
Abstract
Acute toxic lung injury by reactive inhalational compounds is an important and still unresolved medical problem. Hazardous gases or vapors, e. g. chlorine, phosgene, sulfur mustard or methyl isocyanate, are released during occupational accidents or combustion processes and also represent a potential threat in terroristic scenarios. According to their broad-range chemical reactivity, the mechanism of lung injury evoked by these agents has long been described as rather unspecific. Consequently, therapeutic options are still restricted to symptomatic treatment. However, in recent years, ion channels of the transient receptor potential (TRP) family have been identified to act as specific sensor molecules expressed in the respiratory tract and to engage defined signaling pathways upon inhalational exposure to toxic challenges. These pulmonary receptor molecules have been primarily characterized in sensory neurons of the lung. However, chemosensory molecules are also expressed in non-neuronal cells, e.g. in the lung epithelium as well as in the pulmonary vasculature. Thus, activation of respiratory chemosensors by toxic inhalants promotes a complex signaling network directly or indirectly regulating pulmonary blood flow, the integrity of the epithelial lining, and the mucociliary clearance of the bronchial system. This review gives a synopsis on reactive lung-toxic agents and their specific target molecules in the lung and summarizes the current knowledge about the pathophysiological role of chemosensory signaling in neuronal and non-neuronal cells in toxic lung injury. Finally, we describe possible future strategies for a causal, specifically tailored treatment option based on the mechanistic understanding of molecular events ensuing inhalation of lung-toxic agents.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Transient Receptor Potential
- Transient Receptor Potential Channel
- Mucociliary Clearance
- Airway Smooth Muscle Cell
- Sulfur Mustard
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The respiratory system represents a delicate interface of our body with the environment providing a large surface area estimated to amount to about 100 m2 depending on the method of determination (Colebatch and Ng 1992; Stone et al. 1992; Thurlbeck 1967). Since prehistoric times the respiratory tract of vertebrates is challenged with noxious inhalational substances, e.g. dust, fumes of wildfires, or ozone of natural origin. Thus, a multitude of defense and repair mechanisms have evolved protecting the respiratory system from harmful injury (Coleridge and Coleridge 1994; Comhair and Erzurum 2002). With the start of the industrial era a vast number of novel compounds occurred, representing potential toxic inhalation hazards (TIHs). Electrophilic or oxidizing substances like chlorine, phosgene, methyl isocyanate, nitrous gases, or nitrogen or sulfur mustards constitute one of the biggest groups of toxic inhalants (for review: Bessac and Jordt 2010).
There is a significant risk of exposure to TIHs, as most of these substances are produced by the chemical industry, either as main products, intermediates, or industrial pollutants. A famous sad example was the Bophal disaster: On December 3, 1984, more than 40 t of methyl isocyanate gas leaked from a pesticide plant in Bhopal, India, immediately killing at least 3,800 people and causing significant morbidity and premature death for many thousands more (Broughton 2005). Methyl isocyanate is an intermediate chemical in the production of carbamate pesticides. Other examples for omnipresent TIHs are chlorine and phosgene, two substances that are essential for the production of a wide range of industrial and consumer products. Additionally, chlorine is a common household chemical and a component of chlorine-based bleach. Bleach is regarded as one of the strongest airway irritants and has been discussed to play a causal role in the development of airway sensory hyper-reactivity (Quirce and Barranco 2010). Approximately 15 million tons of chlorine are produced annually in the United States alone to be used for water purification, pharmaceutical and disinfectant purposes (Samal et al. 2010).
Apart from the commercial and household use of potential lung damaging chemicals, a threat based on the misuse of lung toxicants cannot be ruled out. As most of the TIHs are widely accessible or are easy to synthesize, they can be considered as a terroristic threat. The use of chemicals, in first line TIHs, as warfare agents can be dated to World War I. On the 22nd of April 1915 German troops deployed chlorine at Ypres which is considered the starting point of chemical warfare in history. Thousands of tons of chemicals including chlorine, phosgene and sulfur mustard have been used by the war parties causing millions of casualties, specially due to lung injury (Kehe and Szinicz 2005). During World War II, large amounts of chemicals (e.g. nerve agents, blistering and lung agents) were produced and stockpiled. Blistering and lung agents were used especially by the Japanese Imperial Forces against Chinese troops and civilians and the remnants may still represent a health hazard (Hanaoka et al. 2006). In the European theater of war, no major operations with chemical weapons were carried out. So, most European casualties of chemical weapons during World War II resulted from accidental liberation of these agents, the most severe incident being the release of sulfur mustard from US freighter John Harvey after a bombing raid by the German air force (Alexander 1947). Of note, many thousands of tons of chemicals have been dumped in the Baltic Sea after World War II and still represent a potential environmental and health risk (HELCOM 1994; Sanderson et al. 2008; Sanderson et al. 2010).
Upon exposure to harmful inhalants, ancestral protective mechanisms mediate danger signals (burning sensations, lacrimation, blepharospasm, cough), induce detoxifying and antioxidant molecules (e.g. glutathione, urate, or metallothioneins), and activate self-cleaning mechanisms in the respiratory tract (cough, mucus production, elevated ciliary motility). These airway responses to TIHs are quite unspecific and uniform, in line with the wide array of potential inhalational threats, which display rather unrelated chemical structures. Thus, the molecular events involved in the recognition of harmful inhalational substances have been assumed to be equally unspecific. However, in recent years it has become clear that distinct receptor molecules expressed on the cell membrane of respiratory cells are involved in the detection of TIHs. Especially, taste and olfactory receptors belonging to the superfamily of G protein coupled receptors and receptor-operated or chemosensory ion channels of the transient receptor potential (TRP) family have been identified in all parts of the airway system and found to play a pivotal role as defined target molecules of TIHs.
Similarly, the recent years have also seen a paradigm shift regarding the proposed mechanisms of acute lung injury evoked by reactive TIHs. Hitherto, the toxic action of TIHs has been mainly attributed to unspecific cell damage caused by reactions of the TIHs with biomolecules. In particular, oxidation of membrane lipids and alteration of DNA bases are scenarios which have been suggested to mediate the toxicity of TIHs. In line with this point of view, therapeutic interventions adopted so far are mainly symptomatic, i.e. application of steroids and β-adrenergic agonists as anti-inflammatory and broncho-spasmolytic agents, respectively, or administration of anti-oxidant molecules like e.g. N-acetyl cysteine (Bobb et al. 2005; de Lange and Meulenbelt 2011; Ghanei et al. 2008; Grainge and Rice 2010; Weinberger et al. 2011). Recently, the identification of molecular down-stream effectors of redox signaling in lung cells was a major step forward in the mechanistic understanding of the action of TIHs and possible pulmonary defense systems and will pave the way for more causal and specific therapeutic options. Especially, redox-regulated transcription factors like AP-1, Nrf2, and NF-κB have been identified as critical players activated upon exposure of airway cells to reactive inhalants and to control cell survival and inflammatory responses. Redox-regulated transcription can be orchestrated in a very complex manner by oxidation of cystein-residues. In the case of AP-1 and NF-κB signaling, oxidative stress in the cytosol can stimulate protein kinase-dependent cascades which ultimately lead to the activation of the transcription factors (Brigelius-Flohe and Flohe 2011). Furthermore, transcription factors like NF-κB and Nrf2 can be disinhibited via oxidative modification of inhibiting proteins directing the transcription factor towards ubiquitin-dependent proteasomal degradation under normoxic conditions (Muller and Hengstermann 2012). In contrast, oxidative stress in the nucleus can inhibit DNA binding of transcription factors, again via modification of critical cystein residues (Brigelius-Flohe and Flohe 2011).
Thus, substantial progress has been made in characterizing these events in the cytosol or in the nucleus directly controlling the redox-sensitive transcriptional regulation of target genes involved in the toxic effects of TIHs. The aim of this review is to point out the interconnections between membrane-signaling pathways engaged by defined chemosensory ion channels of the TRP family and the well-known down-stream effectors. Since different compartments of the respiratory tract are differentially impacted by TIHs, we assign the cellular responses in upper and lower compartments of the airways to the chemosensory structures expressed in these parts and the corresponding signaling pathways involved in the different pathophysiological events. Finally, we describe the perspective of a molecularly targeted therapy in respiratory toxicology based on selective agonists or antagonists of chemosensory molecules.
2 Cellular Events and Toxic Symptoms in Acute and Sub-acute Lung Injury by Reactive Inhalants
Each part of the airway system can be compromised by TIHs resulting in impaired lung function (e.g. toxic inhalation injury). Depending on the chemical properties (e.g. water solubility), chemical compounds affect different compartments of the lung. Particles >5 μm in diameter are filtered by impaction and are trapped in mucus coating the epithelium of the upper airways, thus not reaching deep lung compartments (Bonner 2008; Muhle and McClellan 2003). Particles of lower size, gases and aerosols are able to reach lower lung sections and finally the alveolar space. Deposition of TIHs depends mainly on water solubility (Fig. 1): Hydrophilic substances will dissolve in the mucous lining of the upper airways and will exert their toxic effects proximally (e.g. formaldehyde, ammonia, sulfur dioxide) (Bonner 2008; Muhle and McClellan 2003; Shusterman 1999; Tuorinsky and Sciuto 2008). In contrast, lipophilic substances with low water solubility (e.g. phosgene, ozone, oxides of nitrogen) will predominantly affect the lower respiratory tract, down to the alveolar space (Bonner 2008; Muhle and McClellan 2003; Shusterman 1999; Tuorinsky and Sciuto 2008).

Relationship between water solubility of TIHs and pulmonary target sites. Depending on the water solubility different primary target sites of TIHs in the human respiratory system can be determined. Highly water-soluble substances (e.g. ammonia, formaldehyde, sulfur dioxide) will affect mainly the upper respiratory system (A). Substances of intermediate solubility (e.g. ozone, chlorine) will affect tracheal and bronchial regions (B), whereas water insoluble substances (e.g. phosgene, oxides of nitrogens) will interfere with the lower respiratory system down to the alveolar spaces (C) (Bonner 2008, modified) (Shusterman 1999; Tuorinsky and Sciuto 2008)
Nevertheless, depending on the concentration of the TIH and time of exposure, chemicals can compromise the entire respiratory tract (Miller and Chang 2003; Tuorinsky and Sciuto 2008). As gases or vapors move by convection into the peripheral airways and then by diffusion to the alveolar-capillary membranes, TIHs reaching the alveolar space may have more profound health effects, because clearance due to gas exchange is much slower in this compartment (Urbanetti 1997). Pre-existing diseases of the airway system or damaged lung tissue due to former toxic inhalation injury may already compromise normal physiological lung function. In a mouse model of allergic asthma it was shown that additional inhalation of chemicals can enhance allergic airway inflammation in presensitized mice (Ban et al. 2006). Moreover, obstructive lung diseases (asthma, chronic bronchitis) can further constrain the elimination of TIHs leading to very severe health effects (Urbanetti 1997).
It is important to note that early clinical symptoms of airway irritation or damage do not always correlate with the clinical outcome or long-lasting health effects. Moreover, some TIHs may provoke serious health effects (e.g. pulmonary edema) after a latency period that may last several hours, during which signs and symptoms may not be evident, although cell damage has already occurred. Some chemicals have to undergo intracellular biotransformation processes before causing cell toxicity. It is remarkable that biotransformation is generally considered as a detoxification or defense process, but during this process, toxic metabolites may occur that can enhance lung injury (e.g. transformation of benzo(a)pyrene into reactive metabolites) (Cohen 1990; Smith and Brian 1991). If clinical symptoms arise instantaneously after exposure to certain TIHs, already existing target structures (e.g. cell-surface receptors) are affected and induce a cellular response. The biological effect depends on the activated sensor. In the case of stimulated nerve endings, fast cellular responses are most likely and are a typical example for ion channel activation after a toxic stimulus inducing acute pain.
