
Abstract
Cultivation-independent assessment of bacterial viability is essential when (1) results are required fast and at high throughput, and/or (2) when the specific target or mode-of-action of a certain bactericidal process is of interest, and/or (3) when the organisms under investigation are regarded as “uncultivable”. However, aside from cultivation, there exists no “silver bullet” method that demonstrates with absolute certainty whether an organism is alive or dead, and all currently available methods are prone to produce varying results with different organisms and in different environments. Here we discuss the fundamental concept of viability in bacteria, with specific focus on the main aspects that define it. It is argued that the presence of intact and functional nucleic acids, as well as an intact and polarized cytoplasmic membrane are essential components of cellular viability, while numerous other parameters and processes that are linked to viability are explored. Different methods/approaches are discussed with particular emphasis on the advantages and disadvantages of each approach, the applicability of the methods toward environmental samples, and the underlying link between the various viability parameters.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Knowing what is alive and what is dead, as well as what transpires in between those two extremities, is one of the main challenges that microbiologists are facing across all fields of application on a daily basis. The information arising from this knowledge affects people in their daily lives. For example, the production of wine, beer, cheese and yogurt requires well-defined viable microbial starting cultures as well as quality control during the production process [44]. Similarly, the productivity of industrial fermentations that utilise microbial cultures to synthesize metabolites and recombinant proteins can be controlled and optimised through real-time monitoring and increased understanding of the viability and activity of the vector organism [41, 70]. Drinking water and wastewater treatment systems often utilise disinfection processes as critical steps to safeguard the public against microbiological diseases. The dosages of disinfectants and the constant monitoring of the efficacy of such processes, are directly dependent on accurate viability assessment [23, 53, 72]. In medical-related research, a well-known application of viability assessment is testing the efficacy and mode of action of current and new antibiotics [42, 51]. Furthermore, viability assessment is used in research laboratories worldwide to understand the fundamental processes that drive growth, survival and die-off of bacteria in both carefully controlled laboratory environments [4] as well as in complex natural ecosystems [63].
While accurate viability assessment of bacteria is clearly important, it is far from simple. First and foremost, it boils down to the essential question of what exactly defines “life” and “death” for bacteria? This becomes a near-philosophical discussion, initiated already half a decade ago by Postgate [56] and aptly summarised by Roszak and Colwell [60], in which opinions remain highly subjective. Secondly, viability assessment requires a fundamental understanding of the various parameters that constitute and define life or death for a bacterium, relative to both the individual organism and the environment in which it is studied. Moreover, it requires tools that can determine these viability parameters in an accurate and unambiguous manner, also distinguishing between parameters that change immediately upon cell death, and those that are affected in a time-dependent manner. As will be discussed below, these issues alone already pose daunting challenges to researchers and practitioners. Thirdly, viability assessment is complicated by the fact that bacteria represent a broad and complex group of organisms [66]. Based on 16S rRNA sequences, there are in excess of 50 recognized bacterial phyla [58, 62], which in layman’s terms mean groups of bacteria as genetically distinct from each other as a snail is from a human. There are in these phyla an estimated 1–10 million bacterial species, and these organisms exhibit immense heterogeneity on multiple levels including physical properties (e.g., size, shape and composition), nutritional behaviour (e.g., oligotrophy vs. copiotrophy) and physiological states (e.g., dormancy or exponential growth). For a simple example, some organisms have Gram-positive cell walls, while others have Gram-negative cell walls, S-layers, sheaths, etc. The cell wall is the primary barrier between the cell and the environment, and the composition thereof is known to affect the action, functionality and interpretation of some commonly used viability stains [5, 66]. Some bacteria are able to thrive in a high cell density fast growing environment (e.g., fed-batch bioreactors) while others can persist in low cell concentrations under near-starvation conditions (e.g., groundwater). Different growth conditions are likely to affect the way in which bacteria are perceived in terms of viability and activity (e.g., RNA content or growth rates). Moreover, bacterial heterogeneity is not limited to broad groups, but is known to occur even within a single species growing in the same environment [13, 47], further highlighting the need for meaningful analysis on single-cell level.
Technological advances during the last 30 years, specifically the development of powerful epi-fluorescence microscopes and the accompanying methodologies of fluorescent staining, have largely facilitated the rise of fast, cultivation-independent microbial methods. Moreover, the focus has shifted from analysing bulk sample parameters to analysing individual organisms on single-cell level [13]. In fact, improvements in laser technology and microfluidics have pushed single-cell analysis to the point where basic bench top flow cytometers are common instruments in research laboratories [22], and where inline/online instrumentation is a reality rather than a vision [3, 18]. These practical advances have facilitated fundamental shifts in the perception and understanding of microbial diversity and heterogeneity, both within pure cultures and in natural environments [13, 47].
This paper approaches cultivation-independent viability assessment from the perspective of “what is life or death for a bacterium” and how various viability parameters relate to this. The purpose is to demonstrate the subjective nature of the topic, to provide insights in how this topic can be approached in research and/or routine monitoring, as well as to highlight the practical value gained from such information. Specific focus is given to high throughput single-cell methods, typically requiring microscopy or flow cytometry for analysis. The first section covers the cultivation paradox, highlighting the need for cultivation-independent methods for viability assessment, while the second part explores the fundamental aspects of bacterial viability. In the third section, different bacterial processes related to viability are discussed in the context of tools used for the analysis of these processes. The take home message is that the proverbial “silver bullet” for viability assessment remains (and will probably remain) illusive, but an intelligent combination of available methods can be (and have already been) used to obtain interesting and new information on diverse aspects of bacterial viability.
For additional reading, five recent review papers address the current state-of-the-science with broader focus on flow cytometric applications in general [15, 16, 22, 46, 67], while the history of cultivation-independent viability research is outlined in the thorough review papers of Roszak and Colwell [60], McFeters et al. [45], Kell et al. [34], Nebe-von Caron et al. [49] and Joux and Lebaron [31].
2 Viability Versus Cultivability
2.1 Cultivability: The Gold Standard
While the present paper deals specifically with cultivation-independent viability analysis, the basic fact is that cultivation (including conventional plating, most probable number (MPN), and direct viable counts (DVC) remains the single most solid evidence of viability [34], if the appropriate growth medium and cultivation conditions for the organism are known [60]. All bacteria are in theory cultivable, for the simple reason that they have grown/divided in order to exist. Moreover, at least one school of thought on the topic of viability argues that an organism has to be cultivable in order to be classified as viable [56, 60], and several disciplines (e.g., drinking water analysis) still rely heavily on cultivation as primary method for microbial analysis. However, numerous strong arguments exist in favour of looking beyond cultivation alone when describing viability, and these are explored below.
2.2 Time and Throughput
Cultivation can be a time-consuming analysis method, with growth to detectable colony formation taking from 1 day to well over 1 month, depending on the organisms and the growth media. Notably, some advanced cultivation methods (e.g., micro-colony counting and DVC) improve the time problem to some extent [2]. Nonetheless, there are numerous cases, particularly in industrial and hygienic applications, where a long time lapse would not suffice the purpose of the analysis. For example, if a microbiological problem is detected in a drinking water disinfection step only 3 days after the actual event, any corrective action will only occur after the water has already reached the consumer. Similar examples can be found in food and beverage industries. Indeed, the aim for microbiological analysis should be to achieve accurate measurements in a matter of minutes, rather than hours or days. Moreover, cultivation is a labour intensive method that does not allow high throughput detection similar to automated microscopy, solid-phase cytometry or flow cytometry.
