
Abstract
Asthma is one of the highest incidences and disbursed respiratory diseases the world over. There is incomprehensible knowledge of pathophysiological epigenetic pathways and causal interaction in asthma. Hereby, with a focus on DNA methylation, we examine human investigations on the epigenetic processes of asthma. On analysis, it was found that epigenetic research on childhood asthma has uncovered distinct methylation profiles linked to allergic inflammation in immune cells and the airways, demonstrating that methylation plays a regulatory function in the pathogenesis of asthma. Considering these ground-breaking findings, additional research is needed to understand how epigenetic pathways contribute to the endotypes of asthma. Studies on histone alterations in asthma are very few. Future investigations into the epigenetic causes of asthma will be aided by the inclusion of data from groups with accurate phenotyping.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


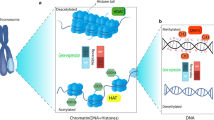
5.1 Introduction
Waddington coined the term “epigenetics,” which is formed by the Greek words “epi” and “genome.” Epigenetics means phenotype changes without a corresponding change in genotype [1]. Although the mechanisms behind were not known when the term was first used, advancement in genetics and genomics has revealed significant epigenetic mechanisms involved in cell function, including both controlling the homeostasis of healthy cells and in the pathophysiology of the disease [2]. Numerous studies on asthma have demonstrated that epigenetic pathways affect the disease’s numerous manifestations, including asthma in children and asthma in adults. Epigenetic regulatory mechanisms, whether directly affected by inflammation and the modulation of respiratory function or indirectly influenced by pharmaceutical therapies or environmental factors, can provide insights into the substantial heterogeneity observed in asthma [3, 4]. This chapter highlights recent advancements in the perception of the epigenetics of asthma in individual research by framing the topic with well-known instances of epigenetic mechanisms in asthma (Fig. 5.1).

Allergic airway sensitization
5.2 Overview of Epigenetic Pathways
5.2.1 DNA Methylation
DNA methylation involves the addition of methyl group on the cytosine C5 site to form 5-methylcytosine (5mC). The existing 5mC in specific genome sites causes alteration in organic phenomenon by the binding transcription factor [5]. Near the gene transcription start site (TSS), abundant CG sequences are found at the DNA regions which is called CpG island (CGI). As a result, both increased and decreased gene expression are correlated with methylation alterations in CGIs [6]. Hence, gene suppression and CGI methylation are frequently linked; moreover, the role of DNA methylation is complex and includes methylation in the gene and CGIs [7]. Heritable modifications to the DNA that affect gene function are known as epigenetic alterations. DNA methylation is a sophisticated gene regulatory system that is passed along cellular division through generations. It also plays a main role in the transcriptional elongation formation of the permutable elements, and the creation and slicing of mutation elements [8, 9].
5.2.2 Histone Alteration
Most of the proteins in the DNA-protein complex chromatin are histones. It is crucial for packing, ensuring genomic integrity, and controlling gene expression so that proteins are closely associated with DNA [10]. A crucial epigenetic mechanism is the regulation of histone function through post-translational changes [11]. Histone acetyltransferases (HATs) and histone deacetylases (HDACS) are the two major categories among the numerous enzymes that are involved in histone modifications. HATs and HDACs are differentiated according to where they are found and how particular they are and can be distinguished accordingly [12]. The contrasting effects on lysine acetylation make it simple to comprehend the functions of HATs and HDACs in histone modifications [13]. HDACs function as transcriptional suppressors by maintaining the chromatin structure once lysine acetylation is reversed. Additionally, gene expression is regulated by events like histone acetylation, methylation, and phosphorylation: by DNA effectiveness histone methylation and nucleosome unfolding [14]. DNA histone interaction efficiency is altered by nucleosome unwrapping. Histone methylation causes modification of histone protein either by adding or deleting a methyl group which is catalyzed by a group of demethylase and methyltransferase. It is important to remember that histone methylation affects DNA methylation in a similar manner [11, 15, 16].
