
Abstract
Pulmonary arterial hypertension (PAH) is a fatal and enigmatic disease of the pulmonary circulatory system for which there is currently no cure. The intricate pathogenesis of PAH poses a barrier to identifying novel therapeutic targets, resulting in high morbidity and mortality rates. To identify potential drugs or biomarkers from the bench to the bedside, there is a need to expand the current knowledge of the pathogenesis of PAH. Alternative therapies and treatment strategies may slow down the progression of this disease, but a complete cure requires uncovering the underlying novel mechanisms. Emerging epigenetics-based studies are paving the way for understanding the pathophysiology of several complicated disorders, such as cancer, peripheral hypertension, and asthma. Thus, epigenetic studies may help to comprehend the multifaceted nature of PAH. This chapter compiles the current knowledge and emerging therapeutic and biomarker potential of epigenetic factors, such as DNA methylation, histone modifications, and noncoding RNAs, in PAH. As no animal models can fully recapitulate human PAH features, we highlight the emerging microfluidic lab-on-a-chip (LoC) technology as an excellent model for the disease and testing therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
14.1 Introduction
Pulmonary arterial hypertension (PAH) is a chronic cardiovascular condition characterized by vascular remodeling that ultimately results in right heart failure and death [1, 2]. Current therapeutics have primarily concentrated on addressing issues with the pulmonary vasculature and abnormal signaling pathways such as those involving prostacyclin, endothelin 1, and nitric oxide. The goal of these therapies is to alleviate the burden on the right side of the heart by reducing afterload [3, 4], yet a universally effective drug is under investigation. The leading causes of PAH are multifactorial and intricate, such as endothelial cell dysfunction [5], smooth muscle cell hyperproliferation [6], vascular inflammation, and immune dysregulation [7], germline mutations [8], and many others. Existing combination therapies can slow down the progression of the disease, but remain incurable [9]. Thus, there is an unmet need to understand this disease mechanism to discover effective drugs for therapeutic purposes.
Pathobiological research, diagnostic, and treatment strategies for PAH have advanced significantly, although the most precise mechanism is still enigmatic [10]. The involvement of aberrant cell signaling, genetics (mutations), and environmental variables, such as hypoxia, viral infections, and anorectic agents in the pathogenesis of PAH has been the subject of much exploration. But less emphasis has been paid to the relationship between epigenetics and PAH [11]. Epigenetics may be a potential pharmacological target since compelling data indicates that epigenetic mechanisms play a critical role in the pathogenesis of PAH [11,12,13,14]. Therefore, targeting epigenetic processes may offer a promising avenue for pharmacological interventions. However, the field of epigenetics investigates alterations in gene expression that can be inherited without changes to the DNA sequence [15]. The three primary mechanisms of epigenetics are DNA methylation, histone modification, and microRNA (miRNA) regulation. These processes play significant roles in modulating gene expression and regulating cellular activity [16]. Some miRNAs including miR-34a [17], miR-17–92 [18], and miR-212-5p [19] were identified as potential therapeutic targets in PAH. Despite the crucial role of epigenetics in the pathogenesis of PAH, the role of histone modification and DNA methylation in this condition has received relatively little research attention [11]. Moreover, a new class of RNAs called circular RNAs (CircRNAs) is emerging as diagnostic and therapeutic agents in various diseases including PAH [20]. Thus, targeting epigenetics in PAH could open the door to addressing many unanswered questions. However, understanding disease mechanisms and evaluating therapeutic effectiveness rely heavily on the fidelity of animal models. Unfortunately, no animal models have been identified that can replicate fully the pathophysiology of PAH in humans [21]. Modern scientific advancements have produced a rapid and resilient microfluidics-based technology that has the potential to connect traditional cell cultures, animal models, and human subjects, thereby bridging the gap between them [22]. One of the most promising advancements in this field is the lab-on-a-chip (LoC) technology, combined with tissue engineering and organ-on-a-chip (OoC) technology [23]. Together, these technologies can accurately emulate human pathobiology and evaluate the efficacy of drugs [24, 25]. In concert with a state-of-the-art microscope imaging system, this might be a revolutionary approach for unraveling the mystery of a putative PAH mechanism in humans. This chapter aims to provide an overview of the functions and mechanisms of epigenetic regulators in the development of PAH, and their potential therapeutic targets. Furthermore, this chapter will discuss the potential of microfluidics-based lab-on-a-chip (LoC) technologies for PAH research.
14.2 Targeting Epigenetics in PAH
Epigenetics refers to the investigation of heritable changes in phenotype that do not involve changes in DNA sequences. There are three primary epigenetic mechanisms: histone modifications (such as acetylation and methylation), noncoding RNA (including microRNA and long noncoding RNA), and DNA methylation, all of which can activate or deactivate genes [26, 27]. A brief overview of these three mechanisms will be discussed later in this chapter.
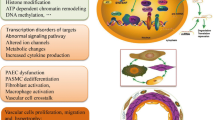
We depicted the role of epigenetic mechanisms in the pathogenesis of PH in Fig. 14.1. DNMT (DNA methyltransferase) is used to catalyze and maintain DNA methylation. The TET (ten-eleven translocation) proteins can block DNMT. Histones found in the nucleosome’s N-terminal tails can be acetylated, deacetylated, methylated, and other posttranslational changes. The epigenetic reader BRD4 (bromodomain-containing protein 4) can identify the acetylated lysine residues on histone tails. Mature miRNA is incorporated into the RISC (RNA-induced silencing complex) to mediate target mRNA destruction or translation inhibition. These three epigenetic variables have a significant impact on PAH and pulmonary vascular remodeling (dysfunction of key vascular cells such as pulmonary artery endothelial cells (PAECs), smooth muscle cells (PASMCs), and fibroblasts). PAECs play a critical role in regulating thrombosis and inflammation. PASMCs contribute to increase in vascular resistance in pulmonary hypertension [27, 28]. Besides, vascular extracellular matrix (ECM) remodeling and homeostasis are regulated by fibroblasts [29].

The role of epigenetic mechanisms in the pathogenesis of PH
14.2.1 DNA Methylation
In 1948, the major epigenetic DNA methylation was first discovered by Rollin Hotchkiss while preparing calf thymus. He found a modified version of the nucleotide cytosine and stated that it existed naturally in DNA [30]. This finding led to the hypothesis that this fragment of the DNA was 5-methylcytosine (5 mC) [30]. In 1980s, numerous studies showed that DNA methylation, which involves a methyl group addition to the DNA molecule at the fifth carbon of the cytosine ring in a CpG dinucleotide sequence, is a critical mechanism for gene regulation and a major epigenetic factor controlling gene activities in eukaryotes [31]. The process of DNA methylation is catalyzed by a family of enzyme known as DNA methyltransferases (DNMTs), during which a methyl group from S-adenyl methionine (SAM) is transferred to fifth carbon of cytosine [32]. The enzymes DNMT3a and DNMT3b are known as de novo DNMT as these can transfer the methyl group into a naked DNA molecule while the DNMT1 functions by copying the methylation pattern of the DNA during replication [33]. There are three classes of enzymes known as writers, erasers, and readers, which are involved in establishment, recognition, and removal of DNA methylation, respectively. The addition of methyl groups to cytosine residues is catalyzed by writers, whereas the modifications and removal events are done by erasers, and lastly, readers are responsible for recognizing and binding these methyl groups to induce subsequent gene expressions [33]. The three DNMT members (DNMT1, DNMT3a, and DNMT3b) catalyze the methylation event. These enzymes have unique expression patterns even though they are structurally similar [34]. The native DNA methylation pattern is conserved by DNMT1, often referred to as the maintenance DNMT. During DNA replication, DNMT1 binds to the replication fork and localizes to the newly synthesized hemimethylated DNA strand, where it mimics the original methylation pattern [35, 36]. DNMT3a and DNMT3b are unique when compared to DNMT1, as these when overexpressed can methylate both the synthetic and native DNA [37], and thus they are also known as de novo DNMT due to their capability of methylating naked DNA. The key property that differentiates DNMT3a from DNMT3b is the pattern of their gene expression [33]. DNA demethylation can be categorized into either passive or active forms. During cell division, inhibition of the DNMT1 leads to low levels of methylation overall, as the newly synthesized cytosine residues remain unmethylated [33]. However, active demethylation can occur in both dividing and non-dividing cells, where the 5mC is regressed back to a naked cytosine by enzymatic reactions [38, 39]. During demethylation, a series of enzymatically catalyzed chemical reactions occur, with subsequent oxidation or deamination of the 5mC to a product, which is further identified by the base excision repair (BER) pathway [40]. There are some other proposed mechanisms of active demethylation, for example, one of the suggested mechanisms is driven by the TET enzymes, in which these enzymes catalyze the addition of a hydroxyl group to the methyl group of 5 mC forming 5 hmC [41, 42], which is reverted back to a naked cytosine by two other separate processes such as iterative oxidation of 5 hmC or 5 hmC deamination by AID/APOBE complex (activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme) [43].
There are three different protein families known as the MBD protein, the UHRF (ubiquitin-like, containing PHD and RING finger domain) proteins and the zinc-finger proteins that can recognize DNA methylation. Some of the proteins of the MBD family are the MBD1, MBD2, MBD3, MBD4, and MeCP2 [44]. There is a methyl CpG binding (MBD) domain conserved in the MBD proteins. This domain has a high affinity for single methylated CpG sites [45]. MBD3 and MBD4 exhibit atypical characteristics among the MBD proteins. MBD3 lacks the ability to directly bind to DNA due to a mutation in the MBD domain [46], while MBD4 binds when it recognizes a mismatched guanine residue with thymine, uracil, or 5-fluorouracil [47,48,49]. Conversely, MeCP2 plays a double function by acting as both a transcriptional repressor and participating in the maintenance of DNA methylation [33]. The UHRF family has multi-domain proteins such as UHRF1 and UHRF2 which uses a SET- and RING-connected DNA binding domain to bind the methylated cytosine residues [50]. During DNA replication, this protein family primarily binds to the DNMT1 and maintains DNA methylation by projection it to a hemimethylated DNA [51, 52]. The final set of methyl binding proteins uses a zinc-finger domain to bind the methylated DNA. This family consists of Kaiso, ZBTB4, and ZBTB38 [53, 54]. These domains are unique as both Kaiso and ZBTB4 have binding affinities for a sequence motif that lacks methylcytosine [55], where Kaiso can also bind to two successive methylated CpG sites [56].
