
Abstract
Perovskite materials show immense potential as an electrolyte in intermediate temperature SOFCs. Recently, a few hexagonal perovskite-based materials demonstrate high oxide ion conductivity at intermediate temperatures in wide range of oxygen partial pressure. In the present work, a prominent hexagonal perovskite-related oxide material, Ba7Nb4MoO20 (BNMO), was synthesized via solid-state reaction method and characterized by X-ray Diffraction, Raman spectroscopy and Fourier-Transform Infrared (FTIR) spectroscopy for phase formation, structural and spectroscopic analysis. The surface morphology and elemental concentration of the synthesized samples was examined using SEM and EDAX spectroscopy. Impedance spectroscopy (EIS) measurement was performed to study the electrical behavior of the sample. At ~650 °C, the electrical conductivity of the composition in the air has been found to be ~1.54 × 10–2 S-cm−1. This demonstrates its possible application as a solid electrolyte for electrochemical devices such as solid oxide fuel cells.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
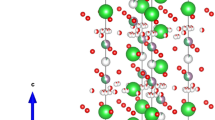
Nowadays, clean energy storing and conversion technologies such as fuel cells, solar cells, super-capacitors, and batteries have emerged as viable options for addressing the world’s growing energy needs. While each of these technologies offers unique advantages out of these fuel cells currently being used to generate green and clean energy, which offers promising alternative to traditional power generation [1]. Fuel cells are the devices that convert energy from chemical reactions into electrical energy [1,2,3]. The fuel cell was first demonstrated by RW Grove in 1838 but it was not commercialized at that time [3,4,5,6]. Among various types of fuel cells, SOFCs have gained significant attention due to their ability to function at intermediate to high temperatures, resulting in higher energy conversion efficiency compared to other fuel cell types. This makes them a promising alternative for power generation applications. In Solid oxide fuel cells (SOFCs), as air flows along the cathode, the oxygen molecules acquire electrons and split into oxide ions at the cathode-electrolyte interface [6,7,8,9]. Yttrium-stabilized-zirconia (YSZ) is highly effective electrolyte material in SOFCs, however due to its high operating temperature, it faces substantial challenges such as high manufacturing cost, low lifetime, and reduced chemical stability [10,11,12]. These issues limit the widespread use of SOFCs. Bismuth oxide-based electrolyte is another option for SOFCs, but it has a limited temperature range of stability, (around 700–820 ℃). When the oxygen pressure is low, this electrolyte can be reduced to bismuth metal [10,11,12]. Efforts are being made to find new electrolyte materials for SOFCs that can reduce operating temperatures, provide longer lifetimes, increase chemical and structural stability, and have lower manufacturing costs. Hexagonal perovskite materials show immense potential as an electrolyte in Intermediate temperature SOFCs [12, 13]. These structures consist of hexagonal \(A{X}_{3}\) layers that are closely packed together, or a combination of cubic and hexagonal \(A{X}_{3}\) layers in close proximity [14]. Hexagonal perovskites may prove advantageous in creating new ionic conductors for various energy-related technologies. Several crystal structures are associated with hexagonal perovskite oxides. However, it is challenging to find oxide ion conductors that have hexagonal perovskite-like structures. Sacha Fop et al. and M. Yashima et al. have reported hexagonal perovskites with high oxide ion conductivity at intermediate temperatures at broad range of oxygen partial pressure [14,15,16]. Bezdorozhev et al. have reported the characteristics and phase composition of hexagonal perovskite are influenced by the amount of strontium present [17]. Sakuda et al. have reported the effect of doping Cr6+ on conductivity enhancement in Ba7Nb4MoO20 hexagonal perovskite [18]. The properties of the H-perovskite-based electrolytes Ba3NbMoO8.5 and Ba7Nb4MoO20 have been reported by Nakayama [19]. Recently, Yasui et al. have published their work on hidden chemical order in Ba7Nb4MoO20 by using XRD and NMR. [20, 21] Therefore, in worldwide a lot of work is going on this special hexagonal oxide. As for structure concern, Ba7Nb4MoO20 crystallizes in the three-dimensional trigonal