TIHs with similar chemical properties (e.g. chemical reactivity) are likely to interact with similar cellular targets. The cellular response and the resulting tissue lesions are based upon the molecular function of the affected cellular targets. Thus, TIHs with similar reactivity will provoke comparable or identical clinical symptoms, although their chemical structure may differ. Moreover, if specific cellular targets and the cellular response of certain TIHs are known, the pathophysiology of related substances can be predicted to a certain degree. Numerous studies have been conducted to assess the mode of action of toxic reactive inhalants. Unfortunately, the mode of action of most toxicants is largely unknown and the clinical picture of intoxication is generally nonspecific, which complicates diagnosis and therapy. Therefore, detailed knowledge about the molecular toxicology of lung irritants is required to develop specific therapeutic strategies.
The biological response of the respiratory system after exposure to TIHs is a result of complex interactions of multiple factors, including the intensity of exposure (concentration and time), condition of exposed tissues and also protective and regenerative mechanisms (Urbanetti 1997). Toxic effects can be mediated by direct interaction of the TIHs and respiratory cells leading to cellular dysfunction or cell death. Moreover, chemical substances may activate afferent nerve endings immediately provoking clinical symptoms. Inflammatory mediators, chemokines and interleukins released from affected lung epithelial cells, stimulated macrophages or other cell types will trigger additional biological effects such as cellular infiltration, vasodilation and leakage, pulmonary edema, mucus production, repair and remodeling processes.
3 Clinical Symptoms of TIH Exposure Related to the Nasopharynx
Usually the nasal mucosa, nasopharynx and larynx are exposed to the highest concentrations of toxicants as these structures represent the first line of defense after inhalation. Typical first symptoms will occur due to mucosal irritation like a tickle of the throat and cough which are both biological alert signs with the aim of initiating an escape reaction from the dangerous environment. Coughing and sneezing are triggered by activation of peripheral sensory nerve endings in the nasal airway lining. These nerve endings can be considered as a first defense mechanism against chemical challenges (Bessac and Jordt 2008, 2010). The purpose of this initial reflex-response is the removal of irritants from the nasal epithelial surface. After activation of nasal trigeminal, glossopharyngeal or vagal chemosensory neurons (type C and small diameter myelinated Aδ fibres) by TIHs, sensory-autonomic pathways elicit parasympathetic reflexes, mediated via cholinergic efferent neurons, resulting in secretions from nasal, lacrimatory and salivary glands in combination with the dilatation of vessels in the nasal mucosa and sinuses (Bessac and Jordt 2008, 2010). Mucosal swelling and leakage are common results leading to narrowing or obstruction of the nasal passage (Baraniuk and Kim 2007). The resulting edema of the larynx and glottis can be life-threatening due to acute obstruction of the upper airways.
4 Clinical Symptoms of TIH Exposure Related to the Tracheobronchial System
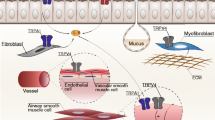
When irritants reach the tracheobronchial section, sensory neuronal activity can trigger tracheal and bronchial constriction via contraction of bronchial smooth muscle cells, mucus secretion, and neurogenic inflammation (Fig. 2), events which are considered as typical reactions after exposure to lung irritants (Miller and Chang 2003). Rhonchus is the typical diagnostic finding resulting from mucous secretion and constriction of the bronchial musculature. TIHs can be removed from the respiratory tract by different mechanisms. The airways of the lower respiratory tract are lined with cells that are predominantly ciliated or mucus producing. TIHs are trapped within the mucus layer and are removed by ciliated cells via mucociliary transport. Macrophages, either integrated in the epithelium or macrophages in the alveoli, are capable of phagocytosis and thereby clearing inhaled particles. Macrophages migrate to upper parts of the respiratory tract, where they are trapped in the mucociliar transport and are cleared from the airways. Moreover, activated macrophages release cytokines, chemokines, growth factors, proteolytic enzymes and ROS. These mediators recruit and activate other cells that either participate in tissue regeneration or may contribute to tissue remodeling processes. After exposure to lung irritants, mucociliary clearance may increase when small or moderate concentrations of toxic chemicals are inhaled. However, high concentrations or very aggressive chemical substances may also interfere with the cilia of the respiratory epithelium or influence the epithelial integrity itself. As shown in an in vitro human bronchial co-culture cell culture model, sulfur mustard induced a widening of intercellular spaces and moreover, a loss in cell-matrix adhesion molecules was observed resulting in a decrease of transepithelial resistance (Pohl et al. 2009). Mucin production increased with resultant cessation of ciliar beat.

Schematic section of a bronchus. Left part shows the physiological state, right part illustrates the (patho)physiological response after TIH exposure. Exposure with TIHs can lead to smooth muscle contraction (A) resulting in a reduction of airway diameter. Moreover, the epithelium lining the respiratory system and endothelium of pulmonary vessels can be affected, resulting in dysfunction, increased production of mucus (B) and fluid leakage into the aerial lumen (C), enhancing the reduction of airway diameter
5 Clinical Symptoms of TIH Exposure Related to the Alveolar System
Chemical substances with little water solubility or high concentrations of TIHs are able to reach deep lung compartments, e.g. the alveoli. The complex damaging processes of TIHs in the alveolar space are depicted in Fig. 3 and include liberation of inflammatory mediators, invasion of inflammatory cells, hypersecretion, and disturbance of cell-cell contacts leading to the destruction of the endothelial-epithelial barrier.

Schematic illustration of the alveolar space and the cellular effects after TIH exposure. After exposure to TIHs, complex injury to lung tissue may occur. Damage to endothelial cells accompanied with epithelial damage, leads to alveolar edema and release of chemo-attractants. Inflammatory cells (e.g. monocytes, neutrophils) migrate from the blood vessels into the alveolar space, secreting interleukins thus inducing and maintaining inflammation. Proteinases are activated, enhancing cellular damage. Mucin production by secretory cells is enhanced. Growth factors are secreted, inducing repair and/or remodeling processes
Apparently, type I alveolar epithelial cells (covering 90–95 % of the alveolar surface) appear to be most vulnerable to many lung toxicants probably because of their large surface area. Dysfunctional alveolar type I cells have to be replaced by new type I cells originating from type II alveolar cells. Alternatively, fibroblasts are activated to repair tissue lesions resulting in the formation of connective tissue and scars. Chemicals that affect both alveolar type I and type II cells have to be considered as extremely harmful, as regenerative cells are compromised and tissue damage cannot be adequately repaired. As the blood-air barrier is extremely thin (~0.4–1 μm), toxic substances may penetrate into lung capillaries (especially after damage to alveolar type I cells) and affect the endothelium, which is comparably vulnerable as epithelial cells type I. This damage will result in progressive impairment of the blood-air barrier and permeability will increase. Tight and adherence junctions between cells are loosened, facilitating loss of epithelial resistance. As a result, blood plasma enters from lung capillaries into the lung interstitium. The resulting interstitial edema increases the diffusion distance through the blood-air barrier leading to disturbance of gas exchange. First, edema fluid is evacuated from the lung interstitium by the lymphatic system. Ongoing loss of epithelial and endothelial resistance will increase plasma leakage, overstraining the lymphatic system and resulting in alveolar edema. A pathological cycle is initiated: formation of edema foam, ascending from the alveolar space to the upper parts of the respiratory system, blocking bronchioles and impairing oxygenation. These events then cause increased pulmonary resistance which leads to further leakage into the alveoli. The whole process may result in a toxic lung edema that may be fatal.
Toxic lung edema due to increased alveolar permeability is hard to control, because tissue architecture is affected or even destroyed. Recovery of the integrity of the alveolar epithelial surface is an essential strategy after toxic inhalation injury. Especially, alveolar type I cells are vulnerable and may undergo cell death mediated either by direct TIH effects or indirectly by specific cytokines released from affected cells, residual structural cells or activated macrophages. Various acute inflammatory mediators are expressed in the lung during acute lung injury (e.g. IL-1α, IL-8, TNF-α, C3b, leukotrienes). In parallel, proteases are released from infiltrating neutrophils and mononuclear cells and degrade the extracellular matrix. Resulting debris is subsequently cleared by macrophages (attracted and differentiated from bone marrow and circulatory pools). Next, growth factors (e.g. TGF-ß, CTGF, PDGF) stimulate basal cells (type II alveolar cells) to differentiate into type I cells and other cell types reconstitute connective tissue, vascular tissue, and nerve endings. A careful balance of catalytic and anabolic processes is mandatory for a complete physiological repair. If one of the processes is gaining an upper hand, fibrosis due to enhanced production of collagen and connective tissue or emphysema due to enhanced protease activity may occur (Bonner 2008).
6 Members of the TRP Channel Family as Chemosensory Molecules in the Airways
Virtually all pathophysiological processes mentioned above are regulated or modulated by activation of chemosensory molecules in the plasma membrane of cells in the respiratory system. In particular chemosensory TRP channels play a pivotal role in the detection of TIHs, in the control of adaptive responses, and in the initiation of detrimental signaling cascades.
TRP channels represent one of the largest superfamilies of ion channels in the human genome (Clapham et al. 2001; Montell et al. 2002; Nilius and Owsianik 2011; Pedersen et al. 2005). Mammalian TRP channels can be subdivided into six families, i.e. TRPC, TRPM, TRPV, TRPP, TRPML, and TRPA channels as shown in Fig. 4. The common structural features of the members of the TRP superfamily are six transmembrane-spanning domains (TM1-6) and a putative pore region (P) located between transmembrane domains 5 and 6 (see Fig. 5). TRP channels are permeable to cations with striking differences in ion selectivity. Many TRP channels conduct monovalent cations like Na+ or K+ as well as bivalents like Ca2+, Mg2+ or bivalent cations of transition metals. Some TRP channels like TRPM4 and TRPM5 show high selectivity for monovalent cations, other channels like TRPV5 or TRPV6 are highly selective for Ca2+ (for review: Owsianik et al. 2006).

