, Che C-M, Yan S-C (2009) J Am Chem Soc 131:1835
Wu P, Ma D-L, Leung C-H, Yan S-C, Zhu N, Abagyan R, Che C-M (2009) Chem Eur J 15:13008
Yu C, Chan KH-Y, Wong KM-C, Yam VW-W (2009) Chem Commun 3756
Michael JC (2002) Coord Chem Rev 232:69
Jenkins Y, Friedman AE, Turro NJ, Barton JK (1992) Biochemistry 31:10809
Friedman AE, Chambron JC, Sauvage JP, Turro NJ, Barton JK (1990) J Am Chem Soc 112:4960
Shi S, Geng X, Zhao J, Yao T, Wang C, Yang D, Zheng L, Ji L (2010) Biochimie 92:370
Shi S, Zhao J, Geng X, Yao T, Huang H, Liu T, Zheng L, Li Z, Yang D, Ji L (2010) Dalton Trans 39:2490
Sun J, An Y, Zhang L, Chen HY, Han Y, Wang YJ, Mao ZW, Ji LN (2011) J Inorg Biochem 105:149
Rajput C, Rutkaite R, Swanson L, Haq I, Thomas JA (2006) Chem Eur J 12:4611
Pourtois G, Beljonne D, Moucheron C, Schumm S, Kirsch-De Mesmaeker A, Lazzaroni R, Brédas J-L (2003) J Am Chem Soc 126:683
Gill MR, Garcia-Lara J, Foster SJ, Smythe C, Battaglia G, Thomas JA (2009) Nat Chem 1:662
Shi S, Liu J, Yao T, Geng X, Jiang L, Yang Q, Cheng L, Ji L (2008) Inorg Chem 47:2910
Xu L, Zhang D, Huang J, Deng M, Zhang M, Zhou X (2010) Chem Commun 46:743
Goncalves DPN, Rodriguez R, Balasubramanian S, Sanders JKM (2006) Chem Commun 2006:4685
Ren L, Zhang A, Huang J, Wang P, Weng X, Zhang L, Liang F, Tan Z, Zhou X (2007) ChemBioChem 8:775
Alzeer J, Vummidi BR, Roth PJC, Luedtke NW (2009) Angew Chem Int Ed 48:9362
Qin H, Ren J, Wang J, Luedtke NW, Wang E (2010) Anal Chem 82:8356
Ren J, Qin H, Wang J, Luedtke N, Wang E, Wang J (2011) Anal Bioanal Chem 399:2763
Alzeer J, Luedtke NW (2010) Biochemistry 49:4339
Cha A, Snyder GE, Selvin PR, Bezanilla F (1999) Nature 402:809
Caravan P, Ellison JJ, McMurry TJ, Lauffer RB (1999) Chem Rev 99:2293
Bünzli JC (2010) Chem Rev 110:2729
Fu PKL, Turro C (1999) J Am Chem Soc 121:1
Zhang H, Yu H, Ren J, Qu X (2006) Biophys J 90:3203
Lis S, Elbanowski M, Mąkowska B, Hnatejko Z (2002) J Photochem Photobiol, A 150:233
Lehn J-M (1990) Angew Chem Int Ed 29:1304
Galezowska E, Gluszynska A, Juskowiak B (2007) J Inorg Biochem 101:678
Worlinsky JL, Basu S (2009) J Phys Chem B 113:865
Xu H, Zhang H, Qu X (2006) J Inorg Biochem 100:1646
Knobloch B, Linert W, Sigel H (2005) Proc Natl Acad Sci USA 102:7459
Lee J, Park M, Son HS, Lee SB, Lee HC, Ku JK, Park JW (2002) Biopolymers 67:413
Xie H, Yang D, Heller A, Gao Z (2007) Biophys J 92:L70
Delaney S, Barton JK (2003) Biochemistry 42:14159
Teulade-Fichou M-P, Carrasco C, Guittat L, Bailly C, Alberti P, Mergny J-L, David A, Lehn J-M, Wilson WD (2003) J Am Chem Soc 125:4732
Mergny JL, Lacroix L, Teulade-Fichou MP, Hounsou C, Guittat L, Hoarau M, Arimondo PB, Vigneron JP, Lehn JM, Riou JF, Garestier T, Helene C (2001) Proc Natl Acad Sci USA 98:3062
Peduto A, Pagano B, Petronzi C, Massa A, Esposito V, Virgilio A, Paduano F, Trapasso F, Fiorito F, Florio S, Giancola C, Galeone A, Filosa R (2011) Bioorg Med Chem 19:6419
Cummaro A, Fotticchia I, Franceschin M, Giancola C, Petraccone L (2011) Biochimie 93:1392
Franceschin M, Ginnari-Satriani L, Alvino A, Ortaggi G, Bianco A (2010) Eur J Org Chem 2010:134
Petraccone L, Fotticchia I, Cummaro A, Pagano B, Ginnari-Satriani L, Haider S, Randazzo A, Novellino E, Neidle S, Giancola C (2011) Biochimie 93:1318
