
Abstract
Genome editing systems have emerged as an advanced bioengineering tool capable of targeting and editing the genomes of almost all organisms in a sequence-specific manner. This chapter presents an overview of the leading developments in the modern tool armory for genome editing that meet the high standards of efficiency, safety, and accessibility in genome engineering, i.e., ZFNs, TALENs, and CRISPR. These novel tools, primarily based on engineered nucleases, have proved to be one of the most effective and reliable tools for genome engineering. The engineered nucleases have enabled the alteration of targeted DNA sequences in a wide range of organisms and cell types. We will cover the mechanism and application of these methods for genome editing in current biology, functional genome screening, healthcare, agriculture, gene therapy, biological sciences, drug development, etc. General strategies used for designing specific ZFNs, TALENs, and CRISPR/Cas9 systems and analyzing their activity have been indicated. The therapeutic applications of these tools in controlling disease and their potential usage in the development of agricultural and industrial products, environmental protection, food development, immunotherapy, and treatment of genetic diseases, neurodegenerative diseases, and cancer are also briefly touched upon.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
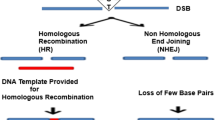
The modifications (like insertions, deletions, and substitutions) in the genomes of organisms are commonly referred to as genome engineering (El-Mounadi et al. 2020). Genome or gene editing techniques are used for genome engineering to incorporate site-specific modifications into any genomic DNA, making use of different DNA repair mechanisms found endogenously. Gene editing usually deals with one target gene, i.e., a single gene is modified, whereas genome editing refers to large-scale modifications of the complete genomic DNA (Robb 2019). This technology has addressed the unmet need for the tools to introduce different types of genetic modifications that can cause a change in the physical as well as genomic traits of an individual or a population. Currently, scientists are making headway in developing gene-therapy treatment strategies by employing these advanced genome editing tools to prevent and treat various diseases in humans and animals. A breakthrough in the field occurred when Capecchi in 1989 for the first time demonstrated that the introduction of a segment of DNA, having homologous arms at both ends, into embryonic stem cells allowed its integration into the host genome via homologous recombination, causing inheritable changes in the cell (Capecchi 1989). Later, the discovery and development of methods to introduce an artificial DNA restriction enzyme into cells, which can cut genomic or dsDNA and generate a double-stranded break (DSB) at specific recognition sites, increased our ability to use cellular repair systems for genome engineering (Zhang et al. 2011). The mechanism of action for site-directed nucleases is based on the site-specific cleavage of the DNA or induction of a double-stranded break/nick (DSB) at targeted regions of DNA sequence by nucleases followed by triggering the two prominent DNA repair pathways of the cell, i.e., homology-directed repair (HDR) and nonhomologous end joining (NHEJ) (Fig. 9.1). The HDR repair mechanism uses homologous donor DNA to repair DNA damage, whereas NHEJ is an error-prone mechanism in which broken ends of DNA are joined together, often resulting in a heterogeneous pool of insertions or deletions. Though one of the efficient repair mechanisms active in the cells, NHEJ has a high rate of mutation and results in frequent nucleotide insertions or deletions (indels). HDR has low efficiency as it requires higher sequence similarity between the template and donor DNA strands. In the HDR mechanism, there is always a chance for reversion if the used DNA template is identical to the original undamaged DNA (Jasin and Rothstein 2013; Ghezraoui et al. 2014). The development of various techniques for genome engineering has focused on the use of different endonucleases (to create DSB with high precision) followed by employing these repair mechanisms to develop an engineered genome with new properties (Mandip and Steer 2019; Khalil 2020) (Fig. 9.1).

An overview of the mode of action of ZFNs, TALENs, and CRISPR/Cas9 system: ZFNs TALENs, and CRISPR/cas9 nucleases will induce double-strand breaks (DSBs) in a gene at a targeted location and can be repaired by either NHEJ or HDR. NHEJ-mediated repair leads to the introduction of variable-length indel mutations. HDR with double-stranded DNA “donor templates” can lead to the introduction of precise nucleotide substitutions or insertions
2 Tools/Methods for Genome Engineering
The invention of genome editing tools opened up a whole set of opportunities for assisting the treatment of various diseases at the genome level (National Human Genome Research Institute 2019). Though various tools or methods for DNA modification have already existed for several decades, the development of more precise methods has made genome editing much cheaper, faster, and more efficient. Every genome editing tool being employed so far is based on one mechanism in which a targeted broken portion of DNA sequence in a gene or genome activates the cell repair mechanism that repairs the break in DNA sequence, i.e., HDR and NHEJ for DNA repair (National Institute of Health 2020). These tools and techniques allow efficient and accurate changes in genomic DNA by introducing DSB at a specific or targeted site in DNA followed by known modifications (i.e., insertion, deletion, indels, etc.). Currently, several genome engineering or genome/gene editing techniques exist which are primarily based on the following tools (nucleases) to target-specific sequences (molecular scissors): (1) zinc finger nucleases (ZFNs) (Porteus and Baltimore 2003; Miller et al. 2007; Sander et al. 2011; Wood et al. 2011), (2) transcription activator-like effector nucleases (TALENs) (Boch et al. 2009; Moscou and Bogdanove 2009; Christian et al. 2010; Hockemeyer et al. 2011; Wood et al. 2011; Zhang et al. 2011; Reyon et al. 2012; Sanjana et al. 2012), and (3) the RNA-guided CRISPR-Cas nuclease system (Deveau et al. 2010; Horvath and Barrangou 2010; Bhaya et al. 2011; Makarova et al. 2011; Cho et al. 2013; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013) (Table 9.1). The tools used for making sequence-specific cuts in the genome for the genome editing tool are briefly described below.
2.1 Zinc Finger Nucleases (ZFNs)
The first endonucleases used for genome engineering were Zinc finger nucleases (ZFNs), which were composed of zinc finger domains fused with FokI endonuclease (Kim et al. 1996). ZFNs are members of the zinc finger protein (ZFP) family, in which the zinc fingers (ZF) are novel DNA-binding domains that can bind to discrete base sequences. These ZFs have Cys2-His2 fingers and each ZF can recognize a triplet (3 bp) of DNA sequence (Miller et al. 1985; Wolfe et al. 2000). The ZFNs used for genomic engineering are comprised of a tandem array of ZFs, also known as the ZF array that confers unique nucleotide sequence-binding specificity. The dimerization of FokI endonuclease of ZFNs on the binding of two ZFNs to the opposite DNA strands allows the cleavage of the dsDNA at the target sites (Fig. 9.2). For genome editing, two recombinant ZFNs recognizing two different (one each) closely located nucleotide sequences within the target DNA sequence are employed, which with the help of FokI, creates a double-strand break (DSB) at desired target DNA sequence. Since the series of linked ZF domains (ZF arrays) determine the specificity of the target nucleotide sequence, by changing the array of ZFs, any desired sequence may be targeted. A certain degree of off-target effects (nonspecific/desired sequence cleavage) sometimes occurs when the employed pair of ZFNs is not able to recognize the desired target sequence for cleavage. The addition of more fingers per ZFN is recommended to minimize off-target effects and successfully specify rarer and longer target cleavage sites.

Illustration of a pair of ZFNs bound to targeted nucleotide sequence: Zinc fingers are shown as ZF, with short circles indicating binding with the DNA base pairs. FokI cleavage domains are shown as shaded boxes, with common cleavage sites, spaced by N bp, and indicated by vertical arrows as ZFN-induced DSB. Zinc fingers are numbered from the N-terminus. The linker between the binding and cleavage domains of one protein is labeled. The spacer between the zinc finger-binding sites is 5–7 bp in this case
The FokI domains of ZFNs are key to their successful application as they carry features that help in the cleavage of a complex genome at a specific target. FokI dimerization is crucial for the cleavage of the dsDNA. The lower strength of the interaction between FokI monomer domains causes the cleavage of DNA by FokI of ZFNs, requiring independent and appropriately placed two adjacent binding occurrences of ZFNs in correct orientations to allow catalytically active dimer formation (Miller et al. 1985; Vanamee et al. 2001; Szczepek et al. 2007) (Fig. 9.2). ZFNs-based genome editing is mainly dependent on the ability of endonuclease to create site-specific double-strand break (DSB) onto the locus of interest. In all eukaryotic cells, the DSBs generated by ZFNs are efficiently repaired by the NHEJ or HDR pathway (Szczepek et al. 2007; Lieber 2010; Moynahan and Jasin 2010) (Fig. 9.1).