structure having space group \(P\bar{3}m\textit{1}\). The crystal structure Ba7Nb4MoO20 is shown in Fig. 1. This structure comprises seven inequivalent Ba2+ sites in which first and second Ba2+ site Ba2+ is separately bonded to ten O2− atoms in a 10-coordinate geometry with varying Ba–O bond distances in these two inequivalent sites. In 3rd Ba2+ site, Ba2+ is bonded to six O2− atoms in a 6-coordinate geometry. There are three shorter and three longer Ba–O bond lengths. Ba2+ forms BaO12 cuboctahedra at the fourth Ba2+ site by bonding with twelve O2− atoms, which share corners with nine other BaO12 cuboctahedra, faces with six other BaO12 cuboctahedra, and faces with seven NbO6 octahedra. Ba2+ forms BaO12 cuboctahedra at the fifth Ba2+ site by bonding with twelve O2− atoms, which share corners with nine other BaO12 cuboctahedra, faces with six other BaO12 cuboctahedra, and faces with seven other NbO6 octahedra. Ba2+ is bound to twelve O2− atoms in the sixth Ba2+ site to create the BaO12 cuboctahedra, which also include nine equivalent BaO12 cuboctahedra, three equivalent MoO4 tetrahedra, three equivalent BaO12 cuboctahedra on their faces, and four equivalent NbO6 octahedra on their faces. Ba2+ forms BaO12 cuboctahedra at the seventh Ba2+ site by bonding with twelve O2− atoms. These cuboctahedra share faces with four NbO6 octahedra, three comparable NbO4 tetrahedra, and corners with nine other cuboctahedra. A detailed crystal structure of Ba7Nb4MoO20 was reported in the literature [1, 14,15,16,17,18,19,20, 22].

Crystal structure of Ba7Nb4MoO20
In this work, we synthesized the Ba7Nb4MoO20 (BNMO) hexagonal perovskite by using the solid-state reaction technique and investigated the phase formation of the material via XRD analysis and pattern was indexed with standard ICSD database. Compositional concentration and its microstructure were examined using SEM–EDX technique. The sample's chemical composition and molecular structure were analyzed and identified through Raman spectroscopy. Additionally, FT-IR was conducted to explore the molecular structure and examine the vibrations and rotations of chemical bonds. The band-gap energy was calculated by Ultraviolet–Visible (UV–Vis) spectroscopy via Tauc’s plot. Electrochemical Impedance spectroscopy measurement was carried out in order to check the conductive behavior of the sample.
2 Experimental
2.1 Synthesis
In this paper, we prepared the Ba7Nb4MoO20 sample by solid-state reaction method. Barium carbonate (BaCO3, \(99.99\%\)), Niobium pentoxide (Nb2O5, 99.99%), and Molybdenum trioxide (\(\text{Mo}{\rm O}_{3}\), 99.99%) were used as the starting materials [1, 15]. The powders of these materials were mixed in stoichiometric ratio as described below:
The molecular weights of the precursor components are as follows:
It is important to ensure that the stoichiometric ratio of the precursor powders was thoroughly mixed by using an agate mortar and pestle for 2 h, and ball milled for 2 h in order to obtain a homogeneous mixture. The precursor sample powder is then calcined in a muffle furnace, maintaining the heating rate of 5º/min, at a temperature of 1050 ℃ for 48 h in static air [1]. The calcined sample was then ground thoroughly for 2 h using an agate-mortar and pestle. And hence obtained calcined sample was pelletized by using hydraulic press. The average thickness and diameter of the samples were 1.89 mm and 9.49 mm, respectively. The pelletized samples were sintered in a furnace, maintaining a heating rate of 5º/min, at 1200 ℃ for 5 h [1]. Then, further characterizations were done from the calcined powders and sintered pellets of the sample.
2.2 Characterizations
2.2.1 XRD Analysis
The XRD study has been performed to evaluate phase purity and stability of the samples by using Rigaku Miniflex desktop diffractometer at room temperature. While XRD analysis the Cu-Kα1 radiation was used, and the resulting XRD patterns were collected with a step size of 0.02° in the range of 20°–80°, using a wavelength (λ) of 1.5415 Å [16, 20, 21].
2.2.2 SEM-EDX Analysis
The Scanning Electron Microscope (SEM) micrograph was recorded at room temperature for the surfaces of BNMO (JEOL Japan SEM, JSM 6610LV) and the image was taken at resolution of 2 μm.
2.2.3 RAMAN Analysis
The sample's chemical composition and molecular structure were analyzed and identified through Renishaw Invia II Raman microscopy at room temperature in the range of 200–1000 \({\rm cm}^{-1}\).