2.3 Dealing with So-called “Uncultivable” Bacteria
One of the strongest arguments in favour of cultivation-independent analysis is the analysis of indigenous environmental bacteria. Firstly, for the vast majority of environmental bacteria, appropriate cultivation methods are simply not known [58, 69], thus nullifying the use of conventional cultivation-based approaches. Secondly, it is highly unlikely that all organisms in the same environmental sample would share the same synthetic cultivation medium, rendering this approach inappropriate for analysis of a diverse indigenous microbial community. Notably, some recent studies have addressed this problem by cultivating bacteria in sterile media prepared from the same aqueous environments from which the targeted organisms originated (e.g., [69]). Thirdly, several reasons such as extremely slow growth of specific organisms might render cultivation impractical to detect [49]. Organisms that do not grow readily on conventional growth media are broadly (and most probably erroneously) referred to as “uncultivable/unculturable” bacteria. In the case of environmental samples, it is clear that only cultivation-independent analysis can provide the much-needed information when viability is assessed, which makes it the most appropriate approach for analysis of samples from e.g., wastewater, drinking water and natural surface waters [6, 63].
2.4 The Viable-But-Not-Cultivable (VBNC) Paradigm
The possibility of VBNC bacteria is often highlighted as a key reason for doing cultivation-independent viability analysis [28]. In short: the argument is that normally cultivable bacteria under certain conditions become uncultivable on media that they previously grew on, while maintaining other measurable viability signs [34, 52]. However, it would not be over-critical to state that the VBNC term is used very liberally in peer-reviewed literature, often with scant regard for the basic concepts of viability, which it apparently describes. The most common VBNC example is the use of temperature shifts to regulate cultivability of Vibrio vulnificus, with cold-stored cells losing their ability to grow on standard media, and regaining it through a temperature upshift [52]. Another example was shown by Berney et al. [4] with E. coli exposed to low levels of solar radiation stress, no longer growing on the standard cultivation media, but still appearing viable with cultivation-independent parameters. At first sight, these two examples seem to underpin the VBNC concept. However, in the latter case of the stressed E. coli cells, it was demonstrated that a considerable percentage of the “uncultivable” cells were capable of growing anaerobically on medium that was specifically supplemented with sodium pyruvate (which scavenges reactive oxygen species) [4], showing that the cells were indeed cultivable if the appropriate medium is used. Similarly, Bogosian and Bourneuf [7] described a control experiment that showed that the cold-stored uncultivable V. vulnificus cells were probably injured cells that were merely sensitive to the high concentrations of hydrogen peroxide found in rich cultivation media. In fact, the brilliant critical review by Kell et al. [34], later mirrored by Bogosian and Bourneuf [7], needs to be illuminated in the VBNC context. This has to serve as a blueprint for VBNC advocates for the experimental standard to which this concept should be held, and the importance of careful use of this terminology. The somewhat critical opinion of VBNC data expressed herein is not meant to undermine the efforts of understanding different physiological states in bacteria, but rather to emphasize the care that should be taken with viability analysis tools, and the expression of data generated with these methods.
2.5 A Description of Specific Injuries and Slow Death
There is another clear advantage of cultivation-independent methods over cultivation-based viability methods. Cultivation provides only a binary (presence/absence) result, i.e. the organisms are either detected as cultivable or not, a result which is then usually interpreted as meaning alive or dead. But this type of result limits the researcher in his/her understanding of the process that brought about the result. The fundamental aspects of cell death, such as the sequence of damage or type of injury caused by a certain bactericidal agent (e.g., [8]), or the specific targets of different bactericidal agents and the kinetics of damage to these targets (e.g., [51]), all requires specific tools that can explore individual cellular processes and mechanisms in more detail than a simplistic yes/no answer. The fact that cultivation-independent viability analysis targets a variety of individual cellular processes renders this approach meaningful and suitable to address such complex questions.
3 How Dead is Dead?
In the absence of cultivation-based methods, the determination of viability becomes a difficult and contentious topic. Argumentatively, three main cellular components are prerequisites for life, namely: (1) the presence of functional nucleic acids, allowing transcription/translation and DNA replication, (2) the presence of minimum cellular energy, allowing basic functioning of cellular processes, and (3) the presence of an intact and functional cellular membrane, maintaining the unique intracellular environment (Fig. 1). These three components are inherently linked to one another as well as with other cellular components and viability parameters, and without them, a cell cannot be considered as viable.

For all bacteria, one of the core processes essential for life is the maintenance of a membrane potential. The membrane potential is the voltage difference between the interior and exterior of a cell (usually between 70 and 200 mV in bacteria) and in microorganisms this is typically generated by the electron transport chain (proton translocation) or enzymes like oxaloacetate decarboxylase [37] that can translocate sodium ions (Fig. 2). The membrane potential (∆φ) and the pH gradient (Z∆pH) produce the proton motive force (also called the electrochemical gradient), which drives ATP synthesis via the F1FO ATP synthase in most bacteria [17]. Membrane potential is used for (1) generating energy (ATP synthase), (2) driving active transport of molecules across the membrane (such as the active efflux/uptake of potassium ions), (3) enabling motility (flagella), and (4) keeping the cytoplasm from equilibrating with the environment (like maintaining a specific intracellular pH) (Fig. 2). Technically, a bacterial cell that cannot maintain a membrane potential can be considered dead, since this will lead to an inhibition of selective exchange of molecules across the cytoplasmic membrane like ions, metabolites, amino acids or carbon and energy sources. For example, the maintenance of a high intracellular concentration of potassium is essential for protein and nucleic acid synthesis and turgor pressure. The difference between intracellular and extracellular potassium concentration can be up to 100-fold (5 M intracellular, 0.05 M extracellular) as for the bacterium Halobacterium salinarium [43]. Likewise, sodium ion homeostasis is essential for numerous sodium/solute symporters present in bacterial membranes [32]. Generally speaking, without a membrane potential, cells cannot maintain an intracellular environment that enables the functioning of life-supporting metabolic processes and the cells become more vulnerable to their environment. This additionally means that most other cellular functions that are measured with viability indicators (e.g., efflux pump activity, substrate uptake, ATP synthesis) are dependent or linked to a membrane potential. Hence, measuring the membrane potential, or the dissipation thereof, is in theory a good discriminator between living and dead cells.

The importance of membrane potential for bacterial cells. Membrane potential is produced by the electron transport chain comprising of, e.g. complex I–IV (I NADH dehydrogenase, II succinate dehydrogenase, III cytochrome bc 1 complex, IV terminal oxidase, e.g. cytochrome c oxidase) and/or sodium-pumping decarboxylases (e.g., oxaloacetate decarboxylase). Membrane potential powers processes like the ATP synthase, solute-ion symporter, e.g. (porline/Na+), ion antiproters (e.g., H+/K+), efflux pumps (e.g., ethidium) and motility
However, a cornerstone to the above argument in favour of membrane potential as a main viability indicator is the principle that a bacterium should be able to “maintain” its membrane potential: a bacterium that experiences a lethal stress does not necessarily disintegrate immediately, nor does it lose its membrane potential on the spot. In most cases, death occurs with a time-dependent shutdown of cellular processes (in minutes or hours) that is dependent on the type of lethal agent (e.g., antibiotics, H2O2, chlorine, UV light or starvation), the extent of the cellular damage, and the ability of the cell to recover form injury (Fig. 1). For example, cells of Salmonella typhimurium that have been exposed to UV-A light for 2 h, retained a membrane potential and the ability to actively transport glucose immediately after irradiation but the majority of the cells lost these abilities after 24 h dark storage [8]. These same authors demonstrated in a subsequent study that a major cause of cell death is irreversible damage to cellular proteins [9]. This is a clear example of lethal injury requiring a certain timeframe to manifest in detectable viability parameters, highlighting the dangers of rash interpretations of viability staining.