Different strategies have been reported for the synthesis of ZFNs of desired DNA-binding specificity by “modular assembly” of different ZFs that have unique triplet base specificities (Segal et al. 2003; Sander et al. 2010; Thakore and Gersbach 2015). The ZFs developed for the modular assembly had been mostly for triplet sequences only (Choo and Klug 1994; Jamieson et al. 1994; Rebar and Pabo 1994; Segal et al. 1999, 2003; Dreier et al. 2001, 2005; Bae et al. 2003; Thakore and Gersbach 2015). The modular assembly of ZF components led to the generation of active ZFNs with specificity to a large number of endogenous sequences (Kim et al. 2009; Remy et al. 2010; Gaj et al. 2013b; Gupta and Musunuru 2014; Shiva and Suma 2019). Apart from the modular assembly approach, several other alternative strategies have also been developed for making ZFPs (Wu et al. 2007, 2013; Chandrasegaran and Carroll 2016; Paschon et al. 2019). These new approaches were focused on accommodating the deviation from strict functional modularity (like many natural and designed fingers can only contact with the adjacent ZF and to bases present outside of their proximal DNA triplet) which was observed for many of the ZF and making them specific (Fairall et al. 1993; Pavletich and Pabo 1993; Houbaviy et al. 1996; Nolte et al. 1998; Wolfe et al. 2001; Segal et al. 2006). These approaches could permit more selective binding and reduce the complications and wasted efforts that occur in modular designing for producing new ZFPs (Ramirez et al. 2008; Chandrasekharan et al. 2009; Chandrasegaran and Carroll 2016; Paschon et al. 2019).
Whatever may have been the methods used for designing ZFNs module, they were always first evaluated in vitro for their affinity and specificity toward the target DNA sequence followed by their application in vivo system. It is done as there is always a possibility that ZFNs/ZFPs which are validated in vitro could fail in performing the genome editing in vivo (Urnov et al. 2010; Wang et al. 2013a; Paschon et al. 2019). Many times, it may arise from the complexity of the genome which sometimes contains multiple copies of sequences that are identical or highly related (paralogues or pseudogenes) to the intended targeted sequences which can act as an additional target for ZFNs. The researchers have tried to address this problem by focusing on DNA-protein interactions and creating minor sequence divergence to reduce the chances of nonspecific targeting of related genomic regions (Carroll 2011; Urnov et al. 2010; Laoharawee et al. 2018). The specificity, recognition, and cleavage of desired sites by ZFNs are determined by the amino acid sequence of each ZF, nuclease (FokI) domain interactions, and quantity of the ZFs. The structure of both the functional domains of ZFNs, i.e., a catalytic domain and binding domains, can be optimized to increase specificity and enhance the affinity for the novel models developed by genome engineering (Jackson and Bartek 2009; Paschon et al. 2019). For improving the accuracy of targeting by ZFN, the “selection-based methods” have been also developed to optimize its cleavage specificity and reduce the nonspecific toxicity (Rahim et al. 2021).
2.2 Transcription Activator-Like Effector Nucleases (TALENs)
The second tool developed for genome editing or genome engineering is Transcription Activator-Like Effector Nucleases (TALENs) which display better specificity and functionality than ZFNs. Similar to ZFNs, TALENs also consist of an endonuclease, i.e., DNA cleavage domain, and a site-specific DNA-binding domain derived from transcription activator-like effectors (TALEs) proteins which together allow the creation of DSBs at specific sites. The DNA cleavage domain used for TALENs is primarily the FokI nuclease. The DNA-binding domains of TALENs, i.e., TALEs originated from a repeated sequence of highly conserved proteins of “phytopathogenic Xanthomonas” (Boch et al. 2009; Boch and Bonas 2010; Chandrasegaran and Carroll 2016). In Xanthomonas, the transcription activator-like effectors (TALEs) proteins are present in the cytoplasm where they promote the modification of genes that help in transcription. TALE proteins are capable of localization to the nucleus, DNA binding, and transcription activation of the target gene (Schornack et al. 2006). The studies conducted on the mechanism of action of these effector proteins showed that these proteins can mimic the functioning of eukaryotic transcription factors in binding with DNA and activating gene expression (Becker and Boch 2021).
Soon after the realization of the TALE domains simplicity, i.e., one monomer binds/recognizes one nucleotide, the first chimeric TALE domain-fused nuclease (TALEN) was constructed (Joung and Sander 2013; Gaj et al. 2013b; Nemudryi et al. 2014; Becker and Boch 2021). The chimera was developed by inserting the DNA-binding domain of TALE in a plasmid vector which was used for ZFNs (Christian et al. 2010). This ultimately leads to the formation of a genetic construct that has DNA binding and catalytic domain of restriction endonuclease, i.e., FokI. The DNA-binding domain (i.e., TALE) monomers that bind with the single nucleotide in the targeted DNA sequence are repeats of 34 amino acid residues in which amino acids at 12 and 13 positions are highly variable and known as repeat variable domain (RVD). The RVD region of TALE is responsible for the recognition of specific nucleotides. The variation in RVDs allows them to bind to different nucleotides with different efficiencies (Fig. 9.3). The TALEs with different RVDs were combined to form artificial nucleases (i.e., TALENs) which bind and cleave the targeted DNA sequences. The TALENs nucleases contain a half repeat (i.e., 20 amino acid residues of the last tandem repeat that bind to the nucleotide at the 3′ end of the recognition site), N-terminal domain, nuclear localization signals, and FokI catalytic domain (Fig. 9.3). The presence of thymidine at the 5′ end of the target sequence interacts with the N-terminal domain of the TALE and affects its overall binding efficiency (Lamb et al. 2013). TALENs always work in pair, their binding sites are located at the opposite site of DNA strands and are separated by small fragment (i.e., 12–25 bp) known as “spacer sequence.” After the TALENs enter the nucleus, they bind with the targeted sequence and the FokI domains located at the C-terminal of TALE cause the DSBs (Fig. 9.1).

Illustration of a pair of transcription activator-like effector nucleases (TALENs) bound to targeted nucleotide sequence: TALE repeats, i.e., RVD are shown as colored boxes that are responsible for the recognition of specific nucleotides. RVDs bind to different nucleotides with different efficiency. Letters inside each repeat represent the two hypervariable residues. TALE-derived amino (N domain) and carboxy-terminal domains required for DNA-binding activity are shown as pink boxes. The nonspecific nuclease domain from the FokI endonuclease is shown as a larger shaded green box. TALENs bind and cleave as dimers on a target DNA site. The TALE-derived amino- and carboxy-terminal domains flanking the repeats may make some contact with the DNA. Cleavage by the FokI domains occurs in the “spacer” sequence that lies between the two regions of the DNA bound by the two TALEN monomers. The amino acid sequence of a single TALE repeat is expanded below with the two hypervariable residues highlighted in red and bold text. TALE-derived DNA-binding domain aligned with its target DNA sequence is shown in the box indicated as TAL effector RVD DNA-binding codes
Despite simple designing codes as compared to ZFNs, there has been difficulty in the cloning of the designed TALE arrays comprised of large-scale repeats. To overcome this problem, different strategies have been developed such as High-Throughput Solid Phase Assembly, Golden Gate Cloning, and Connection-independent cloning techniques which help in assembling the desired TALE arrays (Schmid-Burgk et al. 2013). Several other modifications have also been made to TALENs to make them a better tool than the ZFNs such as (1) site selection enhancement by varying the length of the spacer sequence (Nemudryi et al. 2014); and (2) development of mutant variants of the TALE’s N-terminal domains that could more specifically bind to A, G, and C nucleotide (Nemudryi et al. 2014; Mak et al. 2012; Lamb et al. 2013).
2.3 Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
After two years of the discovery of TALENS, the discovery of Clustered Regularly Interspersed Short Palindromic Repeats (CRISPR) led to the development of a third genome engineering tool that has revolutionized the field of biotechnology and the health sector tremendously (Nemudryi et al. 2014; Lino et al. 2018; Kaminski et al. 2021). The CRISPR was first time discovered in Escherichia coli (E. coli) in 1987, and later on, found in many other prokaryotes too, e.g., 87% in archaea and 48% in eubacteria (Grissa et al. 2007). The CRISPR system has noncoding RNAs and CRISPR-associated (Cas) protein which has a nuclease activity (Ishino et al. 1987; Jore et al. 2012). In bacteria, the CRISPR-Cas system plays an important role in the adaptive immune response. It helps in protecting bacteria from phage infection by generating memory in the bacterial chromosomes against phage (Barrangou and Marraffini 2014; Renaud et al. 2016; Kim et al. 2021).
