
Abstract
An experimental study is the most powerful design in examining causal relationships. The three major types of experimental study in humans include clinical trial, field trial, and community trial, which differ by objectives, principles, implementations, and target populations. Clinical trial aims to evaluate the treatment effects of new drugs or therapies among patients to improve the prognosis. Field trial aims to examine the potential preventive effect of the intervention in reducing morbidity or mortality among healthy individuals. Community trial implements the intervention among healthy people at the group level instead of at the individual level. By performing experimental studies, researchers make causal inferences, confirm the risk factors and protective factors for diseases, and evaluate the effects of interventions in disease prevention and control.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


-
Follow the ethical principles! Read the Declaration of Helsinki. Always remember: “The health of my patient will be my first consideration.” and “A physician shall act in the patient’s best interest when providing medical care.”
-
Experimental study serves as the “gold standard” in medical studies for causal inference.
-
Different from observational studies, researchers determine the status of exposure of the participants in experimental studies.
-
A successful randomization with a large sample size is powerful in eliminating confounding due to known and unknown confounders at baseline. However, if adherence to treatment is poor, or loss to follow-up is serious, new confounding will arise. Loss to follow-up may also introduce selection bias.
An experimental study is the most powerful design in examining causal relationships. The three major types of experimental study in humans include clinical trial, field trial, and community trial, which differ by objectives, principles, implementations, and target populations. Clinical trial aims to evaluate the treatment effects of new drugs or therapies among patients to improve the prognosis. Field trial aims to examine the potential preventive effect of the intervention in reducing morbidity or mortality among healthy individuals. Community trial implements the intervention among healthy people at the group level instead of at the individual level. By performing experimental studies, researchers make causal inferences, confirm the risk factors and protective factors for diseases, and evaluate the effects of interventions in disease prevention and control.
6.1 Basic Ideas of Experimental Study
An experimental study is a prospective study comparing the effect of an intervention against a control in humans. In experimental studies, participants are assigned to groups with different treatments or agents and followed up for a period of time to see if the outcomes vary across groups.
An experimental study is generally expensive and ethically restricted, usually focusing on a narrow question in a highly selected population with a well-defined protocol. Therefore, the experimental studies are reserved for relatively mature research questions suggested by previous observational studies appealing for further confirmative evidence. The key difference between an experimental study and an observational study is that the status of exposure is decided by researchers in an experiment. Not all research question is amenable to the experimental design. It is not ethical to expose people to harmful substances, and it is not always feasible to study the long-term effect of an intervention.
In a classic two-group experiment, one group receives the treatment of interest, and the other group does not. The experimental groups are expected to be identical with respect to extraneous factors affecting the outcome. Thus, if the treatment of interest does not have any effect on the outcome, an identical outcome frequency with random variation would be observed between the two groups. In other words, if the frequency of the outcome varies across the groups, the difference is attributable to the treatment effect plus random variation. This objective can be achieved if all extraneous factors and conditions which may have an effect on the outcome have been controlled. However, in human studies, it is not possible to create completely identical groups with respect to all extraneous factors. Instead, researchers expect the groups to be comparable and exchangeable, with the net effect of extraneous factors to be minimalized and much less than the effect of the treatment. In a classic experimental study, a control group is always needed to provide a basis for comparison, randomization is often employed to minimize the influence of confounding, and blinding is used when possible, to eliminate the biases that arise from knowing the treatment assignment.
6.1.1 Study Question
Each experimental study should have a specific question stated clearly and in advance. This encourages proper design and enhances the credibility of the findings. The primary question should be the one the researchers are most interested in and the one that could be adequately answered. Generally, the primary question is based on comparing outcomes across treatment groups. The outcome could be a beneficial event including improved prognosis, prolonged survival, increased rate of cure, released symptoms, reduced complications, or improved quality of life. There also may be a series of secondary questions in an experimental study, which can be elucidated by the data collected. The secondary questions may comprise different response variables and subgroup hypotheses. Both primary and secondary questions should be relevant scientific questions, with important implications in medicine or public health. Adverse events or side effects should also be collected through the experimental study. Unlike the primary and secondary questions, adverse events and side effects may not always be specified in advance. Investigators usually monitor a variety of clinical and laboratory measurements and record the reports from participants. The safety and well-being of participants are the most crucial concerns in performing an experiment. Investigators should always monitor the balance of benefits and risks, and be guided by the independent ethical review committees.