4 Specific Processes that can be Measured with Viability Indicators
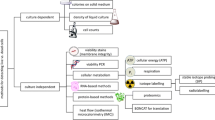
The following section covers a variety of cellular processes, functions and parameters that are linked to bacterial viability, and for which in many cases well-developed methodology is already available (Fig. 3). It deals mostly with fluorescent dyes (or substrates that become fluorescent upon intracellular cleavage or binding) that are used in combination with single-cell methods such as epi-fluorescence microscopy, solid-phase cytometry and particularly flow cytometry. Examples of individual stains, methods and applications are discussed, but it is important that the reader does not regard this information as methodological standard protocols. All too often these stains are used in a manner “according-to-the-manufacturer’s-guidelines”, which completely disrespects the complex interplay of the stains with the target organism, the different environments and the basic concepts of viability. It is strongly encouraged that individual stains/protocols should be tested in detail and rigorously optimised (e.g., stain selection, stain concentrations, staining times, use of specific staining buffers, use of fixation and permeabilisation reagents and appropriate positive and negative controls) in a manner relevant to the sample that is being investigated. In this respect, Sträuber and Müller [66] highlighted the importance of understanding the mechanisms of specific stains and their interaction with different types of bacteria. Table 1 provides an overview of some of the criteria and related information that are required to make an informed decision on a specific stain and staining protocol, with an example of membrane integrity staining provided. While several examples of stains and dyes are discussed in the section below, it is noted that the range of available products is significantly broader and expanding on a regular basis. A good tabular overview of viability stains can be found in Tracy et al. [67].

The different processes are discussed as individual entities for the sake of clarity, but it is emphasized that these separate processes are often linked directly to each other, forming part of a broader viability concept or continuum (Fig. 1) [49]. Therefore, these processes (and the related methodologies) should preferably not be viewed or used in isolation. Moreover, it is important to realise that the parameters measured in typical viability assessments are usually not measuring viability as such, but rather provide information about a specific cellular function that can be related to viability (Fig. 3). Hence, if the mode of action of a bactericidal agent is known (e.g., oxidative membrane damage by chlorine dioxide), specific viability indicators can be used to assess the mode of action of interest. However, if a biocidal agent has multiple target sites (e.g., UV-A light), or if a completely unknown sample is analysed (e.g., natural surface water), viability stains are best used in concert (e.g., [4, 28, 40, 51, 70]). This enables the researcher to derive a meaningful interpretation of the sample in question.
4.1 Presence of (Intact) Nucleic Acids
4.1.1 Principle
Without intact and functional nucleic acids, a cell will not be able to replicate or produce any proteins and thus perform even the most basic cellular functions including repair, stress response and cell division. Cells without nucleic acids are described in literature as ghost cells [31], and are regarded as irreversibly dead (Fig. 1).
4.1.2 Methodology
Well-known total cell count stains such as SYTO 9, SYBR Green I and II, PicoGreen and Acridine Orange can be used to stain the nucleic acids of bacteria [23, 38]. From a practical perspective, it is important to ensure that the outer membrane is properly permeabilised (e.g., by adding EDTA) during nucleic acid staining, since it was shown previously that some Gram-negative bacteria partially reject such stains (Fig. 4) [5]. Schumann et al. [63] makes the relevant point that partially disintegrated dead cells without a chromosome would go undetected with any nucleic acid targeting stains, and this was elucidated by Phe et al. [54], who showed that propidium iodide cannot bind to cellular nucleic acids that were damaged extensively by progressive chlorination, leaving such cells undetected. Argumentatively, cells that are damaged to an extent that nucleic acids can no longer be detected should perhaps not even be recognised as cells, but rather as organic particles. As an alternative, a recent publication by Saint-Ruf et al. [61] proposed the use of highly fluorescent hydrazides that do not bind to nucleic acids but rather to carbonyl groups on intracellular proteins, for the detection of dead cells where nucleic acids might already be absent or damaged to an extent that does not allow binding of conventional nucleic acid stains. However, while the absence of nucleic acids is a certain “death” indicator, the mere presence of nucleic acids is not a measurement of viability. For example, none of the general stains that are mentioned above are sensitive enough to detect the specific nucleic acid damage caused by UV-C disinfection, commonly used in drinking water treatment. The inactivation of bacteria by far-ultraviolet (190–300 nm) radiation results from the absorption of the radiation by the DNA, causing the formation of thymine dimers that distort the conformation of the DNA double helix and interfere with cell replication and transcription [1, 25]. A recent paper describes the use of a monoclonal antibody against cyclobutyl thymine dimers (anti-TDmAb) that results in dimer specific fluorescence [1]. This can be used on a single-cell level and has been demonstrated to detect UV damage in protozoan parasites, using epi-fluorescence microscopy, but a general, broad-based application of this approach has not yet been described. Moreover, it should be emphasized that such an approach is limited to a detection of cellular injury, but does not describe the extent of the injury (lethal or not), or the ability of the cell to repair the damaged nucleic acids. It is also possible to distinguish between the DNA and RNA content of cells by using a specific staining combination such as Hoechst 33342 or DAPI (for DNA content) and Pyronin Y (for RNA content) [64], while general stains such as SYBR Green I and II or Acridine Orange have apparently different affinities or fluorescence emission for DNA and RNA. Additionally, RNA staining in pure cultures can be accomplished with FISH probes [33].

The potential impact of pre-treatment in staining protocols: nucleic acid labelling of stationary phase Gram-negative E. coli cells with SYBR Green I without (a) and with (b) a pre-treatment step using a commercially available lysis buffer. Staining was done for 15 min at 25 °C; analysis was done with a Cyflow Space instrument (Partec)
4.1.3 Applications
Detection of nucleic acids is a basic check to determine if bacterial cells have one of the essential components of life, but is typically used for viability assessment only in situations where extremely aggressive damage to cells is expected. For example, assessment of non-specific damage to nucleic acids has been used to characterise disinfection of indigenous microbial communities during chlorination of drinking water [53, 54]. These authors have shown a considerable reduction in fluorescence intensity of cells stained with SYBR Green II after extensive chlorination, suggesting that this is linked to irreversible oxidative damage of cellular DNA and that this approach can be used as an indicator for complete disinfection. The efficacy of ozonation as disinfection process during drinking water treatment was similarly demonstrated with SYBR Green I and flow cytometry, which showed no detectable nucleic acids or cell structures after ozonation [23]. It has been proposed previously that the mere presence of specific clusters of high (HNA) and low (LNA) nucleic acid content bacteria, as visualised with common nucleic acid staining and flow cytometry (Fig. 5), differentiates between active and inactive bacteria in environmental samples [21, 39]. However, recent studies have directly questioned this interpretation (e.g., [69]), and it should be assumed that such a general approach is probably too simplistic, given the heterogeneity observed in indigenous microbial communities.