There are two types of CRISPR/Cas systems depending on the structural variation that existed in Cas genes (1) Class 1 systems contain multiple protein effectors complexes, and (2) Class 2 has one effector protein. To date, six types of CRISPR/Cas systems and 29 subtypes of Cas-system have been reported (Moon et al. 2019; Liu et al. 2020). CRISPR-Cas9 type II system is one of the most used, advanced, and versatile CRISPR systems for genome engineering or editing because of its specificity which is stemming from the Cas protein. The Cas protein of this CRISPR-Cas9 type II system was extracted from Streptococcus pyogenes (i.e., SpCas9) which targets the specific DNA sequences and is responsible for the advanced specificity of the system (Jiang et al. 2013).
Sequencing of the various bacterial genomes revealed the presence of short unique DNA regions known as spacers DNA, which are separated from one another by short palindromic sequence repeats (Deshpande et al. 2015; Lino et al. 2018). These structures are found to be located in the proximity of Cas genes. The cas gene gives rise to protein products that have nuclease and helicase activity (Haft et al. 2005). Spacer DNA is a homologous DNA found in several phages and plasmids (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005; Barrangou et al. 2007) (Fig. 9.4). Cas9 protein is polyfunctional, it interferes with the foreign DNA and pre-crRNA processing (Sapranauskas et al. 2011). The processing of crRNA depends on small noncoding RNA known as transactivating RNA (tracrRNA). The tracrRNA forms a duplex after binding with the complementary repeat sequence present in the pre-rRNA. RNase III (present in the host cell) in the presence of Cas9 cleaves this duplex and leads to the formation of mature crRNA (Makarova et al. 2006; Marraffini and Sontheimer 2010). The CRISPR-Cas9 system employs two main components, i.e., Cas9 endonuclease and a single-stranded guide RNA (sgRNA) or tracrRNA-crRNA chimera (Cong et al. 2013). The sgRNA recognizes and binds with the targeted sequence, and Cas9 cleaves the DNA causing DSB (Fig. 9.4). The site of cleavage for Cas9 endonuclease is 3 bp upstream of an “NGG” PAM located on genomic DNA. This DSB generated gets repaired by NHEJ or HDR (Fig. 9.1) (Pawelczak et al. 2018).

Illustration of clustered regularly interspaced short palindromic repeats (CRISPR-Cas9 System) bound with targeted DNA sequence: CRISPR system has a single chimeric sgRNA (crRNA and tracrRNA) to which introduces a DSB into the target nucleotide sequence. A protospacer is a site that is recognized by the CRISPR/ Cas9 system. A spacer is a sequence in sgRNA that is responsible for complementary binding to the target site. PAM is a short motif (NGG in the case of CRISPR/Cas9) whose presence at the 3′-end of the protospacer is required for introducing a break. A Cas9 nuclease is capable of introducing DSB into selected DNA site
The mechanism of genome editing with the help of the CRISPR system both inside the prokaryotic cells and in vitro is divided into three stages, i.e., adaptation, transcription, and intervention. In adaptation, a small fragment of foreign DNA entering the bacterial cell gets inserted into the CRISPR locus of the bacterial genome leading to the formation of the new spacer or protospacer (i.e., a viral genome fragment). Viral protospacer is complementary to the spacer present in the host cell and these protospacers are flanked by a short, conserved sequence (2–5 bp) which is known as a protospacer adjacent motif (PAM) (Mojica et al. 2009a). The PAM is inserted at the AT-rich side of the sequence that also has a promoter element and a landing site for regulatory proteins present just before the CRISPR cassette (Deltcheva et al. 2011; Jinek et al. 2012). In the transcription step, the complete CRISPR locus formed is transcribed into a long poly-spacer precursor crRNA (pre-crRNA) (Fig. 9.4). The Cas6 endonucleases are responsible for the formation of mature crRNA in most of the CRISPR/Cas systems (Carte et al. 2008; Lillestøl et al. 2009; Mojica et al. 2009b; Hale et al. 2012; Pawelczak et al. 2018). The short nucleotide CRISPR RNA (crRNAs) has one spacer sequence whose repeat ends are involved in the formation of a stem-like loop structure. The 5′ end with eight nucleotide repeats has an OH group and forms a stem whereas the 3′ end with 2′,3′-cyclic phosphate (hairpin structure) forms a loop (Haurwitz et al. 2010; Gesner et al. 2011).
During the intervention step, the viral DNA or RNA interacts with the crRNA and Cas proteins. The crRNA identifies the complementarily of the protospacer sequence whereas Cas protein leads to its degradation (Marraffini and Sontheimer 2010; Rath et al. 2015; Shabbir et al. 2019). The coevolution of viruses/phages with their host over time has led to the formation of a wide range of CRISPR/Cas system in prokaryotes (Hale et al. 2009; Sashital et al. 2011; Richter et al. 2012; Bondy-Denomy et al. 2013; Newsom et al. 2021).
3 Applications of Genome Engineering/Editing Methods
The development of genome editing tools has given possibilities of directly targeting and modifying genome sequences in eukaryotes. The recent progress in the development of programmable nucleases such as ZFNs, TALENs, and CRISPR-Cas-associated nucleases has significantly accelerated the progress of genome engineering in different fields ranging from basic research to biomedical and applied biotechnological research. The application of different gene editing tools in different fields of biological sciences and their future possibilities are briefly indicated below and summarized in Table 9.2.
3.1 In Genetic Engineering of Cell Lines and Animal Models
Before the development of engineered nucleases, the study of the genetically modified mammalian cell line was costly, labor-intensive, time-limited, and required specialized expertise. However, with the introduction of cost-effective and user-friendly gene editing technologies, the custom cell line bearing any genome modifications can now be generated easily in a few days, e.g., gene deletion (Lee et al. 2010), gene inversion (Xiao et al. 2013), gene knockout (Santiago et al. 2008; Mali et al. 2013), gene addition (Moehle et al. 2007; Hockemeyer et al. 2011; Hou et al. 2013), gene correction (Urnov et al. 2005; Ran et al. 2013), gene addition as well as chromosomal translocation (Torres et al. 2014). Along with cell line engineering, the targeted nucleases have also accelerated the generation of genetically modified organisms, such as the accelerated creation of transgenic zebrafish (Doyon et al. 2008; Sander et al. 2011; Hwang et al. 2013), livestock (Hauschild et al. 2011; Carlson et al. 2012), monkeys (Liu et al. 2014), mice (Cui et al. 2011; Wang et al. 2013a, b; Wu et al. 2013), rats (Geurts et al. 2009; Tesson et al. 2011; Li et al. 2013), etc.
3.2 In Genetic Engineering of Plant Cells
These engineered nucleases have also emerged as a dominant tool for plant engineering (Baltes and Voytas 2015). For example, both CRISPR-Cas9 and TALENs have been used for the modification of multiple alleles in the haploid breed of wheat to create resistance variety against powdery disease (Wang et al. 2014b). Moreover, TALENs were used for soybean to knock out the nonessential gene that is used for fatty acid metabolism and thus produce simple plant cells with reduced metabolic constituents (Haun et al. 2014). The purified proteins comprised of various genomic engineering tools can be directly injected into the plant protoplast to effect germline-transmissible changes which are almost indistinguishable from the natural variety (Luo et al. 2015; Woo et al. 2015). The technological advancement of these tools could be very much helpful to reduce some regulatory problems which are associated with the use of transgenic plants. The targeted nucleases have been also used for the inactivation of pathogenic genes that help in the prevention of parasitic or viral infections and knock out specific factors leading to the development of pathogens resistance varieties (Ghorbal et al. 2014; Lin et al. 2014; Wu et al. 2015).