2.2.4 FTIR analysis
FT-IR analysis was conducted at room temperature to explore the molecular structure and to examine the vibrations and rotations of chemical bonds by using Nicolet iS50 FTIR Tri-detector. The spectra of the sample were obtained in the range of 400–4000 \({\rm cm}^{-1}\).
2.2.5 UV-Vis’s Spectroscopy Analysis
UV–Vis’s spectroscopy analysis of the sample was conducted by using JASCO V 670 Spectrometer. The spectrum was recorded in absorption mode within the wavelength range of 200–2000 nm (with 400 nm/min scan speed). Data from the absorbance spectra were used to determine the sample's band gap energy.
2.2.6 Impedance Spectroscopy Analysis
Electrical impedance spectroscopy for the synthesized system was carried out in the temperature range of 573–923 K using 6500 P Wayne Kerr Impedance analyzer. The pellets were coated with a layer of Platinum paint and dried for 30 min in a muffle furnace at 1000 ̊C. The measurements were then performed within the frequency range 20 Hz to 1 MHz and temperature range of 573–923 K with an interval of 50 K.
3 Results and Discussion
3.1 XRD Study
In order to determine the physical properties of the calcined sample for the composition, X-ray diffraction (XRD) was conducted (by using Rigaku Miniflex desktop) at room temperature. [6] While XRD analysis the Cu-Kα1 radiation was used, and the resulting XRD patterns were collected with a step size of 0.02° in the range of 20°–80°, using a wavelength (λ) of 1.5415 Å [16, 20, 21]. The XRD pattern of the Ba7Nb4MoO20 sample is shown in Fig. 2 and unit cell parameters are given in Table 1. The obtained diffractogram was analyzed to confirm that the samples whether in a pure phase or not, and their compositions were identified with the space group \(P\bar{3}m\textit{1}\) [1, 15]. Crystal structure of the composition has 7H polytype hexagonal phase. The peaks observed are very sharp in all samples of XRD pattern which suggests that the samples prepared are highly crystalline. The peaks with maximum intensity are observed at 27.91° and 30.61°. A few minor peaks marked as * are also observed revealing some impurity phases (Such as Ba5Nb4O15) present in the sample.

XRD pattern of calcined BNMO sample at room temperature
3.2 SEM and EDAX Analysis
The Scanning Electron Microscope (SEM) micrograph was recorded at room temperature for the surfaces of BNMO (JEOL Japan SEM, JSM 6610LV). Before the SEM investigations, the pellets were coated with a gold Palladium alloy to prevent charging of the surface (This coating provides a uniform surface of the pellets for analysis and helps to increase the secondary electron emission). Figure 3 shows the SEM image of the surface of dense Ba7Nb4MoO20 pellet sample.

The SEM micrographs of the surface of the dense \(\text{Ba}_{7}{\rm Nb}_{4}\text{Mo}{\rm O}_{20}\) sample at 2 µm scale
From the SEM micrographs, it is observed that primary phase formation has taken place. The grains show cored morphology with good contact, which implies that the pellet is dense. The typical grain size of the sample was measured using Image-J software [6]. The results show that the average grain size for Ba7Nb4MoO20 ranges from 0.4 to 3.0 μm. Archimedes’ density of the composition was found to be around 95% to theoretical one.
To verify the presence of all constituent elements in the sample of Ba7Nb4MoO20, we conducted an EDS mapping of a dense sample surface (Fig. 4). The resulting EDS analysis data is presented in Table 2, which shows the atomic% and weight% of elements present in the composition for the Ba7Nb4MoO20 sample. 5–10% variation was seen in the theoretical and experimental data of elemental concentration.

EDS scan for the BNMO sample
3.3 Raman Analysis
The Raman scattering spectra of the Ba7Nb4MoO20 sample were obtained at room temperature in the range of 200–1000 \({\rm cm}^{-1}\), as shown in Fig. 5. Ba7Nb4MoO20 sample belongs to the \({D}_{{3}_{d}}\) crystallographic point group with a \(P\bar{3}m1\) space group [23]. The intense peak is observed at 300 \({\rm cm}^{-1}\). Observations from the Raman spectra indicate that Mo exhibits a tetrahedral \({\left[\text{Mo}{\rm O}_{4}\right]}^{2-}\) mode at around 900 \({\rm cm}^{-1}\) [23].