5.2.3 Silencing of Transcriptional Genes and Non-coding RNA
Non-coding RNA (ncRNAs) are transcription that regulates the biological process. Based on their nucleotide size, ncRNAs are divided into two classes, i.e., small nucleotide has a size of less than 200 and long nucleotide has a size of more than 200 [17]. According to research, ncRNAs are known to contribute to cell differentiation and organogenesis. Despite being categorized as ncRNAs, small and big ncRNAs are different and display distinctive characteristics that define their function [18]. Non-coding RNA includes transfer RNA (tRNAs), ribosomal RNA (rRNAs), and small RNAs (sRNAs) such as microRNA, small nucleolar RNA (snRNAs), small interfering RNA (siRNA), small RNA derived from transfer RNA (tsRNA), etc. These small RNAs play numerous roles in regulating cellular processes [19].
Long non-coding RNAs (lncRNAs) are frequently used to control nearby protein-coding genes. Further complicating their role in the epigenetic regulation of cell activity is the fact that lncRNAs target histone methyltransferases and demethylases [20]. This chapter deals with asthma in adults, children, and its vulnerability. Since DNA methylation has been the subject of the bulk of recent high-quality human research on asthma, this study focuses on epigenetic mechanisms and provides an overview of microRNAs and histone alterations [18, 21] (Fig. 5.2).

Epigenetic level
5.2.4 Asthma in Adults
There are few investigations into DNA methylation in adult asthma. In an asthmatic study involving smokers, it was demonstrated that Protocadherin-20 (PCDH20) exhibited higher levels of methylation in sputum cells [22]. Even after taking environmental factors into account, this connection remained strong. A connection between CGIs and particular asthma endotypes was found after methylation alterations in the airway epithelium were assessed [23]. Interleukins induced alterations in DNA methylation have been linked to eosinophils. The inhaled corticosteroids block IL-13 signaling through the IL-13Ra1 receptor and result in the reduction of FENO [24]. Separate research on the methylation of blood found methylation variations in gene networks linked to asthma subtypes, with purine metabolism and calcium signaling genes being enriched in eosinophilic asthma and neutrophil asthma had significant accumulation for SUMOylation, basal cell carcinoma signaling, and Wnt/−catenin pathways [25, 26].
5.2.5 Asthma in Children
DNA methylation serves as a biomarker in all asthma investigations during human studies, involving the sampling of various biological compartments, such as epithelial and immunological, and utilizing individual genes and genomes [27, 28]. These studies showed a relationship between methylation patterns in the blood and various processes and features of pediatric asthma [29]. Studies on candidate genes have demonstrated that nitrogen dioxide (NO2) and particulate matter can affect the methylation of the adrenergic β2 receptor agonist (ADRB2) in the gene associated with asthma [30]. Moreover, the methylation of ARDB2 was connected to lessened dyspnea in young asthmatics, according to a related investigation. The idea that these variances are caused by cohort-specific traits must be further investigated [31]. Hypomethylation at CpG (cytosine guanine) dinucleotide site of arachidonate12-lipoxygenase (ALOX12) gene associated with persistent wheezing. Genome-wide methylation investigations have demonstrated that the presence of CGIs in blood samples is associated with an elevated risk of chronic wheezing. Asthma is associated with hyper-responsiveness mediated by hypomethylation of important cytokine IL-13, transcription factor RUNX3 modulates Th1/Th2 balance, microRNA, and TIGIT in blood [32,33,34]. Like this, asthma in children was connected to hypomethylation of CGIs linked to IL5RA. Cell-specific and epigenetic methylation in pediatric asthma is interrelated; the DNA of 14 CpG sites in eosinophils has been found to have methylated cytosine and adenosine residues [35]. PCSK6 was hypomethylated in children with atopic and atopy asthma. Assessments of blood spots and umbilical blood have been carried out to evaluate links between allergic disease and asthma [36]. There was a reduced incidence of asthma in toddlers by higher methylation of the GATA3 CpGs transcription factor. In mid-childhood, DNA methylation of cord blood is linked to IgE in genes involved in cell signaling, growth, and development. After accounting for the difference in DNA methylation between birth and mid-childhood, two methylation sites, i.e., C7orf50 and ZAR1, remained steady [37, 38]. Two methylation sites (C7orf50 and ZAR1) remained stable after adjusting for the change in DNA methylation from birth to mid-childhood. Methylation of DNA sequence on AXL receptor at birth was linked to an increased incidence of wheezing, demonstrating that methylation has an impact on gender [39].
