
Abstract
Compounds containing 4-aminoquinoline moiety are known to exhibit various pharmacological properties such as antimalarial, anticancer, antitubercular and antivirus. Chloroquine, hydroxyquinoline and amodiaquine are a few well-known 4-aminoquinoline-based medications. Many new derivatives of this compound have also been synthesized and developed as lead molecules in the drug development process. The already existing 4-aminoquinoline-based drugs are also explored for the treatment of more recent diseases like coronavirus disease 2019 (COVID-19). Drug resistance also gives scope for testing newer molecules against various other pathogenic infections. This molecule has always drawn the attention of medicinal chemists for the synthesis of new derivatives. This article gives an overview of the recent development of 4-aminoquinoline derivatives and their pharmacological activities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Nitrogen-containing heterocyclic compounds are present in several biologically active natural products which are being used in traditional medicines or approved prescribed drugs. Many of their synthetic counterparts are also available in the clinical market for the treatment of various diseases (Kerru et al. 2020). Some of the notable synthetic nitrogen heterocyclic-based drugs are chlorpromazine, diazepam, metronidazole, azidothymidine, isoniazid, barbituric acid, chloroquine, captopril etc. (Parashar and Negi 2015). In addition to serving as the foundation of medicinal chemistry, the nitrogen heterocyclic compounds are also a crucial component of the majority of vitamins, enzymes, coenzymes, hormones, nucleic acids and alkaloids (Al-Ghorbani et al. 2015; Heravi and Zadsirjan 2020). Due to these reasons, nitrogen heterocycles have always been attractive targets to synthetic organic and medicinal chemists.
Among various nitrogen-containing heterocyclic compounds, 4-aminoquinoline pharmacophores have gained a lot of interest from researchers to design and develop biologically important molecules (Matada et al. 2021; Yadav and Shah 2021). In 1810, Gomes et al. isolated quinine from Cinchona bark and it was the first 4-aminoquinoline-based drug that was used for the treatment of malaria (Meshnick and Dobson 2001). In 1856, William Henry Perkins attempted to synthesize quinine but he isolated the first synthetic textile dye called ‘mauveine’. However, later in 1944, the first total synthesis of quinine was accomplished (Woodward and Doering 1944). This sparked the interest of medicinal chemists in the 4-aminoquinoline pharmacophore that resulted in many drugs including chloroquine (Deshpande and Kuppast 2016; Manohar et al. 2014). Further research demonstrated that the 4-aminoquinoline class of compounds exhibits a wide range of biological activities such as antimalarial (Hu et al. 2017; Narula et al. 2019; Raj et al. 2015; Vandekerckhove and D’hooghe 2015), anticancer (Musiol 2017), antimicrobial (Musiol et al. 2017), antifungal (Musiol et al. 2010), antiviral (Kaur and Kumar 2021), antitubercular (Rangappa and Siddappa 2014; Rawat and Beena 2013; Singh et al. 2015), anti-inflammatory (Ambatkar and Khedekar 2019; Mukherjee and Pal 2013) and anti-HIV (Chokkar et al. 2019). The main objective of this article is to update the literature regarding the recent development of 4-aminoquinoline-based compounds and their biological activities.
2 Antimalarial Activity
Among all biological activities, the antimalarial properties of 4-aminoquinolines have received the greatest attention. Chloroquine, a 4-aminoquinoline-based drug, was first synthesized by Hans Andersag in 1934 (Krafts et al. 2012). Initially, it was considered to be toxic, but subsequent studies demonstrated its potential antimalarial properties, making it one of the most popular medications for the treatment of malaria. Later on, a number of analogues, namely amodiaquine, isoquine, sontoquine, piperaquine and primaquine were discovered (Fig. 10.1). Although the prevalence of malarial infection was reduced by these medications, the development of drug resistance, particularly against Plasmodium falciparum, prompted medicinal chemists to look for other safer medications. This led to the discovery of artemisinin (Wang et al. 2019). Currently, the first-line treatment for malarial infection caused by Plasmodium falciparum is artemisinin-based combination therapy (ACT), wherein the artemisinin and its derivatives are co-administered with some drug from a different class of antimalarials such as amodiaquine, mefloquine and piperaquine (Kremsner and Krishna 2004). However, in 2008, the first case of resistance in P. falciparum against artemisinin and its derivatives was observed in Western Cambodia (Dondorp et al. 2009; Noedl et al. 2008). The emergence of multidrug resistance and the parasite’s increasing capacity to tolerate the artemisinin-based combination therapy urges researchers to develop novel treatments and antimalarial medications to further reduce malaria occurrences and fatalities.