Phylogenetic tree of TRP channels. The relationships between the TRP channels are illustrated by the branch lengths

Common structural features of TRP channels. TRP channels have six transmembrane regions connected by intra- and extracellular loops. A loop between transmembrane domain 5 and 6 is essential for the formation of a pore in a functional TRP channel tetramer at the plasma membrane. PM plasma membrane, TM1-6 transmembrane-spanning domains 1–6, P pore region
The archetypal TRP channel, eponymous for the whole super-family, is the product of the trp gene of the fruit fly, Drosophila melanogaster. Cloning of the Drosophila TRP protein was preceded by the characterization of a mutant fruit fly with a visual defect. In this mutant strain, termed transient receptor potential (TRP) (Minke 1977; Montell et al. 1985), exposure to light only led to a short-lived depolarizing current due to an inactivating mutation of the TRP channel, which is an indispensable component of the phototransduction cascade in Drosophila (Hardie and Minke 1992; Montell et al. 1985; Montell and Rubin 1989). In some analogy to Drosophila TRP, many mammalian TRP channels exert sensory functions, although mostly outside of the visual system. Thus, TRP channels like TRPV1 (Caterina et al. 1999; Cesare et al. 1999; Tominaga et al. 1998), TRPV2 (Caterina et al. 1999), TRPV3 (Peier et al. 2002b; Smith et al. 2002; Xu et al. 2002), and TRPV4 (Guler et al. 2002; Watanabe et al. 2002) were reported to be activated by elevated temperature and are involved in thermo-sensing (for review: Tominaga 2007), whereas TRPM8 is activated by cold temperatures (McKemy et al. 2002; Peier et al. 2002a). Other TRP channels like TRPM5 are part of the signal transduction pathway engaged by taste receptors (Perez et al. 2002; Zhang et al. 2003). Of note, TRPM5 channel activity also depends on temperature (Talavera et al. 2005), a fact that explains why sweet or bitter food has a more intense taste in higher temperatures. Furthermore, TRP channels like TRPV4 react to mechanical stimuli in various cell types and organs (for review: Liedtke 2007; Plant and Strotmann 2007; Yin and Kuebler 2010) including the lung vasculature (Jian et al. 2008).
The most important function of TRP channels in the context of this review is their role as chemosensory molecules. In the respiratory system, especially TRPV1, TRPV4, TRPA1, and TRPM8 are involved in the detection of potentially harmful inhalants. However, these channels display a remarkable promiscuity with regard to their activators. For example, TRPA1 is activated by a large number of chemically unrelated substances (Table 1). Furthermore, TRP channels of the TRPC family are expressed in the lung and form part of redox-regulated signaling pathways. The activation of these TRP channels by TIHs results either from covalent modification of cystein residues of the ion channel or from phospholipase C (PLC)-regulated changes in lipid messengers (diacylglyercol, phosphatidyl-inositolphosphate) or intracellular calcium levels. In the first scenario, the TRP channel can be activated directly by reaction with an electrophilic TIH (Brone et al. 2008; Hinman et al. 2006; Macpherson et al. 2007a) or the reaction of the TIH with constituents of the plasma membrane leading to the generation of secondary TRP activators (Taylor-Clark et al. 2009; Trevisani et al. 2007). Regarding PLC-promoted signaling events leading to the activation of TRP channels, either stimulation of Gq/11-coupled protease-activated receptors (PARs) or purinergic receptors with subsequent activation of PLC-β or stimulation of redox-sensitive PLC isoforms is involved (see below).
Of note, some pulmonary TRP channels are involved in O2 sensing by detecting hypoxic as well as hyperoxic states (for review: Takahashi et al. 2012). Hypoxia can lead to an activation of TRPA1- or TRPC6-promoted calcium responses (Fuchs et al. 2011; Takahashi et al. 2011; Weissmann et al. 2006). TRPA1 acting as an O2 sensor is expressed in vagal neurons in the lung and its activation by hypoxia occurs via reduced activity of proline hydroxylases (PHDs). In normoxia PHDs oxidize a proline residue in TRPA1 (Pro394) thus inhibiting the channel (Takahashi et al. 2011). Hyperoxic states lead to the direct oxidation of cystein residues of TRPA1 (Cys633 and Cys856), a modification, which overrides the inhibition of TRPA1 by proline hydroxylation (Takahashi et al. 2011). Whereas TRPA1 is directly modified by the ambient O2 concentration, TRPC6 is activated by hypoxic states via the enhanced generation of reactive oxygen species during hypoxia, which in turn lead to an accumulation of diacyl glycerol (DAG) (Weissmann et al. 2006). Thus, TIHs leading to obstruction of the airways via bronchoconstriction or increased mucus secretion can indirectly activate TRPA1 or TRC6. In contrast, oxidizing TIHs could potentially mimick hyperoxic conditions and by this means directly activate TRPA1.
To explain the role of TRP channels in pulmonary responses upon exposure to TIHs in more detail, this review will first focus on the involvement of TRP channels in irritative responses of the upper respiratory tract. Afterwards, we will discuss the special role of TRPA1 and TRPV1 in the cough reflex. Thereafter, the impact of TRP channels, especially TRPV4, in the regulation of mucociliary clearance will be addressed. Finally, the role of members of the canonical family of TRP channels, TRPCs, in redox-regulated pathways will be discussed.
7 TRP Channels and Irritative Responses in the Upper Respiratory Tract
As stated above, the mucosa of the eyes and the mucosa of the nasopharynx are first-line target of TIHs, especially those with high water solubility. Of note, all features of mucosal responses after exposure to TIHs, i.e. lacrimation, sternutation, nasal discharge, and irritative pain, are regulated by TRP channels.
7.1 Irritant-Induced Lacrimation
With regard to lacrimation responses several TRP channels are involved. TRPM8, TRPA1, and TRPV1 are known to be functionally expressed in nerve terminals in the cornea (Murata and Masuko 2006) and prototypical TRP agonists like menthol (TRPM8), allyl isothiocyanate (TRPA1), and capsaicin (TRPV1) lead to increased lacrimation (Parra et al. 2010). Of note, TRPM8, which is activated by cool temperatures, appears not to be involved in lacrimation caused by irritants, but to regulate basal tearing rate via a cooling effect of the evaporating tear film (Parra et al. 2010). Consequently, low concentrations of the TRPM8 agonist menthol led to increased tear flow in mice without inducing nocifensive effects, whereas high concentrations of menthol induced nocifensive behavior in a TRPM8-independent manner (Robbins et al. 2012). Apart from the aforementioned TRP channels expressed in corneal nerve endings, other thermo- and osmosensitive TRPV channels, i.e. TRPV2, TRPV3, and TRPV4 have also been described in corneal epithelial cells (Mergler et al. 2011). However, their (patho)physiological functions in the eye still await further clarification. Of note, a non-TRPM8 and non-TRPA1 component is involved in tear flow excited by drying of the cornea, which leads to increased osmolarity of the tear fluid (Hirata and Oshinsky 2012). Thus, especially the osmosensitive TRPV4 channel could be involved in the regulation of basal tear flow. Regarding the role TRP channels in irritant-induced lacrimation, TRPV1 and TRPA1 play the predominant role. Thus, well known lachrymators like constituents of tear gas (e.g. 2-chlorobenzalmalononitrile, CS), pepper spray (capsaicin) or onions (allicin) exert their irritative effects via activation of TRPA1 or TRPV1 (Salazar et al. 2008).
7.2 Role of TRPA1 and TRPV1 in Nasal Irritation by TIHs
Irritants acting on the nasal mucosa lead to a stimulation of trigeminal C fiber nerve endings with subsequent irritative responses. Many well-known trigger substances of these trigeminal reflexes, like capsaicin, menthol, or mustard oil directly target TRP channels. Capsaicin, for instance, has been extensively characterized as a tool to investigate airway reflexes mediated via C fibers (Coleridge and Coleridge 1977; Coleridge et al. 1965; Fuller et al. 1985). Application of capsaicin or other TRPV1 agonists lead to the release of substance P or CGRP by nerve endings in the nasal mucosa (Lundberg et al. 1984; Lundblad et al. 1983; Malis et al. 2001; Petersson et al. 1989) and thus, contributes to neurogenic inflammation in the nasopharynx. Functional studies have suggested that TRPV1, TRPA1, and TRPM8 are expressed in trigeminal neurons innervating the nasal cavity (Damann et al. 2006; Frasnelli et al. 2011; Karashima et al. 2007). Thus, these chemosensory channels have been postulated as critical sensor molecules in the nasopharynx involved in the detection of oxidizing or electrophilic TIHs (Bessac et al. 2008; Gerhold and Bautista 2009).
Interestingly, TRPV1 expression in the nasal mucosa has been also demonstrated in non-neuronal cells, e.g. in epithelial cells, vascular endothelium, and submucosal glands (Seki et al. 2006). Furthermore, stimulation of human cells from nasal or bronchial epithelium with capsaicin led to the production of interleukin-6 (IL-6) which could be blocked by co-treatment with TRPV1 antagonist capsazepine (Seki et al. 2007).
7.3 Cough Reflex: Role of TRPA1 and TRPV1
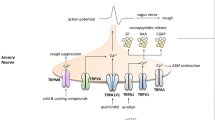
The cough reflex is an important danger signal upon exposure to harmful inhalants and protects the airways by removing ingested stimuli. However, chronic cough severely impacts the quality of life and represents an important, but often intractable reason for patients to consult a physician. Therefore, novel therapeutic options to treat chronic cough are urgently needed. In this context, it is important to note that cough can be induced via two distinct mechanisms, i.e., either by chemical irritation stimulating a reflex mediated via C fibers or by mechanical stimuli, which produce a cough reflex via Aδ fibers (for review: Brooks 2011). Whereas the role of TRP channels in mechanical induced cough is still not clearly defined, the role of these channels in chemically-induced cough is well established. Especially, TRPA1 and TRPV1 channels represent pivotal factors in regulating irritant-induced cough (for review: Bessac and Jordt 2008; Geppetti et al. 2010; Materazzi et al. 2009) and are promising therapeutic targets (for review: Banner et al. 2011; Preti et al. 2012).
Thus, TRPA1 agonists like allyl isothiocyanate (AITC) and TRPV1 agonists like capsaicin are potent inducers of cough reflex. Vice versa, TRPA1- and TRPV1- inhibitors have been shown to block cough responses provoked by exposure to the respective agonist in animal models (Brozmanova et al. 2012; Grace et al. 2012). In a comparative study using the guinea pig model, the TRPV1 activator capsaicin was more efficient to induce cough than the TRPA1 activator AITC (Brozmanova et al. 2012). Regarding potential endogenous activators of the TRP-promoted cough reflex, it has been shown that prostaglandin E2 (PGE2) and bradykinin (Bk) can induce a tussive response in animal models and are able to activate TRPA1 and TRPV1 in human, mouse and guinea pig airway cells (Grace et al. 2012). These findings are commensurate with previous results showing that Bk can activate TRPA1 via a PLC- and cAMP-dependent protein kinase (PKA)-dependent mechanism (Bandell et al. 2004; Wang et al. 2008). Interestingly, the contribution of these channel in Bk- or PGE2-promoted cough was additive: thus, the sole use of TRPV1- and TRPA1-blockers led to partial inhibition of the tussive response to both stimuli, whereas combination of TRPV1 and TRPA1 inhibitors completely abrogated cough responses (Grace et al. 2012). While the exact activation mechanism of TRPA1 and TRPV1 by PGE2 and Bk is not fully elucidated, these findings indicate that a number of exogenous or endogenous processes leading to the release of inflammatory mediators including Bk and PGE2 could potentially lead to an activation of these cough-associated TRP channels.
The importance of human TRPs in the pathogenesis of cough has been recently investigated in a genetic analysis of single nucleotide polymorphisms (SNPs) of TRPV1 and their relationship with cough symptoms in 844 asthmatic and 2,046 non-asthmatic patients (Smit et al. 2012). Of note, seven SNPs of TRPV1 showed a statistically significant association with cough in non-asthmatics (six SNPs with nocturnal cough and one with usual cough). Furthermore, four SNPs of TRPV1 were associated with an increased risk of cough in persons (asthmatics and non-asthmatics) with exposure to irritants (Jugg et al. 2011). For other TRP channels like TRPV4 and TRPA1 no significant relationship of SNPs with cough could be detected in this study.