Raman spectra of BNMO sample at room temperature
The low-intensity peaks between 400 and 700 \({\rm cm}^{-1}\) at 420, 500, 550, and 650 \({\rm cm}^{-1}\) provide evidence for octahedrally coordinated Nb or Mo sites [19]. The modes between 400 and 700 \({\rm cm}^{-1}\) are attributed to \(\text{Nb}{\rm O}_{6}\) or \(\text{Mo}{\rm O}_{6}\) in several different compounds. The peak splitting occurs at 750 to 900 \({\rm cm}^{-1}\) because the stretching vibration mode has various frequencies. Two bands at approximately 300 and 800 \({\rm cm}^{-1}\) correspond to [\(\text{Nb}{\rm O}_{4}\)] or [\(\text{Mo}{\rm O}_{4}\)] tetrahedral groups [23]. Minor changes in the spectra could be attributed to the unique structural properties of various compounds, such as the presence of face-sharing octahedra in the structure.
3.4 FTIR Analysis
FTIR spectra of the Ba7Nb4MoO20 was (Nicolet iS50 FTIR Tri-detector) obtained in the range of 400–4000 \({\rm cm}^{-1}\) at room temperature as shown in Fig. 6. In IR plot analysis, the peak near 820 \({\rm cm}^{-1}\) is attributed to the [Mo–O] stretching vibration mode of the tetrahedral group of \({\left[\text{Mo}{\rm O}_{4}\right]}^{2-}\) [24]. In the range of 440–460 \({\rm cm}^{-1}\), weak infrared absorption bands are observed, this can be caused by vibration modes associated to bending in the medium frequency range [24].

FT-IR spectra of Ba7Nb4MoO20 sample is obtained at room temperature
3.5 UV–Vis’ Spectroscopy Analysis
In Fig. 7a UV–visible spectra of the sample Ba7Nb4MoO20 is plotted. The spectrum was recorded between 200 and 2000 nm wavelength range. Data from the absorbance spectra were used to determine the sample's band gap energy. Tauc’s plot for BNMO sample is shown in Fig. 7b. The Tauc’s equation was used to calculate the value of Band gap [25, 26]. The Tauc’s equation is:

a UV–Visible spectra of the sample BNMO and b Energy band gap was determined by Tauc’s plot of BNMO sample
In order to calculate the optical band-gap energy of the Ba7Nb4MoO20 sample, a plot of “\({\left(\alpha hv\right)}^\frac{1}{n} Vs hv\)” was formed using the above relation, “A” is a constant that is independent of energy, “\(\alpha\)” is the incident light's absorption coefficient and “\(hv\)” represents the energy of the light. Depending on the kind of electron transition, the value of “n” is assumed to be either 2 or 1/2 for indirect or direct inter-band transitions respectively. The linear portion of the plot was extrapolated to zero, and the resulting value for the energy band gap was determined to be 3.3870 eV. In contrast, the Ba7Nb4MoO20 sample from the Materials Project website exhibits an energy band gap of 2.64 eV [28]. A little bit higher energy gap was obtained for the investigated sample possibly due to the presence of some impurity phases in the sample.
3.6 Impedance Spectroscopy Analysis
Electrical spectroscopy for the synthesized system was carried out in the temperature range of 573–923 K using 6500 P Wayne Kerr Impedance analyzer. Figure 8a shows the Arrhenius plot for the BNMO sample. The conductivity values at different temperature were calculated using \(\sigma =\frac{L}{RA}\) relation where “σ” is the conductivity and “R” is the resistance, “A” is the area and “L” denotes the length. The resistance “R” is calculated using the fitted Cole–Cole plot [1, 15, 27].

a Representative complex impedance spectra of the BNMO sample at 500 °C. b Arrhenius plot for the BNMO sample
From plot (a), it has been found that as the temperature rises, the conductivity of the sample increases, which may be explained by an increase in charge carrier concentration. The values of conductivity exhibited by the sample at different intermediate temperature ranges are shown in Table 3. The Arrhenius graphic clearly shows that conductivity is significantly influenced by temperature. At 510 °C, the BNMO sample's conductivity was found to be 3.54 \({\rm mScm}^{-1}\). Putting this beside the conductivity reading provided by Sacha Fop et al., which is approximately equivalent to 4.0 \({\rm mScm}^{-1}\) [1, 16]. The maximum conductivity for the BNMO sample comes out to be at 650 ̊C with a value of 0.01547 \({\rm Scm}^{-1}\). This value of electrical conductivity can be considered decent for electrolytes of intermediate temperature solid oxide fuel cells and for the other electrochemical devices.