4-Aminoquinoline-based antimalarial drugs
To tackle drug-resistant issues, the concept of hybrid molecules has been the most fascinating topic in medicinal chemistry, where two or more pharmacophores are connected covalently to create one single molecule (Meunier 2008; Muregi and Ishih 2010). The two units of these compounds may exert dual pharmacological action by acting on different targets, or one molecule may be able to balance off the side effects caused by the other molecule. With this aim, many research groups have prepared a large variety of aminoquinoline derivatives by modifying the side chain of chloroquine or combining chloroquine with new pharmacophores acting on different targets.
A series of 4-aminoquinoline-linked Mannich bases (1a-1i and 2a-2c) were synthesized and tested for in vitro antimalarial activity against P. falciparum chloroquine-sensitive strain (3D7) (Singh et al. 2021). All of the compounds demonstrated mild activity, with MIC values ranging from 15.6 to 125 μg/mL. Compounds 1b and 1c were found to be the most active. Both compounds had MIC values of 15.6 μg/mL when compared to chloroquine (MIC = 0.4 μg/mL).

The hybrid molecules 3a-3e were synthesized by conjugating 4-aminoquinoline with pyrano[2,3-c]pyrazole and tested against P. falciparum chloroquine susceptible (3D7) and chloroquine-resistant (K1) strains (Shamsuddin et al. 2021). Compounds 3a, 3b, 3c and 3e displayed very good antimalarial activity against the sensitive strain 3D7, with EC50 values of 0.19 μM, 0.0130 μM, 0.113 μM and 0.026 μM, respectively. Aside from their potent antimalarial activity against the chloroquine susceptible strain, compounds 3a, 3b and 3d also had potent activity against the resistant strain K1, with EC50 values of 0.25 μM, 0.02 μM and 0.30 μM, respectively. When the cytotoxicity of compounds was evaluated against the Vero mammalian cell line, it was discovered that they exhibited modest cytotoxic activities and high selectivity index values ranging from 355 to 1354.

1,2,3,4-Tetrahydro-β-carbolines (THβC) are present in a variety of pharmacologically active substances with putative antimalarial properties. Keeping this in mind, a series of hybrid molecules 4a-4b, 5a-5d, 6a-6h and 7a-7b were synthesized by coupling this moiety with 4-aminoquinoline where the two pharmacophoric groups were linked with either a triazole or an acyl hydrazide linker (Sharma et al. 2020). The antimalarial activity of these compounds was tested against the chloroquine-resistant W2 strain of P. falciparum. In general, the acyl hydrazide hybrids 6 and 7 were found to be less active than the triazole-linked hybrids 4 and 5. The insertion of a flexible alkyl chain between the triazole and quinoline moiety in the 1H-1,2,3-triazole tethered THβC-4-aminoquinoline conjugates significantly improved the antimalarial activity. Compounds 5a-5d were more active than 4a-4b (IC50 = 4.02–9.28 μM), having an IC50 value of 0.49–1.37 μM. The antimalarial activity of the aliphatic acyl hydrazide linked hybrids 6a-6h was improved by lengthening the alkyl chain; however, the kind of substituent at the C-1 position of THβC did not appear to affect the antimalarial activity. Cytotoxic activity of the most active compounds 5a, 5c (triazole as linker) and 7a, 7b (acyl-hydrazide as linker) was assessed on mammalian Vero cell lines. As anticipated, compounds 7a and 7b were mildly cytotoxic, whereas 5a and 5c were found to be noncytotoxic, with SI > 300.