These findings suggest that TRPV1 plays a crucial role in mediating chemically-induced cough-responses. However, with regard to reactive, electrophilic inhalants like acrolein, croton aldehyde, methyl isocyanate, or AITC, TRPA1 may be more important as primary target. As stated above, electrophilic and oxidizing compounds are able to activate TRPA1 as well as TRPV1 via covalent modification of cystein residues (Hinman et al. 2006; Macpherson et al. 2007a; Salazar et al. 2008). However, the electrophilic α, β-unsaturated aldehyde acrolein activated C fibers in wild-type and TRPV1-knockout mice, a response that could not be modified by additional treatment with TRPV1 blocker iodoresiniferatoxin indicating that at least in this murine model TRPV1 is not the primary target of a prototypical electrophilic inhalant (Symanowicz et al. 2004). The importance of TRPA1 in response to electrophilic inhalants is further supported by findings in guinea pigs, which presented pronounced coughing upon challenge with AITC, cinnamaldehyde, acrolein, or croton aldehyde, which could be blocked by inhibition of TRPA1 using the selective antagonist HC-030031 but not by inhibition of TRPV1 using the selective blocker capsazepine (Andre et al. 2009).
7.4 TRPV4 and Mucociliary Clearance
As mentioned before, a critical function of the bronchial system involved in adaptive responses to exposure to TIHs is related to the mucociliary clearance system. The driving force for increased ciliary activity in the bronchial system is an increase of the intracellular calcium concentration in ciliary cells (Salathe and Bookman 1995). Therefore, chemosensory, calcium-permeable channels of the TRP family are plausible candidates to modulate ciliary beat frequency after chemical irritation. In fact, TRPV4 expression has been described in many ciliary cells, e.g. in the ovarian duct (Andrade et al. 2005), in cholangiocytes (Gradilone et al. 2007), and also in bronchial epithelial cells (Lorenzo et al. 2008; Seminario-Vidal et al. 2011).
In ciliated cells of the oviduct treatment with high viscosity fluid increased ciliar activity, an effect that could be blocked by loading cells with TRPV4 antibodies and was mimicked by TRPV4 agonists (Andrade et al. 2005) indicating that TRPV4 is a major component of mechanically induced ciliary activity in these cells. In contrast, TRPV4 appeared not to be critically involved in ciliary activity upon viscous challenge in bronchial epithelium, since highly viscous fluid did not provoke different effects in tracheal epithelial cells from wild-type or TRPV4-knock-out mice (Lorenzo et al. 2008). However, stimulation of tracheal cells with ATP, a well-known activator of ciliary beat frequency in the bronchial system, led to a receptor-operated calcium signal strongly dependent on TRPV4 (Lorenzo et al. 2008). Intriguingly, activation of TRPV4 has been described to stimulate the release of ATP by airway epithelial cells induced by cell swelling in a pathway involving pannexin 1, RhoA and myosin light chain phosphorylation (Seminario-Vidal et al. 2011).
These findings suggest that TRPV4 and ATP may form part of a self-amplifying system with TRPV4 responsible for ATP-promoted calcium signaling and subsequently triggering ATP release after activation (Fig. 6). Moreover, ATP release by airway cells has been demonstrated after treatment with the lung toxicant bleomycin and has been proposed to form a universal danger signal (Riteau et al. 2010).

Involvement of TRPV4 in ATP-signaling in bronchial epithelium. Stimulation of metabotropic purinoceptors (e.g. P2Y2) leads to the activation of TRPV4 which in turn can trigger the release of ATP. By this means, the interplay of ATP-dependent G protein coupled receptors and TRPV4 may sustain a self-amplifying signaling cascade with high extracellular levels of ATP representing a danger signal in lung epithelial cells
8 Pulmonary TRPC Channels
In recent years evidence for an involvement of TRPC channels in lung function and toxicity became evident. Although TRPC channels are not the primary sensor molecules in different lung tissues, they are essential members of signal transduction cascades mediating toxicity in the targeted cells. TRPC channel activity is induced after ligand binding to a receptor (e.g. a Gq/11 protein-coupled receptor) and complex intracellular signaling cascades (Dietrich et al. 2005).
The seven TRPC family members can be grouped into subfamilies on the basis of their amino acid sequence similarity. While TRPC1 and TRPC2 are almost unique, TRPC4 and TRPC5 share ~65 % identity. TRPC3, 6 and 7 form a structural and functional subfamily with 70–80 % identity at the amino acid level and their ability to be activated by DAG (Dietrich et al. 2005; Hofmann et al. 1999; Okada et al. 1999), a product of phosphatidylinositol 4,5-bisphosphate cleavage by activated phospholipases C. In all eukaryotic cells, activation of phospholipase C (PLC)-coupled membrane receptors by hormones or neurotransmitters leads to an increase in the intracellular Ca2+ concentration [Ca2+]i. Both activation of PLCβ and ε-isozymes by G protein coupled receptors and of PLCγ isoforms by receptor tyrosine kinases results in the hydrolysis of phosphatidyinositol 4,5-bisphosphate and the generation of inositol 1,4,5-trisphosphate (IP3) and DAG. While it seems clear that TRPC3/6/7 channels are directly activated by DAG and are responsible for receptor-operated calcium entry (ROCE) (Hofmann et al. 1999; Okada et al. 1999), the regulation of other members and their involvement in store operated calcium entry (SOCE) is discussed controversially. SOCE occurs when inositol 1,4,5-trisphosphate (IP3) or some other signal discharges Ca2+ from intracellular stores in the endoplasmic reticulum (ER). The subsequent fall in the ER Ca2+ concentration then signals to the plasma membrane and activates store-operated channels. A major breakthrough in understanding SOCE was the identification of the calcium sensor STIM (Liou et al. 2005; Roos et al. 2005; Zhang et al. 2005) and the Orai channels (Cahalan 2009; Feske et al. 2006; Vig et al. 2006) in 2005 and 2006, respectively. STIM 1 which is predominantly located in the ER, is a single transmembrane domain protein containing two N-terminal Ca2+-binding EF hands in the ER lumen and is therefore ideally situated to detect calcium levels in the ER. Upon Ca2+ release reduced calcium levels induce STIM redistribution to punctae and opening of store-operated channels in the plasma membrane. The so called Orai proteins with only four predicted transmembrane domains and intracellular C- and N-termini, were identified as the pore forming unit of store-operated channels by the analysis of T cells from patients with severe combined immunodeficiency (SCID) reviewed in Cahalan (2009). Although Orai 1 channels can multimerize with themselves to form pore forming units, it was also suggested that Orai molecules might interact with TRPC channels heterologously expressed in HEK293 cells to form store-operated channels (Liao et al. 2009). Moreover, evidence supporting the interaction of the calcium sensor STIM1 with TRPC proteins was presented for the same heterologous expression system (Yuan et al. 2009a, b).
TRPCs are also regulated by phosphorylation. TRPC3 channels can be inactivated by direct phosphorylation of amino acids threonine 11 (Thr 11) and serine 263 (Ser 263) by protein kinase G (PKG) (Kwan et al. 2004) and by protein kinase C (PKC)-mediated phosphorylation of Ser 712 (Trebak et al. 2005). The receptor tyrosine kinase Src phosphorylates TRPC3 at Tyrosine 226 (Tyr 226) and inhibits TRPC3 activity (Kawasaki et al. 2006), while Fyn another member of the Src family increases TRPC6 activity (Hisatsune et al. 2004). It has been shown that TRPC6 is negatively regulated after phosphorylation of Thr 69 by PKG (Takahashi et al. 2008b). Most interestingly, this phosphorylation is essential for anti-hypertrophic effects of phosphodiesterase 5 inhibitors (Kinoshita et al. 2010; Koitabashi et al. 2010; Nishida et al. 2010), which are also important therapeutics for pulmonary hypertension (Reichenberger et al. 2007).
TRPC channels can form functional homo- and heterotetramers within two defined subgroups (TRPC1/4/5 and TRPC3/6/7) excluding TRPC2 which is a non-functional pseudogene in humans (Goel et al. 2002; Hofmann et al. 2002). Novel combinations of TRPC1- with TRPC4 or 5 together with TRPC3, 6 or 7 in tetrameric complexes with three different TRPC family members have also been identified in HEK293 cells, as well as in embryonic brain, but not in adult rat tissues (Strubing et al. 2003). Evidence for the importance of the highly conserved pore region in the TRPC tetramer is derived from site-directed mutagenesis which results not only in the complete loss of channel activity upon heterologous expression, but also in a dominant-negative effect of a mutated channel monomer on functional homo- or heteromeric channel tetramers (Hofmann et al. 2002).
The TRPC family includes seven members (TRPC1-7), but only six are functional in humans, because TRPC2 is a pseudogene in the human genome. TRPC7 expression in lung is sparse and is predominantly identified in alveolar macrophages (Finney-Hayward et al. 2010). Expression of the other TRPC channels in pulmonary tissues and recent evidence concerning their potential function is summarized in the next paragraph.
9 Role of TRPC1, TRPC3, TRPC4, and TRPC5 in the Lung
Although TRPC1 was the first mammalian TRP channel to be cloned, its physiological functions are still elusive. It is not even clear, if TRPC1 is an ion channel or merely an adaptor protein regulating ion channel activity of other TRPC members (Storch et al. 2012). Homomeric TRPC1 complexes do not translocate to the plasma membrane (Hofmann et al. 2002), but TRPC1 as a member of heteromeric TRPC1/5 or TRPC1/4 complexes has a high impact on TRPC4/5 currents (Strubing et al. 2001) and decreases calcium permeability in such channel complexes (Storch et al. 2012). It has been reported that TRPC1 is important for the proliferation of bronchial smooth muscle cells, which may induce thickening of the airways facilitating airway hyperreactivity (summarized in Li et al. 2003). Along these lines, TRPC1 is up-regulated in proliferating airway smooth muscle cells while TRPC1 down-regulation inhibits cell proliferation (Sweeney et al. 2002a, b). However, it is not clear, if proliferation is influenced exclusively by TRPC1 channels or by TRPC1/4 channel complexes, because heteromeric TRPC1/4 channel complexes seem to be more important for the lung extra-alveolar endothelial barrier than for the capillary endothelial barrier (summarized in Cioffi et al. 2009). TRPC3 expression is up-regulated by tumour necrosis factor (TNF)-α in human airway smooth muscle cells (White et al. 2006). TNF-α is a pro-inflammatory cytokine which has been implicated in asthma and COPD because it increases airway hyperresponsiveness (AHR) by increasing airway smooth muscle Ca2+. Therefore, TNF-α blockage might represent a promising pharmacological target for the treatment of asthma and COPD (summarized in Amrani 2007). The function of TRPC4 in lung vascular endothelial cells was analysed using TRPC4−/− mice and wild-type controls. A defect in thrombin-induced Ca2+ influx is associated with reduced actin stress fiber formation and reduced endothelial cell retraction response. In isolated perfused lungs thrombin receptor activation increased the microvessel filtration coefficient (Kfc) by 2.8-fold in wild-type lungs but only 1.4 in TRPC4−/− lungs. This data provides evidence for a role of TRPC4 together with other ion channels in lung endothelial permeability (Tiruppathi et al. 2002). Recently TRPC5 was detected in the apical membrane of neuroepithelial bodies in the intrapulmonary airway epithelium by TRPC5 specific antibodies. However, it remains elusive if and how TRPC5 is involved in signal transduction cascades of these cells (Lembrechts et al. 2012).