4 Conclusions
In this investigation, Ba7Nb4MoO20 sample was prepared via solid-state reaction method. The prepared sample’s XRD patterns are indexed with the MATCH program. The obtained diffractograms identify the structure of the sample belonging to the space group P3̅m1. By using SEM technique, it is observed that highly dense sample formation has taken place. To analyze the phase formation, FT-IR analysis was performed, and it additionally verified the hexagonal structure. UV–visible spectra wavelengths were obtained for Ba7Nb4MoO20 in the range of 200–2000 nm. UV–visible method was used to calculate the optical band-gap energy, and the result was 3.3870 eV. Electrical conductivity of Ba7Nb4MoO20 sample was measured by using impedance spectroscopy. The Arrhenius plot clearly shows that conductivity is significantly influenced by temperature. The maximum conductivity for the Ba7Nb4MoO20 sample was found to be 0.0154 S.cm−1 at 650 \(^\circ{\rm C}\). Based on the investigation, it can be concluded that the hexagonal perovskite-based Ba7Nb4MoO20 system may be considered as a potential candidate solid electrolyte for electrochemical devices such as SOFCs.
References
O’Hayre R, Cha SW, Colella W, Prinz FB (2016) Fuel cell fundamentals. Wiley. https://doi.org/10.1002/9781119191766
Dicks AL, Rand DAJ (2018) Fuel cell systems explained. Wiley. https://doi.org/10.1002/9781118706992
Barbir F (2013) PEM fuel cells. Theory and practice. Academic Press
Grove WRXXIV (1839) On voltaic series and the combination of gases by platinum. London Edinburgh Dublin Philos Mag J Sci 14(86–87):127–130. https://doi.org/10.1080/14786443908649684
Tarique H, Shahid R, Singh P et al (2022) Effect of potassium substitution at Sr-site on the structural, morphological and electrical properties in Sr3 − 3x K3x Si3O9 (0.0 ≤ x ≤ 0.2) trimer strontium meta-silicate. J Mater Sci: Mater Electron 33:14961–14971. https://doi.org/10.1007/s10854-022-08413-47
Saravanan R (2018) Solid oxide fuel cell (SOFC) materials. Mater Res Found 23:182. https://doi.org/10.21741/9781945291517
Ormerod RM (2003) Solid oxide fuel cells. Chem Soc Rev 32(1):17–28. https://doi.org/10.1039/b105764m
Singhal S (2000) Advances in solid oxide fuel cell technology. Solid State Ionics 135(1–4):305–313. https://doi.org/10.1016/S0167-2738(00)00452-5
Kilner JA, Burriel M (2014) Materials for intermediate-temperature solid-oxide fuel cells. Annu Rev Mater Res 44(1):365–393. https://doi.org/10.1146/annurev-matsci-070813-113426
Hussain S, Yangping L (2020) Review of solid oxide fuel cell materials: cathode, anode, and electrolyte. Energy Transitions 4(2):113–126. https://doi.org/10.1007/s41825-020-00029-8
Minh NQ (1993) Ceramic fuel-cells. J Am Ceram Soc 76(3):563–588. https://doi.org/10.1111/j.1151-2916.1993.tb03645.x
Skinner SJ (2001) Recent advances in Perovskite-type materials for solid oxide fuel cell cathodes. Int J Inorg Mater 3(2):113–121. https://doi.org/10.1016/S1466-6049(01)00004-6
Fop S, McCombie KS, Wildman EJ et al (2020) High oxide ion and proton conductivity in a disordered hexagonal perovskite. Nat Mater 19(7):752–757. https://doi.org/10.1038/s41563-020-0629-4
Fop S, McCombie KS, Wildman EJ, Skakle JMS, Mclaughlin AC (2019) Hexagonal perovskite derivatives: a new direction in the design of oxide ion conducting materials. Chem Commun 55(15):2127–2137. https://doi.org/10.1039/C8CC09534E
Yashima M, Tsujiguchi T, Sakuda Y, et al (2021) High oxide-ion conductivity through the interstitial oxygen site in Ba7Nb4MoO20-based hexagonal perovskite related oxides. Nat Commun 12(1). https://doi.org/10.1038/s41467-020-20859-w