3.3 In Genetic Engineering for Insect-Borne Disease
Interestingly, the targeted nuclease has been also used to limit mosquito or insect-borne diseases (Burt 2003; Sinkins and Gould 2006). Genome editing enables the introduction of a particular gene or mutation in the host that can also get transferred to its progeny (Windbichler et al. 2011). This gene editing technique has been used in the vector of malaria, i.e., Aedes aegypti, Anopheles stephensi, and Anopheles gambiae for disease control and prevention (Gantz and Bier 2015; Hammond et al. 2016). Countries like Saudi Arabia, Turkey, Korea, Philippines, India, USA, Europe, China, and Japan are using the CRISPR technique for combating vector-borne diseases (Mahto et al. 2022). Smidler et al. reported the targeted disruption of the thioester-containing protein1 (TEP1) gene using TALEN in Anopheles gambiae mosquitos, which transmit malaria. The TEP1 gene of An. gambiae has been identified as a key gene for immunity against plasmodium infection (Miller et al. 2011). Gene editing in Ae. aegypti and An. stephensi using ZFNs and TALENs was reported in 2013 (Degennaro et al. 2013). De Gennaro et al. investigated the involvement of the odorant receptor coreceptor (orco) gene and the odorant receptor pathway in host identification and susceptibility to the chemical repellent N,N-diethyl-meta-toluamide (DEET) in Ae. aegypti (Christian et al. 2010). The developed ZFN was injected into embryos of Ae. aegypti in this experiment with promising results.
3.4 In Genetic Engineering of Industrially Important Microorganisms
The targeted nucleases also offer a convenient means for developing modified bacterial and yeast strains for synthetic biology such as metabolic pathway engineering. For example, the bacterial species belonging to the order Actinomycetales are one of the key sources of industrially relevant secondary metabolites. However, the large numbers of Actinomycetales species are historically resistant to genetic manipulation and had severely hindered their use for metabolic engineering. Now, CRISPR-Cas9 has been used to deactivate several genes of actinomycetes (Tong et al. 2015). This indicates the ability of the CRISPR/Cas9 system to create designer bacteria with enhanced secondary metabolite production capabilities. The CRISPR has also helped in metabolic pathway engineering in yeast by creating random mutagenesis in yeast chromosomal DNA at high efficacy (Jakočiūnas et al. 2015), allowing rapid screening of the desired mutants (Ryan et al. 2014).
3.5 In Genetic Engineering for Functional Genomics
The CRISPR-based knockout strategy has been playing an important role in functional genomics (Hilton and Gersbach 2015), e.g., facilitated the discovery of genomic loci that make cells drug-resistant (Koike-Yusa et al. 2014; Shalem et al. 2014; Wang et al. 2014a; Zhou et al. 2014; Blancafort et al. 2008). The genome editing tools also uncovered how the cells can initiate host immune response (Parnas et al. 2015), as well as keep giving new insights into the genetic basis of cell fitness (Hart et al. 2015; Wang et al. 2015). The genome editing tools have also increased the understanding of how certain viruses affect cell death (Ma et al. 2015). The genome-wide application of the CRISPR strategy has helped in the discovery of functional noncoding elements (Kim et al. 2013; Korkmaz et al. 2016) and understanding of their role in the structure and evolution of the human genome (Findlay et al. 2014). The CRISPR has also helped in identifying the factors key to zebrafish development (Shah et al. 2015) as well as disease development in mice (Chen et al. 2015).
3.6 In Genetic Engineering for Therapeutics
Genome editing technologies have great potential to treat/cure various diseases at genetic levels (Cox et al. 2015; Porteus 2015; Maeder and Gersbach 2016). For example, the ZFN-mediated disruption of HIV co-receptor CCR5 allowed the development of resistance against HIV in both CD4+ T cells (Perez et al. 2008) and CD34 hematopoietic stem/progenitor cells (HSPCs) (Holt et al. 2010; Tebas et al. 2014). Along with the introduction of gene modifications that enhance autologous cell therapies, targeted nucleases can also mediate genome editing in situ through combining viral vector, such as AAV (Gaj et al. 2016a). The delivery of an AAV vector designed to target a defective copy of the factor IX gene and provide a repair template had led to effective gene correction in mouse liver increasing factor IX protein production in both neonatal (Li et al. 2011) and adult (Anguela et al. 2013) mice. Recently, the in vivo gene editing tool has been used for the restoration of expression of the dystrophin gene allowing the rescue of muscle function in mouse models of Duchenne muscular dystrophy (Long et al. 2016; Nelson et al. 2016; Tabebordbar et al. 2016). A therapeutic gene editing tool has been successfully used in a mouse model of human hereditary tyrosinemia disease (Yin et al. 2014). This approach has been also used for the correction of disease-causing mutations in the ornithine transcarbamylase gene in the liver in a neonatal model of disease (Yang et al. 2016).
3.7 In Genetic Engineering: Epigenome Editing (Modulating Gene Expression)
Along with the DNA recognition ability of CRISPR-Cas9, the flexibility associated with constructing arrays of ZFs and TALEs proteins capable of binding to specific sequences allows their fusion with transcriptional activator and expression protein domains to modulate the expression of any gene from its promoter or enhancer sequences. The fusion of engineered zinc finger proteins either with the transcriptional domain derived from herpes-simplex or Kruppel-associated box (KRAB) repression protein had been used for the generation of the first fully synthetic transcriptional effector protein (Sadowski et al. 1988; Margolin et al. 1994; Beerli et al. 1998). Several other types of effector domains were extended and featured over the next 15 years using the zinc finger-based transcriptional modulators (Beerli and Barbas 2002). For example, modulation of transcription through targeted methylation or demethylation was done using the Dnmt3a methyltransferase domain (Rivenbark et al. 2012; Siddique et al. 2013) and the ten-eleven translocation methylcytosine dioxygenase 1 (TET1) (Chen et al. 2014). The TALE transcription factor has also emerged as an effective platform to achieve modulation of targeted transcription (Miller et al. 2011; Zhang et al. 2011). Similar to zinc finger, the TALE is also compatible with several modifiers such as the TET1 hydroxylase catalytic domain which is used for targeted CpG demethylase domains (Maeder et al. 2013), and the lysine-specific histone demethylase domain (LSD1) which has been used for targeted CpG histone demethylation (Beerli et al. 2000; Pollock et al. 2002; Magnenat et al. 2008; Polstein and Gersbach 2012; Maeder et al. 2013; Mendenhall et al. 2013; Perez-Pinera et al. 2013). TALE activators have also been effectively engineered to regulate gene expression in response to endogenous chemical stimuli (Li et al. 2012), proteolytic cues (Copeland et al. 2016; Lonzarić et al. 2016), external stimuli (Mercer et al. 2014), and optical signals (Konermann et al. 2013). The potential is immense.
3.8 Genome Engineering for Transcription Modulator
Because of the excellent ease, the CRISPR-Cas9 system has been also used for transcriptional modulation via fusion of a particular effector domain with the catalytically disabled variant of Cas9 protein (Qi et al. 2013). The mutant form is unable to cleave DNA and is referred to as dCas9 (Dead Cas9 Endonuclease) because of its ability to bind to the DNA in an RNA-directed manner. The carboxyl domain of dCas9 protein fused with the effector domain can modulate the gene expression from either strand of the targeted DNA sequences (Farzadfard et al. 2013; Maeder et al. 2013; Perez-Pinera et al. 2013; Gilbert et al. 2014; Hu et al. 2014). Moreover, dCas9 can inhibit gene expression by simply blocking the transcription initiation or elongation via the process known as CRISPR interference (Qi et al. 2013), whereas the fusion of transcriptional repressor domains with dCas9 can also be used to effectively silence a gene from the promoter region (Gilbert et al. 2013; Balboa et al. 2015; Zalatan et al. 2015). The light-inducible dCAs9-based system has been shown to be capable of allowing optical control of gene expression or achieving altered conditional control of gene expression (Nihongaki et al. 2015; Polstein and Gersbach 2015). The first-generation dCas9 activators were found to display a sub-optimal level of activation (Karlson et al. 2021). The development of second-generation CRISPR activators has rapidly emerged and expanded as a hugely promising area of research (Vora et al. 2016; Chen and Qi 2017).
Even though a lot has been accomplished using these genetic engineering tools still many challenges remain to limit the realization of the full potential of the genome editing tool. Most importantly are the development of new techniques which are capable of introducing gene modifications without DNA breaks such as Oligonucleotide-Directed Mutagenesis (ODM) and Base Editing (Komor et al. 2018). These methods can convert one target base pair to a different base pair without requiring DSBs and in the future can be promising technologies for the study of potential treatments for genetic diseases (Komor et al. 2018). The targeted recombinases that can recognize specific DNA sequences and incorporate desired therapeutic factors into the human genome can be designed and developed (Akopian et al. 2003; Pruett-Miller et al. 2009; Mercer et al. 2012; Gaj et al. 2013a; Sirk et al. 2014; Wallen et al. 2015). This could very well herald the era of the beginning of the union between regenerative medicines and genome engineering (see Table 9.2). However, despite the existence of substantial knowledge gained from genome editing in immortalized cell lines, its application in regenerative medicine that requires genetic manipulation of the progenitor or stem cell populations is still in its infancy as their epigenome as well as the organization of the genome and its functional regulation is inherently different from the transformed cell lines. It is important to fully explore and understand the functional landscape of the potential role and usage of these technologies in progenitor cells and stem cells before their large-scale usage in designer therapeutic applications that could mean reprograming the cell fate and behavior for the next generation of advancement in gene therapy and synthetic biology.