Therefore, the risk that infants would develop asthma, wheeze, and high IgE levels are correlated with the methylation patterns at birth seen in cord blood and blood spots [40]. According to studies on the methylation of buccal DNA, increased methylation of arginase 2 (ARG2) has been associated with a decrease in the fraction of exhaled nitric oxide (FeNO) in asthmatic children [41]. Similarly, to this, reducing allergen exposure resulted in less methylation of the FOXP3 promoter. Together, these findings point to a function for methylation in the regulation of immune signaling, and airway obstruction CGIs in the STAT5A transcription factor were differently methylated in asthmatics’ airway epithelial cells (AECs), which caused STAT5A expression to be downregulated [42]. Following stimulation by certain ligands like Interleukin-2,3,7 and GM-CSF, this pathway is important in the regulation of downstream signaling. Asthma in nasal epithelial is associated with hypomethylation of ALOX15 and POSTN genes. Th2 cytokines induce the upregulation of the POSTN gene in asthmatic airway epithelium, which can be exploited as a biomarker for long-term prediction in the treatment of severe asthma [34, 43, 44].
5.2.6 Methylated Differentially in the Nasal Epithelial of African American Newborns
This study also demonstrated that in children of African American, European, and Hispanic heritage, a nasal epithelial classifier based on methylation may be able to differentiate between atopy and allergic asthma. This methylation pattern is applied in the epigenetic regulation of FeNO (fractional exhaled nitric oxide) and Immunoglobulin E [45, 46].
5.2.7 Asthma Risk and Vulnerability
Environmental factors have a big impact on asthma and they also affect DNA methylation. Acyl-CoA synthetase (ACSL3) long-chain family member methylation has been linked to asthma symptoms in children under the age of five, according to research on prenatal exposure to polycyclic organic matter(POM) from traffic [47]. An association between elevated FOXP3 methylation and defective regulatory T cells was found in distinct research on asthmatic children exposed to higher levels of ambient pollution [48]. Multiple studies have demonstrated a link between air pollution, variable DLG2 methylation, and corresponding changes in blood expression [49]. Like how prenatal tobacco exposure is linked to significant changes in children’s blood DNA methylation, tobacco smoke affects DNA methylation, as shown by differential methylation of placental and fetal lung tissue [50]. These differentially methylated sites show the importance of environmental factors in methylation patterns, although this is unclear how they are connected to asthma. SMAD3 methylation at birth was linked to a higher risk of childhood asthma in the offspring of women with asthma, who also had higher levels of it [51]. In another study, the blood methylome of children showed a comparable impact on maternal asthma. In children, maternal blood eosinophils, FeNO, and total IgE all had a negative correlation with MAPK8IP3 methylation. According to this research, exposure to an asthmatic mother while the child is still in the uterus may influence DNA methylation, which may have an impact on how asthma develops [52, 53].
5.3 Role of MicroRNAs in Asthma
MiRNAs play a significant role in the control of both healthy and unhealthy cellular responses. Let-7 family miRNAs have been linked to regulating Th2 inflammation in human, animal, and in vitro research. MiR-21 controls the cytokine IL-12, which is involved in the polarization of Th1 cells [54]. MiR-146a has been identified as a potential asthmatic molecule in studies on the effects of genetic variation on miRNA function and their relationship to asthma; variation in HLA-G in children influences asthma risk through interaction with miR-152. Several cellular elements of the allergic response, including eosinophils, macrophages, and mast cells in both asthma and allergic rhinitis, have been linked to Th2 responses by two miRNAs, miR-155 and miR-221 [55, 56]. Several miRNAs, including miR-26, 133a, 140, 206, and 221, have been linked to a role in smooth muscle cell proliferation and function. Few studies have examined the impact of miRNAs on severe asthma despite their crucial role [57]. These studies on patients with severe asthma showed that miR-221 governs the proliferation of airway smooth muscle cells, miR-28-5p and miR-146a/b led to the activation of circulating CD8+ T cells in severe asthma, and miR-223-3p, miR-142-3p, and miR-629-3p were linked to serious neutrophilic asthma [58]. In the Childhood Asthma Management Program (CAMP) cohort, eight serum microRNAs, including miR-296-5p, were connected to PC20 [59]. Inhaled corticosteroid budesonide, used to treat asthma, had a negligible impact on the miRNA profile in steroid-naive asthmatics’ bronchial epithelium after therapy. These findings on the activity of several miRNAs in asthma show their impact on the regulation of inflammatory events, Th1/Th2 polarization, cell function, disease severity, and treatment response [55, 60, 61].