6.1.2 Choice of Intervention
The intervention techniques employed by an experimental study may be single or a combination of diagnostic, preventive, or therapeutic biologics, drugs, regimens, devices, or procedures. In an experimental study, the intervention a participant receives is assigned by the investigator for the purpose of a study instead of the subject’s need. Ethical constraints severely limit the types of interventions and circumstances for an experiment to be performed on human subjects. Adherence to the scientific protocol should not conflict with the subject’s best interests. Any exposure given to participants should cause no known harm and should be limited to potential prevention or cure of disease.
6.1.3 Choice of Control
The choice of the control group is an important design issue in experimental studies, for it provides the basis to make a valid comparison. The methodological principle of choice of control is that the distribution of extraneous factors is the same between the intervention group and the control group to make the two groups comparable. The ethical principle of choice of control is that if there is an optimal, known best therapy or standard, usual care, the new intervention should be compared against it, or added to it. The commonly used types of control groups include standard control, placebo control, self-control, and cross-over control.
6.1.3.1 Standard Control
Standard control is the most commonly used control in clinical trials. The optimal or standard treatment is assigned to the control group or to both groups, while a new treatment or new therapy is assigned or added to the intervention group. The effect of the new treatment should be compared against the standard care when the latter is available, instead of against a placebo. Although comparing a new treatment to a placebo or blank control might provide a larger effect size, the goal of the study is to determine whether the new treatment is better than the one currently used, but not if the new treatment has any effect.
6.1.3.2 Placebo Control
When a new intervention is added to standard care or usual care, it is compared against that care plus a placebo. Or when there is no standard care available, the new intervention is compared against a placebo. Placebo control is also commonly used in field trials including the trial of vaccines. Using a placebo control has two major benefits: helps in keeping the blinding, and helps in controlling the “placebo effect.”
For keeping the blinding successful, the formulation, size, and appearance of the placebo should be identical to the new drug. Thus, the participants would not know which study group they are in, improving their adherence to taking the treatment, and preventing them from dropping out once they know they are not in the intervention group. The researchers would also have no idea about which groups of participants are taking the intervention therapy, preventing them from making differential observations and data collection.
The placebo itself has no treatment or preventive effect at all. However, using any form of the drug may induce certain “effect” in both the intervention group and the control group. This kind of psychological benefit is called the placebo effect, even if it occurs among participants in the intervention group. By using a placebo in the trial, the psychological effects in both groups cancel out, and the real effect of the intervention can be observed. However, if the drug or the treatment of intervention has a certain side effect, the subjects might gradually realize the assigned group, and blinding might be broken, thus the compliance and the control of the placebo effect might be weakened.
6.1.3.3 Self-Control
The subjects themselves may serve as the control group before the intervention is given. Or the contralateral body or organ may serve as the control when intervention is assigned to one side. But researchers still have to pay attention if the extraneous factors change before and after the intervention is given. If so, the estimate of the effect might still be confounded.
6.1.3.4 Cross-Over Control
The cross-over design also allows the subject to serve as his or her own control, while in this case, the study has more than one period. In the first period, each subject receives either intervention or control treatment, and in the second period the alternative. The order in which intervention or control is given to the subject is usually randomized. Depending on the characteristics of the intervention, a wash-out period is required between the two periods. The use of the cross-over design is thus limited to those interventions that the effects during the first period can be washed out. A cross-over control may have more than two periods and may have more than two arms.
6.1.4 Randomization
Observational studies are often used to compare the effects of different treatments given to patients in clinical settings. However, when some of the manifestations affect both the outcome and the treatment allocation, the effect estimates can be biased. The patients in different treatment groups differ in many ways, and the groups might be incomparable. For example, the general condition of a patient has a definite impact on the disease progression and prognosis, and the general condition also determines whether the doctor would choose surgery or a more conservative treatment. In this kind of situation, the differences observed in the outcome between groups may contribute to not only the potential treatment effect but also the confounding brought by the severity of the disease. And this type of confounding is called “confounding by indication.” Thus, the effect estimates gained from observational studies are faced with uncontrolled confounding when the different treatment groups are not comparable, since not all confounders can be realized, identified, measured, and controlled.