10 TRPC6: Involvement in Redox-Sensitive Pathways in the Lung
Many studies were already published on the role of TRPC6 channels in the airways. Indeed, TRPC6 function in the lung might be particularly important, because it is the most prominently expressed TRPC channel in lungs (Hofmann et al. 2000). Therefore, we set out to analyze allergic responses in a TRP6-deficient mouse model expecting a reduced response in comparison to wild-type (WT) mice. However, we found an increased methacholine-induced AHR in TRPC6-deficient mice compared to WT mice. This surprising finding is most probably due to compensatory up-regulation of TRPC3 in airway smooth muscle cells (Sel et al. 2008). By experimental inflammation induced by intraperitoneal ovalbumin (OVA) sensitization followed by OVA aerosol challenges we were able to detect a decreased level of T-helper type 2 (Th2) cytokines (IL-5 and IL-13) as well as reduced IgE levels in the blood of TRPC6−/− mice compared to WT mice. Mucus production in goblets cells from challenged mice however was not altered in TRPC6-deficient mice (Sel et al. 2008). This data points to an important role of TRPC6 in the immune responsive rather than in airway smooth muscle tissues. Because neutrophil numbers are increased in the sputum of COPD and asthma patients, TRPC6 function in these cells might be also important for the progress of the diseases. Migration of TRPC6−/− neutrophils in response to macrophage inflammatory protein-2 (MIP2 also known as CXCL2) was reduced compared to WT neutrophils (Damann et al. 2009). In the same report an involvement of TRPC6 in cytoskeletal rearrangements during neutrophil migration was demonstrated suggesting an important role of TRPC6 in the migration of lung neutrophils. TRPC6 was also identified in other immune cells of the lung like alveolar macrophages (Finney-Hayward et al. 2010). Most interestingly, TRPC6 mRNA expression was significantly increased in macrophages obtained from COPD patients compared to healthy controls, while TRPC3 and TRPC7 levels remained unchanged (Finney-Hayward et al. 2010).
Under conditions of alveolar hypoxia induced by obstruction of the lung by particulate matter the pulmonary arterial vessels constrict diverting blood flow to the well ventilated areas in the lung to ensure maximal oxygenation of the venous blood by adequately matching ventilation with perfusion. This alveolar hypoxia-mediated vasoconstrictive phenomenon is known as acute hypoxic pulmonary vasoconstriction (HPV) and was first described by von Euler and Liljestrand in 1946. Sustained pulmonary vasoconstriction (chronic HPV) is often accompanied by vascular remodelling, i.e. the muscularization of smaller arteries and arterioles due to SMC proliferation and migration. In severe forms of pulmonary hypertension (such as idiopathic and familial PAH), pulmonary artery remodelling is extensive resulting from intimal fibrosis and medial hypertrophy. Such occlusion of the pulmonary arterioles is associated with the formation of plexiform lesions resulting from the proliferation of endothelial cells, migration and proliferation of SMC, and accumulation of circulating cells (including macrophages and endothelial progenitor cells). In both pulmonary and systemic circulation, the blood flow and intraluminal pressure are mainly regulated by changes in vessel diameters.
TRPC6 was found to be up-regulated in hypoxia-induced, as well as idiopathic chronic pulmonary hypertension (IPAH) (Lin et al. 2004; Yu et al. 2004). IPAH is caused by excessive smooth muscle proliferation and results in right heart failure. IPAH is associated with up-regulated expression of TRPC6 and TRPC3, but not TRPC1 genes at the mRNA and protein levels (Kunichika et al. 2004; Yu et al. 2004). Most interestingly, in patients with IPAH the allele frequency of a C/G single nucleotide polymorphism (SNP) at position −254 upstream of the transcription start of the TRPC6 gene is significantly higher (12 %) than in normal subjects (6 %). This polymorphism creates a binding site for nuclear factor-κB (Yu et al. 2009). Therefore, TRPC6 might also be induced at least in patients with the C/G polymorphism during inflammation caused by toxic irritants in the lung.
During an extensive analysis of TRPC6−/− mice, we set out to determine pulmonary arterial pressure (PAP) in isolated lung during acute (<20 min) and prolonged (60–160 min) hypoxia. Much to our surprise, acute HPV was completely absent in TRPC6−/− mice, while vasoconstriction after prolonged hypoxia was not significantly different in TRPC6−/− mice compared to wild type mice. These data show for the first time, that the acute hypoxic vasoconstrictor response and the prolonged phase are regulated by different molecular mechanisms. Only for acute HPV TRPC6 is indispensable and cannot be compensated by up-regulation of other TRPC channels. Moreover, the lack of acute HPV in TRPC6−/− mice has profound physiological relevance, because partial occlusion of alveolar ventilation provoked severe hypoxemia in TRPC6−/− but not in WT mice (Weissmann et al. 2006).
We recently identified a novel mechanism of TRPC6 channel activation by redox signalling which is important for cellular toxicity by hypoxia in the lung (Weissmann et al. 2012). Lung ischemia-reperfusion-induced injury (LIRI) is a life-threatening condition that causes pulmonary edema in lungs intended for transplantation and can be induced by ischemia in isolated perfused mouse lungs from wild-type mice. Most interestingly, after 90 min of ischemia, edema was completely absent in isolated lungs from TRPC6-deficient mice. Edema did also occur in lungs deficient in NADPH oxidase 2 (NOX2), raising the possibility that the two proteins are members of the same signal transduction pathway. After identifying other proteins in pulmonary endothelial cells, we postulate a novel mechanistic model comprising Nox2-derived production of superoxide, activation of phospholipase C-γ, inhibition of DAG kinase, and DAG-mediated activation of TRPC6 which is essential in increasing endothelial permeability. In later stages fluid and immune cells can then enter the alveolar space inducing LIRI.
The NOX family consists of seven members, NOX1-5, and two dual oxidases (DUOX) DUOX1 and 2 (Altenhofer et al. 2012; Lassegue et al. 2012). After activation they produce superoxide anions which can form multiple forms of reactive oxygen species (ROS). NOX1 is most highly expressed in colon epithelium, while NOX2 is also known as gp91phox the enzymatic complex responsible for the phagocyte respiratory burst. NOX3 is not expressed in lung tissues but in the inner ear and the cochlea of mice (Paffenholz et al. 2004). The NOX4 isoform is abundant in kidney cortex where it was originally discovered and NOX5 is the only isoform which can be activated by Ca2+ through its Ca2+-binding EF hands at the amino terminus. Unfortunately NOX5 is not expressed in rodents and cannot be analyzed in mouse models. In the lung, NOX isoforms are expressed in invading inflammatory cells (NOX1, 2 and 4) epithelial cells (NOX1, 4 and DUOX1 and 2), endothelial cells (NOX1, 2, 4), smooth muscle cells (NOX2 and 4) and mesenchymal cells (NOX2 and 4) (Hecker et al. 2012). It is tempting to speculate that NADPH oxidases may also represent important cellular sensors for TRPC activation by inhaled toxins. Along these lines, recent evidence suggest that NOX isoforms are activated not only by inflammatory mediators, but also by toxic substances like cisplatin (Rybak et al. 2012), bleomycin (Carnesecchi et al. 2011) and paraquat (Cristovao et al. 2009). Cisplatin induced hearing loss is associated with higher levels of ROS and down-regulation of NOX3 prevents cisplatin ototoxicity (Rybak et al. 2012). NOX4-deficient mice are protected from bleomycin-induced lung fibrosis while inflammatory mediators are not changed in the mouse model (Hecker et al. 2012). The activation of NOX isoforms by toxins is best known for paraquat, a toxic herbicide in various tissues, including lung. Paraquat increased the expression of NOX1 in rat dopaminergic N27 cells (Bokemeyer et al. 1998). NOX1 isoforms transfer an electron to paraquat enabling the paraquat-cation to donate an electron to molecular oxygen, generating a superoxide anion (Bielefeld et al. 2005; Bus and Gibson 1984). Subsequently, the accumulation of ROS induces neurotoxicity in the substantia nigra which may result in Parkinson’s disease (Cristovao et al. 2012).
Superoxide anions not only convert nitric oxide (NO) to ONOO- decreasing vasodilation in the vasculature, but also induce multiple signaling cascades in different cell types initiating transcription of inflammatory target proteins which can induce asthma, acute lung injury, lung fibrosis, COPD and lung cancer (Lee and Yang 2012). Moreover, superoxide anions produced by NOX isoforms are able to form hydrogen peroxide (H2O2) in a reaction with H2O catalyzed by extracellular superoxide dismutase (SOD). Hydrogen peroxide is able to permeate the cell membrane to activate PLC isoforms which produce DAG from PIP2. Furthermore, inhibition of DAG kinases which degrade DAG to phosphatidic acid (PA) by hydrogen peroxide will induce DAG accumulation stimulating TRPC3/6/7 channels whereby increasing intracellular Ca2+ concentrations. While moderate elevated Ca2+ levels increase cellular permeability by actin fiber formation resulting in cell shape changes, Ca2+ overload will induce cell death by necrosis or apoptosis (see Fig. 7).

Putative signalling mechanisms for toxin activation of TRPC channels. See text for details. NOX NADPH oxidase, SOD superoxide dismutase, H 2 O 2 hydrogenperoxide, PLC phospholipase C, DAG diacyl glycerol, PIP 2 phosphatidylinositol 4,5-bisphosphate, DAGK diacyl glycerol kinase, PA phosphatidic acid, Ex extracellular, In intracellular (Modified from Weissmann et al. 2012)
Possible signal transduction pathways from NOX isoforms as toxicity sensor to TRPC3/6/7 activation and the resulting cellular effects are summarized in Fig. 7. In the future, it will be essential to dissect these signal transduction cascades in more detail to identify pharmacological targets to be exploited in acute and chronic toxicity.
11 Outlook: Respiratory Chemosensors and Targeted Therapy in Lung Toxicology
To summarize, the present synopsis describes the role of chemosensory TRP channels in the airways with regard to the detection of TIHs and the mediation of TIH-promoted adaptive or noxious effects. The fact that TRP channels represent specific targets of TIHs broadens our mechanistic understanding of toxic lung injury and has implications on novel therapeutic approaches in pulmonary toxicology.
TRP channels are expressed throughout the airways from nasal mucosa to the alveolo-capillary system. They have been found in neuronal cells, especially in nerve endings of C fibers, but also in non-neuronal cells, i.e. in the pulmonary epithelium, in smooth muscle cells of bronchi and vasculature as well as in pulmonary endothelial cells. Regarding the function of TRP channels (in particular TRPA1) in neuronal cells, a critical role as toxicant sensor has been established and activation of TRP channels in nerve endings of the airways leads to the stimulation of protective reflexes, but also to neurogenic inflammation. With respect to the role of pulmonary TRP channels in non-neuronal cells, TRPC channels expressed in vascular smooth muscle cells or pulmonary endothelium are the best characterized members. Pulmonary TRPC channels fulfill important functions in regulating pulmonary blood flow and are not directly activated by TIHs but subsequently to the activation of G protein coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs), in turn leading to the stimulation of phospholipase C. Furthermore, a role of TRPV channels (in particular TRPV4) in the respiratory epithelium has been elucidated. These channels are activated following stimulation of Gq coupled receptors (e. g., protease activated receptors) and promote the release of inflammatory cytokines upon exposure towards TIHs thus contributing to perturbations of the epithelial barrier function.