5.4 Role of Histone Modifications in Asthma
A poorly understood mechanism for asthma is an epigenetic modification of the histones [62]. The amount of cellular acetylation activity was related to the severity of bronchial hyperresponsiveness in allergic asthmatic children, and the ratio of HDAC/HAT activity was biased toward greater histone acetylation in those cases [63]. Adults with asthma who had severe asthma had lower nuclear HDAC and HAT activity in their mononuclear cells than those who had milder cases of the condition [64]. An analysis of the genome-wide histone changes in T cell subsets from asthma patients and healthy controls revealed variations in cell enhancers involved in T cell development that were specific to asthma. Furthermore, compared to cells from healthy people, human bronchial epithelial cells (HBECs) from adult asthmatics showed impaired tight junction integrity [65, 66]. In HBECs from asthmatic patients, there was greater expression of Sirtuins 6 and 7, HDACs 1 and 9, and HDACs. Tight junction molecules increased to levels like those reported in healthy controls when HDAC was inhibited. Together, these findings point to an overlap that is particular to cells and asthma and is caused by various histone changes [67].
5.5 Aspects of the Future
By combining omics data from microRNAs, genome-wide variation, methylome, and transcriptome cohorts with known traits, it may be feasible to better comprehend these associations. The importance of histone changes in asthma, environmental data, and the intensity of relationships with certain asthma endotypes all lay considerable limitations on our understanding of asthma. In light of these findings, it is necessary to create better prediction models, biomarkers, and medicines that target dysregulated pathways.
References
Abdel-Aziz MI, et al. Omics for the future in asthma. Semin Immunopathol. 2020;42(1):111–26.
Agache I. Severe asthma phenotypes and endotypes. Semin Immunol. 2019;46:101301.
Agache I, et al. Advances and highlights in asthma in 2021. Allergy. 2021;76(11):3390–407.
Alashkar Alhamwe B, et al. The role of epigenetics in allergy and asthma development. Curr Opin Allergy Clin Immunol. 2020;20(1):48–55.
Alashkar Alhamwe B, et al. Extracellular vesicles and asthma-more than just a co-existence. Int J Mol Sci. 2021;22:9.
Alizadeh Z, et al. Role of epigenetics in the pathogenesis of asthma. Iran J Allergy Asthma Immunol. 2017;16(2):82–91.
Bélanger É, Laprise C. Could the epigenetics of eosinophils in asthma and allergy solve parts of the puzzle? Int J Mol Sci. 2021;22:16.
Benincasa G, et al. Epigenetics and pulmonary diseases in the horizon of precision medicine: a review. Eur Respir J. 2021;57(6):2003406.
Benjamin S, et al. Phthalates impact human health: epidemiological evidences and plausible mechanism of action. J Hazard Mater. 2017;340:360–83.
Boulet LP. Airway remodeling in asthma: update on mechanisms and therapeutic approaches. Curr Opin Pulm Med. 2018;24(1):56–62.
Brook PO, et al. Epigenome-modifying tools in asthma. Epigenomics. 2015;7(6):1017–32.
Camoretti-Mercado B, Lockey RF. Airway smooth muscle pathophysiology in asthma. J Allergy Clin Immunol. 2021;147(6):1983–95.
Castro-Rodríguez JA, et al. Epigenetics in allergic diseases and asthma. Rev Chil Pediatr. 2016;87(2):88–95.