References
Akopian A, He J, Boocock MR, Stark WM (2003) Chimeric recombinases with designed DNA sequence recognition. Proc Natl Acad Sci 100:8688–8691
Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, Haurigot V et al (2013) Robust ZFN-mediated genome editing in adult hemophilic mice. Blood J Am Soc Hematol 122:3283–3287
Bae K-H, Do Kwon Y, Shin H-C, Hwang M-S, Ryu E-H, Park K-S et al (2003) Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol 21:275–280
Balboa D, Weltner J, Eurola S, Trokovic R, Wartiovaara K, Otonkoski T (2015) Conditionally stabilized dCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Rep 5:448–459
Baltes NJ, Voytas DF (2015) Enabling plant synthetic biology through genome engineering. Trends Biotechnol 33:120–131
Barone G, Arora A, Ganesh A, Abdel-fatah T, Moseley P, Ali R et al (2018) The relationship of CDK18 expression in breast cancer to clinicopathological parameters and therapeutic response. Oncotarget 9:29508–29524
Barrangou R, Marraffini LA (2014) CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54(2):234–244
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. https://doi.org/10.1126/science.1138140
Basu S, Adams L, Guhathakurta S, Kim Y (2017) A novel tool for monitoring endogenous alpha-synuclein transcription by NanoLuciferase tag insertion at the 3′ end using CRISPR-Cas9 genome editing technique. Nat Publ Gr:1–11. https://doi.org/10.1038/srep45883
Becker S, Boch J (2021) TALE and TALEN genome editing technologies. Gene Genome Edit 2:100007
Beerli RR, Barbas CF (2002) Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol 20:135–141
Beerli RR, Segal DJ, Dreier B, Barbas CF (1998) Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci 95:14628–14633
Beerli RR, Schopfer U, Dreier B, Barbas CF (2000) Chemically regulated zinc finger transcription factors. J Biol Chem 275:32617–32627
Bhaya D, Davison M, Barrangou R (2011) CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273–297
Blancafort P, Tschan MP, Bergquist S, Guthy D, Brachat A, Sheeter DA et al (2008) Modulation of drug resistance by artificial transcription factors. Mol Cancer Ther 7:688–697
Boch J, Bonas U (2010) Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561
Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR (2013) Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493:429–432
Burt A (2003) Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proc R Soc London Ser B Biol Sci 270:921–928
Capecchi MR (1989) Altering the genome by homologous recombination. Science 244:1288–1292
Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M et al (2012) Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci 109:17382–17387
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188(4):773–782
Carte J, Wang R, Li H, Terns RM, Terns MP (2008) Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22:3489–3496
Chandrasegaran S, Carroll D (2016) Origins of programmable nucleases for genome engineering. J Mol Biol 428:963–989
Chandrasekharan S, Kumar S, Valley CM, Rai A (2009) Proprietary science, open science and the role of patent disclosure: the case of zinc-finger proteins. Nat Biotechnol 27(2):140–144
Charpentier E, Doudna JA (2013) Rewriting a genome. Nature 495:50–51
Chattopadhyay A, Purohit J, Mehta S, Parmar H, Achary VMM, Reddy MK (2022) Precision genome editing toolbox: applications and approaches for improving rice’s genetic resistance to pathogens. Agronomy 12:565
Chen M, Qi LS (2017) Repurposing CRISPR system for transcriptional activation. Adv Exp Med Biol 983:147–157
Chen H, Kazemier HG, de Groote ML, Ruiters MHJ, Xu G-L, Rots MG (2014) Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res 42:1563–1574
Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X et al (2015) Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160:1246–1260
Chen Y, Farzadfard F, Gharaei N, Chen WCW, Cao J, Lu TK et al (2017) Randomized CRISPR-Cas transcriptional against alpha-synuclein toxicity randomized CRISPR-Cas transcriptional perturbation screening reveals protective genes against alpha-synuclein toxicity. Mol Cell 68:247–257.e5. https://doi.org/10.1016/j.molcel.2017.09.014
Chen JS, Chen JS, Ma E, Harrington LB, Da Costa M, Tian X et al (2018) CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 6245:1–8
Cho SW, Kim S, Kim JM, Kim J-S (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Choo Y, Klug A (1994) Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc Natl Acad Sci 91:11163–11167
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A et al (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761
Cohen J (2017) The Birth of CRISPR Inc. Science 355:680–684
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Copeland MF, Politz MC, Johnson CB, Markley AL, Pfleger BF (2016) A transcription activator–like effector (TALE) induction system mediated by proteolysis. Nat Chem Biol 12:254
Cox DBT, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ (2011) Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol 29:64–67
Davis D, Stokoe D (2010) Zinc Finger Nucleases as tools to understand and treat human diseases. BMC Med 8:1–11
Degennaro M, McBride CS, Seeholzer L, Nakagawa T, Dennis EJ, Goldman C et al (2013) orco mutant mosquitoes lose strong preference for humans and are not repelled by volatile DEET. Nature 498:487–491
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
Deshpande K, Vyas A, Balakrishnan A, Vyas D (2015) Clustered regularly interspaced short palindromic repeats/Cas9 genetic engineering: robotic genetic surgery. Am J Robot Surg 2(1):49–52
Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C et al (2019) Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol 37(3):252–258. https://doi.org/10.1038/s41587-019-0016-3
Deveau H, Garneau JE, Moineau S (2010) CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64:475–493
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE et al (2008) Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26:702–708
Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF (2001) Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 276:29466–29478
Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A et al (2005) Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 280:35588–35597
El-Mounadi K, Morales-Floriano ML, Garcia-Ruiz H (2020) Principles, applications, and biosafety of plant genome editing using CRISPR-Cas9. Front Plant Sci 11:56
Fairall L, Schwabe JWR, Chapman L, Finch JT, Rhodes D (1993) The crystal structure of a two zinc-finger peptide reveals an extension to the rules for zinc-finger/DNA recognition. Nature 366:483–487
Farzadfard F, Perli SD, Lu TK (2013) Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol 2:604–613
Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J (2014) Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513:120–123
Gaj T, Mercer AC, Sirk SJ, Smith HL, Barbas CF III (2013a) A comprehensive approach to zinc-finger recombinase customization enables genomic targeting in human cells. Nucleic Acids Res 41:3937–3946
Gaj T, Gersbach CA, Barbas CF III (2013b) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405
Gaj T, Epstein BE, Schaffer DV (2016a) Genome engineering using adeno-associated virus: basic and clinical research applications. Mol Ther 24:458–464
Gaj T, Sirk SJ, Shui SL, Liu J (2016b) Genome-editing technologies: principles and applications. Cold Spring Harb Perspect Biol 8(12):a023754
Gantz VM, Bier E (2015) The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348:442–444
Gesner EM, Schellenberg MJ, Garside EL, George MM, MacMillan AM (2011) Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol 18:688–692
Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM et al (2009) Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325:433
Ghezraoui H, Piganeau M, Renouf B, Renaud J-B, Sallmyr A, Ruis B et al (2014) Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell 55:829–842
Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio J-J (2014) Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32:819–821
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE et al (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451
Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH et al (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661
Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform 8:1–10
Guo M, Chen H, Dong S, Zhang Z, Luo H (2022) CRISPR - Cas gene editing technology and its application prospect in medicinal plants. Chin Med 17:1–19. https://doi.org/10.1186/s13020-022-00584-w
Gupta RM, Musunuru K (2014) Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest 124(10):4154–4161
György B, Lööv C, Zaborowski M, Takeda S, Kleinstiver B, Commins C et al (2018) CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s disease. Mol Ther Nucleic Acid 1–31. https://doi.org/10.1016/j.omtn.2018.03.007
Haft DH, Selengut J, Mongodin EF, Nelson KE (2005) A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1:e60
Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L et al (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956
Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S et al (2012) Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45:292–302
Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, Katsanos D et al (2016) A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34:78–83
Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G et al (2015) High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163:1515–1526
Haun W, Coffman A, Clasen BM, Demorest ZL, Lowy A, Ray E et al (2014) Improved soybean oil quality by targeted mutagenesis of the fatty acid desaturase 2 gene family. Plant Biotechnol J 12:934–940
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA (2010) Sequence-and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358
Hauschild J, Petersen B, Santiago Y, Queisser A-L, Carnwath JW, Lucas-Hahn A et al (2011) Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci 108:12013–12017
Hilton IB, Gersbach CA (2015) Enabling functional genomics with genome engineering. Genome Res 25:1442–1455
Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP et al (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29:731–734
Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V et al (2010) Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28:839–847
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–171
Hou Z, Zhang Y, Propson NE, Howden SE, Chu L-F, Sontheimer EJ et al (2013) Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci 110:15644–15649
Houbaviy HB, Usheva A, Shenk T, Burley SK (1996) Cocrystal structure of YY1 bound to the adeno-associated virus P5 initiator. Proc Natl Acad Sci 93:13577–13582
Hu X (2016) CRISPR/Cas9 system and its applications in human hematopoietic cells. Blood Cells. Mol Dis 62:6–12. https://doi.org/10.1016/j.bcmd.2016.09.003
Hu J, Lei Y, Wong W-K, Liu S, Lee K-C, He X et al (2014) Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res 42:4375–4390
Huang C, Sun H, Xu D, Chen Q, Liang Y, Wang X et al (2017) ZmCCT9 enhances maize adaptation to higher latitudes. PNAS 10:1–8. https://doi.org/10.1073/pnas.1718058115
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD et al (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31:227–229
Imran M, Waheed Y, Ghazal A, Ullah S, Safi SZ, Jamal M et al (2017) Modern biotechnology-based therapeutic approaches against HIV infection (Review). Biomed Rep 7:504–507. https://doi.org/10.3892/br.2017.1006
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Jakočiūnas T, Bonde I, Herrgård M, Harrison SJ, Kristensen M, Pedersen LE et al (2015) Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng 28:213–222
Jamieson AC, Kim S-H, Wells JA (1994) In vitro selection of zinc fingers with altered DNA-binding specificity. Biochemistry 33:5689–5695
Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5:a012740
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA-programmed genome editing in human cells. elife 2:e00471
Jore MM, Brouns SJJ, van der Oost J (2012) RNA in defense: CRISPRs protect prokaryotes against mobile genetic elements. Cold Spring Harb Perspect Biol 4:a003657
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14(1):49–55
Kachhawaha N (2021) CRISPR edited microbes and their industrial potential review. Res J Biotechnol 16:223–228. https://doi.org/10.6084/m9.figshare.14216363.v1
Kaminski MM, Abudayyeh OO, Gootenberg JS, Zhang F, Collins JJ (2021) CRISPR-based diagnostics. Nat Biomed Eng 5(7):643–656
Karlson CKS, Mohd-Noor SN, Nolte N, Tan BC (2021) CRISPR/dCas9-based systems: mechanisms and applications in plant sciences. Plan Theory 10(10):2055
Khalil AM (2020) The genome editing revolution. J Genet Eng Biotechnol 18:1–16
Khalil K, Elayat M, Khalifa E, Daghash S, Elaswad A, Miller M et al (2017) Generation of myostatin gene- edited channel catfish (Ictalurus punctatus) via zygote injection of CRISPR/Cas9 system. Sci Rep 7:7301. https://doi.org/10.1038/s41598-017-07223-7
Kim Y-G, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci 93:1156–1160
Kim HJ, Lee HJ, Kim H, Cho SW, Kim J-S (2009) Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19:1279–1288
Kim Y-K, Wee G, Park J, Kim J, Baek D, Kim J-S et al (2013) TALEN-based knockout library for human microRNAs. Nat Struct Mol Biol 20:1458–1464
Kim S, Hupperetz C, Lim S, Kim CH (2021) Genome editing of immune cells using CRISPR/Cas9. BMB Rep 54(1):59
Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K (2014) Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32:267–273
Komor AC, Badran AH, Liu DR (2018) Editing the genome without double-stranded DNA breaks. ACS Chem Biol 13:383–388
Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L et al (2013) Optical control of mammalian endogenous transcription and epigenetic states. Nature 500:472–476
Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K et al (2016) Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol 34:192–198
Lamb BM, Mercer AC, Barbas CF III (2013) Directed evolution of the TALE N-terminal domain for recognition of all 5′ bases. Nucleic Acids Res 41:9779–9785
Laoharawee K, DeKelver RC, Podetz-Pedersen KM, Rohde M, Sproul S, Nguyen HO et al (2018) Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol Ther 26(4):1127–1136
Lee HJ, Kim E, Kim J-S (2010) Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20:81–89
Lee HG, Kim DH, Choi Y-R, Yu J, Hong S-A, Seo PJ et al (2021) Enhancing plant immunity by expression of pathogen-targeted CRISPR-Cas9 in plants. Gene Genome Ed. 1:100001. https://doi.org/10.1016/j.ggedit.2021.100001
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS et al (2011) In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475:217–221
Li Y, Moore R, Guinn M, Bleris L (2012) Transcription activator-like effector hybrids for conditional control and rewiring of chromosomal transgene expression. Sci Rep 2:1–7
Li W, Teng F, Li T, Zhou Q (2013) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31:684–686
Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X (2020) Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther 5:1–23. https://doi.org/10.1038/s41392-019-0089-y
Liao Y, Bai Q, Xu P, Wu T, Guo D, Peng Y et al (2018) Mutation in rice abscisic acid2 results in cell death, enhanced disease-resistance, altered seed dormancy and development. Front Plant Sci 9:405. https://doi.org/10.3389/fpls.2018.00405
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211
Lillestøl RK, Shah SA, Brügger K, Redder P, Phan H, Christiansen J et al (2009) CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol 72:259–272
Limsirichai P, Gaj T, Schaffer DV (2016) CRISPR-mediated activation of latent HIV-1 expression. Mol Ther 24:499–507
Lin S-R, Yang H-C, Kuo Y-T, Liu C-J, Yang T-Y, Sung K-C et al (2014) The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Acids 3:e186
Lino CA, Harper JC, Carney JP, Timlin JA (2018) Delivering CRISPR: a review of the challenges and approaches. Drug Deliv 25(1):1234–1257
Liu Z, Zhou X, Zhu Y, Chen Z-F, Yu B, Wang Y et al (2014) Generation of a monkey with MECP2 mutations by TALEN-based gene targeting. Neurosci Bull 30:381–386
Liu Z, Dong H, Cui Y, Cong L, Zhang D (2020) Application of different types of CRISPR/Cas-based systems in bacteria. Microb Cell Factories 19(1):1–14
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E et al (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351:400–403
Lonzarić J, Lebar T, Majerle A, Manček-Keber M, Jerala R (2016) Locked and proteolysis-based transcription activator-like effector (TALE) regulation. Nucleic Acids Res 44:1471–1481
Luo S, Li J, Stoddard TJ, Baltes NJ, Demorest ZL, Clasen BM et al (2015) Non-transgenic plant genome editing using purified sequence-specific nucleases. Mol Plant 8:1425–1427
Lynch VJ, Bedoya-Reina OC, Ratan A, Perry GH, Miller W, Schuster SC (2015) Elephantid genomes reveal the molecular bases of woolly mammoth adaptations to the arctic. Cell Rep 12:217–228. https://doi.org/10.1016/j.celrep.2015.06.027
Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J et al (2015) A CRISPR-based screen identifies genes essential for West-Nile-virus-induced cell death. Cell Rep 12:673–683
Maeder ML, Gersbach CA (2016) Genome-editing technologies for gene and cell therapy. Mol Ther 24:430–446. https://doi.org/10.1038/mt.2016.10
Maeder ML, Linder SJ, Reyon D, Angstman JF, Fu Y, Sander JD et al (2013) Robust, synergistic regulation of human gene expression using TALE activators. Nat Methods 10:243–245
Magnenat L, Schwimmer LJ, Barbas CF (2008) Drug-inducible and simultaneous regulation of endogenous genes by single-chain nuclear receptor-based zinc-finger transcription factor gene switches. Gene Ther 15:1223–1232
Mahto KK, Prasad P, Kumar M, Dubey H, Ranjan A (2022) Role of CRISPR technology in gene editing of emerging and re-emerging vector borne disease. In: Puerta-Guardo H, Manrique-Saide DP (eds) Mosquito research - recent advances in pathogen interactions, immunity, and vector control strategies. IntechOpen Limited. https://doi.org/10.5772/intechopen.104100
Maiti P, Manna J, Dunbar GL (2017) Current understanding of the molecular mechanisms in Parkinson’s disease: targets for potential treatments. Transl Neurodegener 6:1–35. https://doi.org/10.1186/s40035-017-0099-z