DNA methylation plays a crucial role in gene regulation and transcriptional silencing [57]. In particular, when DNA is hypermethylated in regions of the genome that contain a high density of CpG sites (known as CpG islands), this is often associated with transcriptional repression [58]. In a study of PH, where the pulmonary arterial smooth muscle cells (PASMCs) were isolated from fawn hooded rats, a diminish in the SOD2 gene expression was observed. These observations were also seen in the plexiform lesions of PH [14, 16]. It was later demonstrated that when CpG islands are selectively hypermethylated in both the promoter and the intronic regions of the SOD2 gene, the expression is reduced by approximately 50% [14]. Another study claimed that epigenetic regulation was influenced by the hypermethylation of the GNLY genes, specifically in the case of pulmonary veno-occlusive disease (PVOD) [59]. Some of the key biological pathways, where DNA methylation is involved in epigenetic regulations are the sympathetic nervous system (SNS), renin–angiotensin–aldosterone system (RAAS), and renal sodium retention system (RSRS) [60]. The SNS system plays a key role in the pathophysiology and pathogenesis of hypertension [61], whereas the RAAS contributes to the occurrence of the disease [62].
A vast amount of data suggests that DNA methylation has an integral contribution in pulmonary vascular remodeling in PAH. In a study, where some of the genes such as the ATP binding cassette 1 (ABCA1), adiponectin (ADIPOQ), and apolipoprotein A4 (APOA4) were detected in PAH patients, the authors saw that ABCA1 was majorly hypermethylated, indicating that the metabolism of cholesterol might be related with PAH [63]. Some of the key genetic risk factors of heritable PAH (HPAH) is the heterozygous loss of function (LOF) mutation in the BMPR2 gene [64]. A study performed bisulfite sequencing to determine the DNA methylation profile of the BMPR2 gene and found that this gene was hypermethylated in the HPAH patients, whereas the wild type allele was more hypermethylated when compared to the aberrant allele [64]. In another study, a group investigated and demonstrated that when rodents with PAH were administered with monocrotaline or exposed to hypoxia, the global DNA methylation increased in their lungs [65]. Their data shows the upregulation of the enzyme DNMT3b in both rodent models and PAH patients. Also, the authors showed that knockout DNMT3b−/− rats, suffered from severe pulmonary vascular remodeling and regular inhibition of DNMT3b caused proliferation of PASMCs as a response to deprivation of the platelet-derived growth factor-BB. Overall, their study reveals that DNMT3b mediates the pathogenesis of PAH coupling epigenetic regulations with vascular remodeling [65].
Pulmonary hypertension shares common risk factors with hypertension, such as age, obesity, alcohol consumption, drugs, etc. Evidence suggests that variations DNA methylation is associated with these factors [66, 67]. A study demonstrated that alcohol consumption enhances the DNA methylation of the ADD1 gene promoter, thus increasing the hypertension susceptibility [68]. Furthermore, a study claimed that differential variability in methylation can influence obesity. After analyzing genome wide methylation profiles of CpG sites in both obese and lean individuals, the authors found a significant number of differential methylated CpG in obese cases [66]. Age is also a common risk factor in both PH and hypertension, and genome wide methylation studies of CpG sites showed that approximately 28.8% of the CpG sites were associated with age and around 7.9% CpG sites were capable to predict age, making this association between DNA methylation strong and ubiquitous [67]. In addition, a cross-over trial, with human exposure to concentrated ambient particles (CAP) showed that these particles diminished methylation of toll-like receptors (TLR4) and Alu [69].
14.2.2 Histone Modification
Histones are protein that is basic in nature and contains high abundance of arginine and lysine residues, located in nuclei of eukaryotes [70]. These proteins play essential roles in protecting DNA from becoming tangled and in process being damaged [71]. There are five known histone proteins, among which H1 and H5 are the linker histones, while H2, H3, and H4 are core histones [70]. Nucleosomes are higher order structures that wrap 146 base pairs of DNA around core histone proteins [16]. Histones are key proteins that maintain the chromatin structure and influence long-term gene regulations [72]. Even though the chromatin exhibits a compact structure, histone modifications result in the chromatin losing its compactness [73]. Histone acetylation, ubiquitylation, and histone methylation process take place at the N-terminal where acetylation and methylation processes are carried out by acetyltransferases and methyltransferases, respectively [73]. The discovery of histone H1 and histone H3 phosphorylation was made in regard to the condensation of the chromosomes during meiosis [73].
Histone modifying enzymes (HME) carry out the posttranslational modifications of histone tails and thus can regulate gene expression. These enzymes (Table 14.1) can be categorized in two ways, as writers (induce modifications) and as erasers (revert modifications). Apart from the common modifications stated above, some of the other histone modifications including SUMOylation, ADP ribosylation, O-GlcNAcylation, and proline isomerization [74,75,76,77].
In epigenetic mechanisms, it is integral for the HMEs to collaborate with other cofactors which involve numerous protein–protein interactions (PPI). Furthermore, these histone modifying enzymes can form functional complexes by interacting with non-histone proteins [78]. The activity of these enzymes is crucial, as any imbalance in their activity along with the change in the compactness of the chromatin is associated with several diseases including the vascular remodeling in pulmonary arterial hypertension [79]. Some of these enzymes can also be used as potential drug targets against PAH. For example, SAHA, a broad spectrum histone deacetylase (HDAC) inhibitor drug, has demonstrated the ability to decrease the migration of monocyte induced by fibroblasts, suggesting that such HDAC inhibiting drugs reduce vascular inflammation [13].
Amongst the five histone proteins, H3 maintains the chromatin structure in eukaryotes. Modifications of the N-terminal H3 tail by adding acetyl or methyl groups to arginine or lysine residues can be done post-translations. Also, phosphorylation of the serine or threonine residues is possible [72]. Gene expression can affect the methylation pattern of lysine-9, as this residue is hypermethylated when the gene expression decreases while it gets monomethylated when the gene gets activated [65]. The acetylation of H3 lysine residues is carried out by the enzyme histone acetyltransferase. Such acetylation events may associate with hypertension as they induce the glial cell line-derived neurotrophic factor [80]. This factor is key for the survival of dopaminergic neurons and can be followed up with melatonin treatment [80]. The neurons expressing melatonin can give input to the neurons of the rostral ventrolateral medulla (RVLM), which can regulate the sympathetic outflow to blood vessels. In humans, a dysfunctional RVLM can act as a mechanism to promote hypertension [81]. Post acetylation, the histone proteins are more accessible to the RNA polymerase due to losing structural compactness [82]. Thus, this enables the adjacent genes to get transcribed. Furthermore, bromodomain-containing proteins (BRDPs) can bind with these acetylated histones and recruit chromatin-modifying factors and induce transcription [83].
There are three families of enzymes that regulate histone modifications, out of which the histone deacetyl transferases (HDACs) family is the most researched in regard to PAH [84]. This enzyme family contains a highly conserved domain and maintains the equilibrium of lysine acetylation by removing acetyl groups in histones. One of the very first studies on HDACs and their association with PAH reported that when PAH was induced by hypoxia in bovine models, the phenotype of the pulmonary adventitial fibroblasts showed abnormality in the activity of class I HDACs. The authors also reported that these fibroblasts had an increased levels of proteins due to an elevation in the activity of the class I HDAC [85]. In another study, observation of the human idiopathic PAH lung homogenates showed that out of six screened HDACs, the expression levels of HDAC1 and HDAC5 were elevated [13]. Boucherat and colleagues demonstrated that in PAH patients, the HDAC6 gene is upregulated in the lungs, in the pulmonary endothelial cells, distal pulmonary arteries, and PASMCs [86]. The MEF2 transcription factor family is known to be linked with class II HDACs and plays a crucial part in cardiovascular development, wherein HDACs maintain transcriptional inactivity of MEF2s [87]. In PAH, the nucleus of pulmonary arterial endothelial cells experiences an abundant accumulation of HDAC4 and HDAC5, which causes a suppression of MEF2’s transcriptional activity [88].
14.2.3 Crosstalk of DNA Methylation with Histone Modification
It is difficult to find concrete data on how epigenetic mechanisms influence pulmonary hypertension (PH), but several hypotheses claim that they have substantial roles in initiation, advancement, and establishment stages of PH [89]. Usually, in case of eukaryotes, histone proteins are bound with DNA to help package the molecule into small nuclear pockets. The N-terminal of the amino acids in the histones is chemically modified by process like acetylation, methylation, phosphorylation, and ubiquitination [33]. The DNMTs usually target the CpG sites and methylate the DNA to repress gene expression [33]. The histone modifications that lead to tighter packaging of DNA induce gene repression. DNMT1 and DNMT3a enzymes are associated with the histone methyltransferase SUV39H1 that inhibits gene expressions [90], while DNMT1 and DNMT3b interact with histone deacetylases to condense the DNA more tightly represses transcription [91]. In areas with active transcription, DNA methylation is removed by the TET enzyme, and inhibition of DNMT binding to the unmethylated CpG sites is done by the histone tails containing H3K4me3 [33]. DNA methylation and histone modifications are crucial epigenetic mechanisms involved in the development of PAH. The subsequent section provides a detailed explanation of their roles in this context.
14.2.4 DNA Methylation and Histone Modifications as Therapeutic Targets of PAH
PAH becomes severe as it progresses, eventually causing right ventricular failure and death. The therapies available at present are not effective enough to reverse the disease and therefore result in high mortality and morbidity [92]. Hence, it is an utmost challenge and requirement to develop newer therapeutics for treating PAH. There are several therapeutic targets to treat PAH, such as drugs targeting estrogen signaling, drugs against growth factors, or even drugs targeting properties like epigenetic modifications and DNA damage [92]. PAH can influence DNA damage as it is associated with oxidative stress and inflammation [93]. Recently, the drug Olaparib which is approved for ovarian cancer has been in clinical trials to treat PAH [92]. This study is sponsored by Laval University, and their drug is under a preclinical study targeting to inhibit DNA damage and poly (ADP-ribose) polymerases (PARP) where they have claimed that PARP1 inhibition is cardio protective and can also reverse PAH disease conditions in an animal model [94]. It has been reported that PARP1 expression and activation levels are elevated in PAH-PASMCs and PAH distal PAs [95]. This particular study also showed that in PH rats induced with both hypoxia and MCT, the PARP1 inhibiting drug veliparib was successful in reversing the PAH conditions [95]. Another drug that is being clinically studied is Apabetalone, which targets epigenetic modifications. This drug targets the inhibition of bromodomain-containing protein 4 (BRD4), which plays a significant role in the pathogenesis of PAH. Their results indicate that BRD4 inhibition can also reverse the disease conditions in animal models [96]. According to a study conducted in 2015, patients with PAH have more expression of the BRD4 gene in their distal PAs and PASMCs [97]. They also claimed that such overexpression of the BRD4 gene results in multiplication of the PASMC [97].