Double-staining of indigenous high (HNA) and low (LNA) nucleic acid content bacteria from river water with SYBR Green I and propidium iodide before (A1, A2) and after (B1, B2) cell wall permeabilisation using a commercially available lysis buffer. Staining was done for 15 min at 25 °C; analysis was done with a Cyflow Space instrument (Partec)
4.1.4 Opinion
Indigenous bacterial cells have widely different amounts of nucleic acids (both DNA and RNA), typified by the HNA and LNA bacteria found in nearly all natural water samples (Fig. 5) [69]. So the amount of cellular nucleic acids is per se not a useful indicator of life or death, especially when indigenous microbial communities are considered. While the complete lack of nucleic acids would be a clear indicator of cell death [63], it should be regarded as an extremely conservative approach to viability assessment. Firstly, only aggressive disinfection processes result in DNA damage that is detectable with general methods. Secondly, small but lethal damage to DNA (e.g., UV-C disinfection) would not be detected with this approach. The RNA content of bacteria is at best an indicator of cellular activity and not viability [60]. Moreover, when slow-growing or dormant cells are present in a sample, it is likely that RNA content of such cells is beneath the detection limit of most methods, while the cells remain essentially viable and even active.
4.2 Membrane Integrity
4.2.1 Principle
Bacterial membranes provide a highly regulated physical barrier between the intracellular and extracellular environment. Severe structural/physical damage to the cytoplasmic membrane of bacteria is usually irreversible and most likely leads to cell death [31]. An intact membrane can be detected through exclusion of molecules based on their molecular size, charge, hydrophobicity and presence of groups that cause steric hindrance for membrane diffusion [26, 63, 66].
4.2.2 Methodology
Membrane integrity staining was made famous by the unfortunately named “LIVE/DEAD” kit [10], containing propidium iodide (PI) as the selective molecule for permeabilised membranes. The name is “unfortunate” because it reflects hypothetical concepts of “life” or “death”, rather than a methodological concept that is assessed (membrane permeability) [63], and results gained from the application of this method are often wrongly interpreted as meaning “alive or dead”. PI is a large (668 Da), double-charged, red-fluorescent dye that intercalates with double-stranded DNA and normally only enters cells of which the cytoplasmic membrane of the cell is permeabilised [5, 10]. PI is commonly used in combination with a total nucleic acid stain such as SYBR Green I or SYTO 9 (Table 1; Fig. 5) [5, 19], and the specific flow cytometric patterns that can be detected in this manner are indicative of different degrees of cellular damage [5]. Several alternative stains are available, which provide the user with additional information of membrane damage. These include SYTOX Green, which is an asymmetrically triple-charged cyanine dye and ethidium homodimer-2 (Eth-D2) that consists of two phenanthridinium fluorophores and which is quadruply charged [36, 63]. In an interesting test, Schumann et al. [63] used these three stains together on natural aquatic communities and found decreasing dye permeability in the order of (1) PI, (2) SYTOX Green, and (3) Eth-D2, ascribing the result to the molecular structure of the dyes. These results suggest that the magnitude of membrane damage can be measured with the use of different dyes, but conclusive information in this regard is still required. Positive controls for membrane integrity staining include treatment with heat (90 °C, 3–5 min), ethanol, or with a membrane damaging detergent (Fig. 5).
4.2.3 Applications
Analysis of membrane damage is best used for viability assessment in situations where aggressive physico-chemical damage to cells is expected. This includes disinfection by heat [6], oxidants [40], several antibiotic compounds [51] and physical processes like sonication or electroperforation. PI has been used more than any other viability stain on both pure cultures and natural microbial communities from environmental samples [10, 19, 31]. For example, Berney et al. [6] showed with flow cytometric analysis that 70–80% of indigenous communities in drinking water displayed intact cytoplasmic membranes, while Schumann et al. [63] used epi-fluorescence microscopy to demonstrate that between 50 and 60% of indigenous surface water communities had intact membranes. However, these former examples are snapshot analysis of complex communities that reveals little about the processes leading to the membrane damage. From controlled laboratory scale experiments from the authors’ group, Berney et al. [4] and Bosshard et al. [8] described die-off of E. coli, Salmonella and Shigella cells from solar (UV-A) radiation with a range of viability parameters, demonstrating that cell membrane damage is typically the last level of damage to become detectable. Similarly, Lisle et al. [40] described the impact of chlorination on E. coli cells with multiple parameters, and also showed that membrane integrity was the last viability indicator to be disabled in the disinfection process.
4.2.4 Opinion
Most membrane integrity stains are applicable for pure cultures as well as indigenous communities, and analysis with epi-fluorescence microscopy and flow cytometry is typically yielding clear results. Membrane integrity analysis is, similar to nucleic acid damage, a conservative indicator for viability [4, 40]. It can be assumed that a cell with a severely damaged cytoplasmic membrane can be considered as “dead”, due to an inability to maintain a unique intracellular environment [5, 49], although some exceptions to this have been noted and discussed in some detail [47, 65]. However, the reverse argument (that cells with intact membranes are “alive”) is not necessarily true [5, 31]. A straightforward example is UV-C disinfection (discussed above), which would result in inactivation of bacteria without any immediate detectable damage to the cytoplasmic membrane. It is of importance to note that the value of membrane integrity stains is not to measure “life” or “death”, but to assess a particular location-specific damage to cells. Hence, both the application/purpose and the interpretation of data from membrane integrity analysis, as well as the manner and nomenclature in which such data are reported, should be considered with the utmost care.
4.3 Membrane Potential
4.3.1 Principle
As discussed in detail in Sect. 3 (above), only living cells are in theory capable of maintaining a membrane potential. An irreversible loss of membrane potential would expose the cell to potentially lethal stress factors (e.g., pH, salts) in its environment. The processes shown in Fig. 2 demonstrate that a loss of membrane potential would have profound impacts on several vital cellular functions and processes, eventually leading to cell death.
4.3.2 Methodology
The presence of a membrane potential selectively regulates the passage of lipophilic cationic and anionic charged molecules through the cytoplasmic membrane. Depending on the charge of the dye, such molecules can either accumulate in polarized (cationic dyes) or depolarised cells (anionic dyes) [11, 31]. For example, uptake of anionic bis-(1,3-dibutylbarbituric acid) tri-methine oxonol (DiBAC4(3)), also known as BOX, is limited to cells that are depolarized, upon which non-specific binding to intracellular proteins occurs [4, 26]. Alternatively, Rhodamine 123 (Rh123) is a polar cationic dye with a single delocalised positive charge, which crosses polarized membranes and accumulates in viable cells [20, 51]. Additional membrane potential dyes include 3,3′-dihexyloxacarbocyanine (DiOC6(3)), 3,3′-diethyloxacarbocyanine (DiOC2(3)) and 3,3′-dipropylthiadicarbocyanine (DiSC3(5)) [11, 50, 51]. Anionic dyes like DiBAC4(3) are typically used to obtain a yes/no answer about the membrane potential of the cell and are thus suitable as viability stains. Other stains like DiOC2(3) have been used to accurately measure the membrane potential and changes thereof in bacterial cells on a single-cell level [50]. For many of the membrane potential dyes, a pre-treatment step with EDTA is often needed to permeabilize the outer membrane [6, 31], but this treatment may by itself affect the cell’s membrane potential. One drawback is that these dyes are usually fluorescent as such, which means background fluorescence can be a problem when analysing such samples. A dilution or washing step can be useful to avoid this problem. The appropriate control for membrane potential staining is to use uncoupling agents such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or ionophores (e.g., nigericin, valinomycin) [31, 47, 51], while heat-killed cells (3 min, 90 °C) serve as a usable absolute negative control [6].