Mak AN-S, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL (2012) The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335:716–719
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:1–26
Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P et al (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477
Makarova KS, Wolf YI, Koonin EV (2013) The basic building blocks and evolution of CRISPR – Cas systems. Biochem Soc Trans 41:1392–1400. https://doi.org/10.1042/BST20130038
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Mandip KC, Steer CJ (2019) A new era of gene editing for the treatment of human diseases. Swiss Med Wkly
Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen H-J, Rauscher FJ (1994) Krüppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci 91:4509–4513
Marraffini LA, Sontheimer EJ (2010) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571
Martinez-Lage M, Torres-Ruiz R, Rodriguez-Perales S (2017) CRISPR/Cas9 technology: applications and human disease modeling. Elsevier Inc. https://doi.org/10.1016/bs.pmbts.2017.09.002
Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK et al (2013) Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 31:1133–1136
Mercer AC, Gaj T, Fuller RP, Barbas CF III (2012) Chimeric TALE recombinases with programmable DNA sequence specificity. Nucleic Acids Res 40:11163–11172
Mercer AC, Gaj T, Sirk SJ, Lamb BM, Barbas CF III (2014) Regulation of endogenous human gene expression by ligand-inducible TALE transcription factors. ACS Synth Biol 3:723–730
Miller J, McLachlan AD, Klug A (1985) Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J 4:1609–1614
Miller JC, Holmes MC, Wang J, Guschin DY, Lee Y-L, Rupniewski I et al (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF et al (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148
Moehle EA, Rock JM, Lee Y-L, Jouvenot Y, DeKelver RC, Gregory PD et al (2007) Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci 104:3055–3060
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C (2009a) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C (2009b) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. https://doi.org/10.1099/mic.0.023960-0
Moon SB, Kim DY, Ko JH, Kim YS (2019) Recent advances in the CRISPR genome editing tool set. Exp Mol Med 51(11):1–11
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11:196–207
National Human Genome Research Institute (2019) What is genome editing? US Dep Heal Hum Serv, 1. Available at: https://www.genome.gov/about-genomics/policy-issues/what-is-Genome-Editing
National Institute of Health (2020) Gene editing – digital media kit. US Dep Heal Hum Serv, 1. Available at: https://www.nih.gov/news-events/gene-editing-digital-press-kit
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RMC et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–407
Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM (2014) TALEN and CRISPR/Cas genome editing systems: tools of discovery. Acta Nat 6(3 (22)):19–40
Newsom S, Parameshwaran HP, Martin L, Rajan R (2021) The CRISPR-Cas mechanism for adaptive immunity and alternate bacterial functions fuels diverse biotechnologies. Front Cell Infect Microbiol 10:619763
Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M (2015) CRISPR-Cas9-based photoactivatable transcription system. Chem Biol 22:169–174
Nolte RT, Conlin RM, Harrison SC, Brown RS (1998) Differing roles for zinc fingers in DNA recognition: structure of a six-finger transcription factor IIIA complex. Proc Natl Acad Sci 95:2938–2943
Nymark M, Sharma AK, Sparstad T, Bones AM, Winge P (2016) OPEN A CRISPR / Cas9 system adapted for gene editing in marine algae. Nat Publ Gr, 6–11. https://doi.org/10.1038/srep24951
Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ et al (2015) A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162:675–686
Paschon DE, Lussier S, Wangzor T, Xia DF, Li PW, Hinkley SJ et al (2019) Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat Commun 10:1–12
Pavletich NP, Pabo CO (1993) Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science 261:1701–1707
Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ (2018) Modulating DNA repair pathways to improve precision genome engineering. ACS Chem Biol 13:389–396
Perales M, Kebriaei P, Kean LS, Sadelain M (2018) Reprint of: building a safer and faster CAR: seatbelts, airbags, and CRISPR. Biol Blood Marrow Transplant 24:S15–S19. https://doi.org/10.1016/j.bbmt.2017.12.789
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O et al (2008) Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26:808–816
Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F et al (2013) Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 10:239–242
Pollock R, Giel M, Linher K, Clackson T (2002) Regulation of endogenous gene expression with a small-molecule dimerizer. Nat Biotechnol 20:729–733
Polstein LR, Gersbach CA (2012) Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. J Am Chem Soc 134:16480–16483
Polstein LR, Gersbach CA (2015) A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol 11:198–200
Porteus MH (2015) Towards a new era in medicine: therapeutic genome editing. Genome Biol 16:1–12
Porteus M (2016) Genome editing: a new approach to human therapeutics. Annu Rev Pharmacol Toxicol 56:163–190. https://doi.org/10.1146/annurev-pharmtox-010814-124454
Porteus MH, Baltimore D (2003) Chimeric nucleases stimulate gene targeting in human cells. Science 300(5620):763
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663
Pruett-Miller SM, Reading DW, Porter SN, Porteus MH (2009) Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet 5:e1000376
Qasim W (2021) Genome editing of therapeutic T cells. Gene Genome Ed 2:100010. https://doi.org/10.1016/j.ggedit.2021.100010
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP et al (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183
Rager JE, Carberry C, Fry RC (2011) Use of genome editing tools in environmental health research. Physiol Behav 176:139–148. https://doi.org/10.1016/j.cotox.2019.02.007.Use
Rahim J, Gulzar S, Zahid R, Rahim KA (2021) Systematic review on the comparison of molecular gene editing tools. Int J Innov Sci Res Tech 6:1–8
Ramirez CL, Foley JE, Wright DA, Müller-Lerch F, Rahman SH, Cornu TI et al (2008) Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5:374–375
Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE et al (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Raquel D, Rodríguez R, Solís RR, Alberto H, Saldaña B (2019) Genome editing: a perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int J Mol Med 43:1559–1574. https://doi.org/10.3892/ijmm.2019.4112
Rath D, Amlinger L, Rath A, Lundgren M (2015) The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117:119–128
Rebar EJ, Pabo CO (1994) Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science 263:671–673
Remy S, Tesson L, Ménoret S, Usal C, Scharenberg AM, Anegon I (2010) Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res 19(3):363–371
Renaud J-B, Boix C, Charpentier M, De Cian A, Cochennec J, Duvernois-Berthet E et al (2016) Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR-Cas9 nucleases. Cell Rep 14:2263–2272
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30:460–465
Ricci R, Colasante G (2021) CRISPR/dCas9 as a therapeutic approach for neurodevelopmental disorders: innovations and limitations compared to traditional strategies. Dev Neurosci 43(3–4):253–261
Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L (2012) Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res 40:9887–9896
Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD et al (2012) Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 7:350–360
Robb GB (2019) Genome editing with CRISPR-Cas: an overview. Curr Protoc Essent Lab Tech e36 19:1–20
Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S et al (2014) Selection of chromosomal DNA libraries using a multiplex CRISPR system. elife 3
Sadowski I, Ma J, Triezenberg S, Ptashne M (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature 335:563–564
Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D (2010) ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res 38(suppl_2):W462–W468
Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D et al (2011) Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8:67–69
Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F (2012) A transcription activator-like effector toolbox for genome engineering. Nat Protoc 7:171–192
Santiago Y, Chan E, Liu P-Q, Orlando S, Zhang L, Urnov FD et al (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci 105:5809–5814
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39:9275–9282
Sashital DG, Jinek M, Doudna JA (2011) An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol 18:680–687
Schmid-Burgk JL, Schmidt T, Kaiser V, Höning K, Hornung V (2013) A ligation-independent cloning technique for high-throughput assembly of transcription activator–like effector genes. Nat Biotechnol 31:76–81