14.2.5 MicroRNAs (miRNAs)
MicroRNAs (miRNAs), firstly discovered in 1990s, are type of small regulatory molecules consisting of 18 to 24 nucleotides [98, 99]. The involvement of miRNAs in development and diseases is being perceived firmly over time due to their gene regulatory roles, conservation among species, and clear understanding of their mechanism of functions based on sequence complementarity [100,101,102]. The development of high-throughput technologies, computational methods, and experimental techniques enhanced the discovery process of miRNAs [98]. According to an estimation, there is around 1000 miRNAs in human genome with targeting potency of one-third of the entire human genes [103, 104]. MiRNAs can be generated singly or as a cluster from any part of the human genome through transcriptional machinery, irrespective of coding or noncoding regions [98]. A single miRNA can have multiple targets while sometimes multiple miRNAs can work together to regulate the same target, indicating their gene regulatory network attribute [98]. MiRNAs are generated and processed in a multi-step fashion, taking together several molecular players [105]. After originating from nucleus, miRNAs come to the cytoplasm and exert their functions [105]. MiRNAs are known to play a role in the development and progression of various human diseases, such as different types of cancers, neuronal development disorders, cardiovascular diseases, and skin diseases. Particularly, miRNAs have been widely studied in pulmonary hypertension [106,107,108].
MicroRNAs are associated with PAH [109, 110]. Aberrant miRNA expressions contribute to the abnormal remodeling of vascular cells, including adventitial fibroblast (AdvFB) migration, PASMC proliferation, and PAEC dysfunction in PAH [111]. Incomprehension of miRNAs role in complex cellular networks blocks the way to developing new drugs targeting pathological pathways [112]. The miRNA, miR-34a is a promising candidate for regulating the development of PAH. Reducing of its expression leads to elevated proliferation of PASMC, while overexpression produces the opposite effect [17]. Silencing of the miR-17–92 in smooth muscle cells (SMCs) induces PAH in mice [18]. Recently, miR-212-5p, an anti-proliferative miRNA, was suggested as a potential therapeutic target. Here, miR-212-5p was shown to be consistently and selectively elevated in both animal and human models of PAH [19]. Another study reported that miR-30a could be a promising miRNA for therapeutic targets [113]. Overexpression of miR-483 can suppress the PAH gene, resulting in the development of PAH. This finding indicates that miR-483 may serve as a potential disease biomarker and a target for therapeutic interventions [114]. In a recent review, it was found that miR-29, miR-124, miR-140, and miR-204 are crucial miRNAs that share a common expression pattern in both human and animal models [112]. However, several studies have previously documented the list of miRNAs that are dysregulated in PAH [106, 112, 115,116,117].
14.2.5.1 MiRNAs as a Therapeutic Target for PAH
In PAH, miRNA-based therapy is still in its infancy due to several limitations, such as miRNA mimics/antagonist, animal models, and instability of RNA molecules [117, 118]. However, miRNAs have a shorter length with known and conserved sequences and can target multiple genes within a network. Also, miRNAs have potential preclinical evidence against human diseases. So miRNA could be a promising therapeutic target molecule [106, 112]. Currently, four clinical trials related to miRNA and PAH studies have been discovered while searching https://clinicaltrials.gov/ (searched on November 9, 2022). The study (NCT00806312, completed) evaluated the expression and implications of miRNA profiles and inflammation markers in PAH patients. The study (NCT04489251, recruiting) looked into the miRNA and TGF pathways in pediatric PAH patients. The results of the other two studies were either withheld or unknown.
14.2.6 Long Noncoding RNAs (lncRNAs)
In the past, some DNA was regarded as “junk DNA” because it was ubiquitously transcribed but did not encode proteins; its functions were also unknown [119]. With the advent of the human genome project, however, the activities of these proteins were uncovered. This essential protein is referred to as “non-coding RNAs” (ncRNAs), which play a key role in protein-coding gene expression as well as several complex biological processes and disease progressions [120]. However, ncRNAs have been classified into two heterogenous subclasses: small ncRNAs with 15–200 nucleotides (nt) and long ncRNAs (lncRNAs) >200 nt long. There are thousands of ncRNA have been identified in the eukaryotic genome [121,122,123]. This section will give an overview of lncRNAs, what role they play in PAH, and how they might be used to find potential therapeutic targets. In the 1990s, the first lncRNAs were discovered using deep-sequencing and microarray technology. The importance of understanding the role of lncRNAs in epigenetic silencing and the evolution of multicellular organisms readily became increasingly apparent [124]. LncRNAs vary in size, structure (which resembles a flexible scaffold), and functions [125]. Their synthesis takes place in the nucleus via similar processes to mRNA transcription. LncRNAs are transcribed mostly by RNA polymerase II, but also by other polymerases [126]. Interestingly, lncRNAs can act in either the nucleus or the cytoplasm; hence, they are also classed as nuclear or cytoplasmic lncRNAs [127].
Based on recent classification, lncRNAs include long intergenic noncoding RNAs (lincRNAs) [128], natural antisense transcripts (NATs) [129], intronic lncRNAs, bidirectional transcripts [130, 131], sense lncRNA, enhancer RNA (eRNA) [132], and circular RNA (circRNAs) [133]. Nearly all recognized lncRNAs are similar to mRNA, but they have fewer exons, are less abundant, are nuclear-localized, and are not evolutionarily conserved [134,135,136]. They are capable to interact with DNA, RNA, and proteins [127]. Recent research has demonstrated that lncRNAs play a wide range of functions in cellular processes, including regulation of mRNA stability/turnover, chromatic architecture, and interference with posttranslational modification in the cytoplasm [137]; some of them can also translate into short polypeptides [138, 139]. A recent study also indicated that lncRNAs have a role in the development of various human diseases, including neurological, cancerous, and cardiovascular disorder [140].
14.2.6.1 LncRNAs in PAH
The pathogenesis of PAH is significantly influenced by lncRNAs, which exhibit differential expression patterns in pulmonary vascular cells such as PAECs and PASMCs. This differential expression leads to vascular remodeling and the subsequent development of PAH [141]. PASMC dysfunction is primarily attributed to various signaling pathways, including but not limited to PDGF, TGF-β, Wnt, hedgehog, estrogen, Notch, PI3K/AKT/mTOR, and MAPK signaling, as well as apoptotic pathways, hypoxia, and the influence of noncoding RNAs (lncRNAs and miRNAs) [142]. The platelet-derived growth factor B (PDGF-BB), a pro-inflammatory cytokine, was shown to be elevated in the lungs of PAH patients [143, 144]. Intriguingly, PDGF has a strong link with PASMCs proliferation [145]. However, lncRNA H19, a highly evolutionarily conserved maternally transcribed lncRNA, has been observed to be overexpressed in cancer patients cells when stimulated by TNF- α, IL-1, IL-6, TGF-1, and PDGF-BB [146, 147]. Several studies suggest that H19 plays a crucial role in the origin of PAH by increasing the proliferation of PASMCs, whereas decreasing H19 expression inhibits the development of PAH [148]. How precisely PDGF modulates H19 is currently unknown. Therefore, additional research is required to establish H19 as an efficacious PAH therapeutic target [141].
PAH’s pathogenesis also involves endothelial dysfunction, where lncRNAs play a crucial role in the dysregulation process. In particular, dysregulation of lncRNAs has been implicated in the pathogenesis of chronic thromboembolic pulmonary hypertension (CTEPH), a subtype of PAH that is caused by chronic pulmonary embolism. Analysis of microarrays has identified 185 differentially expressed lncRNAs in CTEPH tissue when compared to healthy controls. Among these lncRNAs, NR_001284, NR_036693, NR_033766, and NR_027783 were significantly altered [149]. The MIR22 host gene (MIR22HG), MIR210HG, H19, MALAT1, and MEG9 lncRNAs were differentially expressed in endothelial cells in response to hypoxia, based on NGS studies [150]. Another lncRNA (TYKRIL) can stimulate the growth of PASMC. An ex vivo study using precision-cut lung slices (PCLS) demonstrated that TYKRIL knockdown can reverse pulmonary vascular remodeling. These findings suggest that TYKRIL may serve as a promising therapeutic candidate [151]. Smooth muscle enriched long noncoding RNA (SMILR) was also identified as a promising treatment target, as it is substantially expressed in PAH patients [152]. However, understanding the role of lncRNAs in the pathogenesis of PAH and CTEPH may lead to the identification of new diagnostic and therapeutic targets for these conditions. However, further research is needed to fully elucidate the molecular mechanisms underlying the dysregulation of lncRNAs in these diseases.
14.2.6.2 Therapeutic Application of lncRNAs in PAH
LncRNAs have a functional correlation with many human diseases, in particular PAH. Dysregulation of lncRNAs induces cell proliferation, leading to disease formation. As a result, this could be used as a promising biomarker or as a tool for personalized disease treatment. A study reported that, in the Chinese population, lncRNA MALAT1 (rs619586 A > G) has been found to be associated with a reduced risk of developing PAH, indicating its potential as a biomarker [153]. Studies have reported that BC-819 (BioCancell Therapeutics Inc.), which is a double-stranded DNA plasmid containing diphtheria toxin under the H19 gene promoter regulation, can decrease the size of bladder tumors in mice [154].
A phase II clinical study involving patients with non-muscle-invasive bladder cancer reported that combining BC-819 with Bacillus Calmette-Guerin (BCG) led to an improvement in patient condition [141]. However, there are some potential drawbacks to studying lncRNAs as a promising drug target identification for cardiovascular diseases, specifically PAH. Although much research has been implemented in search of potential lncRNAs, therapeutic development is still in the early phases. RNA-based drug stability, an effective mode of delivery, establishing the secondary structure of lncRNAs, regulating off-target effects, and altering patient-specific dosages are typical challenges [155].
14.2.7 Circular RNAs (circRNAs)
The earliest evidence of any form of circular RNA (circRNA) molecule was reported when Diener found genome of viroids causing disease in potato plants in 1971 [156, 157]. The viroids, consisting of single-stranded and closed RNA molecules, were further confirmed by several studies in the 1970s. Interestingly, various types of circular RNAs were reported so far across different life forms (from plant viruses to humans) [158]. Nigro et al. confirmed a type of circular RNA transcript in human cells for the very first time while studying a tumor suppressor gene [159]. As experimental and computational challenges existed during the earlier studies, circRNAs coming from transcription process were thought to be splicing artifacts in most of the cases [160]. But the advancement of next-generation sequencing technologies and bioinformatics tools in the recent times made it possible to identify large numbers of circRNAs in different species [20, 158, 160]. Salzman et al. were the first to apply high-throughput RNA sequencing technology (RNA-seq) to detect circRNAs in the human body in 2012 [133]. These current approaches of circRNA discovery are further equipped with advanced experimental techniques for validation, functional characterization, and mechanistic study of circRNAs [20].