The artemisinin-quinoline hybrids 8–20 were synthesized and tested for their antimalarial efficacy against chloroquine susceptible strain (3D7) and chloroquine-resistant strains (Dd2 and K1) of P. falciparum (Çapci et al. 2019). All the compounds displayed extremely interesting activity with EC50 values from 0.78 to 27.5 nM and were discovered to be more effective against the resistant strains. Compounds 8–11, in which the carbon chain length between artesunic acid and quinoline moieties was varied from one to four carbon atoms, demonstrated better antimalarial activity than the standard drug chloroquine (EC50 = 12.7 nM), with EC50 values in the nanomolar range (5.3, 8.0, 5.4 and 6.2 nM, respectively). The compound with the shortest linker demonstrated the highest antimalarial activity against all the strains. In the case of the resistant strains, hybrids 8–11 were found to be 34–39 times more potent (EC50 = 4.2–4.9 nM) against the Dd2 strain and 116–168 times more potent (EC50 = 1.8–2.6 nM) against K1 strain than chloroquine. Furthermore, artemisinin-quinoline hybrids 12–16 demonstrated excellent activity with EC50 values between 2.0 nM and 27.5 nM. These compounds were more effective against resistant strains than sensitive strains. When tested towards three distinct strains, compounds 12, 14, 15 and 16 showed nearly identical potency. These compounds had EC50 values of 9.0 nM, 8.8 nM, 9.4 nM and 8.4 nM towards 3D7 strain; 5.9 nM, 4.5 nM, 5.1 nM and 6.1 nM towards Dd2 strain; and 4.3 nM, 2.0 nM, 3.8 nM and 3.7 nM against K1 strain. The EC50 values for the antimalarial activity of compounds 17–19 ranged from 0.78 nM to 14.8 nM. Compound 18 with NH at C4 of the 7-chloroquinoline unit was found to be more active against all three strains than compound 17 with O-atom at C4 of the 7-chloroquinoline unit. Compound 19, obtained by replacing the second oxygen atom in the linker with a nitrogen atom, demonstrated higher activity than compounds 17 and 18 with EC50 values of 1.0 nM, 0.78 nM and 2.7 nM towards Dd2, K1 and 3D7 strains, respectively. Two compounds 16 and 18 were examined for in vivo activity in mice infected with P. berghei. Unfortunately, compound 16 failed to suppress parasitaemia when given subcutaneously for 4 days at a concentration of 105 μmol/kg and hence did not receive attention for additional studies. However, compound 18 reduced parasitaemia by more than 99% at a concentration of 105 μmol/kg for 4 days, while artesunate was unable to treat any animal at this concentration. To investigate the mechanism of action, compound 18 was tested in vitro and in vivo for its capability to inhibit the formation of β-hematin. The investigations demonstrated a 71.9% reduction in β-hematin formation in vivo and the IC50 value of 0.56 mM was observed for in vitro inhibition of the formation of β-hematin. The in vitro inhibition was comparable to chloroquine (IC50 = 0.89 mM), although artesunate did not show any significant activity.
Eighteen 4-aminoquinoline derivatives 21a-21f, 22a-22f and 23a-23f were synthesized and evaluated against chloroquine-sensitive 3D7 strain and chloroquine-resistant K1 strain of P. falciparum (Kondaparla et al. 2017). Four of the 18 compounds examined (21b, 22b, 22c and 22e), with corresponding IC50 values of 28.49, 22.21, 27.16 and 73.05 nM, demonstrated extremely good activity against the 3D7 strain. Eleven compounds were discovered to be more effective (IC50 = 31.19–252.28 nM) than chloroquine (IC50 = 255 nM) against the chloroquine-resistant K1 strain. The cytotoxic activity of the compounds was determined using the MTT assay against the Vero cell line, and the majority of the compounds demonstrated a modest cytotoxic effect with a high selectivity index. Furthermore, some of the compounds (21b-21f, 22b-22f and 23f) were tested for in vivo activity against chloroquine-resistant Plasmodium yoelii (N-67 strain) in Albino mice of the Swiss strain, and all the tested compounds completely suppressed parasitaemia on day 4 when administered orally at a dose level of 100 mg/kg.