Schornack S, Meyer A, Römer P, Jordan T, Lahaye T (2006) Gene-for-gene-mediated recognition of nuclear-targeted AvrBs3-like bacterial effector proteins. J Plant Physiol 163:256–272
Segal DJ, Dreier B, Beerli RR, Barbas CF (1999) Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci 96:2758–2763
Segal DJ, Beerli RR, Blancafort P, Dreier B, Effertz K, Huber A et al (2003) Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry 42:2137–2148
Segal DJ, Crotty JW, Bhakta MS, Barbas CF III, Horton NC (2006) Structure of Aart, a designed six-finger zinc finger peptide, bound to DNA. J Mol Biol 363:405–421
Shabbir MAB, Shabbir MZ, Wu Q, Mahmood S, Sajid A, Maan MK et al (2019) CRISPR-cas system: biological function in microbes and its use to treat antimicrobial resistant pathogens. Ann Clin Microbiol Antimicrob 18(1):1–9
Shah AN, Davey CF, Whitebirch AC, Miller AC, Moens CB (2015) Rapid reverse genetic screening using CRISPR in zebrafish. Nat Methods 12:535–540
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS et al (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87
Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR (2015) Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol Adv 1:1–9. https://doi.org/10.1038/nbt.3235
Shiva KG, Suma K (2019) Gene editing nucleases-ZFNs, TALENS and CRISPR: a review. Chettinad Health City Med J 8:130–135
Siddique AN, Nunna S, Rajavelu A, Zhang Y, Jurkowska RZ, Reinhardt R et al (2013) Targeted methylation and gene silencing of VEGF-A in human cells by using a designed Dnmt3a–Dnmt3L single-chain fusion protein with increased DNA methylation activity. J Mol Biol 425:479–491
Singina GN, Sergiev PV, Lopukhov AV, Rubtsova MP, Taradajnic NP, Ravin NV et al (2021) Production of a cloned offspring and CRISPR/Cas9 genome editing of embryonic fibroblasts in cattle. Dokl Biochem Biophys 496:48–51. https://doi.org/10.1134/S1607672921010099
Sinkins SP, Gould F (2006) Gene drive systems for insect disease vectors. Nat Rev Genet 7:427–435
Sirk SJ, Gaj T, Jonsson A, Mercer AC, Barbas CF III (2014) Expanding the zinc-finger recombinase repertoire: directed evolution and mutational analysis of serine recombinase specificity determinants. Nucleic Acids Res 42:4755–4766
Song S, Yang Y, Liu M, Liu B, Yang X, Yu M et al (2018) MiR-125b attenuates human hepatocellular carcinoma malignancy through targeting SIRT6. Am J Cancer Res 8:993–1007
Su X, Cui K, Du S, Li H, Lu F, Shi D et al (2018) Efficient genome editing in cultured cells and embryos of Debao pig and swamp buffalo using the CRISPR/Cas9 system. Vitr Cell Dev Biol Anim:1–9
Sui T, Xu L, Lau YS, Liu D, Liu T, Gao Y et al (2018) Development of muscular dystrophy in a CRISPR-engineered mutant rabbit model with frame-disrupting ANO5 mutations. Cell Death Dis Artic 9:609. https://doi.org/10.1038/s41419-018-0674-y
Szczepek M, Brondani V, Büchel J, Serrano L, Segal DJ, Cathomen T (2007) Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25:786–793
Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–411
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G et al (2014) Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370:901–910
Tesson L, Usal C, Ménoret S, Leung E, Niles BJ, Remy S et al (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29:695–696
Thakore PI, Gersbach CA (2015) Genome engineering for therapeutic applications. In: Translating gene therapy to the clinic. Academic Press, pp 27–43
Tong Y, Charusanti P, Zhang L, Weber T, Lee SY (2015) CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029
Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S (2014) Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR–Cas9 system. Nat Commun 5:1–8
Universitesi B (2020) Advance genome editing technologies in the treatment of human diseases: CRISPR therapy (Review). Int J Mol Med 46:521–534. https://doi.org/10.3892/ijmm.2020.4609
Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S et al (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11(9):636–646
van Diemen FR, Kruse EM, Hooykaas MJG (2016) CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog 12:e1005701. https://doi.org/10.1371/journal.ppat.1005701
Vanamee ÉS, Santagata S, Aggarwal AK (2001) FokI requires two specific DNA sites for cleavage. J Mol Biol 309:69–78
Vora S, Tuttle M, Cheng J, Church G (2016) Next stop for the CRISPR revolution: RNA-guided epigenetic regulators. FEBS J 283(17):3181–3193
Wallen MC, Gaj T, Barbas CF III (2015) Redesigning recombinase specificity for safe harbor sites in the human genome. PLoS One 10:e0139123
Waltz E (2015) Gene-edited CRISPR mushroom escapes US regulation. Nature 532:293
Wang L, Lin J, Zhang T, Xu K, Ren C, Zhang Z (2013a) Simultaneous screening and validation of effective zinc finger nucleases in yeast. PLoS One 8(5):e64687
Wang H, Hu Y-C, Markoulaki S, Welstead GG, Cheng AW, Shivalila CS et al (2013b) TALEN-mediated editing of the mouse Y chromosome. Nat Biotechnol 31:530–532
Wang T, Wei JJ, Sabatini DM, Lander ES (2014a) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C et al (2014b) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951
Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ et al (2015) Identification and characterization of essential genes in the human genome. Science 350:1096–1101
Wang SW, Gao C, Zheng YM, Yi L, Lu JC, Huang XY et al (2022) Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer 21:1–27. https://doi.org/10.1186/s12943-022-01518-8
Windbichler N, Menichelli M, Papathanos PA, Thyme SB, Li H, Ulge UY et al (2011) A synthetic homing endonuclease-based gene drive system in the human malaria mosquito. Nature 473:212–215
Wolfe SA, Nekludova L, Pabo CO (2000) DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29:183–212
Wolfe SA, Grant RA, Elrod-Erickson M, Pabo CO (2001) Beyond the “recognition code”: structures of two Cys2His2 zinc finger/TATA box complexes. Structure 9:717–723
Woo JW, Kim J, Kwon SI, Corvalán C, Cho SW, Kim H et al (2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33:1162–1164
Wood AJ, Lo T-W, Zeitler B, Pickle CS, Ralston EJ, Lee AH et al (2011) Targeted genome editing across species using ZFNs and TALENs. Science 333:307
Wu J, Kandavelou K, Chandrasegaran S (2007) Custom-designed zinc finger nucleases: what is next? Cell Mol Life Sci 64(22):2933–2944
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S et al (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13:659–662
Wu H, Wang Y, Zhang Y, Yang M, Lv J, Liu J et al (2015) TALE nickase-mediated SP110 knockin endows cattle with increased resistance to tuberculosis. Proc Natl Acad Sci 112:E1530–E1539
Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z et al (2013) Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41:e141–e141
Yang G, Huang X (2019) Methods and applications of CRISPR/Cas system for genome editing in stem cells. Cell Regen 8:33–41. https://doi.org/10.1016/j.cr.2019.08.001
Yang Y, Wang L, Bell P, McMenamin D, He Z, White J et al (2016) A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol 34:334–338
Yang J, Yi N, Zhang J, He W, He D, Wu W et al (2018) Generation and characterization of a hypothyroidism rat model with truncated thyroid stimulating hormone receptor. Sci Rep 8:4004. https://doi.org/10.1038/s41598-018-22405-7
Yang Y, Xu J, Ge S, Lai L (2021) CRISPR/Cas: advances, limitations, and applications for precision cancer research. Front Med 8:1–11. https://doi.org/10.3389/fmed.2021.649896
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M et al (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32:551–553
Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M et al (2015) Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160:339–350
Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P (2011) Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol 29:149–153
Zhang Z, Cheng T, Lui W (2017) Suppression of Epstein-Barr virus DNA load in latently infected nasopharyngeal carcinoma cells by CRISPR/Cas9. Virus Res. https://doi.org/10.1016/j.virusres.2017.04.019
Zhao J, Lai L, Ji W, Zhou Q (2019) Genome editing in large animals: current status and future prospects. Natl Sci Rev 6:402–420. https://doi.org/10.1093/nsr/nwz013
Zhao X, Nie J, Tang Y, He W, Xiao K, Pang C et al (2020) Generation of transgenic cloned buffalo embryos harboring the EGFP gene in the Y chromosome using CRISPR/Cas9-mediated targeted integration. Front Vet Sci 7:1–14. https://doi.org/10.3389/fvets.2020.00199
Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y et al (2014) High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509:487–491
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kaur, R., Singh, A.K., Singh, D.K., Singh, S. (2023). Modern Tools of Genome Engineering and Their Applications. In: Sobti, R., Kuhad, R.C., Lal, R., Rishi, P. (eds) Role of Microbes in Sustainable Development. Springer, Singapore. https://doi.org/10.1007/978-981-99-3126-2_9
Download citation
DOI: https://doi.org/10.1007/978-981-99-3126-2_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-3125-5
Online ISBN: 978-981-99-3126-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)