CircRNAs, unlike their linear counterparts, have covalently closed loop and single-stranded structure generated from a kind of non-canonical splicing called back splicing [20, 158, 160]. Majority of these circRNAs are widely and abundantly expressed in eukaryotes including humans and other mammals [161, 162]. This circularized chemical structure also provide higher stability, causing greater abundance compared to the linear mRNAs [163]. CircRNAs are mainly endogenous and show high level of conservation and specificity in tissue types and development stages [164,165,166,167,168]. Circular RNAs are classified into three primary categories based on the biochemical processes involved in their formation: exonic circRNAs (EcircRNAs), intronic circRNAs (CiRNAs), and exon-intron circRNAs (EIciRNAs). The major biogenesis mechanisms include intron pairing-driven circularization, lariat-driven circularization, RBP/trans-factor-driven circularization, cis-regulation based on variable circularization [133, 162, 167,168,169,170,171,172,173,174,175]. Though EcircRNAs are found mostly in cytoplasm, EIciRNAs and CiRNAs are predominantly reside in nucleus [20, 158, 173, 176]. Several groups also studied circRNAs degradation mechanisms in the recent times [176,177,178,179,180]. CircRNAs act as a novel class of genomic regulators by playing significant roles in various biological and physiological processes [20, 158, 160]. The most predominant function of circRNAs is to regulate target gene expression by competing with a type of noncoding RNA called micro RNAs (miRNAs) [165, 181]. Moreover, they can interact with RNA binding proteins (RBP) or other proteins; work as translational regulators or protein scaffolds, regulate transcription, post transcription or alternate splicing; participate in translation of peptides or proteins; and involve in N6-methyladenosine (m6A) modification of RNAs [20, 160, 182, 183].
14.2.7.1 CircRNAs in PAH
The dysregulated expression of circRNAs has been linked to the pathogenesis, progression, and development of numerous human diseases, including cancer, diabetes, neurodegenerative disorders, and cardiovascular diseases [20, 157, 183]. Specifically, circRNAs showed significant level of involvement in different forms of pulmonary hypertension (PH), e.g., pulmonary arterial hypertension (PAH), idiopathic pulmonary arterial hypertension (IPAH), hypoxia-induced pulmonary hypertension (HPH), and chronic thromboembolic pulmonary hypertension (CTEPH). CircRNA profiling studies have been conducted on clinical samples such as blood and lungs of PH patients, as well as PH rodent and in vitro models, resulting in identification of some differentially expressed circRNAs related to PH [184,185,186,187,188,189,190,191]. Pulmonary vascular cells, e.g., PASMCs, are impacted by regulatory effects of circRNAs which eventually lead to pulmonary vascular remodeling [187,188,189,190,191,192,193,194,195,196,197,198]. Furthermore, circRNAs have been shown to cause functional abnormalities in PAECs and result in right ventricular (RV) remodeling in PH [20, 158, 160]. CircRNAs are found to be overexpressed in PH condition in most of the instances leading to mechanistic effects at molecular level [20, 158, 160]. One example of a differentially expressed circRNA related to PH is circ-CALM4, which has been found to be upregulated in the lungs of HPH mice. It regulates PASMC pyroptosis induced by hypoxia through circ-Calm4/miR-1243p/programmed cell death protein 6 signaling and also regulates the proliferative properties of PASMCs through the miR-337-3p/Myo10 signaling axis [192, 196]. Another circRNA, hsa_circ_0016070, has been found to be overexpressed in the lungs of COPD-PAH patients and enhances PASMC proliferation through miR-942/CCND1 [197]. In contrast, hsa_circNFXL1_009 is downregulated in blood samples of COPD-PAH patients and hypoxic PASMCs and has been shown to play regulatory roles in the proliferation, migration, apoptosis, and potassium channel activation of PASMCs [199]. Additionally, hsa_circ_0046159 is upregulated in the blood of CTEPH patients and exerts regulatory roles through the predicted mechanism of the hsa_circ_0046159/miR-1226-3p/ATP2A2 axis. Circ-GSAP has been demonstrated to be linked with the development and unfavorable outcomes of IPAH and is downregulated in PBMCs and lung tissues of IPAH patients, as well as in lung tissues of monocrotaline-PAH rats, SU5416/hypoxia-induced PAH rats, and hypoxic PMECs [195].
CircRNAs are considered promising biomarkers in different disease conditions due to their stability and abundance [20]. Circ-006848 and circ-GSAP are examples of circRNAs that have been identified as potential diagnostic biomarkers for PH. For instance, Circ-006848 is overexpressed in PAH patients serum and has been shown to be a useful tool in predicting the diagnosis of right ventricular hypertrophy (RVH) in these patients [200]. RVH is a common complication of PAH and is associated with a poor prognosis. Therefore, identifying biomarkers that can accurately predict RVH in PAH patients is important for early diagnosis and management of the disease. On the other hand, circ-GSAP is downregulated in the PBMCs of IPAH patients, and this has been found to correlate with the development of IPAH as well as poor clinical outcomes associated with IPAH [195]. IPAH is a severe form of PAH, identifying diagnostic and prognostic biomarkers for IPAH is crucial for early diagnosis, appropriate treatment, and improved outcomes. Current progress in the field of RNA therapeutics and the relevant attractive properties (stability, conservation, specificity, and abundance) make circRNAs a very promising group of therapeutically potential molecules [20, 158, 160]. Though there is still no evidence of any clinical trials regarding circRNA therapeutics, this promise is being further strengthened by different layers of studies (in vitro, in vivo, and clinical) [20].
14.2.8 Microfluidic Based LoC Technology
Uncovering disease mechanisms is key to developing diagnostic tools and new therapeutic targets. Rapid and resilient technology is needed to simplify complex disease phenomena. Microfluidics technology has emerged together with tissue engineering, microfabrication, and Lab-on-a-Chip (LoC) methods to build ultra-precise models to study various aspects of molecular biology, cell, and synthetic biology research [201]. This emerging technology reduces chemical consumption, minimizes cost, and provides rapid, reliable, and high-throughput screening [202, 203]. This technology can potentially fill the gaps between conventional cell cultures, animal models, or human subjects in various fields, particularly in biomedical research and drug development [22]. Furthermore, recently developed organ-on-a-chip (OoC) systems combine biology with microtechnology [23] that replicates the organ’s physiological environment, allowing researchers to study organ-level responses to drugs, toxins, or diseases without the need for animal models [24, 25] The advent of OoC technology facilitates the building of on-a-chip devices: heart-on-a-chip [204], lung-on-a-chip [205, 206], animal-on-a-chip [207], and body-on-a-chip [208]. These on-a-chip systems mimic biological function and allow for precision sciences. For example, Michas and colleagues recently engineered a microfabricated living cardiac pump on a chip mimicking the human ventricular chamber [209]. This new generation of sophisticated systems leverages the benefit of studying a replicated human organ in a tissue-chip model with structurally, biologically, and biomechanically similar functions [209].
14.2.8.1 Promising LoC Study with PAH
Advancement of modern science integrated micro total analysis system (μTAS), microfluidic LoC, and bio-microelectromechanical systems to develop new tools to investigate complex disease mechanisms. Several research groups focused on designing microfluidics devices to study pulmonary hypertension for potential therapeutic intervention. One such device, constructed by Lee and colleagues, is a microfluidic diagnostic tool integrated with an electrochemical assay system to rapidly identify pulmonary hypertension-associated biomarkers including fibrinogen, adiponectin, low-density lipoprotein, and 8-isoprostane [210]. To understand the vascular phenomenon, such as endothelial cell morphology and proliferation, Bogorad et al., described a 3D printed tissue-engineered microvessel model [211]. A microfluidic cell stretch device showed stretch stress that facilitates cell proliferation in PAH [212]. One elegant study described the fabrication process to create a near-ideal miniature vascular structure mimicking human’s healthy and stenotic blood vessels using 3D printing system [213]. This method can overcome the limitation of conventional 2D wafer-based soft lithography or other biofabrication processes [214,215,216]. However, an advanced ultra-precise 3D printing fabrication process and a conceptual “PAH-chip” design is described later in this section.
Some other promising microfluidics devices have recently been developed to study PAH. Al-hilal and colleagues recently reported PAH-on-a-chip, simulating five human pulmonary artery layers: luminal, intimal, medial, adventitial, and perivascular. PAH-on-a-chip resembles human PAH pathophysiology enabling the growth of different types of pulmonary arterial cells (PAC) including endothelial, smooth muscle, and adventitial cells without altering their phenotypes. Disease-affected PAC cells generally migrate to their designated layers, this phenomenon exactly showed in an experiment with “PAH-on-a-chip” [217], which validates the device’s accuracy and ability to recapitulate human pathophysiology. However, a critical phenomenon was observed in PAH patients where women are affected more by PAH but survive longer than men, known as “sex-paradox” [218,219,220]. The key mechanism of this sex disparity is unknown; however, it is interesting that both sexes follow the same treatment plan but vary in response. The reason could be the unavailability of screening tools and animal models. Moreover, the gold standard in vitro assays is insufficient to recapitulate the intricate cellular interaction [221]. In one study, male and female cells were separately loaded into a PAH-on-a-chip and demonstrated that the chip accurately mimics human PAH. So, this chip might be useful for detecting sex hormones and therapeutic impacts on PAH progression [217, 222]. This PAH-chip has also demonstrated promising capabilities in testing the effectiveness of the anti-PAH drug including fasudil (a Rho kinase inhibitor). The results were shown consistency with the in vivo studies using both Sugen-5416-plus-hypoxia (SuHx) and monocrotaline (MCT) rat model of PAH [223] as well as in patients with PAH [224]. This indicates that the chip could serve as a valuable tool to investigate the effectiveness of other anti-PAH drugs such as ASK-1 inhibitor [225], GS-44421731, or TGF-β trap therapeutics [226].
Another PAH-chip was developed to study pulmonary endothelial and smooth muscle cells by monitoring functional and transcriptomic changes [21]. The study was performed using a combination of BMPR2 (bone morphogenetic protein type II receptor) knockdown and hypoxia (2% O2). BMPR2 is a type of rare genetic mutation that can be initiated by various factors (e.g., hypoxia, inflammation) [227] and can increase the susceptibility to PAH [228]. PAH-chip data showed that combination treatment significantly increased the proliferation of pulmonary artery smooth muscle cells (PASMCs) compared to separate treatments, similar to the effect observed in static cell culture [229]. A receptor tyrosine kinase inhibitor (10 μM of imatinib mesylate) was used to inhibit the proliferation. This demonstrates PAH-chip can measure therapeutic effectiveness. This sophisticated PAH-chip has the potential to trigger PAH diseases and evaluating their response to various possible treatments, leveraging studies on pulmonary vascular remodeling and drug target analysis [21]. By providing a platform to investigate cellular interactions and responses to potential therapies, this advanced chip technology can significantly improve our understanding of PAH and potentially lead to more effective treatment strategies in the future.