Thus, according to a simplified model, pulmonary TRP channels can be subdivided into sensor TRP and effector TRP channels. The former (e.g. TRPA1) are mainly expressed in neuronal cells and are directly activated by TIHs. The latter (e.g. TRPV4) are mainly expressed in non-neuronal cells and represent a more distal effector of GPCR- or RTK-promoted signaling pathways. Of note, prototypical sensor TRPs, like TRPA1 have been recently also described in non-neuronal cells. However, their function in this cellular context is still less defined.
The point of view that TIHs act via defined signaling pathways originating from membrane receptors contrasts with previous models describing the action of TIHs merely as unspecific damages of critical biomolecules like membrane lipids or the DNA. According to the general opinion of unspecific effects promoted via TIHs the therapy of toxic lung injury relies on symptomatic efforts or the attempt to inactivate reactive inhalants by applying antioxidative substances or radical scavengers. Based on the view that specific molecules and signaling events are involved in toxic effects in the respiratory system, the potential of targeted therapeutic interventions arises. TRP channels are especially suited for targeted therapies since these molecules are exposed at the plasma membrane and thus, easily accessible for drugs. In fact, a number of modulators of TRP channels with relatively low systemic toxicity have been identified and are available for in vivo testing. Whereas the concept of targeted therapies in toxicology is relatively new, this approach is well established in cancer therapy. In analogy to the experience in oncology, specific drugs targeting TRP-promoted signaling events should be combined with symptomatic and unspecific approaches.
References
Alexander SF (1947) Medical report on the Bari harbor mustard casualties. Mil Surg 101(1):1–17
Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH (2012) The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci: CMLS 69(14):2327–2343. doi:10.1007/s00018-012-1010-9
Amrani Y (2007) TNF-alpha and calcium signaling in airway smooth muscle cells: a never-ending story with promising therapeutic relevance. Am J Respir Cell Mol Biol 36(3):387–388. doi:36/3/387 [pii]
Andersson DA, Gentry C, Moss S, Bevan S (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci: The Official J Soc Neurosci 28(10):2485–2494. doi:10.1523/JNEUROSCI.5369-07.2008
Andrade YN, Fernandes J, Vazquez E, Fernandez-Fernandez JM, Arniges M, Sanchez TM, Villalon M, Valverde MA (2005) TRPV4 channel is involved in the coupling of fluid viscosity changes to epithelial ciliary activity. J Cell Biol 168(6):869–874. doi:10.1083/jcb.200409070
Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P, Patacchini R (2008) Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest 118(7):2574–2582. doi:10.1172/JCI34886
Andre E, Gatti R, Trevisani M, Preti D, Baraldi PG, Patacchini R, Geppetti P (2009) Transient receptor potential ankyrin receptor 1 is a novel target for pro-tussive agents. Br J Pharmacol 158(6):1621–1628. doi:10.1111/j.1476-5381.2009.00438.x
Ban M, Langonne I, Huguet N, Pepin E, Morel G (2006) Inhaled chemicals may enhance allergic airway inflammation in ovalbumin-sensitised mice. Toxicology 226(2–3):161–171. doi:10.1016/j.tox.2006.06.012
Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41(6):849–857
Bang S, Kim KY, Yoo S, Kim YG, Hwang SW (2007) Transient receptor potential A1 mediates acetaldehyde-evoked pain sensation. Eur J Neurosci 26(9):2516–2523. doi:10.1111/j.1460-9568.2007.05882.x
Banner KH, Igney F, Poll C (2011) TRP channels: emerging targets for respiratory disease. Pharmacol Ther 130(3):371–384. doi:10.1016/j.pharmthera.2011.03.005
Baraniuk JN, Kim D (2007) Nasonasal reflexes, the nasal cycle, and sneeze. Curr Allergy Asthma Rep 7(2):105–111
Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM (2005) Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA 102(34):12248–12252. doi:10.1073/pnas.0505356102
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124(6):1269–1282. doi:10.1016/j.cell.2006.02.023
Bessac BF, Jordt SE (2008) Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology 23:360–370. doi:10.1152/physiol.00026.2008
Bessac BF, Jordt SE (2010) Sensory detection and responses to toxic gases: mechanisms, health effects, and countermeasures. Proc Am Thorac Soc 7(4):269–277. doi:10.1513/pats.201001-004SM
Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118(5):1899–1910. doi:10.1172/JCI34192
Bessac BF, Sivula M, von Hehn CA, Caceres AI, Escalera J, Jordt SE (2009) Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J: Official Publication Federation Am Soc Exper Biol 23(4):1102–1114. doi:10.1096/fj.08-117812
Bielefeld EC, Hu BH, Harris KC, Henderson D (2005) Damage and threshold shift resulting from cochlear exposure to paraquat-generated superoxide. Hear Res 207(1–2):35–42. doi:10.1016/j.heares.2005.03.025
Bobb AJ, Arfsten DP, Jederberg WW (2005) N-acetyl-L-cysteine as prophylaxis against sulfur mustard. Mil Med 170(1):52–56
Bokemeyer C, Berger CC, Hartmann JT, Kollmannsberger C, Schmoll HJ, Kuczyk MA, Kanz L (1998) Analysis of risk factors for cisplatin-induced ototoxicity in patients with testicular cancer. Br J Cancer 77(8):1355–1362
Bonner C (2008) Respiratory toxicity. In: Smart RC, Hodgson E (eds) Molecular and biochemical toxicology, 4th edn. Wiley, Hoboken, pp 639–670
Brigelius-Flohe R, Flohe L (2011) Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15(8):2335–2381. doi:10.1089/ars.2010.3534
Brone B, Peeters PJ, Marrannes R, Mercken M, Nuydens R, Meert T, Gijsen HJ (2008) Tear gasses CN, CR, and CS are potent activators of the human TRPA1 receptor. Toxicol Appl Pharmacol 231(2):150–156. doi:10.1016/j.taap. 2008.04.005
Brooks SM (2011) Perspective on the human cough reflex. Cough 7:10. doi:10.1186/1745-9974-7-10
Broughton E (2005) The bhopal disaster and its aftermath: a review. Environ Health: Global Access Sci Source 4(1):6. doi:10.1186/1476-069X-4-6
Brozmanova M, Mazurova L, Ru F, Tatar M, Kollarik M (2012) Comparison of TRPA1-versus TRPV1-mediated cough in guinea pigs. Eur J Pharmacol 689(1–3):211–218. doi:10.1016/j.ejphar.2012.05.048
Bus JS, Gibson JE (1984) Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect 55:37–46
Cahalan MD (2009) STIMulating store-operated Ca(2+) entry. Nat Cell Biol 11(6):669–677. doi:ncb0609-669 [pii]10.1038/ncb0609-669
Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, Krause KH (2011) A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15(3):607–619. doi:10.1089/ars.2010.3829
Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398(6726):436–441. doi:10.1038/18906
Cesare P, Moriondo A, Vellani V, McNaughton PA (1999) Ion channels gated by heat. Proc Natl Acad Sci USA 96(14):7658–7663
Cioffi DL, Lowe K, Alvarez DF, Barry C, Stevens T (2009) TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal 11(4):765–776. doi:10.1089/ARS.2008.2221
Clapham DE, Runnels LW, Strubing C (2001) The TRP ion channel family. Nat Rev Neurosci 2(6):387–396. doi:10.1038/35077544
Cohen GM (1990) Pulmonary metabolism of foreign compounds: its role in metabolic activation. Environ Health Perspect 85:31–41
Colebatch HJ, Ng CK (1992) Estimating alveolar surface area during life. Respir Physiol 88(1–2): 163–170
Coleridge HM, Coleridge JC (1977) Impulse activity in afferent vagal C-fibres with endings in the intrapulmonary airways of dogs. Respir Physiol 29(2):125–142
Coleridge HM, Coleridge JC (1994) Pulmonary reflexes: neural mechanisms of pulmonary defense. Ann Rev Physiol 56:69–91. doi:10.1146/annurev.ph.56.030194.000441
Coleridge HM, Coleridge JC, Luck JC (1965) Pulmonary afferent fibres of small diameter stimulated by capsaicin and by hyperinflation of the lungs. J Physiol 179(2):248–262
Comhair SA, Erzurum SC (2002) Antioxidant responses to oxidant-mediated lung diseases. Am J Physiol Lung Cell Mol Physiol 283(2):L246–L255. doi:10.1152/ajplung.00491.2001
Cristovao AC, Choi DH, Baltazar G, Beal MF, Kim YS (2009) The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 11(9):2105–2118. doi:10.1089/ARS.2009.2459
Cristovao AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS (2012) NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson’s disease. J Neurosci: Official J Soc Neurosci 32(42):14465–14477. doi:10.1523/JNEUROSCI.2246-12.2012
Damann N, Rothermel M, Klupp BG, Mettenleiter TC, Hatt H, Wetzel CH (2006) Chemosensory properties of murine nasal and cutaneous trigeminal neurons identified by viral tracing. BMC Neurosci 7:46. doi:10.1186/1471-2202-7-46
Damann N, Owsianik G, Li S, Poll C, Nilius B (2009) The calcium-conducting ion channel transient receptor potential canonical 6 is involved in macrophage inflammatory protein-2-induced migration of mouse neutrophils. Acta Physiol (Oxf) 195(1):3–11. doi:APS1918 [pii]10.1111/j.1748-1716.2008.01918.x
de Lange DW, Meulenbelt J (2011) Do corticosteroids have a role in preventing or reducing acute toxic lung injury caused by inhalation of chemical agents? Clin Toxicol 49(2):61–71. doi:10.3109/15563650.2011.553196
Dhaka A, Uzzell V, Dubin AE, Mathur J, Petrus M, Bandell M, Patapoutian A (2009) TRPV1 is activated by both acidic and basic pH. J Neurosci: Official J Soc Neurosci 29(1):153–158. doi:10.1523/JNEUROSCI.4901-08.2009
Dietrich A, Kalwa H, Rost BR, Gudermann T (2005) The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflugers Archiv: Eur J Physiol 451(1):72–80. doi:10.1007/s00424-005-1460-0
Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441(7090):179–185
Finney-Hayward TK, Popa MO, Bahra P, Li S, Poll CT, Gosling M, Nicholson AG, Russell RE, Kon OM, Jarai G, Westwick J, Barnes PJ, Donnelly LE (2010) Expression of transient receptor potential C6 channels in human lung macrophages. Am J Respir Cell Mol Biol 43(3):296–304. doi:10.1165/rcmb.2008-0373OC
Frasnelli J, Albrecht J, Bryant B, Lundstrom JN (2011) Perception of specific trigeminal chemosensory agonists. Neuroscience 189:377–383. doi:10.1016/j.neuroscience.2011.04.065
Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Gudermann T, Dietrich A, Weissmann N (2011) Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res 12:20. doi:10.1186/1465-9921-12-20
Fujita F, Uchida K, Moriyama T, Shima A, Shibasaki K, Inada H, Sokabe T, Tominaga M (2008) Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J Clin Invest 118(12):4049–4057. doi:10.1172/JCI35957
Fuller RW, Dixon CM, Barnes PJ (1985) Bronchoconstrictor response to inhaled capsaicin in humans. J Appl Physiol 58(4):1080–1084
Geppetti P, Patacchini R, Nassini R, Materazzi S (2010) Cough: the emerging role of the TRPA1 channel. Lung 188(Suppl 1):S63–68. doi:10.1007/s00408-009-9201-3
Gerhold KA, Bautista DM (2009) Molecular and cellular mechanisms of trigeminal chemosensation. Ann New York Acad Sci 1170:184–189. doi:10.1111/j.1749-6632.2009.03895.x
Ghanei M, Shohrati M, Jafari M, Ghaderi S, Alaeddini F, Aslani J (2008) N-acetylcysteine improves the clinical conditions of mustard gas-exposed patients with normal pulmonary function test. Basic Clin Pharmacol Toxicol 103(5):428–432. doi:10.1111/j.1742-7843.2008.00318.x