Xu Y, Yamazaki S, Osuga H, Sugiyama H (2006) Nucleic Acids Symp Ser 50:183
Shinohara K-I, Sannohe Y, Kaieda S, Tanaka K-I, Osuga H, Tahara H, Xu Y, Kawase T, Bando T, Sugiyama H (2010) J Am Chem Soc 132:3778
Çetinkol ÖP, Engelhart AE, Nanjunda RK, Wilson WD, Hud NV (2008) ChemBioChem 9:1889
Xu W, Tan JH, Chen SB, Hou JQ, Li D, Huang ZS, Gu LQ (2011) Biochem Biophys Res Commun 406:454
Granzhan A, Ihmels H, Jager K (2009) Chem Commun 2009:1249
Cheng MK, Modi C, Cookson JC, Hutchinson I, Heald RA, McCarroll AJ, Missailidis S, Tanious F, Wilson WD, Mergny JL, Laughton CA, Stevens MF (2008) J Med Chem 51:963
Kerwin SM, Chen G, Kern JT, Thomas PW (2002) Bioorg Med Chem Lett 12:447
Samudrala R, Zhang X, Wadkins RM, Mattern DL (2007) Bioorg Med Chem 15:186
Kern JT, Kerwin SM (2002) Bioorg Med Chem Lett 12:3395
Mazzitelli C, Brodbelt J, Kern J, Rodriguez M, Kerwin S (2006) J Am Soc Mass Spectrom 17:593
Manet I, Manoli F, Zambelli B, Andreano G, Masi A, Cellai L, Monti S (2011) Phys Chem Chem Phys 13:540
Manet I, Manoli F, Zambelli B, Andreano G, Masi A, Cellai L, Ottani S, Marconi G, Monti S (2011) Photochem Photobiol Sci 10:1326
Sun H, Xiang J, Zhang Y, Xu G, Xu L, Tang Y (2006) Chin Sci Bull 51:1687
Tong C, Hu Z, Wu J (2010) J Fluoresc 20:261
Nakayama S, Kelsey I, Wang J, Sintim HO (2011) Chem Commun 47:4766
Thomes JC, Weill G, Daune M (1969) Biopolymers 8:647
Yan S, Huang R, Zhou Y, Zhang M, Deng M, Wang X, Weng X, Zhou X (2011) Chem Commun 47:1273
Xu L, Liao GL, Chen XA, Zhao CY, Chao H, Ji LN (2010) Inorg Chem Commun 13:1050
Rickling S, Ghisdavu L, Pierard F, Gerbaux P, Surin M, Murat P, Defrancq E, Moucheron C, Kirsch-De Mesmaeker A (2010) Chem Eur J 16:3951
Tera M, Iida K, Ikebukuro K, Seimiya H, Shin-Ya K, Nagasawa K (2010) Org Biomol Chem 8:2749
Largy E, Hamon F, Teulade-Fichou M-P Methods, in press (DOI: 10.1016/j.ymeth.2012.02.008)
Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP (2012) Nat Chem Biol 8:301
Jao CY, Salic A (2008) Proc Natl Acad Sci USA 105:15779
Salic A, Mitchison TJ (2008) Proc Natl Acad Sci USA 105:2415
Rodriguez R, Müller S, Yeoman JA, Trentesaux C, Riou J-F, Balasubramanian S (2008) J Am Chem Soc 130:15758
Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, Lim JKC, Von Hoff D, Anderes K, Rice WG (2009) Cancer Res 69:7653
Wilson T, Williamson MP, Thomas JA (2010) Organic & Biomolecular Chemistry 8:2617
Acknowledgments
The authors acknowledge the Centre National de la Recherche Scientifique (CNRS), the Institut Curie, and the Fondation Pierre Gilles de Gennes for PhD and post-doctoral fellowships (E.L. and D.V.) We also thank the Agence Nationale de la Recherche (ANR-09-BLAN-0355 “G4 Toolbox”) for funding.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Largy, E., Granzhan, A., Hamon, F., Verga, D., Teulade-Fichou, MP. (2012). Visualizing the Quadruplex: From Fluorescent Ligands to Light-Up Probes. In: Chaires, J., Graves, D. (eds) Quadruplex Nucleic Acids. Topics in Current Chemistry, vol 330. Springer, Berlin, Heidelberg. https://doi.org/10.1007/128_2012_346
Download citation
DOI: https://doi.org/10.1007/128_2012_346
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-34742-9
Online ISBN: 978-3-642-34743-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)