4.3.3 Applications
While some antibiotic compounds are thought to specifically target membrane potential [51], this parameter is often used as a sensitive and general indicator of the viability state of bacteria. Multiple reports have investigated the use of membrane potential stains for the analysis of bacterial pure cultures during fed-batch fermentations, with the general conclusion that this method can provide fast and useful information for monitoring industrial processes [27, 70]. Novo et al. [51] have used a combination of membrane integrity and membrane potential dyes to analyse and describe the mode of action of a range of antibiotics on pure cultures. A typical example is streptomycin, which causes K+ efflux and an inhibition of respiration in bacterial cells, both of which are factors that would contribute to membrane depolarisation. In contrast, chloramphenicol, which targets protein synthesis mechanisms, had no detectable short/mid-term impact on bacterial membrane potential [51]. The same authors also noted that fluorescence intensity of membrane potential stains (e.g. DiBAC4(3)) can be influenced by cell size, which may hamper application and interpretation when natural communities are analysed. In this regard, Müller and Nebe-von Caron [46] note that a typical approach is to create a ratio between fluorescence intensity and cell size when membrane potential is analysed (see also [66]). The application of membrane potential dyes to natural communities has been largely limited [31]. In one such application, Berney et al. [6] have demonstrated the use of DiBAC4(3) on indigenous drinking water communities, showing 60–80% of cells in non-chlorinated drinking water showing “viable” polarized membranes. This was, however, not in relation to specific bactericidal processes, but rather a general “snapshot” assessment of an indigenous community. For accurate application of membrane potential methods on natural communities, additional information is needed about the accuracy of staining small cells that are prevalent in natural environments, and the interaction of the stains with charged compounds in the water samples [31].
4.3.4 Opinion
Membrane potential is a rather sensitive viability parameter and during general disinfection processes (e.g., solar disinfection), the loss of the membrane potential occurs in conjunction with a failure in cellular energy systems (ATP formation and the energy-dependent uptake of molecules), often well before membrane permeabilisation occurs [4, 8]. As a result, extensively damaged cells that have permeable membranes (Sect. 4.2) would typically display a lack of membrane potential as well [6]. However, the loss of membrane potential as a result of cellular inactivation may be time-dependent. For example, many bacteriostatic agents work through interference with protein synthesis, and therefore an immediate response in membrane potential is not expected [51]. Hence, it is essential to assess not only the presence of membrane potential, but also the time-dependant maintenance thereof. Of the available methods, the anionic dyes seem to have broader applicability, but it should always be remembered that the experimental outcome is not a direct measurement of membrane potential, but rather the specific behaviour of a given dye in a cell. While extensive work has been done with membrane potential staining of pure cultures, the application in natural samples is largely limited [6, 20, 31].
4.4 Efflux Pump Activity
4.4.1 Principle
Some molecules that can cross intact cytoplasmic membranes by passive diffusion are pumped out of active cells via specific or non-specific proton antiport transport systems. The pumping is ATP-independent but directly dependent on a transmembrane electrochemical gradient. A loss of this pumping activity is therefore indicative of a change in membrane potential or damage to the membrane.
4.4.2 Methodology
The most commonly used stain for efflux pump activity analysis is ethidium bromide (EB) [30, 31, 41]. The small EB molecules (394 Da) with a single positive charge enter the cell through passive uptake. If the membrane pumps are no longer functioning it accumulates in the cell and binds to nucleic acids. A common approach is the use of EB in combination with a green fluorescent nucleic acid stain (e.g. SYBR Green I), which allows dynamic detection of small changes and intermediate cell states [41]. An appropriate control for EB-staining has been described as using a combination of sodium azide (NaN3) and Tween-20, which apparently halts EB extrusion without compromising the membrane integrity [48], while treatment with verapamil, and m-chlorophenyl hydrazone also inhibit efflux pump activity [46].
4.4.3 Application
Efflux pump activity has been used successfully in the monitoring of industrial fermentation processes with pure cultures of bacteria. For example, Looser et al. [41] demonstrated that the damage caused to E. coli membranes through the expression of a heterologous membrane protein could be followed most accurately with EB-staining, and this can be used for real-time monitoring of the process. In several studies the group of Hewitt [27, 70] have used a protocol combining EB with PI (membrane integrity) and DiBAC4(3) (membrane potential) to describe various sub-populations occurring during industrial fed-batch fermentations.
4.4.4 Opinion
The absence of a pumping activity suggests potential stressful conditions, but not necessarily cell death. Müller and Nebe-von Caron [46] suggested that active transport is obviously low in that are viable but with very low activity, which may lead to false interpretations of staining results. Nebe-von Caron et al. [48] as well as Looser et al. [41] described EB as more sensitive than either membrane potential dyes (e.g., DiBAC4(3)) or membrane integrity dyes (e.g., PI) for the detection of physiological changes in cells, and a similar observation was also made for solar irradiated cultures [4]. However, while EB staining works fairly well for assessment of pure cultures, Joux and Lebaron [31] opined that the current methodology for detection of pumping activity is not universal enough for analysis of environmental samples—an opinion that is shared by the present authors. As discussed below, efflux pump activity can also have a negative impact on other staining methods (e.g., enzyme activity staining), where either the fluorochromes or the cleavage products are in some cases actively exported from viable cells.
4.5 Respiratory Activity
4.5.1 Principle
Respiratory activity in bacteria depends on a functioning electron transport chain, which is the main process for maintaining a membrane potential (Fig. 2). The presence of respiratory activity is therefore an indicator of viability in cells, linking membrane potential and the recycling of reducing equivalents (e.g. NAD+) that are produced in many catabolic reactions.
4.5.2 Methodology
Respiratory activity in bacteria can be detected by the use of artificial electron acceptors, specifically tetrazolium salts, which are reduced to insoluble formazan products [71]. The reduction of tetrazolium salts are indirectly linked with the enzymes that form part of the electron transport chain (NADH dehydrogenase I & II, succinate dehydrogenase, cytochrome c reductase and the terminal oxidases), which is why respiratory activity is also often described together with enzyme activity protocols [67]. The two most commonly used tetrazolium salts are INT (2-(p-iodophenyl)-3-(p-nitrophenyl)-5-phenyltetrazolium chloride), which is reduced to INT-formazan, and CTC (5-cyano-2,3-ditolyl tetrazolium chloride), which is reduced to red-fluorescent 3-cyan-1,5-ditolyl formazan (CTF) [71]. Of these two dyes, CTC seems to be the preferred choice based on the easy detection of the fluorescent product with fluorescence microscopy and/or flow cytometry. Different staining protocols include differences in staining times (4–24 h), the addition of intermediate electron carriers, changing the oxygen concentrations during staining, and also the inclusion of additional substrates in the medium during staining [14, 71]. For negative controls, the dissipation of the membrane potential can be achieved by the addition of a combination of nigericin and valinomycin, or similar reagents as described above in Sect. 4.3 [12, 57].
4.5.3 Application
Respiratory activity staining is one of the older viability assays, with already numerous applications in both pure cultures and environmental microbiology during the last two decades [59, 71]. For example, Schumann et al. [63] found reasonably good correlations between CTC reduction and esterase activity (discussed below) in a number of environmental samples. For a comprehensive review of various CTC applications and comparisons with other methods, the reader is referred to the comprehensive discussion in the study of Creach et al. [14].