The 4-aminoquinoline-imidazole hybrids 24a-24r were synthesized and tested for in vitro antimalarial efficacy against both chloroquine-sensitive 3D7 and chloroquine-resistant K1 strain of P. falciparum (Kondaparla et al. 2018). All of the substances showed moderate to high activity, with IC50 values against the sensitive strain 3D7 ranging from 0.079 to 5 μM and against the resistant strain K1 from 0.291 to 5 μM. Compounds (24a, 24b, 24c, 24 g and 24j) with a heterocyclic moiety attached to an imidazole ring demonstrated promising activity against the K1 strain of P. falciparum, with IC50 values of 1.15, 0.46, 1.70, 1.33 and 0.97 μM, respectively. Compounds with bulky aromatic substituents on the imidazole moiety (compounds 24h and 24p) were found to be inactive against both P. falciparum strains (3D7 and K1). Compound 24m with cyclopentyl substitution demonstrated stronger activity against chloroquine-resistant K1 strain (IC50 = 0.34 μM) than compound 24q with cyclohexyl substitution (IC50 = 0.52 μM). Furthermore, among the various substitutions at the phenyl ring attached to N-(2-(1H-imidazole-1-yl)ethyl)-7-chloroquine in 4-amine moiety, isopropyl group containing compound 24e was shown to be the most effective in the series against the chloroquine-resistant strain with IC50 value of 0.29 μM (IC50 of chloroquine = 0.255 μM). Compound 24i with tert-butyl group substitution also demonstrated substantial efficacy against the resistant strain (IC50 = 0.50 μM). Overall, the author concluded that alkyl groups and alkyl-substituted phenyl rings are the best substitutions which result in compounds with increased antimalarial activity against both strains. Furthermore, no compounds significantly displayed cytotoxicity against the mammalian Vero cell line. To ascertain the most likely mode of action, heme binding assays were also conducted. All of the compounds showed a strong complex formation with hematin and inhibited the formation of β-hematin, implying the same mode of action as that of chloroquine.

Due to the shown potential of pyrimidine-based substances, such as pyrimethamine and trimethoprim, to block dihydrofolate reductase (DHFR), an enzyme involved in the folate pathway of the Plasmodium parasite, pyrimidine is an essential component in designing of antimalarial drugs (Singh and Kaur 2016). As a result, the synthesis of quinoline-pyrimidine hybrids is an appealing strategy to find antimalarial agents.
Compounds 25a-25 l and 26a-26f, the triazole-tethered hybrids of 7-chloroquinoline and pyrimidine-5-carboxylate, were synthesized and evaluated for antimalarial activity against the NF54 strain (chloroquine sensitive) of P. falciparum (Chopra et al. 2018). Compounds 25a-25 l, having an alkyl chain between triazole and quinoline rings, were found more potent than hybrids 26a-26f having triazole moiety directly linked to the quinoline ring. The compounds 25a-25l, which were found to be the most effective against NF54, were also investigated against the Dd2 strain (chloroquine resistant) of P. falciparum. With IC50 values of less than 1 μM, 9 of the 12 analogues (25a-25 l) demonstrated very promising antimalarial activity. In general, compounds with a propylene linker were more effective than those with an ethylene linkage against both strains. Compound 25d, which was found to be the most effective among all hybrids against the sensitive strain (NF54), proved to be less effective against the resistant strain (Dd2), whereas compound 25c was proven to be the most potent against the resistant strain. Furthermore, there were no cytotoxic effects observed against the mammalian Vero cell line. The most active hybrid 25d against the sensitive strain NF54 had a high value (317.50) of the selectivity index. Heme binding and DNA binding studies were conducted to determine the likely mode of action of these hybrids. The complex formation of the compound 25d with heme was observed with binding constant values log K = 4.679 (monomeric) and log K = 5.434 (μ-oxo dimeric), implying the same mechanism of action as that observed with chloroquine. The high values of the binding constant, log K = 3.74 and 3.92 for CT DNA and pUC 18, respectively, further suggest that the antimalarial activity of the synthesized compounds may also be mediated by interaction with parasite DNA in addition to the inhibition of heme polymerization.