The observed association in an observational study comprises the treatment effect, systematic bias, and random error. An experimental study aims to eliminate the part of systematic bias. First of all, it is crucial to reduce to the best extent of incomparability in different treatment groups by balancing the extraneous factors affecting the outcome. Ronald Aylmer Fisher and others developed the practice of randomization to account accurately for extraneous variability in experimental studies. A random assignment mechanism is used to assign treatments to subjects, and the mechanism is unrelated to those extraneous factors that affect the outcome. Thus, the difference in the outcomes across groups that is not attributable to treatment effects could be attributed to chance. A study with random assignment of exposure allows computing the probability of the observed association under the hypothesis and making a statistical inference based on the compatibility between the observation and the hypothesis. Randomization guarantees that statistical tests have valid rates of false positive error.
Successful randomization with a sufficient sample size generates comparable groups at baseline. Not only known confounding factors but also those unknown confounders are balanced across groups. However, compliance with the follow-up and adherence to treatment is critical during the study period to make the effect estimate valid. If adherence to treatment is influenced by extraneous factors affecting the outcome, confounding will arise and affect the effect estimate between exposure received and the outcome. If the loss to follow-up is severe, it would not only affect the study efficiency but also introduce selection bias and confounding if the loss to follow-up is differential with regard to exposure, outcome, and extraneous factors. Therefore, it is important to maintain a low rate of loss to follow-up and high adherence to assigned treatments during the study period.
6.1.4.1 The Randomization Process
Randomization is a mechanism or process by which each subject has the same chance of being assigned to either the intervention group or the control group. Several methods of randomly assigning subjects are introduced here. The commonly used methods for randomization include simple randomization, blocked randomization, and stratified randomization.
6.1.4.2 Simple Randomization
Simple randomization or complete randomization is the most elementary form of randomization. To toss an unbiased coin when a participant is eligible for randomization is an example. One might also use a random digit table on which the equally likely digits from zero to nine are arranged by rows and columns. For larger studies, one may use a random number-producing algorithm provided by most statistical software to generate random numbers in the interval from 0.0 to 1.0 for each subject. The procedure might assign subjects to group A with probability p and subjects to B with probability 1-p. If the random number is between 0 and p, the subject would be assigned to group A, otherwise to group B. And this procedure can be adapted to more than two groups. The advantage of simple randomization is that it is easy to implement. Simple randomization generates an anticipated proportion for the number of subjects in each arm in the long run with a large sample size. However, at any point in the process of randomization, or when the sample size is small, there could be a substantial imbalance. Although this kind of imbalance would not cause the statistical tests to be invalid, it harms the statistical efficiency.
6.1.4.3 Blocked Randomization
Blocked randomization is used to avoid serious imbalance in the number of subjects assigned to each group when the sample size is small. It also helps to have balanced numbers at any point in the randomization procedure during enrollment. If participants are randomly assigned with equal probability to groups A or B, then for blocks with even size, one-half of the participants would be assigned to each group. The order in which the treatments are assigned in each block is randomized. For example, if a block of size 4 is used, there are six possible combinations of treatment assignments: AABB, ABAB, ABBA, BAAB, BABA, and BBAA. Select one from these arrangements randomly and apply it accordingly to the four participants entering the study. Repeat for every consecutive group of four participants until all are randomized. Advantage of the block randomization is that the number of participants in each group is always balanced during the process of randomization, at any time point, and with any sample size. The disadvantage is that strictly speaking, data analysis is more complicated for blocked randomization than for simple randomization. And the use of blocked randomization should be taken into consideration during data analysis.
6.1.4.4 Stratified Randomization
Randomization balances extraneous factors that affect the outcome in studies with large sample sizes and for small studies on average. However, for one single study especially with a small sample size, it is possible that not all baseline characteristics distribute evenly across groups. When there is the concern of imbalance for major prognostic factors, one might employ stratified randomization within the strata of those factors considered. If several factors are considered, the number of strata is the product of the number of subgroups for each factor. Within each stratum, the randomization process could be a simple randomization or a blocked randomization.
6.1.5 Blinding
Blinding is one of the solutions to reduce systematic biases in experimental studies. Not knowing which group the participant is in, the adherence to the exposure, the compliance to the follow-up, and the measurement of outcomes can be improved. Thus, blinding is often employed when it is possible. Some kinds of trials can only be conducted without blinding, including those that have surgical procedures, changes in lifestyle, or behavioral interventions. The main disadvantage of an unblinded experiment is that participants may report symptoms and side effects differentially between intervention and control groups. Also, researchers may measure and collect these data differentially when knowing which group the subjects are from. Moreover, the participants in the control group may have a higher possibility of leaving the study, when knowing that there would not be any extra benefits.