4.5.4 Opinion
Considerable disagreement exists to the exact value of this method for the analysis of environmental samples [14, 68]. One of the shortcomings of the CTC assay is that it often requires long staining times (up to 24 h) [71], which makes it less interesting for a rapid microbiological assay. Such a long staining time could result in other viability changes (e.g., die-off, regrowth) in the samples. Also, for flow cytometric analysis it is essential that the crystals are formed inside the cells, which is not always the case. Moreover, it has been noted that not all bacteria are capable of reducing formazan salts (because not all bacteria have a functioning electron transport chain), which seriously questions the general applicability of the method on indigenous environmental communities [14, 68, 71].
4.6 Enzymatic Activity
4.6.1 Principle
Bacteria maintain a number of housekeeping enzymes with relative general functions (e.g., esterases, dehydrogenases, peptidases). The absence of these enzymes from a cell suggests an inability of the cell to synthesise and maintain new proteins, which is indicative of inactivity and potential death [49]. Enzyme activity is usually measured through the detection of specific cleavage products in cells.
4.6.2 Methodology
All bacteria contain relative unspecific esterases, and the presence thereof can be detected through the addition of an uncharged, non-fluorescent and lipophilic substrate, which upon cleavage, becomes a fluorescent polar product that is retained to varying degrees in cells. For this purpose, a very wide variety of substrates have been tested rather extensively, including fluorescein diacetate (FDA), carboxyfluorescein diacetate (CFDA), carboxyfluorescein diacetate acetoxymethyl ester (CFDA-AM), 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein-AM (BCECF-AM) and calcein-AM [11, 31, 36]. Some of the above-mentioned dyes tend to be pH-dependent, which influences their efficacy [46]. Additionally, some of the fluorogenic substrates or cleavage products can be actively exported from bacterial cells [16, 46], again hampering the interpretation of staining data. As a result, various different staining protocols exist in which different substrates are favoured. It is, however, essential that any chosen protocol should be carefully controlled, e.g., by inhibition of efflux pumps (Sect. 4.4.2) and also by complete heat-inactivation of enzymes (negative control). Apart from esterases, at least two other enzyme systems are often targeted in viability assays. Firstly, the reduction of tetrazolium dyes (e.g. CTC, see section above) is indirectly linked to the enzymes of the electron transport chain (see Sect. 4.5.). Secondly, Schumann et al. [63] describe the use of 7-amino-4-chloromethylcoumarin, l-leucine amide hydrocloride (CMAC-Leu) for the measurement of peptidase activity in bacteria.
4.6.3 Application
Specific disinfection processes like heat-killing would directly affect enzymatic activity in bacterial cells through denaturation of the enzymes. Therefore, loss of specific enzyme activity can be indicative of a specific location or type of cell damage (e.g., proteins). Schumann et al. [63] assessed both esterase and peptidase activity in a number of natural surface water samples. In general, higher esterase activity was detected in all samples, but the exact interpretation from studies based on such grab-samples is difficult to make. In a recent study in the authors’ research group, esterase activity was measured along with several other viability parameters in different drinking water samples. What was particularly interesting was that a relatively good correlation was found between the concentration of esterase positive cells and the concentration of ATP in the water samples (Fig. 6). However, as discussed below, there is sufficient reason to believe that esterase data should be regarded with care, particularly during disinfection experiments.

A general correlation between the concentration of ATP and esterase positive cells (CFDA staining) in water samples (n = 80) including groundwater, tap water, surface water and wastewater effluent. Samples were analysed as described in Berney et al. [6]
4.6.4 Opinion
Most of the substrates used for enzyme activity measurements enter bacterial cells through passive diffusion. Moreover, the actual cleavage (substrate–enzyme reaction) is independent of cellular energy [49]. Hence, the detection of enzyme activity does not necessarily suggest cell viability, and recently killed cells are more than likely to still display enzymatic activity. In fact, Breeuwer and Abee [11] reported several cases where dead cells displayed enhanced esterase activity, probably due to better transport of the dyes into dead cells while enzymes remain active. Therefore, a key aspect again is the time-dependent maintenance of enzymatic activity in cells, rather than the mere presence thereof in a snapshot analysis of a sample. On the other hand, the absence of enzymatic activity is by no means an indicator of dead cells, but can also be associated with a low activity state in bacteria. Moreover, Diaz et al. [16] describes problems with dye uptake as well as active dye extrusion. Heat-killing is an often-used control for esterase stains [6, 55]. However, it has to be recognized that heat would denature proteins, thereby damaging and inactivating the esterase enzymes. Heat-killing is therefore only a control for enzyme presence and activity, but it is not a control for the response of enzyme activity relative to cell viability (or the lack thereof). Hence, it is the authors’ opinion that enzymatic activity should be considered with care, and reported accurately to the experimental or environmental conditions that were studied.
4.7 Cellular Energy: Adenosine Tri-Phosphate (ATP)
4.7.1 Principle
All microorganisms require a minimum amount of energy to maintain life-sustaining processes. Energy in bacteria is generally present in the form of ATP, which can be generated either through oxidative phosphorylation (by creating a proton motive force that drives the ATP synthase) or substrate level phosphorylation (e.g., glycolysis). Additionally, bacteria can accumulate compounds like phosphoenolpyruvate, glycogen or triacylglyceride, which can be converted to ATP under energy starvation conditions. ATP is commonly referred to as the energy currency of microbial cells [29]. It is turned over rapidly in cells due to the coupling of anabolic and catabolic reactions, and as a result, ATP concentrations often respond to physiological states in microorganisms.
4.7.2 Methodology
Khlyntseva et al. [35] presents an extensive review of ATP determination methods (not limited only to bacterial cells). Although bacterial ATP measurements on single-cell level are not commonly used [29], bulk ATP analysis has evolved during the last decade into easy and straightforward measurements [24]. In standard laboratory applications, ATP measurements are typically accomplished with commercially available ATP kits, based on detection of luminescence generated from the interaction of extracted ATP with the luciferin–luciferase enzyme complex. Intracellular and extracellular ATP can be distinguished from each other by using 0.1 μm filtration.
4.7.3 Application
In a recent paper from the authors’ group, it was demonstrated that a good general correlation was obtained between the concentration of intact cells (measured with SGPI-staining) and the concentration of bacterial ATP for a variety of indigenous aquatic microbial communities [24]. Similarly, a good correlation was obtained with esterase-positive cells following CFDA-staining (Fig. 6). However, it should be emphasized that this data set looked specifically at freshwater environments where no dramatic stress conditions could be expected. During sunlight disinfection experiments, it was demonstrated that ATP is sensitive to cellular stress and often displayed decreases considerably earlier than parameters such as cultivability and substrate uptake [4, 8].
4.7.4 Opinion
The advantage of ATP analysis in the context of viability is that it provides the user with an independent tool to compliment the flow cytometry or microscopy data (Figs. 3, 6). Unfortunately, this is in most applications only a bulk parameter and not a single-cell measurement. This is indeed problematic, since the ATP content of bacterial cells is not constant. For example, HNA cells contain on average about 10-fold more ATP than LNA cells [69]. This means that bulk ATP measurements can easily lead to erroneous conclusions when the data are compared to single-cell data. Also, ATP measurements do not take into account the rapid changes in cellular ATP levels, and specific responses to environmental conditions, e.g., increased catabolism or decreased anabolism. It is a known fact that the energy in a cell depends on the balance between the various adenosine phosphates [ATP, adenosine di-phosphate (ADP) and adenosine monophosphate (AMP)] [35]. In addition, it was shown that extracellular ATP can have an influence on total ATP measurements, and should be considered during analysis [24].