14.2.8.2 Microfluidic Chromatin Immunoprecipitation (ChIP) Assays for Epigenetic Study
Epigenetics refers to altering gene expression without changing the DNA sequence. Epigenetic factors play a key role in regulating cellular processes like stem cell pluripotency/differentiation and tumorigenesis [15]. Epigenetic regulators such as DNA methylation, histone modifications, and noncoding RNAs (micro or miRNA) are the potential PAH drug targets [230]. Epigenetic mechanisms can be effectively investigated using chromatin immunoprecipitation (ChIP), which require millions of cells, making it challenging to conduct large-scale studies due to the need for many human or animal models [15]. Presently, the common approach is to use animal models to assess a drug’s efficacy or probe the possible disease mechanism. Various rodent models, including genetically modified mice and those induced by chronic hypoxia, Sugen-5416-plus-hypoxia (SuHx), and monocrotaline (MCT), are frequently employed in PAH research [217, 231]. However, these animal or cellular models have limitations in fully replicating the pathophysiology of human PAH, hindering their ability to demonstrate disease severity, histopathology, and sex-related differences in response to therapy [222, 231,232,233,234]. A new approach has emerged, shifting the paradigm in PAH research. Microfluidic ChIP assays offer the potential to study PAH without the reliance on animal models [235]. Recent studies have shown that microfluidics device enable quick, sensitive, and high-throughput testing with a significantly reduced number of cells (only 2000 cells are required) [236]. Additionally, combining sonication and immunoprecipitation (IP) in the same device has improved the accuracy of the assay. This method enables the testing of just 100 cross-linked cells for ChIP or 500 pg of genomic DNA for methylated DNA IP in only one hour [237]. In PAH research, miRNAs have been identified as promising therapeutic target molecules (as discussed earlier). However, the challenge lies in the divergence of miRNA expression patterns between human PAH and animal models [112]. To overcome all the barriers, future miRNA based therapies and interventions might rely on organ-chip systems that better mimic the human PAH phenotype.
14.2.8.3 Quantification of Endothelial Cells of PAH by LoC Method
Early diagnosis of PAH reduces disease progression and identifies effective treatments. Developing early diagnostic tools will save lives and increase the quality of life. Various molecular and cellular biomarkers have been studied in PAH patients to gain insights into disease progression and potential therapeutic interventions. Among these biomarkers, cellular biomarkers, such as endothelial progenitor cells (EPC) and circulating endothelial cells (CEC), are potential candidates to study the progression of PAH disease and therapeutic intervention as they contribute to vascular repair and remodeling [238]. There are some challenges to study of these cells such as the endothelial dysfunction and EPC number are inversely correlated [239, 240]; how the EPC promotes vascular repairment is still unclear [241]. Therefore, cell isolation and accurate quantification are necessary to explore PAH disease mechanisms and therapeutic targets. Standard techniques, such as enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR), make it relatively simple to measure molecular biomarkers; nevertheless, it is difficult and time-consuming to isolate and quantify cellular biomarkers (EPCs and CECs) using standard methods such as flow cytometry, magnetic bead-based techniques, and colony-forming assays [242, 243]. Furthermore, the quantification of EPC in PAH patients is highly inconsistent [244, 245], showing either an increase [244, 246], decrease [247,248,249], or no-change [248, 250] compared to the healthy controls. To overcome this problem, disposable microfluidics chips, “EPC capture chip” [242] and “CEC capture chip” [251], were designed to trap and count the EPCs and CECs with amazingly small amounts of whole blood (200–400 μL) without sample pre-preprocessing. The reliability of these chips was validated using flow cytometry, resulting in a strong correlation (EPCs, R = 0.83; CECs, R = 0.89). These innovative capture chips could serve as a robust method for measuring, monitoring, and screening samples from PAH patients, as well as those with other cardiovascular and neurodegenerative diseases [252] and cancer [253]. The development of accurate and reliable microfluidic chips represents a significant advancement in PAH research and has the potential to revolutionize disease diagnosis, monitoring, and the discovery of targeted treatments. These microfluidic devices offer new possibilities for understanding disease mechanisms, identifying biomarkers, and eventually improving patient outcomes. As technology continues to evolve, we can expect even more sophisticated and precise microfluidic solutions that will continue to drive progress in PAH and other areas of medicine.
14.2.8.4 Conceptual Model Design for a PAH-Chip
A straightforward conceptual “PAH-chip” model (Fig. 14.2a) is presented to describe how a microfluidic chip is designed and also briefly explains the printing and fabrication process using cutting-edge 3D printing technology. This model “PAH-chip” contains four chambers, each of which has the same size (radius, r = 100 μm and depth, d = 50 μm). However, the dimensions of other components (such as the channel, reservoirs, cell loading zone, and outlet to release waste) are not shown, as this is simply a conceptual model. Based on the experimental model and other requirements, design customization is feasible. These chips can be made for simultaneous loading and screening. For example, Fig. 14.2b design includes four chips under platform (or one chip) that can be simultaneously used for four different sample/cell types. The “PAH-chips” device allows the growth of three major types of PAC cells simultaneously in one chamber. These PAC cells are endothelial cells, smooth muscle cells, and adventitial cells [217].

A conceptual PAH-chip model. (a) A prototype design displaying the various components of a chip, such as a cell loading chamber, reservoirs, cell growth and counting chambers, channels, and waste chamber. (b) Multiple chips can be combined into one for concurrent sample loading. (c) Multiple replicate analyses are possible on a single chip; here showed 12 chips
One advantage of using the “PAH-chips” device is that it allows researchers to study the interactions between these three types of cells in a controlled environment that closely mimics the in vivo conditions of the pulmonary arterial system. Additionally, the device allows for one chamber to be used as a control, which enables researchers to compare the behavior of cells in the experimental chamber to that of cells in the control chamber. However, multiple replicates analysis is also possible using the model design in Fig. 14.2c, which reduces time and provides consistent and rapid experiment. By using ‘PAH-chips’, researchers can better understand the complex interactions between different types of PAC cells and identify promising therapeutic targets for pulmonary arterial diseases. Notably, conventional and standardized approaches (such as flow cytometry and colony-forming assay) are employed to isolate and quantify cells, which is challenging and time-consuming [242, 243]. Advanced microfluidics-based approaches can solve this limitation, as discussed earlier in this section.
Making a “PAH-chip” starts with the design of a 3D model. The “PAH-chip” prototype can be designed using an open-source CAD (computer-aided design) program (e.g., AutoCAD, Fusion 360). An ideal microfluidics device comprises of an inlet (cell loading), reservoir (flow turbulence control), chambers, and an outlet (waste) to expel by-products. Here, the design is similar to Fig. 14.2a. Next, the designed CAD model (e.g., stereolithography file format) needs to be transferred to a slicer software (e.g., DeScribe, Nanoscribe, Germany) in order to make the 3D printing compatible file format (e.g., GWL, Nanoscribe, Germany). Recently, an ultra-high precision two-photon polymerization (2PP)-based 3D printer (e.g., Nanoscribe, Germany) offers a powerful platform to print micro- to meso-level structures. However, after printing, polydimethylsiloxane (PDMS) can be used to construct a PDMS-based microchip. PDMS (a prepolymer and cross-linker at a 10:1 (w/w) ratio) is a widely used elastomer in the field of microfluidics and lab-on-a-chip devices. However, the ratio could be changed based on necessity. These devices are produced based on experimental specifications, testing, and they often require modification (multiple generations of a device) for the desired outcome. The general microfabrication process has already been established by Jeff Hasty’s lab [254] and other pioneered microfluidics research groups in pulmonary hypertension [21, 212, 217, 255].
In the last 20 years, most microfluidic devices were designed using photolithography, which was initially developed for the semiconductor industry [254]. In recent years, the 3D printing of microfluidic devices and molds of microfluidic devices have gained popularity because of the preciseness and complexity of structures that can be printed [256]. Microfluidic devices for bacteria are often challenging to produce due to the small size of bacteria. In addition, devices that monitor multiple-sized cells (e.g., bacteria and plant cells) can be challenging to produce accurately using photolithography. However, 3D printing provides the accuracy and speed to make these types of structures at a relatively low cost. For example, the Butzin lab’s 3D microfab facility [257] has recently built several new microfluidic devices to monitor bacteria, bacteria–plant interactions, and accurately and quickly count cells or particles precisely and reliably regardless of size and shape (data not shown).
Recently, microfluidics coupled with an advanced microscope system (e.g., Nikon Eclipse Ti2 inverted fluorescence) has shown great success. Live cell imaging, single cell analysis, and cell tracking have already been implemented in antibiotic resistance studies [258,259,260] and other prominent research including single cell immunology [261], cancer cell trapping [262], and host–microbe interaction at single cell level [263]. This integrated knowledge might help to create a better microfluidic system and data analysis platform to probe disease mechanisms and potential drug target identification.
14.2.8.5 Current Limitation of Microfluidic-Based LoC Study in PAH
Although some microfluidic studies were employed to explore pulmonary hypertension, several notable gaps were highlighted in each study. An organ-chip model can mimic human pathophysiology, albeit there is not much work showing the application of therapeutics using chip methods. Some microfluidic devices were built using conventional 2D wafer-based methods; however, they could not accurately recreate the in vivo 3D model [242]. Sato and Costa’s research team examined shear stress to comprehend the dynamics and nature of hypertension and thrombosis. They proposed further customization of their device may aid in clearly understanding the PAH mechanism [212, 213]. Numerous microfluidics research concentrated on certain aspects of cardiovascular function, including angiogenesis [264, 265], transport properties [266,267,268], shear stress [269], cancer metastasis [270], and hemodynamics [271]. Based on the current knowledge, an integrated organ-on-a-chip model with cutting-edge tissue engineering, 3D printing, and microfabrication technologies may facilitate the exploration of the complex processes behind pulmonary hypertension and the identification of effective therapeutic targets.
14.3 Conclusion
Epigenetic studies in concert with PAH have steadily increased in recent times. Clinical and experimental data suggested that the plasticity of epigenetic changes in PAH could be a plausible candidate for future research and therapeutic development. Indeed, microfluidic LoC technology has the potential to offer a novel study design mimicking human pathobiology to resolve the current constraints in PAH studies. Epigenetics, in conjunction with microfluidic LoC technology, is perhaps the most useful research in unraveling the mystery of PAH pathogenesis and identifying novel drugs to treat this chronic progressive cardiopulmonary disease.