There are at least four levels of blinding in a trial. First, the participant does not know which treatment group he or she is in. The adherence and compliance to the study would not be influenced, and the accuracy of the report of symptoms would not be affected. Second, the staff assigning participants to different treatment groups do not know which group the participants would be assigned to. This avoids participant assignment based on the staff’s willingness. Third, the physicians taking care of participants during the whole process do not know which treatment group the participants are in. This ensures the health care provided and symptoms and clinical manifestations recorded would not be affected. Fourth, the researchers including statisticians do not know which treatment group the participants are in. This makes sure that the measurements of treatment effects, the record of side effects, and data analyses would not be affected.
There are three common methods for performing blinding in practice: single-blind, double-blind, and triple-blind.
6.1.5.1 Single-Blind
In a single-blind study, the participants do not know which treatment group they are in. Thus, the biased report of symptoms and side effects by subjects can be reduced. However, the researchers can still influence the administration of treatment, data collection, and analysis in a single-blind study.
6.1.5.2 Double-Blind
In a double-blind study, neither the participants nor the researchers know the treatment assignment. The risk of bias is greatly reduced in a double-blind study. The actions of investigators would occur equally to participants from both groups. Double-blind studies are usually more complex to carry out than a single-blind or unblinded study. An effective data monitoring protocol should be set up. And the emergency unblinding procedures must be established.
6.1.5.3 Triple-Blind
A triple-blind study is an extension of a double-blind design. And it may have different definitions under different circumstances. In some designs, it is the committee monitoring response variables that is not aware of the treatment assignment. While in some designs, it is the group performing statistical analyses that has no idea of the treatment assignment. Thus, the bias introduced during statistical analyses can be avoided.
6.1.6 Data Analysis
Data analysis in experimental studies has special strategies. Noncompliance with the assigned treatment results in a discrepancy between the treatment assigned and the treatment actually received. The standard practice of data analysis in the experimental study is making comparisons based on the treatment assigned instead of received. Such a practice is called the intent-to-treat analysis (ITT). Comparisons based on the treatment received are called according-to-protocol (ATP) or per-protocol analysis (PP). If the compliance to treatment is poor, or there is a considerable loss to follow-up during the study, the association between the exposure received and the outcome might be biased. ITT analysis preserves the validity of the test for the null hypothesis of treatment effects.
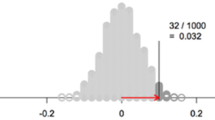
In an intent-to-treat analysis, no matter how the compliance to the assigned treatment is, the analysis takes the assigned treatment as the exposure to test the null hypothesis between the exposure and the outcome. As mentioned earlier, successful randomization is not affected by the extraneous factors affecting the outcome. And randomization has an effect on the outcome through and only through the actual treatment received. Although the association between the treatment actually received and the outcome can be confounded, the association between the treatment assigned (randomization) and the outcome will not be confounded. This makes randomization a valid instrumental variable in examining the association between the treatment and the outcome. The use of the instrumental variable protects the validity of the test of the null hypothesis between treatment and outcome, although the effect estimate might have been biased (Fig. 6.1).

A causal diagram with valid instrument randomization, for the treatment—outcome effect. If 1. Randomization affects outcome; 2. Randomization affects outcome only through treatment; 3. Randomization and outcome share no common causes; and then randomization can be taken as a valid instrumental variable in examining the association between treatment and the outcome. The association between treatment and outcome might have been affected by uncontrolled confounding, however, the association between randomization and outcome has not been confounded
6.1.7 Sample Size
An experimental study should have sufficient statistical power to detect the differences across treatment groups. The sample size of a study is decided by the following aspects:
-
(1)
The significance level, denoted as α. It is the probability of a false positive finding, or Type I error.
-
(2)
The probability of a false negative result, or Type II error, denoted as β. 1 - β is the statistical power of the test.
-
(3)
The difference between the measurements of the outcome across the groups.
6.1.7.1 Sample Size Calculation for Dichotomous Response Variables
Where N = the sample size for each group, Pc is the event rate for the control group, Pe is the event rate for the treatment group, P = (Pc + Pe)/2, Ζα is the critical value which corresponds to the significance level α, and Ζβ corresponds to the power 1 – β.
6.1.7.2 Sample Size Calculation for Continuous Response Variables
Where N = the sample size for each group, σ is the estimated standard deviation, d is the estimated difference of the means, Ζα is the critical value which corresponds to the significance level α, and Ζβ corresponds to the power 1-β.