The 4-aminoquinoline-pyrimidine hybrids 27a-27d, 28a-28d and 29a-29n were synthesized and investigated for in vitro antimalarial efficacy against chloroquine susceptible (D6) and chloroquine-resistant (W2) strains of P. falciparum (Manohar et al. 2012). Out of 22 compounds, 11 compounds (29b, 29c, 29e, 29f and 29h-29n) showed superior antimalarial activity (IC50 = 0.005–0.03 μM) than chloroquine against the sensitive strain, and 12 compounds (29b-29f and 29h-29n) showed better activity (IC50 = 0.01–0.21 μM) than chloroquine against the resistant strain of P. falciparum. Four compounds (29i, 29j, 29l and 29m) showed better effectiveness as compared to pyrimethamine against both strains (D6 and W2). According to the SAR analyses, no clear pattern of activity, with changing the length of the carbon chain, was seen for a specific amino-substituted 4-aminoquinoline-pyrimidine hybrid. However, switching the amino groups for a specific carbon chain length from piperidine to morpholine to 4-methyl piperazine to 4-ethyl piperazine, the antimalarial activity significantly increased. Most of the compounds were found to be nontoxic towards Vero, HepG2 and LLC-PK11 cells up to a concentration of 60 μM, while a few of them showed minor toxicity but had a very high selectivity index. The most effective compounds, 29i and 29m, were chosen for in vivo testing and demonstrated exceptional effectiveness in a P. berghei mice model without any toxic effects. Compound 29i displayed better activity as compared to 29m and chloroquine. Compound 29i provided almost complete parasitaemia suppression and cured 80% of the treated mice at three doses of 30 mg/kg, whereas 29m only cured 20% of the mice, and chloroquine had no effect.

Another series of 4-aminoquinoline-pyrimidine hybrids 30a-30d, 31a-31d, 32a-32l and 33a-33l was synthesized and tested for antimalarial efficacy against chloroquine susceptible (NF54) and resistant (Dd2) strains P. falciparum by the same author (Manohar et al. (2015). Out of 32 compounds, 19 (IC50 = 0.003–0.198 μM) showed superior activity to chloroquine (IC50 = 0.222 μM) against the resistant strain (Dd2). When tested against the chloroquine susceptible strain, six compounds (IC50 = 0.009–0.025 μM) displayed better activity than chloroquine (IC50 = 0.027 μM). Compound 32f was discovered to be the most potent, with an IC50 value of 0.003 μM against the resistant strain (Dd2) and 0.028 μM against the sensitive strain (NF54). The cytotoxicity of the selected active compounds against CHO (Chinese Hamster Ovary) cell lines was also tested, and they were found to be nontoxic. One compound, 32e, was tested for in vivo antimalarial activity against mice infected with P. berghei and it was found that a dose of 30 mg/kg on day 4 suppresses parasites by 93.9%, with a mean survival time of 11 days after infection. Chloroquine, on the other hand, demonstrated 90.3% suppression at a dosage of 15 mg/kg. Furthermore, the complex formation between heme and compound 32f suggested that the heme is the most likely mechanism of action for these hybrids.

A novel series of N-substituted 4-aminoquinoline-pyrimidine hybrids 34a-34j and 35a-35j was synthesized (Maurya et al. 2017). To study the SAR, structural variations were made at two points. The first variation was made in the group that connects the aminoquinoline and pyrimidine moieties, and the second was made in the fourth position of the pyrimidine ring. The majority of the compounds demonstrated potent antimalarial efficacy against chloroquine-sensitive (D6) and resistant (W2) strains of P. falciparum. Ten compounds (IC50 = 0.038–0.044 μM) were shown to be almost as strong as artemisinin (IC50 = 0.045 μM) against the D6 strain, whereas 22 compounds exhibited better activity (IC50 = 0.039–0.257 μM) than chloroquine (IC50 = 0.317 μM) against W2 strain of P. falciparum. In the SAR study, there was no clear pattern of activity observed in altering the carbon chain length or between the two sets of pyrimidine regio-isomers (34a-34j and 35a-35j) but varying the amino substituent for a specific carbon chain length changed the activity. It was discovered that compounds with morpholine, piperidine, or pyrrolidine moieties at the pyrimidine nucleus were less active than those with methyl or ethyl piperazine moieties. Furthermore, none of the compounds were found to be cytotoxic to Vero cell lines. The most active compound, 34b, was studied for heme binding, and good interaction with monomeric heme and μ-oxodimeric heme was observed, implying that heme could be a target of these hybrids.