Cevhertas L, et al. Advances and recent developments in asthma in 2020. Allergy. 2020;75(12):3124–46.
Choi BY, et al. Genetics and epigenetics in allergic rhinitis. Genes (Basel). 2021;12(12):2004.
Chowdhury NU, et al. Sex and gender in asthma. Eur Respir Rev. 2021;30(162):210067.
Clapp PW, Jaspers I. Electronic cigarettes: their constituents and potential links to asthma. Curr Allergy Asthma Rep. 2017;17(11):79.
Conrad LA, Cabana MD, Rastogi D. Defining pediatric asthma: phenotypes to endotypes and beyond. Pediatr Res. 2021;90(1):45–51.
Davidson EJ, Yang IV. Role of epigenetics in the development of childhood asthma. Curr Opin Allergy Clin Immunol. 2018;18(2):132–8.
Devries A, Vercelli D. Epigenetics of human asthma and allergy: promises to keep. Asian Pac J Allergy Immunol. 2013;31(3):183–9.
DeVries A, Vercelli D. Early predictors of asthma and allergy in children: the role of epigenetics. Curr Opin Allergy Clin Immunol. 2015;15(5):435–9.
DeVries A, Vercelli D. Epigenetics in allergic diseases. Curr Opin Pediatr. 2015;27(6):719–23.
DeVries A, Vercelli D. Epigenetic mechanisms in asthma. Ann Am Thorac Soc. 2016;13(Suppl 1(Suppl 1)):S48–50.
Durham A, et al. Epigenetics in asthma and other inflammatory lung diseases. Epigenomics. 2010;2(4):523–37.
Durham AL, Wiegman C, Adcock IM. Epigenetics of asthma. Biochim Biophys Acta. 2011;1810(11):1103–9.
Fang L, Sun Q, Roth M. Immunologic and non-immunologic mechanisms leading to airway remodeling in asthma. Int J Mol Sci. 2020;21(3):757.
Godfrey KM, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017;5(1):53–64.
Gomez JL. Epigenetics in asthma. Curr Allergy Asthma Rep. 2019;19(12):56.
Gruzieva O, et al. An update on the epigenetics of asthma. Curr Opin Allergy Clin Immunol. 2021;21(2):175–81.
Guo C, et al. Serum sphingolipid profile in asthma. J Leukoc Biol. 2021;110(1):53–9.
Han X, et al. LncRNA PTPRE-AS1 modulates M2 macrophage activation and inflammatory diseases by epigenetic promotion of PTPRE. Sci Adv. 2019;5(12):eaax9230.
Harb H, et al. Recent developments in epigenetics of pediatric asthma. Curr Opin Pediatr. 2016;28(6):754–63.
Heijink IH, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. 2020;75(8):1902–17.
Hellings PW, Steelant B. Epithelial barriers in allergy and asthma. J Allergy Clin Immunol. 2020;145(6):1499–509.
Hernandez-Pacheco N, Kere M, Melén E. Gene-environment interactions in childhood asthma revisited; expanding the interaction concept. Pediatr Allergy Immunol. 2022;33(5):e13780.
Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126(3):453–65.
Holgate ST, et al. Asthma. Nat Rev Dis Primers. 2015;1(1):15025.
Hudon Thibeault AA, Laprise C. Cell-specific DNA methylation signatures in asthma. Genes (Basel). 2019;10(11):932.
Isidoro-García M, et al. Pharmacogenetics and the treatment of asthma. Pharmacogenomics. 2017;18(13):1271–80.
Kabesch M. Epigenetics in asthma and allergy. Curr Opin Allergy Clin Immunol. 2014;14(1):62–8.
Kabesch M, Adcock IM. Epigenetics in asthma and COPD. Biochimie. 2012;94(11):2231–41.
Kabesch M, Tost J. Recent findings in the genetics and epigenetics of asthma and allergy. Semin Immunopathol. 2020;42(1):43–60.
Koefoed HJL, Vonk JM, Koppelman GH. Predicting the course of asthma from childhood until early adulthood. Curr Opin Allergy Clin Immunol. 2022;22(2):115–22.