6.1.7.3 Sample Size Calculation for “Time to Failure”
Where N = the sample size for each group, λ is called the hazard rate or force of mortality, Ζα is the critical value which corresponds to the significance level α, and Ζβ corresponds to the power 1 – β.
6.2 Clinical Trial
6.2.1 Basic Ideas of Clinical Trial
Clinical trial is an experimental study with patients as subjects. The goal of a clinical trial is to evaluate a new drug or therapy for a disease to improve prognosis, reduce mortality or improve the quality of life among patients. It also collects information on the adverse effects of a new treatment and provides evidence on the effectiveness and safety of the treatment to enter clinical use.
Subjects in a clinical trial are patients with the disease in question. Participants in a clinical trial should meet the criteria of eligibility well-defined in advance. Patients who do not meet those criteria should not be enrolled. And subjects whose illness is too severe or too mild usually will not be considered eligible since they are less likely to permit the form of treatment or to complete the follow-up. Patients with complicated conditions are usually excluded especially at earlier stages of the trial because of the need to minimalize differences in the extraneous factors affecting the outcome between treatment groups. Therefore, at earlier stages of the trial, the participants are usually a highly selected population with restricted criteria for inclusion, affecting the generalization of the conclusion.
6.2.2 Phases of Clinical Trial
When comparing the effectiveness of a new drug, several phases of clinical research must be performed. Classically the trials of pharmaceutical agents involve phases I to IV.
6.2.2.1 Phase I Studies
Phase I studies collect early data in humans after preclinical information is obtained from in vitro or animal studies. Participants in phase I studies are generally healthy volunteers with sample sizes ranging from 20 to 100. Phase I studies characterize pharmacokinetics and pharmacodynamics and estimate the tolerability in humans. The questions including bioavailability, body compartment distribution, and drug activity are answered by phase I studies. The maximally tolerated dose, the safety range of the dose, and the recommended dose is explored at this stage. Phase I also collects data on side effects.
6.2.2.2 Phase II Studies
Phase II studies evaluate whether the drug has any biological activity or effect once the dose or range of dose is determined with sample sizes ranging from 100 to 300. A phase II study usually employs a randomized control design, compares the effect of the new drug against the standard drug or a placebo, and evaluates the effectiveness and safety of the new treatment. Phase II studies continue to collect side effects data, evaluate the safety, and recommend the dose for clinical use.
6.2.2.3 Phase III Studies
Phase III studies are generally multi-center randomized controlled trials conducted in different countries with sample sizes ranging from 300 to 3000 or more. Phase III studies further evaluate the effectiveness and safety of the new drug or therapy against the standard care, confirming the value in clinical use. Phase III collects data on the adverse effects and the interaction of the drug with other drugs. The treatment approved after phase III can be used in clinical settings.
6.2.2.4 Phase IV Studies
Phase IV studies are conducted after the new treatment is approved and used clinically. All patients who received the new drug can be considered participants. The participants enrolled before phase IV are generally highly selected with restricted criteria for eligibility, which limits the generalizability of the study conclusions. Phase IV studies observe the drug efficacy in the real world, and those patients with complex conditions may also be enrolled. Thus, the limitations of earlier studies can be improved. Phase IV studies are generally open cohort studies, monitoring drug efficacy, side effects, and interaction with other drugs at a large scale in the long run. Phase IV studies can collect data on side effects especially the ones that occur rarely or late.
6.2.3 Case Study of Clinical Trial
A randomized, phase II study examined the efficacy of carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer (NSCLC). The investigators enrolled 123 non-squamous NSCLC stage IIIB or IV patients without former chemotherapy and targetable EGFR or ALK genetic aberrations from 26 medical centers in the USA and Taiwan, China. A 1:1 ratio in blocks of four randomization assigned 60 to the group of pembrolizumab plus chemotherapy, and 63 to the group of chemotherapy alone. The primary endpoint was the proportion of patients who had radiologically confirmed complete or partial responses. Fifty-five percent (95% CI 42–68%) of patients in the pembrolizumab plus chemotherapy group achieved this objective response compared with 29% (18–41%) of patients in the chemotherapy alone group (treatment difference 26% [95% CI 9–42%]; P = 0.0016). The incidence of grade 3 or worse treatment-related adverse events was similar between groups (39% in the pembrolizumab plus chemotherapy group and 26% in the chemotherapy alone group). The most common grade 3 or worse adverse events in the pembrolizumab plus chemotherapy group were anemia (12%) and decreased neutrophil count (5%). In the chemotherapy-alone group, the most common were anemia (15%) and decreased neutrophil count, pancytopenia, and thrombocytopenia (3% each). 2% of patients in the pembrolizumab plus chemotherapy group experienced treatment-related death compared with 3% in the chemotherapy group. These results suggested that the combination of pembrolizumab plus chemotherapy may be an effective treatment for patients with early, advanced non-squamous NSCLC.