5 Conclusions
It is highly unlikely that any single silver-bullet viability staining method exist, due to the heterogeneous nature of microbial life. As a result, the best approach is the use of a combination of cultivation-independent methods that target different cellular processes linked to viability in order to gain specific and meaningful information on different cellular states, types of cell damage and the degree of cellular injury. It is emphasized that the methods should be carefully chosen and optimised prior to application, bearing in mind the particular environment and organisms that are analysed. In this regard, critical points to consider are (1) the cellular process measured with the method (Fig. 3), (2) the specificity of the method towards the target organism, (3) the inherent link between different methods/processes (e.g., Figs. 3, 6), (4) the sequence and time-dependent manner in which cellular processes respond to a specific stress (e.g., Fig. 1), and the ability of cells to recover from a certain injury. The application of cultivation-independent viability assays to environmental samples is a needed and worthwhile endeavour in order to understand bacterial behaviour in complex natural environments. However, pure culture work, specifically using defined organisms under diverse (but controlled) stress conditions, coupled to cell sorting and re-cultivation, has the potential to enhance the actual understanding and interpretation of cultivation-independent methods [46, 49].
References
Al-Adhami BH, Nichols RAB, Kusel JR, O’Grady JO, Smith HV (2007) Detection of UV-induced thymine dimers in individual Cryptosporidium parvum and Cryptosporidium hominis oocysts by immunofluorescence microscopy. Appl Environ Microbiol 73:947–995
Asano S, Iijima K, Suzuki K, Motoyama Y, Ogata T, Kitagawa Y (2009) Rapid detection and identification of beer-spoilage lactic acid bacteria by microcolony method. J Biosci Bioeng 108:124–129
Ateya DA, Erickson JS, Howell PB, Hilliard LR, Golden JP, Ligler FS (2008) The good, the bad, and the tiny: a review of microflow cytometry. Anal Bioanal Chem 391:1485–1498
Berney M, Weilenmann HU, Egli T (2006) Flow-cytometric study of vital cellular functions in Escherichia coli during solar disinfection (SODIS). Microbiol-SGM 152:1719–1729
Berney M, Hammes F, Bosshard F, Weilenmann HU, Egli, T (2007) Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight Kit in combination with flow cytometry. Appl Environ Microbiol 73:3283–3290
Berney M, Vital M, Hülshoff I, Weilenmann HU, Egli T, Hammes F (2008) Rapid, cultivation-independent assessment of microbial viability in drinking water. Water Res 42:4010–4018
Bogosian G, Bourneuf EF (2001) A matter of bacterial life and death. EMBO Rep 2:770–774
Bosshard F, Berney M, Scheifele M, Weilenmann HU, Egli T (2009) Solar disinfection (SODIS) and subsequent dark storage of Salmonella typhimurium and Shigella flexneri monitored by flow cytometry. Microbiol-SGM 155:1310–1317
Bosshard F, Riedel K, Schneider T, Geiser C, Bucheli M, Egli T (2010) Protein oxidation and aggregation in UVA-irradiated Escherichia coli cells as signs of accelerated cellular senescence. Environ Microbiol. doi:10.1111/j.1462-2910.2010.02268.x
Boulos L, Prevost M, Barbeau B, Coallier J, Desjardins R (1999) LIVE/DEAD(R) BacLight (TM): application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J Microbiol Methods 37:77–86
Breeuwer P, Abee T (2000) Assessment of viability of microorganisms employing fluorescence techniques. Int J Food Microbiol 55:193–200
Breeuwer JA, Abee T (2004) Assessment of the membrane potential, intracellular pH and respiration of bacteria employing fluorescence techniques. In: Kowalchuk GA, de Bruijn FJ, Head IM, Akkermans AD, van Elsas JD (eds) Molecular microbial ecology manual, 2nd edn. Springer, pp 1563–1580. ISBN: 978-1-4020-2173-3
Brehm-Stecher B, Johnson EA (2004) Single-cell microbiology: tools, technologies, and applications. Microbiol Mol Biol Rev 68:538–559
Creach V, Baudoux AC, Bertru G, Le Rouzic B (2003) Direct estimate of active bacteria: CTC use and limitations. J Microbiol Methods 52:19–28
Czechowska K, Johnson DR, van der Meer JR (2008) Use of flow cytometric methods for single-cell analysis in environmental microbiology. Curr Opin Microbiol 11:205–212
Diaz M, Herrero M, Garcia LA, Quiros C (2010) Application of flow cytometry to industrial microbial bioprocesses. Biochem Eng J 48:385–407
Dimroth P, Cook GM (2004) Bacterial Na+- or H+-coupled ATP synthases operating at low electrochemical potential. Adv Microb Physiol 49:175–218
Dubelaar GBJ, Gerritzen PL (2000) CytoBuoy: a step forward towards using flow cytometry in operational oceanography. Sci Mar 64:255–265
Falcioni T, Papa S, Gasol JM (2008) Evaluating the flow-cytometric nucleic acid double-staining protocol in realistic situations of planktonic bacterial death. Appl Environ Microbiol 74:1767–1779
Gasol JM, del Giorgio PM (2000) Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Sci Mar 64:197–224
Gasol JM, Zweifel UL, Peters F, Fuhrman JA, Hagstrom A (1999) Significance of size and nucleic acid content heterogeneity as measured by flow cytometry in natural planktonic bacteria. Appl Environ Microbiol 65:4475–4483
Hammes F, Egli T (2010) Cytometric methods for measuring bacteria in water: advantages, pitfalls and applications. Anal Bioanal Chem 397:1083–1095
Hammes F, Berney M, Wang Y, Vital M, Köster O, Egli T (2008) Flow-cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes. Water Res 42:269–277
Hammes F, Goldschmidt F, Vital M, Wang Y, Egli T (2010) Measurement and interpretation of microbial adenosine tri-phosphate (ATP) in aquatic environments. Water Res 44:3915–3923
Harris GD, Adams VD, Sorensen DL, Curtis MS (1987) Ultraviolet inactivation of selected bacteria and viruses with photoreactivation of the bacteria. Water Res 21:687–692
Hewitt CJ, Nebe-von Caron G (2004) The application of multi-parameter flow cytometry to monitor individual microbial cell physiological state. Adv Biochem Eng Biotechnol 89:197–223
Hewitt CJ, Nebe-Von Caron G, Axelsson B, McFarlane CM, Nienow AW (2000) Studies related to the scale-up of high-cell-density E. coli fed-batch fermentations using multiparameter flow cytometry: effect of a changing microenvironment with respect to glucose and dissolved oxygen concentration. Biotechnol Bioeng 70:381–390
Hoefel D, Grooby WL, Monis PT, Andrews S, Saint CP (2003) Enumeration of water-borne bacteria using viability assays and flow cytometry: a comparison to culture-based techniques. J Microbiol Methods 55: 585–597
Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji HKP (2009) Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA 106:15651–15656
Jarnagina JL, Luchsingera DW (1980) The use of fluorescein diacetate and ethidium bromide as a stain for evaluating viability of Mycobacteria. Biotechnic and Histochemistry 55:253–258
Joux F, Lebaron P (2000) Use of fluorescent probes to assess physiological functions of bacteria at single-cell level. Microbes Infect 2:1523–1535