Coduri M, Karlsson M, Malavasi L (2022) Structure-property correlation in oxide-ion and proton conductors for clean energy applications: recent experimental and computational advancements. J Mater Chem A Mater 10(10):5052–5110. https://doi.org/10.1039/d1ta10326a
Bezdorozhev O, Solodkyi I, Ostroverkh A, Morozov I, Ostroverkh Y, Solonin Y (2022) Synthesis and characterization of (Ba1-xSrx)7Nb4MoO20 powders for proton-conducting solid oxide fuel cells. Ukr Chem J 88(4):63–78. https://doi.org/10.33609/2708-129x.88.04.2022.63-78
Sakuda Y, Hester JR, Yashima M Improved oxide-ion and lower proton conduction of hexagonal perovskite-related oxides based on Ba 7 Nb 4 MoO 20 by Cr 6+ doping. https://doi.org/10.2109/jcersj2
Nakayama S (2022) Characteristics of electrolyte-supported SOFC single cells using hexagonal perovskite Ba3MoNbO8.5 and Ba7Nb4MoO20 ceramics. Results in Mater 16. https://doi.org/10.1016/j.rinma.2022.100318
Yasui Y, Tansho M, Fujii K et al (2023) Hidden chemical order in disordered Ba7Nb4MoO20 revealed by resonant X-ray diffraction and solid-state NMR. Nat Commun 14(1):2337. https://doi.org/10.1038/s41467-023-37802-4
Chauhan A (2014) Powder XRD technique and its applications in science and technology. J Anal Bioanal Tech 5(6). https://doi.org/10.4172/2155-9872.1000212
Kumar A, Sharma SK, Grover V, Singh Y, Kumar V, Shukla VK, Kulriya PK (2022) Probing the short-range ordering of ion irradiated Gd2Ti2-yZryO7 (0.0 ≤ y ≤ 2.0) pyrochlore under electronic stopping regime. J Nucl Mater 564:153682. https://doi.org/10.1016/j.jnucmat.2022.153682
Brown Holden AA, Reedyk M, García-González E, Parras M, González-Calbet JM (2000) Raman scattering study of cation-deficient Ba(n)(MoNb)(n-δ)O(3n–x) and related perovskite-like oxides. Chem Mater 12(8):2287–2291. https://doi.org/10.1021/cm000037i
Jena P, Nallamuthu N, Hari Prasad K, Venkateswarlu M, Satyanarayana N (2014) Structural characterization and electrical conductivity studies of BaMoO4 nanofibers prepared by sol–gel and electrospinning techniques. J Sol-Gel Sci Technol 72(3):480–489. https://doi.org/10.1007/s10971-014-3460-z
García-González E, Parras M, González-Calbet JM (1999) Crystal structure of an unusual polytype: 7H-Ba7Nb4MoO20. Chem Mater 11(2):433–437. https://doi.org/10.1021/cm981011i
Redinger A, Siebentritt S (2015) Loss mechanisms in Kesterite solar cells. In Copper zinc tin sulfide-based thin-film solar cells. Wiley :363–386. https://doi.org/10.1002/9781118437865.ch16
Lvovich VF (2012) Impedance spectroscopy. Wiley. https://doi.org/10.1002/9781118164075
Materials Data on Ba7Nb4MoO20 by Materials Project. https://doi.org/10.17188/1704365
Acknowledgements
Authors acknowledge the support of USIC, and DU for providing the experimental facilities. Authors are also appreciative to SERB for providing the funding support through the ECR project [ECR/2016/001152].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Saini, P., Tarique, H., Kumar, H., Singh, A.K., Pandey, R., Singh, P. (2024). Synthesis and Characterizations of Hexagonal Perovskite-Related Oxide for Solid Electrolyte Application. In: Krupanidhi, S.B., Sharma, A., Singh, A.K., Tuli, V. (eds) Recent Advances in Functional Materials and Devices. AFMD 2023. Springer Proceedings in Materials, vol 37. Springer, Singapore. https://doi.org/10.1007/978-981-99-6766-7_1
Download citation
DOI: https://doi.org/10.1007/978-981-99-6766-7_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-6765-0
Online ISBN: 978-981-99-6766-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)