6.3 Field Trial
6.3.1 Basic Ideas of Field Trial
Field trial differs from a clinical trial in the subjects. The participants in a clinical trial are those patients diagnosed with the disease of interest in clinical settings; while the participants in a field trial are those healthy people from the community. Field trial often requires a larger sample size and recruit participants who are not under clinical management. Therefore, they are often more expensive and difficult to conduct. A field trial is limited to studying the prevention of common or severe diseases. The interventions for field trials include health supplements, vaccines, and changes in lifestyle. The principles of study design, control selection, randomization, and blinding for experimental studies apply to field trials. Field trials are used to confirm the causal relationship, risk factors, and preventive factors for diseases and to reduce morbidity.
6.3.2 Design and Implementation
Participants in the field trial are free-living healthy people recruited from the community. The management and conduct of a field trial would be more difficult than a clinical trial. A well-designed feasible protocol on a solid scientific question is crucial for a successful field trial.
6.3.2.1 A Specified Question
A clear scientific question should be stated in advance including the specific intervention and anticipated outcome. The objective of the study should be based on a clear research hypothesis. The intervention should be derived from a risk factor for disease with relatively sufficient evidence from observational studies. And the conclusions gained from the study should have benefits for individuals or public health.
6.3.2.2 Inclusion and Exclusion Criteria
The participants for the field trial are healthy people from the community and are at risk for disease of interest. The inclusion and exclusion criteria should be defined in advance based on the study objective and should be implemented strictly. Participants can be enrolled from those communities with low mobility to avoid a substantial loss to follow-up, otherwise, selection bias may arise and harm the validity of the conclusion. Also, if the disease of interest is of low incidence rate in the population, it is suggested to conduct a field trial in the population at higher risk for the disease to save resources for long-term follow-up. Restricted inclusion criteria and highly selected participants may have an influence on the generalizability of the research conclusion.
6.3.2.3 Choice of Intervention
A clear definition and description of the intervention are necessary. The dose, contents, method, frequency of application, etc. of the intervention should be introduced clearly. Adherence to intervention is critical for field trial participants.
6.3.2.4 Time and Interval of Follow-up
The time of each visit and interval during follow-up are decided by the effect of an intervention. Investigators balance the need for collecting necessary data, maintaining participants in the follow-up, and the cost. During the study, it is important to improve the compliance and adherence of the participants to avoid a loss of follow-up and selection bias.
6.3.3 Case Study of Field Trial
Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomized controlled trial
Genital human papillomavirus (HPV) infection leads to cervical cancer, a major cause of cancer deaths in women worldwide. 230,000 die and 470,000 are diagnosed due to cervical cancer annually. The most prevalent oncogenic HPV strains, HPV-16 and HPV-18, can be vaccinated to prevent up to 70% of cervical cancers from developing. A bivalent HPV-16/18 L1 virus-like particle vaccine was tested for efficacy, safety, and immunogenicity in a randomized, double-blind, controlled trial. Between July and December 2000, 1,113 North American and Brazilian women aged 15–25 were enrolled with an average age of 20. HPV infection was tested using self-obtained cervicovaginal samples and cervical cytology. After randomization, 560 of 1,113 women received the vaccine and 553 received the placebo. 958 women completed the first phase through month 18, with similar rates of vaccination and placebo dropouts. According-to-protocol HPV-16/18 vaccine effectiveness against the incident and persistent infection was 91.6% and 100%, respectively. Intention-to-treat analysis showed 95.1% efficacy against persistent infection. Neither the vaccine nor the placebo groups experienced any vaccine-related adverse effects. In this trial, the bivalent HPV vaccine proved efficacious, safe, well-tolerated, and highly immunogenic.