Jung H (2001) Towards the molecular mechanism of Na(+)/solute symport in prokaryotes. Biochim Biophys Acta 1505:131–143
Karner M, Fuhrman JA (1997) Determination of active marine bacterioplankton: a comparison of universal 16S rRNA probes, autoradiography, and nucleoid staining. Appl Environ Microbiol 63:1208–1213
Kell DB, Kaprelyants AS, Weichart DH, Harwood CR, Barer MR (1998) Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Leeuwenhoek 73:169–187
Khlyntseva SV, Bazel YR, Vishnikin AB, Andruch V (2009) Methods for the determination of adenosine triphosphate and other adenine nucleotides. J Anal Chem 64:657–673
King MA (2000) Detection of dead cells and measurement of cell killing by flow cytometry. J Immunol Methods 243:155–166
Laussermair E, Schwarz E, Oesterhelt D, Reinke H, Beyreuther K, Dimroth P (1989). The sodium ion translocating oxaloacetate decarboxylase of Klebsiella pneumoniae. Sequence of the integral membrane-bound subunits beta and gamma. J Biol Chem 264:14710–14715
Lebaron P, Parthuisot N, Catala P (1998) Comparison of blue nucleic acid dyes for flow cytometric enumeration of bacteria in aquatic systems. Appl Environ Microbiol 64:1725–1730
Lebaron P, Servais P, Agogue H, Courties C, Joux F (2001) Does the high nucleic acid content of individual bacterial cells allow us to discriminate between active cells and inactive cells in aquatic systems? Appl Environ Microbiol 67:1775–1782
Lisle JT, Pyle BH, McFeters GH (1999) The use of multiple indices of physiological activity to access viability in chlorine disinfected Escherichia coli O157:H7. Lett Appl Microbiol 29:42–47
Looser V, Hammes F, Keller M, Berney M, Kovar K, Egli T (2005) Flow-cytometric detection of changes in the physiological state of E. coli expressing a heterologous membrane protein during carbon-limited fedbatch cultivation. Biotechnol Bioeng 92:69–78
Lunde CS, Hartouni SR, Janc JW, Mammen M, Humphrey PP, Benton BM (2009) Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob Agents Chemother 53:3375–3383
Madigan MT, Martinko JM (2006) Brock biology of microorganisms, 11th edn. Pearson Benjamin Cummings, San Francisco
Malacrino P, Zapparoli G, Torriani S, Dellaglio F (2001) Rapid detection of viable yeasts and bacteria in wine by flow cytometry. J Microbiol Methods 45:127–134
McFeters GA, Yu FP, Pyle BH, Stewart PS (1995) Physiological assessment of bacteria using fluorochromes. J Microbiol Methods 21:1–13
Müller S, Nebe-von Caron G (2010) Functional single-cell analyses–flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev. doi:10.1111/j.1574-6976.2010.00214.x
Müller S, Harms H, Bley T (2010) Origin and analysis of microbial population heterogeneity in bioprocesses. Curr Opin Biotechnol 21:100–113
Nebe-von Caron G, Stephens P, Badley RA (1998) Assessment of bacterial viability status by flow cytometry and single cell sorting. J Appl Microbiol 84:988–998
Nebe-von Caron G, Stephens PJ, Hewitt CJ, Powell JR, Badley RA (2000) Analysis of bacterial function by multi-colour fluorescence flow cytometry and single cell sorting. J Microbiol Methods 42:97–114
Novo DJ, Perlmutter NG, Hunt RH, Shapiro HM (1999). Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry 35:55–63
Novo DJ, Perlmutter NG, Hunt RH, Shapiro HM (2000) Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrob Agents Chemother 44:827–834
Oliver JD (2005) The viable but nonculturable state in bacteria. J Microbiol 43:93–100
Phe MH, Dossot M, Guilloteau H, Block JC (2005) Nucleic acid fluorochromes and flow cytometry prove useful in assessing the effect of chlorination on drinking water bacteria. Water Res 39:3618–3628
Phe MH, Dossot M, Guilloteau H, Block JC (2007) Highly chlorinated Escherichia coli cannot be stained with propidium iodide. Can J Microbiol 53:664–670
Porter J, Deere D, Pickup R, Edwards C (1996) Fluorescent probes and flow cytometry: new insights into environmental bacteriology. Cytometry 23:91–96
Postgate JR (1969) Viable counts and viability. In: Norris JR, Ribbons DW (eds) Methods in microbiology, vol 1. Academic Press Inc., London
Rao M, Streur TL, Aldwell FE, Cook GM (2001) Intracellular pH regulation by Mycobacterium smegmatis and Mycobacterium bovis BCG. Microbiology 147:1017–1024
Rappé M, Giovannoni S (2003) The uncultured microbial majority. Annu Rev Microbiol 57:369–394
Rodriquez GG, Phipps D, Ishiguro K, Ridgway HF (1992) Use of a fluorescent redox probe for direct visualisation of actively respiring bacteria. Appl Environ Microbiol 58:1801–1808
Roszak DB, Colwell RR (1987) Survival strategies of bacteria in the natural environment. Microbiol Rev 51:365–379
Saint-Ruf C, Cordier C, Megret J, Matic I (2010) Reliable detection of dead microbial cells using fluorescent hydrazides. Appl Environ Microbiol 76:1674–1678
Schloss PD, Handelsman J (2004) Status of the microbial sensus. Microbiol Mol Biol Rev 68:686–691
Schumann R, Schiewer U, Karsten U, Rieling T (2003) Viability of bacteria from different aquatic habitats. II. Cellular fluorescent markers for membrane integrity and metabolic activity. Aquat Microb Ecol 32:137–150
Shapiro HM (1981) Flow cytometric estimation of DNA and RNA content in intact cells stained with Hoechst 33342 and Pyronin Y. Cytometry 2:143–150
Shi L, Gunther S, Hubschmann T, Wick LY, Harms H, Müller S (2007) Limits of propidium iodide as a cell viability indicator for environmental bacteria. Cytometry A 71:592–598
Sträuber H, Müller S (2010) Viability states of bacteria--specific mechanisms of selected probes. Cytometry A 77:623–634
Tracy BP, Gaida SM, Papoutsakis ET (2010) Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr Opin Microbiol 21:85–99
Ullrich S, Karrasch B, Hoppe HG (1999) Is the CTC dye technique an adequate approach for estimating active bacterial cells? Aquat Microb Ecol 17:207–209
Wang Y, Hammes F, Boon N, Chami M, Egli T (2009) Isolation and characterization of low nucleic acid (LNA)-content bacteria. ISME J 3:889–902
Want A, Thomas ORT, Kara B, Liddel J, Hewitt CJ (2009) Studies related to antibody fragment (Fab) production in Escherichia coli W3110 fed-batch fermentation processes using multi-parameter flow cytometry. Cytometry A 75:148–154
Winding A, Binnerup SJ, Sorensen J (1994) Viability of indigenous soil bacteria assayed by respiratory activity and growth. Appl Environ Microbiol 60:2869–2875
Ziglio G, Andreottola G, Barbesti S, Boschetti G, Bruni L, Foladori P, Villa R (2002) Assessment of activated sludge viability with flow cytometry. Water Res 36:460–468
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin heidelberg
About this chapter
Cite this chapter
Hammes, F., Berney, M., Egli, T. (2010). Cultivation-independent Assessment of Bacterial Viability. In: Müller, S., Bley, T. (eds) High Resolution Microbial Single Cell Analytics. Advances in Biochemical Engineering / Biotechnology, vol 124. Springer, Berlin, Heidelberg. https://doi.org/10.1007/10_2010_95
Download citation
DOI: https://doi.org/10.1007/10_2010_95
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-16886-4
Online ISBN: 978-3-642-16887-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)