Abbreviations
- ABCA1:
-
ATP binding cassette 1
- APOA4:
-
Apolipoprotein A4
- BER:
-
Base excision repair
- BMPR2:
-
Bone morphogenetic protein type II receptor
- BRDP:
-
Bromodomain-containing protein
- CAP:
-
Concentrated ambient particles
- ChIP:
-
Chromatin immunoprecipitation
- COPD:
-
Chronic obstructive pulmonary disease
- DNMT:
-
DNA methyltransferase
- ELISA:
-
Enzyme-linked immunosorbent assay
- HDAC:
-
Histone deacetyl transferases
- HME:
-
Histone modifying enzymes
- IL:
-
Interleukins
- LoC:
-
Lab-on-a-chip
- MBD:
-
Methyl CpG binding
- MCT:
-
Monocrotaline
- OoC:
-
Organ-on-a-chip
- PAC:
-
Pulmonary arterial cells
- PAEC:
-
Pulmonary endothelial cells
- PAH:
-
Pulmonary arterial hypertension
- PARP:
-
Poly (ADP-ribose) polymerases
- PASMC:
-
Pulmonary arterial smooth muscle cell
- PDGF-BB:
-
Platelet-derived growth factor B
- PH:
-
Pulmonary hypertension
- PPI:
-
Protein–protein interactions
- PVOD:
-
Pulmonary veno-occlusive disease
- RAAS:
-
Renin–angiotensin–aldosterone system
- RISC:
-
RNA-induced silencing complex
- RSRS:
-
Renal sodium retention system
- RVLM:
-
Rostral ventrolateral medulla
- SAM:
-
S-adenyl methionine
- SMC:
-
Smooth muscle cell
- SNS:
-
Sympathetic nervous system
- TET:
-
Ten-eleven translocation
- TGF:
-
Transforming growth factor
- TLR:
-
Toll-like receptors
- TNF:
-
Tumor necrosis factor
- UHRF:
-
Ubiquitin-like, containing PHD and RING finger domain
- VEGF:
-
Vascular endothelial growth factors
References
Thenappan T, et al. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492.
Tonelli AR, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;188(3):365–9.
Humbert M, et al. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130(24):2189–208.
Humbert M, Ghofrani H-A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71(1):73–83.
Giaid A, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–9.
Morrell NW, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation. 2001;104(7):790–5.
Perros F, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185(3):311–21.
Ma L, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–61.
Lau EM, et al. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14(10):603–14.
Luna R, et al. Insights on the epigenetic mechanisms underlying pulmonary arterial hypertension. Braz J Med Biol Res. 2018;51:e7437.
Kim J-D, et al. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Exp Mol Med. 2015;47(7):e175.
Tuder RM, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25S):D4–D12.
Zhao L, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126(4):455–67.
Archer SL, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–71.
Ho L, et al. Epigenetic mechanisms as emerging therapeutic targets and microfluidic chips application in pulmonary arterial hypertension. Biomedicine. 2022;10(1):170.
Kim GH, et al. Epigenetic mechanisms of pulmonary hypertension. Pulm Circ. 2011;1(3):347–56.
Wang P, et al. miRNA-34a promotes proliferation of human pulmonary artery smooth muscle cells by targeting PDGFRA. Cell Prolif. 2016;49(4):484–93.
Chen T, et al. Loss of microRNA-17∼ 92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med. 2015;191(6):678–92.
Chen T, et al. MicroRNA-212-5p, an anti-proliferative miRNA, attenuates hypoxia and sugen/hypoxia-induced pulmonary hypertension in rodents. Mol Ther Nucleic Acids. 2022;29:204–16.
Ali MK, et al. The role of circular RNAs in pulmonary hypertension. Eur Respir J. 2022;
Wojciak-Stothard B et al. A microfluidic chip for pulmonary arterial hypertension. 2021.
Low L, Tagle D. Tissue chips–innovative tools for drug development and disease modeling. Lab Chip. 2017;17(18):3026–36.
Leung CM, et al. A guide to the organ-on-a-chip. Nat Rev Methods Primers. 2022;2(1):1–29.
Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32(8):760–72.
Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet. 2022;23(8):467–91.
Cheng X, Wang Y, Du L. Epigenetic modulation in the initiation and progression of pulmonary hypertension. Hypertension. 2019;74(4):733–9.
Bisserier M, et al. Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary hypertension disease. Vasc Biol. 2020;2(1):R17–34.
Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med. 2013;91(3):297–309.
Wang A, et al. Substrate stiffness and stretch regulate profibrotic mechanosignaling in pulmonary arterial adventitial fibroblasts. Cell. 2021;10(5):1000.
Hotchkiss RD. The quantitative separation of purines, pyrimidines, an d nucleosides by paper chromatography. J Biol Chem. 1948;175(1):315–32.
Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development: developmental clocks may depend on the enzymic modification of specific bases in repeated DNA sequences. Science. 1975;187(4173):226–32.
Bestor TH, Verdine GL. DNA methyltransferases. Curr Opin Cell Biol. 1994;6(3):380–9.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38.
Xie S, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236(1):87–95.
Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279(46):48350–9.
Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–26.
Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Mayer W, et al. Demethylation of the zygotic paternal genome. Nature. 2000;403(6769):501–2.
Zhang F, et al. Active tissue-specific DNA demethylation conferred by somatic cell nuclei in stable heterokaryons. Proc Natl Acad Sci. 2007;104(11):4395–400.
Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146(6):866–72.
Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5.
Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–33.
Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
Meehan RR, et al. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. 1989;58(3):499–507.
Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–92.
Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl CpG-binding proteins. Genet Res. 1998;72(1):59–72.
Bellacosa A, et al. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci. 1999;96(7):3969–74.
Petronzelli F, et al. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275(42):32422–9.
Wong E, et al. Mbd4 inactivation increases C→ T transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci. 2002;99(23):14937–42.
Hashimoto H, et al. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455(7214):826–9.
Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450(7171):908–12.
Achour M, et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 2008;27(15):2187–97.
Prokhortchouk A, et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15(13):1613–8.
Filion GJ, et al. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol. 2006;26(1):169–81.
Sasai N, Nakao M, Defossez P-A. Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. 2010;38(15):5015–22.
Daniel JM, et al. The p120 ctn-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 2002;30(13):2911–9.
Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–59.
Gao F, et al. Global analysis of DNA methylation in hepatocellular carcinoma by a liquid hybridization capture-based bisulfite sequencing approach. Clin Epigenetics. 2015;7(1):1–11.
Perros F, et al. Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2013;187(2):189–96.
Han L, et al. DNA methylation and hypertension: emerging evidence and challenges. Brief Funct Genomics. 2016;15(6):460–9.
DeLalio LJ, Sved AF, Stocker SD. Sympathetic nervous system contributions to hypertension: updates and therapeutic relevance. Can J Cardiol. 2020;36(5):712–20.
Ji L, et al. Association between polymorphisms in the renin-angiotensin-aldosterone system genes and essential hypertension in the Han Chinese population. PLoS One. 2013;8(8):e72701.
Hautefort A, et al. Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget. 2017;8(32):52995.
Liu D, et al. Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. Am J Respir Crit Care Med. 2017;196(7):925–8.
Yan Y, et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci Adv. 2020;6(50):eaba2470.
Xu X, et al. A genome-wide methylation study on obesity: differential variability and differential methylation. Epigenetics. 2013;8(5):522–33.
Smith JA, et al. Epigenomic indicators of age in African Americans. Hereditary Genet Curr Res. 2014;3:3.
Han L, et al. The interactions between alcohol consumption and DNA methylation of the ADD1 gene promoter modulate essential hypertension susceptibility in a population-based, case–control study. Hypertens Res. 2015;38(4):284–90.
Bellavia A, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2(3):e000212.
Lehninger AL, Nelson DL, Cox MM. Lehninger principles of biochemistry. Macmillan; 2005.
Redon C, et al. Histone H2a variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12(2):162–9.
Millis RM. Epigenetics and hypertension. Curr Hypertens Rep. 2011;13(1):21–8.
Gupta S, Yel L. Molecular biology and genetic engineering. Middleton’s Allergy: Principles and Practice. 2013;10:162–18.
Sakabe K, Wang Z, Hart GW. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci. 2010;107(46):19915–20.
Nathan D, et al. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006;20(8):966–76.
Wang Y, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306(5694):279–83.
Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126(5):905–16.
Meng F, et al. Discovery and development of small molecules targeting epigenetic enzymes with computational methods. In: Epi-informatics. Elsevier; 2016. p. 75–112.
Gerthoffer W. Epigenetic targets for oligonucleotide therapies of pulmonary arterial hypertension. Int J Mol Sci. 2020;21(23):9222.
Irmak M, Sizlan A. Essential hypertension seems to result from melatonin-induced epigenetic modifications in area postrema. Med Hypotheses. 2006;66(5):1000–7.
Morimoto S, et al. Sympathetic activation and contribution of genetic factors in hypertension with neurovascular compression of the rostral ventrolateral medulla. J Hypertens. 1999;17(11):1577–82.
Cohen I, et al. Histone modifiers in cancer: friends or foes? Genes Cancer. 2011;2(6):631–47.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Chelladurai P, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. 2021;178(1):54–71.
Li M, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187(5):2711–22.
Boucherat O, et al. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci Rep. 2017;7(1):1–14.
Desjardins CA, Naya FJ. The function of the MEF2 family of transcription factors in cardiac development, cardiogenomics, and direct reprogramming. J Cardiovasc Dev Dis. 2016;3(3):26.
Kim J, et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation. 2015;131(2):190–9.
Chelladurai P, Seeger W, Pullamsetti SS. Epigenetic mechanisms in pulmonary arterial hypertension: the need for global perspectives. Eur Respir Rev. 2016;25(140):135–40.
Fuks F, et al. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31(9):2305–12.
Fuks F, et al. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24(1):88–91.
Ali MK, Ichimura K, Spiekerkoetter E. Promising therapeutic approaches in pulmonary arterial hypertension. Curr Opin Pharmacol. 2021;59:127–39.
Federici C, et al. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(2):219–28.
Provencher S. Olaparib for PAH: a multicenter clinical trial. 2018.
Meloche J, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129(7):786–97.
Provencher S. Apabetalone for pulmonary arterial hypertension: a pilot study. 2018.
Meloche J, et al. Bromodomain-containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circ Res. 2015;117(6):525–35.
Sun BK, Tsao H. Small RNAs in development and disease. J Am Acad Dermatol. 2008;59(5):725–37.
Arasu P, Wightman B, Ruvkun G. Temporal regulation of lin-14 by the antagonistic action of two other heterochronic genes, lin-4 and lin-28. Genes Dev. 1991;5(10):1825–33.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–62.
Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–6.
Cummins J, Velculescu V. Implications of micro-RNA profiling for cancer diagnosis. Oncogene. 2006;25(46):6220–7.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Zeng Y. Principles of micro-RNA production and maturation. Oncogene. 2006;25(46):6156–62.