6.4 Community Trial
6.4.1 Basic Ideas of Community Trial
Community trial conducts intervention among healthy people, and the interventions are given at the population level instead of at the individual level. Community trial is used to evaluate the effect of interventions that are not suitable to be given at the individual level. For example, some interventions on dietary factors are easier to be performed at the family level; changing the source of drinking water from the river to tap water is easier to be conducted at the community level. These kinds of interventions are not given individually.
Community trial often uses cluster randomization. The success of cluster randomization depends on the relative sample size within each group compared to the total sample size. If the number of clusters is large, randomization has a higher possibility to be successful. If there are only two communities randomized, the meaning of randomization is limited and the comparability of baseline characteristics of the two communities has a great impact on the results. During the study, investigators need to pay attention to the changes in extraneous factors including mobility, economic changes, medical care conditions, and implementation of other programs in the community.
6.4.2 Case Study
Research on prevention and control strategies of liver cancer in Qidong and the effect of the community trial
Liver cancer is one of the most common malignant tumors in China, which has a serious impact on people’s health. According to a survey from 1990 to 1992, the standardized mortality rate of liver cancer in China was 17.83/100,000 person-years, accounting for about 18.8% of cancer deaths. Nationwide, the mortality rate of liver cancer in the 90s was higher than in the 70s. The incidence of liver cancer increased after the age of 40 and increases with age, and the age of onset was earlier in high-incidence areas. The male-to-female sex ratio was close to 3:1.
The increase in the incidence of liver cancer may come from the improvement of liver cancer diagnosis, the increase in the proportion of middle-aged and elderly people, and the increase in the incidence of liver cancer caused by the increase of environmental carcinogens. In the early 1970s of the twentieth century, the risk factors for liver cancer were not yet clear, and health workers carried out a large number of investigations and studies in Qidong. The earliest case-control study carried out in 1973 included 100 cases of primary liver cancer, 100 cases of other malignant tumors, and 100 cases of healthy people, and explored the association between liver history, tumor history, pesticide exposure and poisoning history, drinking water source and water quality, tobacco, alcohol and eating habits, family history, and other factors and liver cancer. Patients with hepatitis, liver cirrhosis, and respiratory diseases in Qidong People’s Hospital since 1964 were followed up to confirm that patients with liver disease had a high risk of liver cancer. Since 1976, a prospective cohort study has been carried out in Qidong, and long-term follow-up of nearly 15,000 people has been carried out, and the incidence of liver cancer among hepatitis B surface antigen carriers was 361.55/100,000, the incidence rate of non-carriers was 30.90/100,000, and the relative risk was 11.70, confirming the association between hepatitis B virus and liver cancer. The evidence accumulated by years of long-term research suggested and basically clarified that hepatitis B virus, aflatoxin, drinking water pollution, and gene susceptibility were risk factors for liver cancer in this population.
Therefore, the prevention and control strategy of liver cancer in Qidong area was as follows: to carry out intervention research on the suspected causes of liver cancer. By observing changes in the incidence and mortality of liver cancer, the effect of the intervention was evaluated and the etiology was further verified. A range of intervention strategies and specific interventions were identified and implemented. In the early 1970s of the twentieth century, measures of "prevention and control of hepatitis, improvement of drinking water, and prevention of mildew in food" were proposed. Put forward the requirement of "hydration of drinking water wells" to reduce residents’ drinking of ditches and river water, and later formed a "deep well tap water supply network" to improve the quality of drinking water; Corn harvesting adopted "fast harvest and quick drying into the warehouse to remove mold", and then changed the staple food to rice, changing the eating habits of residents and reducing the intake of aflatoxin from corn. Various measures have been taken to cut off the transmission of hepatitis B virus and protect susceptible people, and since 1983, large-scale neonatal hepatitis B vaccination has been carried out in Qidong to reduce the epidemic of hepatitis B virus. The academic views and research decisions based on etiology research have been responded to and supported by the government, forming a comprehensive prevention and control strategy on the spot.
Interventions and on-site implementation of major risk factors for liver cancer include:
-
1.
Anti-mildew and Detoxification
As the main chemo-preventive measure, it was important to reduce the intake of food contaminated with aflatoxin by the population. A number of case-control studies and food testing have found a significant association between mildew in corn and the occurrence of liver cancer. Prevention interventions were implemented at two levels: changing the structure of staple foods at the community level to promote the use of rice, with 96.4% of the population switching to rice by 1986; At the individual level, it was promoted to prevent the intake of mildew corn, and preventive measures are taken in the "harvest, storage, and eating" process. This greatly reduced the aflatoxin exposure of Qidong residents.