Goel M, Sinkins WG, Schilling WP (2002) Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem 277(50):48303–48310
Grace M, Birrell MA, Dubuis E, Maher SA, Belvisi MG (2012) Transient receptor potential channels mediate the tussive response to prostaglandin E2 and bradykinin. Thorax 67(10): 891–900. doi:10.1136/thoraxjnl-2011-201443
Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, Larusso NF (2007) Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA 104(48):19138–19143. doi:10.1073/pnas.0705964104
Grainge C, Rice P (2010) Management of phosgene-induced acute lung injury. Clin Toxicol 48(6): 497–508. doi:10.3109/15563650.2010.506877
Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M (2002) Heat-evoked activation of the ion channel, TRPV4. J Neurosci: Official J Soc Neurosci 22(15):6408–6414. doi:20026679
Hanaoka S, Nomura K, Wada T (2006) Determination of mustard and lewisite related compounds in abandoned chemical weapons (yellow shells) from sources in China and Japan. J Chromatogr A 1101(1–2):268–277. doi:10.1016/j.chroma.2005.10.028
Hardie RC, Minke B (1992) The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron 8(4):643–651
Hecker L, Cheng J, Thannickal VJ (2012) Targeting NOX enzymes in pulmonary fibrosis. Cell Mol Life Sci: CMLS 69(14):2365–2371. doi:10.1007/s00018-012-1012-7
HELCOM (1994) Report on chemical munitions dumped in the Baltic Sea, Report to the 15th meeting of Helsinki Commission. Danish Environmental Protection Agency
Hill K, Schaefer M (2009) Ultraviolet light and photosensitising agents activate TRPA1 via generation of oxidative stress. Cell Calcium 45(2):155–164. doi:10.1016/j.ceca.2008.08.001
Hinman A, Chuang HH, Bautista DM, Julius D (2006) TRP channel activation by reversible covalent modification. Proc Natl Acad Sci USA 103(51):19564–19568. doi:10.1073/pnas.0609598103
Hirata H, Oshinsky ML (2012) Ocular dryness excites two classes of corneal afferent neurons implicated in basal tearing in rats: involvement of transient receptor potential channels. J Neurophysiol 107(4):1199–1209. doi:10.1152/jn.00657.2011
Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K (2004) Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279(18):18887–18894
Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397(6716):259–263. doi:10.1038/16711
Hofmann T, Schaefer M, Schultz G, Gudermann T (2000) Transient receptor potential channels as molecular substrates of receptor-mediated cation entry. J Mol Med (Berl) 78(1):14–25
Hofmann T, Schaefer M, Schultz G, Gudermann T (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99(11): 7461–7466. doi:10.1073/pnas.102596199
Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI (2008) High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am J Respir Cell Mol Biol 38(4):386–392. doi:10.1165/rcmb.2007-0192OC
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427(6971):260–265. doi:10.1038/nature02282
Jugg BJ, Smith AJ, Rudall SJ, Rice P (2011) The injured lung: clinical issues and experimental models. Phil Trans Royal Soc Lond Series B Biol Sci 366(1562):306–309. doi:10.1098/rstb.2010.0235
Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T, Nilius B (2007) Bimodal action of menthol on the transient receptor potential channel TRPA1. The J Neurosci: Official J Soc Neurosci 27(37):9874–9884. doi:10.1523/JNEUROSCI.2221-07.2007
Kawasaki BT, Liao Y, Birnbaumer L (2006) Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc Natl Acad Sci USA 103(2):335–340
Kehe K, Szinicz L (2005) Medical aspects of sulphur mustard poisoning. Toxicology 214(3): 198–209. doi:10.1016/j.tox.2005.06.014
Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, Li Y, Nakagawa Y, Usami S, Fujiwara M, Yamada Y, Minami T, Ueshima K, Nakao K (2010) Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res 106(12): 1849–1860. doi:10.1161/CIRCRESAHA.109.208314
Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA (2010) Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol 48(4):713–724. doi:10.1016/j.yjmcc.2009.11.015
Kunichika N, Yu Y, Remillard CV, Platoshyn O, Zhang S, Yuan JX (2004) Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 287(5):L962–L969
Kwan HY, Huang Y, Yao X (2004) Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Nat Acad Sci USA 101(8):2625–2630. doi:101/8/2625 [pii]
Lassegue B, San Martin A, Griendling KK (2012) Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110(10):1364–1390. doi:10.1161/CIRCRESAHA.111.243972
Lee IT, Yang CM (2012) Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol 84(5):581–590. doi:10.1016/j.bcp. 2012.05.005
Lembrechts R, Brouns I, Schnorbusch K, Pintelon I, Timmermans JP, Adriaensen D (2012) Neuroepithelial bodies as mechanotransducers in the intrapulmonary airway epithelium: involvement of TRPC5. Am J Respir Cell Mol Biol 47(3):315–323. doi:10.1165/rcmb.2012-0068OC
Li S, Westwick J, Poll C (2003) Transient receptor potential (TRP) channels as potential drug targets in respiratory disease. Cell Calcium 33(5–6):551–558. doi:S0143416003000605 [pii]
Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L (2009) A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Nat Acad Sci USA 106(9):3202–3206. doi:0813346106 [pii]10.1073/pnas.0813346106
Liedtke W (2007) TRPV channels’ role in osmotransduction and mechanotransduction. Handb Exper Pharmacol 179:473–487. doi:10.1007/978-3-540-34891-7_28
Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS (2004) Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95(5):496–505. doi:10.1161/01.RES.0000138952.16382.ad
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+−store-depletion-triggered Ca2+ influx. Curr Biol 15(13):1235–1241
Lorenzo IM, Liedtke W, Sanderson MJ, Valverde MA (2008) TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci USA 105(34):12611–12616. doi:10.1073/pnas.0803970105
Lundberg JM, Brodin E, Hua X, Saria A (1984) Vascular permeability changes and smooth muscle contraction in relation to capsaicin-sensitive substance P afferents in the guinea-pig. Acta Physiol Scand 120(2):217–227
Lundblad L, Saria A, Lundberg JM, Anggard A (1983) Increased vascular permeability in rat nasal mucosa induced by substance P and stimulation of capsaicin-sensitive trigeminal neurons. Acta Otolaryngol 96(5–6):479–484
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007a) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445(7127):541–545. doi:10.1038/nature05544
Macpherson LJ, Xiao B, Kwan KY, Petrus MJ, Dubin AE, Hwang S, Cravatt B, Corey DP, Patapoutian A (2007b) An ion channel essential for sensing chemical damage. J Neurosci: Official J Soc Neurosci 27(42):11412–11415. doi:10.1523/JNEUROSCI.3600-07.2007
Malis DD, Rist B, Nicoucar K, Beck-Sickinger AG, Morel DR, Lacroix JS (2001) Modulatory effect of two novel CGRP receptor antagonists on nasal vasodilatatory responses to exogenous CGRP, capsaicin, bradykinin and histamine in anaesthetised pigs. Regul Pept 101(1–3):101–108
Materazzi S, Nassini R, Gatti R, Trevisani M, Geppetti P (2009) Cough sensors. II. Transient receptor potential membrane receptors on cough sensors. Handb Exper Pharmacol 187:49–61. doi:10.1007/978-3-540-79842-2_3
Matta JA, Cornett PM, Miyares RL, Abe K, Sahibzada N, Ahern GP (2008) General anesthetics activate a nociceptive ion channel to enhance pain and inflammation. Proc Natl Acad Sci USA 105(25):8784–8789. doi:10.1073/pnas.0711038105
McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416(6876):52–58. doi:10.1038/nature719
McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM (2007) TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci USA 104(33):13525–13530. doi:10.1073/pnas.0705924104
Mergler S, Garreis F, Sahlmuller M, Reinach PS, Paulsen F, Pleyer U (2011) Thermosensitive transient receptor potential channels in human corneal epithelial cells. J Cell Physiol 226(7): 1828–1842. doi:10.1002/jcp. 22514
Miller K, Chang A (2003) Acute inhalation injury. Emerg Med Clin North Am 21(2):533–557
Minke B (1977) Drosophila mutant with a transducer defect. Biophys Struct Mech 3(1):59–64
Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2(4):1313–1323
Montell C, Jones K, Hafen E, Rubin G (1985) Rescue of the Drosophila phototransduction mutation trp by germline transformation. Science 230(4729):1040–1043
Montell C, Birnbaumer L, Flockerzi V (2002) The TRP channels, a remarkably functional family. Cell 108(5):595–598
Muhle H, McClellan R (2003) Respirationstrakt. In: Marquardt H, Schäfer S (eds) Lehrbuch der Toxikologie, vol 2. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, pp 365–382
Mukhopadhyay I, Gomes P, Aranake S, Shetty M, Karnik P, Damle M, Kuruganti S, Thorat S, Khairatkar-Joshi N (2011) Expression of functional TRPA1 receptor on human lung fibroblast and epithelial cells. J Recept Signal Transduct Res 31(5):350–358. doi:10.3109/10799893.2011.602413
Muller T, Hengstermann A (2012) Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem Res Toxicol 25(9):1805–1824. doi:10.1021/tx300145n
Murata Y, Masuko S (2006) Peripheral and central distribution of TRPV1, substance P and CGRP of rat corneal neurons. Brain Res 1085(1):87–94. doi:10.1016/j.brainres.2006.02.035
Nilius B, Owsianik G (2011) The transient receptor potential family of ion channels. Genome Biol 12(3):218. doi:10.1186/gb-2011-12-3-218
Nishida M, Watanabe K, Sato Y, Nakaya M, Kitajima N, Ide T, Inoue R, Kurose H (2010) Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem 285(17):13244–13253. doi:10.1074/jbc.M109.074104
Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y (1999) Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem 274(39):27359–27370
Owsianik G, Talavera K, Voets T, Nilius B (2006) Permeation and selectivity of TRP channels. Ann Rev Physiol 68:685–717. doi:10.1146/annurev.physiol.68.040204.101406
Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, Bergstrom DE (2004) Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 18(5):486–491. doi:10.1101/gad.1172504
Parra A, Madrid R, Echevarria D, del Olmo S, Morenilla-Palao C, Acosta MC, Gallar J, Dhaka A, Viana F, Belmonte C (2010) Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat Med 16(12):1396–1399. doi:10.1038/nm.2264
Pedersen SF, Owsianik G, Nilius B (2005) TRP channels: an overview. Cell Calcium 38(3–4): 233–252. doi:10.1016/j.ceca.2005.06.028
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002a) A TRP channel that senses cold stimuli and menthol. Cell 108(5):705–715
Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, Story GM, Colley S, Hogenesch JB, McIntyre P, Bevan S, Patapoutian A (2002b) A heat-sensitive TRP channel expressed in keratinocytes. Science 296(5575):2046–2049. doi:10.1126/science.1073140
Perez CA, Huang L, Rong M, Kozak JA, Preuss AK, Zhang H, Max M, Margolskee RF (2002) A transient receptor potential channel expressed in taste receptor cells. Nat Neurosci 5(11): 1169–1176. doi:10.1038/nn952
Petersson G, Malm L, Ekman R, Hakanson R (1989) Capsaicin evokes secretion of nasal fluid and depletes substance P and calcitonin gene-related peptide from the nasal mucosa in the rat. Br J Pharmacol 98(3):930–936