Zhou G, Chen T, Raj JU. MicroRNAs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2015;52(2):139–51.
Lee A, et al. Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB Rep. 2014;47(6):311.
Negi V, Chan SY. Discerning functional hierarchies of microRNAs in pulmonary hypertension. JCI insight. 2017;2(5):e91327.
Chen J-Q, et al. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev. 2016;15(12):1171–80.
Meloche J, et al. Therapeutic potential of microRNA modulation in pulmonary arterial hypertension. Curr Vasc Pharmacol. 2015;13(3):331–40.
Boucherat O, Potus F, Bonnet S. microRNA and pulmonary hypertension. Adv Exp Med Biol. 2015;888:237–52.
Santos-Ferreira CA, et al. Micro-RNA analysis in pulmonary arterial hypertension: current knowledge and challenges. Basic Transl Sci. 2020;5(11):1149–62.
Ma W, et al. Inhibition of microRNA-30a alleviates vascular remodeling in pulmonary arterial hypertension. Mol Ther Nucleic Acids. 2021;26:678–93.
Zhang J, et al. Micro RNA-483 amelioration of experimental pulmonary hypertension. EMBO Mol Med. 2020;12(5):e11303.
Xu J, et al. MicroRNAs in pulmonary hypertension, from pathogenesis to diagnosis and treatment. Biomol Ther. 2022;12(4):496.
Wang Y, et al. Epigenetic regulation and its therapeutic potential in pulmonary hypertension. Front Pharmacol. 2018;9:241.
Carregal-Romero S, et al. MicroRNA nanotherapeutics for lung targeting. Insights into pulmonary hypertension. Int J Mol Sci. 2020;21(9):3253.
Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014;195:49–54.
Ohno S, Much S. Junk. in DNA in our genome. Brookhaven Symp Biol. 1972.
Beermann J, et al. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;
Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14(2):100–12.
Engreitz JM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539(7629):452–5.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–307.
Brown CJ, et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71(3):527–42.
Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–89.
Statello L, et al. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22(2):96–118.
Lesizza P, et al. Noncoding RNAs in cardiovascular disease, In Nucleic acid nanotheranostics. 2019, Elsevier. p. 43–87.
Ulitsky I, et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147(7):1537–50.
Katayama S, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309(5740):1564–6.
Rearick D, et al. Critical association of ncRNA with introns. Nucleic Acids Res. 2011;39(6):2357–66.
Buratowski S. Transcription. Gene expression--where to start? Science (New York, NY). 2008;322(5909):1804–5.
Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–4.
Salzman J, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7(2):e30733.
Guo C-J, et al. Distinct processing of lncRNAs contributes to non-conserved functions in stem cells. Cell. 2020;181(3):621–36. e22
Quinn JJ, et al. Rapid evolutionary turnover underlies conserved lncRNA–genome interactions. Genes Dev. 2016;30(2):191–207.
Hezroni H, et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11(7):1110–22.
Yao R-W, Wang Y, Chen L-L. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–51.
Anderson DM, et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160(4):595–606.
Nelson BR, et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 2016;351(6270):271–5.
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–61.
Han Y, et al. Role of long non-coding RNAs in pulmonary arterial hypertension. Cell. 2021;10(8):1892.
Zahid KR, et al. Pathobiology of pulmonary artery hypertension: role of long non-coding RNAs. Cardiovasc Res. 2020;116(12):1937–47.
Rieg AD, et al. PDGF-BB regulates the pulmonary vascular tone: impact of prostaglandins, calcium, MAPK-and PI3K/AKT/mTOR signalling and actin polymerisation in pulmonary veins of Guinea pigs. Respir Res. 2018;19(1):1–18.
Nogueira-Ferreira R, et al. Exploring the monocrotaline animal model for the study of pulmonary arterial hypertension: a network approach. Pulm Pharmacol Ther. 2015;35:8–16.
Wang Z, et al. Long non-coding RNA MEG3 mediates high glucose-induced endothelial cell dysfunction. Int J Clin Exp Pathol. 2018;11(3):1088.
Stuhlmüller B, et al. Detection of oncofetal h19 RNA in rheumatoid arthritis synovial tissue. Am J Pathol. 2003;163(3):901–11.
Wang S-H, et al. Upregulation of H19 indicates a poor prognosis in gallbladder carcinoma and promotes epithelial-mesenchymal transition. Am J Cancer Res. 2016;6(1):15.
Su H, et al. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir Res. 2018;19(1):1–18.
Gu S, et al. Aberrant expression of long noncoding RNAs in chronic thromboembolic pulmonary hypertension. Mol Med Rep. 2015;11(4):2631–43.
Voellenkle C, et al. Implication of long noncoding RNAs in the endothelial cell response to hypoxia revealed by RNA-sequencing. Sci Rep. 2016;6:24141.
Zehendner CM, et al. Long noncoding RNA TYKRIL plays a role in pulmonary hypertension via the p53-mediated regulation of PDGFRβ. Am J Respir Crit Care Med. 2020;202(10):1445–57.
Lei S, et al. LncRNA-SMILR modulates RhoA/ROCK signaling by targeting miR-141 to regulate vascular remodeling in pulmonary arterial hypertension. Am J Phys Heart Circ Phys. 2020;319(2):H377–91.
Zhuo Y, et al. Functional polymorphism of lncRNA MALAT1 contributes to pulmonary arterial hypertension susceptibility in Chinese people. Clin Chem Lab Med. 2017;55(1):38–46.
Smaldone MC, Davies BJ. BC-819, a plasmid comprising the H19 gene regulatory sequences and diphtheria toxin A, for the potential targeted therapy of cancers. Curr Opin Mol Ther. 2010;12(5):607–16.
Gomes CP, et al. The function and therapeutic potential of long non-coding RNAs in cardiovascular development and disease. Mol Ther Nucleic Acids. 2017;8:494–507.
Diener T. Potato spindle tuber virus: a plant virus with properties of a free nucleic acid: III. Subcellular location of PSTV-RNA and the question of whether virions exist in extracts or in situ. Virology. 1971;43(1):75–89.
Diener T. Potato spindle tuber “virus”: IV. A replicating, low molecular weight RNA. Virology. 1971;45(2):411–28.
Wang Q, et al. Circular RNAs in pulmonary hypertension: emerging biological concepts and potential mechanism. Animal Model Exp Med. 2022;5(1):38–47.
Nigro JM, et al. Scrambled exons. Cell. 1991;64(3):607–13.
Wang J, et al. Circular RNAs: a rising star in respiratory diseases. Respir Res. 2019;20(1):1–10.
Kristensen LS, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
Jeck WR, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453–61.
Salzman J, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777.
Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Dong R, et al. Increased complexity of circRNA expression during species evolution. RNA Biol. 2017;14(8):1064–74.
Werfel S, et al. Characterization of circular RNAs in human, mouse and rat hearts. J Mol Cell Cardiol. 2016;98:103–7.
Chen W, Schuman E. Circular RNAs in brain and other tissues: a functional enigma. Trends Neurosci. 2016;39(9):597–604.
Li X, Yang L, Chen L-L. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428–42.
Starke S, et al. Exon circularization requires canonical splice signals. Cell Rep. 2015;10(1):103–11.
Zhang Y, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51(6):792–806.
Li Z, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–64.
Ashwal-Fluss R, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56(1):55–66.
Conn SJ, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–34.
Zhang X-O, et al. Complementary sequence-mediated exon circularization. Cell. 2014;159(1):134–47.
Gao Y, et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun. 2016;7(1):1–13.
Lee Y, et al. Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 2020;36(3):177–88.
Liu C-X, et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell. 2019;177(4):865–80. e21
Hansen TB, et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–22.
Lasda E, Parker R. Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circRNA clearance. PLoS One. 2016;11(2):e0148407.
Hansen TB, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Yu C-Y, Kuo H-C. The emerging roles and functions of circular RNAs and their generation. J Biomed Sci. 2019;26(1):1–12.
Liu C, et al. A narrative review of circular RNAs as potential biomarkers and therapeutic targets for cardiovascular diseases. Ann Transl Med. 2021;9(7):578.
Xu S-L, et al. Regulation of circular RNAs act as ceRNA in a hypoxic pulmonary hypertension rat model. Genomics. 2021;113(1):11–9.
Miao R, et al. Microarray expression profile of circular RNAs in chronic thromboembolic pulmonary hypertension. Medicine. 2017;96(27):e7354.
Wang J, et al. Characteristics of circular RNA expression in lung tissues from mice with hypoxia-induced pulmonary hypertension. Int J Mol Med. 2018;42(3):1353–66.
Huang CX, et al. Hsa_circ_0016070/micro-340-5p Axis accelerates pulmonary arterial hypertension progression by upregulating TWIST1 transcription via TCF4/β-catenin complex. J Am Heart Assoc. 2022;11(14):e024147.
Jing X, et al. Circular RNA Sirtuin1 represses pulmonary artery smooth muscle cell proliferation, migration and autophagy to ameliorate pulmonary hypertension via targeting microRNA-145-5p/protein kinase-B3 axis. Bioengineered. 2022;13(4):8759–71.
Diao W, et al. Evaluating the effect of Circ-Sirt1 on the expression of SIRT1 and its role in pathology of pulmonary hypertension. Cell Transplant. 2022;31:09636897221081479.
Huang Y, et al. Expression and clinical significance of circular RNA hsa_circ_0003416 in pediatric pulmonary arterial hypertension associated with congenital heart disease. J Clin Lab Anal. 2022;36(4):e24273.
Li S-S, et al. hsa_circWDR37_016 regulates hypoxia-induced proliferation of pulmonary arterial smooth muscle cells. Cardiovasc Ther. 2022;2022:7292034.
Jiang Y, et al. Circular RNA Calm4 regulates hypoxia-induced pulmonary arterial smooth muscle cells pyroptosis via the Circ-Calm4/miR-124-3p/PDCD6 axis. Arterioscler Thromb Vasc Biol. 2021;41(5):1675–93.
Ma C, et al. circRNA CDR1as promotes pulmonary artery smooth muscle cell calcification by upregulating CAMK2D and CNN3 via sponging miR-7-5p. Mol Ther Nucleic Acids. 2020;22:530–41.
Wang Y, et al. Hsa_circ_0002062 promotes the proliferation of pulmonary artery smooth muscle cells by regulating the Hsa-miR-942-5p/CDK6 signaling pathway. Front Genet. 2021;12:1201.
Yuan P, et al. Impact of circGSAP in peripheral blood mononuclear cells on idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2021;203(12):1579–83.
Zhang J, et al. Circ-calm4 serves as an miR-337-3p sponge to regulate Myo10 (Myosin 10) and promote pulmonary artery smooth muscle proliferation. Hypertension. 2020;75(3):668–79.