-
2.
Improve Drinking Water
Based on Qidong’s research, Professor Su Delong proposed that the high incidence of liver cancer was related to drinking water pollution. The incidence and mortality of liver cancer among residents with different types of drinking water differed significantly: the incidence of liver cancer in drinking ditch water could be as high as 141.40/100,000, and the incidence of drinking deep well water was 0.23/100,000. Algal toxins, microcystins, and other substances in ditch water are cancer-promoting factors of liver cancer and may interact with aflatoxin. Although there was no direct evidence of carcinogenesis, the drinking water improvement project has solved the problem of drinking water pollution for Qidong residents, and by 2010, 99% of residents were drinking pipe water.
-
3.
Prevention and Treatment of Hepatitis B
HBsAg was screened in blood donors in Qidong, and positive people were not allowed to donate blood, cutting off the transmission route of the virus and reducing the epidemic. A randomized controlled intervention trial of hepatitis B vaccine immunization for the prevention of liver cancer in nearly 80,000 infants between 1984 and 1990 reported a decrease in HBsAg positivity, reporting a 75.9% immune protection rate and a decrease in HBV carrier rate among vaccinated people. After more than 20 years of follow-up in the second phase, vaccination was found to have sustained immunity against chronic HBV infection.
-
4.
Carry Out Research on Early Diagnosis and Early Treatment
For the secondary prevention of liver cancer prevention and treatment, strategies and research on early diagnosis and treatment were carried out in the area. In the first stage, a large-scale screening of alpha-fetoprotein—a biomarker of liver cancer was carried out in 1.8 million people in the 1970s, and a large number of early cases were detected and treated; The second stage was in the 1980s: the high-risk group of liver cancer in Qidong was defined as HBsAg-positive men aged 30–59 years; In the third stage, in the 1990s, periodic screening of high-risk groups was carried out and the screening effect was evaluated. The screening results showed that the early case detection rate of the screening team was high, and the survival rate was higher than that of the control group; In the fourth stage, Qidong was established as a national sample for early diagnosis and treatment of liver cancer in 2006. Most of the long-term survivors of liver cancer in Qidong were beneficiaries who were found through screening and resected surgically, which shows that screening can detect early cases, and after receiving appropriate treatment, survival can be extended or even cured.
The decrease in morbidity and mortality is the goal of tumor prevention and treatment and an important indicator to test the effect of intervention strategies and measures. After more than 40 years of efforts, the age-standardized incidence and mortality of liver cancer in Qidong have decreased. Although the crude incidence and mortality rate of liver cancer in Qidong have increased in the past 40 years, after controlling for the factors of population growth and the increase in the proportion of the elderly population, the incidence of age-standardized liver cancer decreased from 49.95/100,000 in 1972 to 38.22/100,000 in 1990 and 25.75/100,000 in 2011.
The decline in the incidence and mortality of liver cancer in Qidong was accompanied by significant evidence of changes in risk factors for liver cancer. From 1989 to 2012, the level of aflatoxin adducts representing aflatoxin exposure decreased significantly, from 19.2 pg/mg in 1989 to 2.3 pg/mg in 1999 and undetectable in 2009. The drinking water of residents was changed from the easily polluted house ditch water and the water from the Yangtze River to tap water from deep wells and the Yangtze River, and the quality of drinking water was significantly improved. After 2002, the vaccination rate of hepatitis B among newborns reached 100%, the short-term and medium-term efficacy of the vaccine was confirmed, and the long-term effect and association with the decline in the incidence of liver cancer have yet to be confirmed by long-term follow-up. The above facts and evidence from changes in biomarkers, ecological changes, and changes in population immunoprevention show that even if the mechanism of action of some risk factors for liver cancer has yet to be elucidated, after controlling these risk factors, the incidence and mortality of liver cancer in the population have indeed decreased significantly, which is enough to prove that these preventive measures are effective.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Zhengzhou University Press
About this chapter

Cite this chapter
Liu, X. (2023). Experimental Epidemiology. In: Wang, C., Liu, F. (eds) Textbook of Clinical Epidemiology. Springer, Singapore. https://doi.org/10.1007/978-981-99-3622-9_6
Download citation
DOI: https://doi.org/10.1007/978-981-99-3622-9_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-3621-2
Online ISBN: 978-981-99-3622-9
eBook Packages: MedicineMedicine (R0)