Plant TD, Strotmann R (2007) Trpv4. Handb Exper Pharmacol 179:189–205. doi:10.1007/978-3-540-34891-7_11
Pohl C, Papritz M, Moisch M, Wubbeke C, Hermanns MI, Uboldi C, Dei-Anang J, Mayer E, Kirkpatrick CJ, Kehe K (2009) Acute morphological and toxicological effects in a human bronchial coculture model after sulfur mustard exposure. Toxicol Sci: Official J Soc Toxicol 112(2):482–489. doi:10.1093/toxsci/kfp211
Preti D, Szallasi A, Patacchini R (2012) TRP channels as therapeutic targets in airway disorders: a patent review. Expert Opin Ther Pat 22(6):663–695. doi:10.1517/13543776.2012.696099
Quirce S, Barranco P (2010) Cleaning agents and asthma. J Invest Allergol Clin Immunol: Official Organ Inter Assoc Asthmol 20(7):542–550; quiz 542p following 550
Reichenberger F, Voswinckel R, Enke B, Rutsch M, El Fechtali E, Schmehl T, Olschewski H, Schermuly R, Weissmann N, Ghofrani HA, Grimminger F, Mayer E, Seeger W (2007) Long-term treatment with sildenafil in chronic thromboembolic pulmonary hypertension. Eur Respir J: Official J Eur Soc Clin Respir Physiol 30(5):922–927. doi:10.1183/09031936.00039007
Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I (2010) Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 182(6):774–783. doi:10.1164/rccm.201003-0359OC
Robbins A, Kurose M, Winterson BJ, Meng ID (2012) Menthol activation of corneal cool cells induces TRPM8-mediated lacrimation but not nociceptive responses in rodents. Invest Ophthalmol Vis Sci 53(11):7034–7042. doi:10.1167/iovs.12-10025
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169(3):435–445
Rybak LP, Mukherjea D, Jajoo S, Kaur T, Ramkumar V (2012) siRNA-mediated knock-down of NOX3: therapy for hearing loss? Cell Mol Life Sci: CMLS 69(14):2429–2434. doi:10.1007/s00018-012-1016-3
Salathe M, Bookman RJ (1995) Coupling of [Ca2+]i and ciliary beating in cultured tracheal epithelial cells. J Cell Sci 108(Pt 2):431–440
Salazar H, Llorente I, Jara-Oseguera A, Garcia-Villegas R, Munari M, Gordon SE, Islas LD, Rosenbaum T (2008) A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nat Neurosci 11(3):255–261. doi:10.1038/nn2056
Samal A, Honovar J, White CR, Patel RP (2010) Potential for chlorine gas-induced injury in the extrapulmonary vasculature. Proc Am Thorac Soc 7(4):290–293. doi:10.1513/pats.201001-006SM
Sanderson H, Fauser P, Thomsen M, Sorensen PB (2008) Screening level fish community risk assessment of chemical warfare agents in the Baltic Sea. J Hazard Mater 154(1–3):846–857. doi:10.1016/j.jhazmat.2007.10.117
Sanderson H, Fauser P, Thomsen M, Vanninen P, Soderstrom M, Savin Y, Khalikov I, Hirvonen A, Niiranen S, Missiaen T, Gress A, Borodin P, Medvedeva N, Polyak Y, Paka V, Zhurbas V, Feller P (2010) Environmental hazards of sea-dumped chemical weapons. Environ Sci Technol 44(12):4389–4394. doi:10.1021/es903472a
Sawada Y, Hosokawa H, Matsumura K, Kobayashi S (2008) Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci 27(5):1131–1142. doi:10.1111/j.1460-9568.2008.06093.x
Seki N, Shirasaki H, Kikuchi M, Sakamoto T, Watanabe N, Himi T (2006) Expression and localization of TRPV1 in human nasal mucosa. Rhinology 44(2):128–134
Seki N, Shirasaki H, Kikuchi M, Himi T (2007) Capsaicin induces the production of IL-6 in human upper respiratory epithelial cells. Life Sci 80(17):1592–1597. doi:10.1016/j.lfs.2007.01.037
Sel S, Rost BR, Yildirim AO, Sel B, Kalwa H, Fehrenbach H, Renz H, Gudermann T, Dietrich A (2008) Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin Exp Allergy 38(9):1548–1558
Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, Jones LC, O'Neal WK, Penuela S, Laird DW, Boucher RC, Lazarowski ER (2011) Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem 286(30):26277–26286. doi:10.1074/jbc.M111.260562
Shusterman D (1999) Upper and lower airway sequelae of irritant inhalations. Clin Pulm Med 6(1):18–31
Smit LA, Kogevinas M, Anto JM, Bouzigon E, Gonzalez JR, Le Moual N, Kromhout H, Carsin AE, Pin I, Jarvis D, Vermeulen R, Janson C, Heinrich J, Gut I, Lathrop M, Valverde MA, Demenais F, Kauffmann F (2012) Transient receptor potential genes, smoking, occupational exposures and cough in adults. Respir Res 13:26. doi:10.1186/1465-9921-13-26
Smith BR, Brian WR (1991) The role of metabolism in chemical-induced pulmonary toxicity. Toxicol Pathol 19(4 Pt 1):470–481
Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, Davis JB (2002) TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418(6894):186–190. doi:10.1038/nature00894
Stone KC, Mercer RR, Gehr P, Stockstill B, Crapo JD (1992) Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol 6(2):235–243
Storch U, Forst AL, Philipp M, Gudermann T, Mederos y Schnitzler M (2012) Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J Biol Chem 287(5):3530–3540. doi:10.1074/jbc.M111.283218
Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE (2001) TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29(3):645–655
Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE (2003) Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem 278(40): 39014–39019
Sweeney M, McDaniel SS, Platoshyn O, Zhang S, Yu Y, Lapp BR, Zhao Y, Thistlethwaite PA, Yuan JX (2002a) Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J Appl Physiol 92(4):1594–1602
Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX (2002b) Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283(1):L144–L155
Symanowicz PT, Gianutsos G, Morris JB (2004) Lack of role for the vanilloid receptor in response to several inspired irritant air pollutants in the C57Bl/6J mouse. Neurosci Lett 362(2):150–153. doi:10.1016/j.neulet.2004.03.016
Takahashi N, Mizuno Y, Kozai D, Yamamoto S, Kiyonaka S, Shibata T, Uchida K, Mori Y (2008a) Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2(4):287–298
Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R (2008b) Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol 586(Pt 17):4209–4223. doi:156083 [pii]10.1113/jphysiol.2008.156083
Takahashi N, Kuwaki T, Kiyonaka S, Numata T, Kozai D, Mizuno Y, Yamamoto S, Naito S, Knevels E, Carmeliet P, Oga T, Kaneko S, Suga S, Nokami T, Yoshida J, Mori Y (2011) TRPA1 underlies a sensing mechanism for O2. Nat Chem Biol 7(10):701–711. doi:10.1038/nchembio.640
Takahashi N, Kozai D, Mori Y (2012) TRP channels: sensors and transducers of gasotransmitter signals. Frontiers Physiol 3:324. doi:10.3389/fphys.2012.00324
Talavera K, Yasumatsu K, Voets T, Droogmans G, Shigemura N, Ninomiya Y, Margolskee RF, Nilius B (2005) Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature 438(7070):1022–1025. doi:10.1038/nature04248
Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T (2009) Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 12(10):1293–1299. doi:10.1038/nn.2379
Taylor-Clark TE, Ghatta S, Bettner W, Undem BJ (2009) Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol Pharmacol 75(4):820–829. doi:10.1124/mol.108.054445
Thurlbeck WM (1967) The internal surface area of nonemphysematous lungs. Am Rev Respir Dis 95(5):765–773
Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB (2002) Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res 91(1):70–76
Tominaga M (2007) The role of TRP channels in thermosensation. In: Liedtke WB, Heller S (eds) TRP ion channel function in sensory transduction and cellular signaling cascades. CRC Press, Boca Raton
Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21(3):531–543
Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW Jr (2005) Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol 67(2):558–563
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci USA 104(33): 13519–13524. doi:10.1073/pnas.0705923104
Tuorinsky S, Sciuto A (2008) Toxic inhalation injury and toxic industrial chemicals. In: Lenhart M (ed) Medical aspects of chemical warfare. Office of the General Surgeon, Washington, DC, pp 339–370
Urbanetti J (1997) Toxic inhalation injury. In: Sidell F, Takafuji E, Franz D (eds) Medical aspects of chemical and biological warfare. Office of the Surgeon General, Washington, DC, pp 247–270
Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312(5777):1220–1223. doi:1127883 [pii]10.1126/science.1127883
Wang S, Dai Y, Fukuoka T, Yamanaka H, Kobayashi K, Obata K, Cui X, Tominaga M, Noguchi K (2008) Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain: J Neurol 131(Pt 5):1241–1251. doi:10.1093/brain/awn060
Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B (2002) Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem 277(49):47044–47051. doi:10.1074/jbc.M208277200
Weinberger B, Laskin JD, Sunil VR, Sinko PJ, Heck DE, Laskin DL (2011) Sulfur mustard-induced pulmonary injury: therapeutic approaches to mitigating toxicity. Pulm Pharmacol Ther 24(1): 92–99. doi:10.1016/j.pupt.2010.09.004
Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T (2006) Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Nat Acad Sci USA 103(50):19093–19098
Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A (2012) Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nature Commun 3:649. doi:10.1038/ncomms1660
White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME (2006) Role of transient receptor potential C3 in TNF-alpha-enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol 35(2):243–251. doi:2006-0003OC [pii]10.1165/rcmb.2006-0003OC
Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, DiStefano PS, Curtis R, Clapham DE (2002) TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418(6894):181–186. doi:10.1038/nature00882
Yin J, Kuebler WM (2010) Mechanotransduction by TRP channels: general concepts and specific role in the vasculature. Cell Biochem Biophys 56(1):1–18. doi:10.1007/s12013-009-9067-2
Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX (2004) Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101(38):13861–13866
Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX (2009) A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119(17):2313–2322. doi:10.1161/CIRCULATIONAHA.108.782458
Yuan JP, Kim MS, Zeng W, Shin DM, Huang G, Worley PF, Muallem S (2009a) TRPC channels as STIM1-regulated SOCs. Channels 3(4):9198 [pii]
Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S (2009b) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11(3):337–343. doi:ncb1842 [pii]10.1038/ncb1842
Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, Zuker CS, Ryba NJ (2003) Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell 112(3):293–301
Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437(7060):902–905
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Büch, T., Schäfer, E., Steinritz, D., Dietrich, A., Gudermann, T. (2013). Chemosensory TRP Channels in the Respiratory Tract: Role in Toxic Lung Injury and Potential as “Sweet Spots” for Targeted Therapies. In: Nilius, B., et al. Reviews of Physiology, Biochemistry and Pharmacology, Vol. 165. Reviews of Physiology, Biochemistry and Pharmacology, vol 165. Springer, Cham. https://doi.org/10.1007/112_2012_10
Download citation
DOI: https://doi.org/10.1007/112_2012_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-00998-8
Online ISBN: 978-3-319-00999-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)