Zhou S, et al. Circular RNA hsa_circ_0016070 is associated with pulmonary arterial hypertension by promoting PASMC proliferation. Mol The Nucleic Acids. 2019;18:275–84.
Guo J, et al. CircATP2B4 promotes hypoxia-induced proliferation and migration of pulmonary arterial smooth muscle cells via the miR-223/ATR axis. Life Sci. 2020;262:118420.
Jin X, et al. hsa_circNFXL1_009 modulates apoptosis, proliferation, migration, and potassium channel activation in pulmonary hypertension. Mol Ther Nucleic Acids. 2021;23:1007–19.
Guo HM, Liu ZP. Up-regulation of circRNA_0068481 promotes right ventricular hypertrophy in PAH patients via regulating miR-646/miR-570/miR-885. J Cell Mol Med. 2021;25(8):3735–43.
Duncombe TA, Tentori AM, Herr AE. Microfluidics: reframing biological enquiry. Nat Rev Mol Cell Biol. 2015;16(9):554–67.
Nguyen T, et al. Point-of-care devices for pathogen detections: the three most important factors to realise towards commercialization. TrAC Trends Anal Chem. 2020;131:116004.
Nguyen T, et al. From lab on a chip to point of care devices: the role of open source microcontrollers. Micromachines. 2018;9(8):403.
Jayne RK, et al. Direct laser writing for cardiac tissue engineering: a microfluidic heart on a chip with integrated transducers. Lab Chip. 2021;21(9):1724–37.
Zhang B, et al. Advances in organ-on-a-chip engineering. Nat Rev Mater. 2018;3(8):257–78.
Huh D, et al. Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–8.
Sin A, Baxter GT, Shuler ML. Animal on a chip: a microscale cell culture analog device for evaluating toxicological and pharmacological profiles. In: Microfluidics and BioMEMS. 2001. SPIE.
Sin A, et al. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol Prog. 2004;20(1):338–45.
Michas C, et al. Engineering a living cardiac pump on a chip using high-precision fabrication. Sci Adv. 2022;8(16):eabm3791.
Lee G, et al. Single microfluidic electrochemical sensor system for simultaneous multi-pulmonary hypertension biomarker analyses. Sci Rep. 2017;7(1):1–8.
Bogorad MI, et al. Tissue-engineered 3D microvessel and capillary network models for the study of vascular phenomena. Microcirculation. 2017;24(5):e12360.
Sato K, Nitta M, Ogawa A. A microfluidic cell stretch device to investigate the effects of stretching stress on artery smooth muscle cell proliferation in pulmonary arterial hypertension. Inventions. 2018;4(1):1.
Costa PF, et al. Mimicking arterial thrombosis in a 3D-printed microfluidic in vitro vascular model based on computed tomography angiography data. Lab Chip. 2017;17(16):2785–92.
Westein E, et al. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc Natl Acad Sci. 2013;110(4):1357–62.
Nesbitt WS, et al. A shear gradient–dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15(6):665–73.
Jain A, et al. Assessment of whole blood thrombosis in a microfluidic device lined by fixed human endothelium. Biomed Microdevices. 2016;18(4):1–7.
Al-Hilal TA, et al. Pulmonary-arterial-hypertension (PAH)-on-a-chip: fabrication, validation and application. Lab Chip. 2020;20(18):3334–45.
Humbert M, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–30.
Shapiro S, et al. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141(2):363–73.
Chung L, et al. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Wiley Online Library; 2014.
de Souza Carvalho C, Daum N, Lehr C-M. Carrier interactions with the biological barriers of the lung: advanced in vitro models and challenges for pulmonary drug delivery. Adv Drug Deliv Rev. 2014;75:129–40.
Sarkar T, et al. A protocol for fabrication and on-chip cell culture to recreate PAH-afflicted pulmonary artery on a microfluidic device. Micromachines. 2022;13(9):1483.
Mouchaers K, et al. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur Respir J. 2010;36(4):800–7.
Ruan H, et al. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congenit Heart Dis. 2019;14(4):645–50.
Budas GR, et al. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;197(3):373–85.
Roman BL, St. Hilaire C. Catching a disease: a molecular trap as a therapy for pulmonary arterial hypertension. American Thoracic Society; 2016. p. 1047–9.
Yuan JX-J, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Am Heart Assoc. 2005:534–8.
Southgate L, et al. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol. 2020;17(2):85–95.
Burton VJ, et al. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood, The Journal of the American Society of Hematology. 2011;117(1):333–41.
Barroso M, et al. Inhibition of cellular methyltransferases promotes endothelial cell activation by suppressing glutathione peroxidase 1 protein expression. J Biol Chem. 2014;289(22):15350–62.
Zaiman A, et al. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol. 2005;33(5):425–31.
Heath D. The rat is a poor animal model for the study of human pulmonary hypertension. Cardioscience. 1992;3(1):1–6.
Robbins IM. Advancing therapy for pulmonary arterial hypertension: can animal models help? American Thoracic Society; 2004. p. 5–6.
Schermuly RT, et al. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443–55.
Lu C, Verbridge SS. Microfluidic methods for molecular biology, vol. 1. Springer; 2016.
Wu AR, et al. Automated microfluidic chromatin immunoprecipitation from 2,000 cells. Lab Chip. 2009;9(10):1365–70.
Cao Z, Lu C. A microfluidic device with integrated sonication and immunoprecipitation for sensitive epigenetic assays. Anal Chem. 2016;88(3):1965–72.
Pezzuto B, et al. Circulating biomarkers in pulmonary arterial hypertension: update and future direction. J Heart Lung Transplant. 2015;34(3):282–305.
Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–6.
Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95(4):343–53.
Yan F, et al. Paracrine mechanisms of endothelial progenitor cells in vascular repair. Acta Histochem. 2022;124(1):151833.
Hansmann G, et al. Design and validation of an endothelial progenitor cell capture chip and its application in patients with pulmonary arterial hypertension. J Mol Med. 2011;89(10):971–83.
Kraan J, et al. Clinical value of circulating endothelial cell detection in oncology. Drug Discov Today. 2012;17(13–14):710–7.
Asosingh K, et al. Letter by Asosingh et al regarding article, “circulating endothelial progenitor cells in patients with eisenmenger syndrome and idiopathic pulmonary arterial hypertension”. Circulation. 2009;119(9):e230.
Diller G-P, et al. Response to letter regarding article,“circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension”. Circulation. 2009;119(9):e231.
Toshner M, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180(8):780–7.
Diller G-P, et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117(23):3020–30.
JunHui Z, et al. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med. 2008;102(7):1073–9.
Fadini GP, et al. Depletion of endothelial progenitor cells may link pulmonary fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2007;176(7):724–5.
Smadja DM, et al. Circulating endothelial cells: a new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation. 2009;119(3):374–81.
Sallmon H, et al. Circulating endothelial cell quantification by microfluidics Chip in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2017;56(5):680–2.
Lee S, et al. Reduced circulating angiogenic cells in Alzheimer disease. Neurology. 2009;72(21):1858–63.
Dome B, et al. Circulating endothelial cells, bone marrow-derived endothelial progenitor cells and proangiogenic hematopoietic cells in cancer: from biology to therapy. Crit Rev Oncol Hematol. 2009;69(2):108–24.
Ferry MS, Razinkov IA, Hasty J. Microfluidics for synthetic biology: from design to execution. In: Methods in enzymology. Elsevier; 2011. p. 295–372.
Dani K. An integrated microfluidic platform for vascular studies and more efficient preclinical development. Johns Hopkins University; 2020.
Prabhakar P, et al. 3D-printed microfluidics and potential biomedical applications. Front Nanotechnol. 2021;3:609355.
3D-Microfab. https://www.sdstate.edu/3d-microfabrication-shared-facility.
Deter HS, Hossain T, Butzin NC. Antibiotic tolerance is associated with a broad and complex transcriptional response in E. coli. Sci Rep. 2021;11(1):1–15.
Hossain T, et al. Antibiotic tolerance, persistence, and resistance of the evolved minimal cell, Mycoplasma mycoides JCVI-Syn3B. Iscience. 2021;24(5):102391.
Hossain T, Singh A, Butzin NC. Escherichia coli cells are primed for survival before lethal antibiotic stress. Microbiol Spectr. 2023:e01219-23.
Jammes FC, Maerkl SJ. How single-cell immunology is benefiting from microfluidic technologies. Microsyst Nanoeng. 2020;6(1):1–14.
Hisey CL, et al. A versatile cancer cell trapping and 1D migration assay in a microfluidic device. Biomicrofluidics. 2019;13(4):044105.
Delincé MJ, et al. A microfluidic cell-trapping device for single-cell tracking of host–microbe interactions. Lab Chip. 2016;16(17):3276–85.
Chung S, et al. Cell migration into scaffolds under co-culture conditions in a microfluidic platform. Lab Chip. 2009;9(2):269–75.
Song JW, Munn LL. Fluid forces control endothelial sprouting. Proc Natl Acad Sci. 2011;108(37):15342–7.
Young EW, et al. Technique for real-time measurements of endothelial permeability in a microfluidic membrane chip using laser-induced fluorescence detection. Anal Chem. 2010;82(3):808–16.
Douville NJ, et al. Fabrication of two-layered channel system with embedded electrodes to measure resistance across epithelial and endothelial barriers. Anal Chem. 2010;82(6):2505–11.
Shao J, et al. A microfluidic chip for permeability assays of endothelial monolayer. Biomed Microdevices. 2010;12(1):81–8.
Reinitz A, et al. Human brain microvascular endothelial cells resist elongation due to shear stress. Microvasc Res. 2015;99:8–18.
Song JW, et al. Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS One. 2009;4(6):e5756.
Shevkoplyas SS, et al. Direct measurement of the impact of impaired erythrocyte deformability on microvascular network perfusion in a microfluidic device. Lab Chip. 2006;6(7):914–20.
Acknowledgments
We thank the members of Dr. Butzin’s lab for the critical review of this manuscript and for sharing their experiences. This work is supported by the National Science Foundation Award Numbers 1922542 and 1849206, and by a USDA National Institute of Food and Agriculture Hatch project grant number SD00H653-18/ project accession no. 1015687.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Rahman, K.M.T., Islam, T., Islam, M.F., Carbone, R.G., Butzin, N.C., Ali, M.K. (2023). Targeting Epigenetics in Pulmonary Arterial Hypertension. In: Gupta, G., Oliver, B.G., Dua, K., Ali, M.K., Dave, P. (eds) Targeting Epigenetics in Inflammatory Lung Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-99-4780-5_14
Download citation
DOI: https://doi.org/10.1007/978-981-99-4780-5_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-4779-9
Online ISBN: 978-981-99-4780-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)