Kong LD, Wu QP. Effect of ketogenic diet on obesity asthma. Zhonghua Jie He He Hu Xi Za Zhi. 2022;45(2):222–6.
Koppelman GH, Nawijn MC. Recent advances in the epigenetics and genomics of asthma. Curr Opin Allergy Clin Immunol. 2011;11(5):414–9.
Lebold KM, Jacoby DB, Drake MG. Inflammatory mechanisms linking maternal and childhood asthma. J Leukoc Biol. 2020;108(1):113–21.
Li CY, Guo XJ, Gan LX. The epigenetics in asthma. Zhonghua Jie He He Hu Xi Za Zhi. 2009;32(10):759–61.
Lira G, et al. Psychological stress in asthma: repercussions on epigenetics-genetics, immune responses, and pulmonary function in the pediatric population. Allergol Immunopathol (Madr). 2022;50(2):78–88.
Long A, et al. Epigenetics and the environment in airway disease: asthma and allergic rhinitis. Adv Exp Med Biol. 2020;1253:153–81.
Lovinsky-Desir S, Miller RL. Epigenetics, asthma, and allergic diseases: a review of the latest advancements. Curr Allergy Asthma Rep. 2012;12(3):211–20.
Lu X, Li R, Yan X. Airway hyperresponsiveness development and the toxicity of PM2.5. Environ Sci Pollut Res Int. 2021;28(6):6374–91.
Lu Y, et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol. 2021;23(10):1060–72.
Lynch SV, Vercelli D. Microbiota, epigenetics, and trained immunity. Convergent drivers and mediators of the asthma trajectory from pregnancy to childhood. Am J Respir Crit Care Med. 2021;203(7):802–8.
Maneechotesuwan K. Role of microRNA in severe asthma. Respir Investig. 2019;57(1):9–19.
Martino D, Prescott S. Epigenetics and prenatal influences on asthma and allergic airways disease. Chest. 2011;139(3):640–7.
McKenzie C, et al. The nutrition-gut microbiome-physiology axis and allergic diseases. Immunol Rev. 2017;278(1):277–95.
Mekov E, et al. Update on asthma-COPD overlap (ACO): a narrative review. Int J Chron Obstruct Pulmon Dis. 2021;16:1783–99.
Miller RL, Ho SM. Environmental epigenetics and asthma: current concepts and call for studies. Am J Respir Crit Care Med. 2008;177(6):567–73.
Mims JW. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol. 2015;5(Suppl 1):S2–6.
Murphy SK, Hollingsworth JW. Stress: a possible link between genetics, epigenetics, and childhood asthma. Am J Respir Crit Care Med. 2013;187(6):563–4.
Noutsios GT, Floros J. Childhood asthma: causes, risks, and protective factors; a role of innate immunity. Swiss Med Wkly. 2014;144:w14036.
Ntontsi P, et al. Genetics and epigenetics in asthma. Int J Mol Sci. 2021;22(5):2412.
Ober C. Asthma genetics in the post-GWAS era. Ann Am Thorac Soc. 2016;13(Suppl 1(Suppl 1)):S85-90.
Potaczek DP, et al. Epigenetics and allergy: from basic mechanisms to clinical applications. Epigenomics. 2017;9(4):539–71.
Qi C, Xu CJ, Koppelman GH. The role of epigenetics in the development of childhood asthma. Expert Rev Clin Immunol. 2019;15(12):1287–302.
Rico-Rosillo G, et al. Epigenetics, environment and asthma. Rev Alerg Mex. 2014;61(2):99–109.
Rosa MJ, Lee AG, Wright RJ. Evidence establishing a link between prenatal and early-life stress and asthma development. Curr Opin Allergy Clin Immunol. 2018;18(2):148–58.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Almalki, W.H. (2023). Epigenetics in Asthma. In: Gupta, G., Oliver, B.G., Dua, K., Ali, M.K., Dave, P. (eds) Targeting Epigenetics in Inflammatory Lung Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-99-4780-5_5
Download citation
DOI: https://doi.org/10.1007/978-981-99-4780-5_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-4779-9
Online ISBN: 978-981-99-4780-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)