Further, a new series of 4-aminoquinoline-pyrimidine hybrids 36a-36j, 37a-37j, 38a-38e and 39a-39e were developed and tested for their ability to treat malaria in both chloroquine-sensitive (D6) and resistant (W2) strains of P. falciparum (Maurya et al. 2019). The majority of the compounds were strongly active, with IC50 values ranging from 0.027 to 0.661 μM against the D6 strain and 0.0189 to 1.443 μM against the W2 strain of P. falciparum. Twenty-three compounds were shown to be superior to chloroquine in combating the chloroquine-resistant strain (W2). Among all, compound 36d was discovered to be the most potent compound against the D6 strain (IC50 = 0.027 μM), whereas 39e was the most efficient compound against the W2 strain (IC50 = 0.0189 μM). Regarding the SAR, there was no discernible effect of pyrimidine regio-isomers, alkyl linker chain length, or NH substitution on activity. The most active compound, 36d, was examined for heme binding and found to have good interactions with both monomeric heme and μ-oxodimeric heme, indicating that heme could be a target of these hybrids.

The 4-aminoquinoline and pyrimidine hybrids 40a-40j, 41a-41j and 42a-42j were synthesized and investigated for their antimalarial efficacy against P. falciparum chloroquine-sensitive (D6) and chloroquine-resistant (W2) strains (Kumar et al. 2015a, b). Eight compounds (40a, 40f, 40g, 41d, 41f, 41g, 42b and 42d) showed antimalarial activity with IC50 < 0.05 M, and compound 41f was shown to be just as effective as chloroquine against the D6 strain. Except for two, 40e and 42c, all the compounds demonstrated superior efficacy to chloroquine against the resistant W2 strain. From the activity data, it was shown that compounds that performed better against chloroquine-sensitive strains also performed well against chloroquine-resistant strains. There was no discernible trend of activity for a specific 4-aminoquinoline pyrimidine conjugate with increasing or decreasing carbon chain length. Compound 40f with significant in vitro activity was tested for in vivo antimalarial activity in mice infected with P. berghei via the oral route of administration, and it was found that the compound 40f suppresses parasites by 17.85, 37.62 and 96.42% at doses of 11.1, 33.3 and 100 mg/kg, respectively, on day 5, in contrast to the 100% suppression shown by chloroquine.

A novel series of 4-aminoquinoline-purine hybrids 43a-43t were synthesized and tested for in vitro antimalarial activity against P. falciparum chloroquine-sensitive (D6) and resistant (W2) strains (Reddy et al. 2017). In designing these hybrids, structural variations were made at two points. The first variation was made in the diamine linker that connects the aminoquinoline and purine moieties, and the second was made in the secondary amine substitution at the sixth position of the purine ring. Eighteen compounds demonstrated antimalarial activity with IC50 values less than 1 μM (0.061–0.833 μM) against the sensitive strain (D6), while eight compounds (43a, 43f, 43h-43j, 43r-43t) showed superior activity as compared to chloroquine against the resistant strain (W2) of P. falciparum. The strongest compound in the series was found to be compound 43i, with IC50 values of 0.061 μM (D6 strain) and 0.080 μM (W2 strain). Except for two compounds, all of the compounds were shown to be nontoxic towards mammalian (Vero) cells up to the highest tested concentration of 11.86 μM. Furthermore, strong binding with heme revealed that heme may be the primary target of these hybrids for antimalarial activity.

A library of 4-aminoquinoline-guanylthiourea derivatives was designed and based on molecular docking investigations, and nine compounds were chosen for synthesis and tested for in vitro antimalarial efficacy against chloroquine-sensitive strain (D6) and resistant strain (W2) of P. falciparum (Bhagat et al. 2019). Eight of the nine compounds displayed activity against the D6 strain in the range of 0.61–7.55 μM and the W2 strain in the range of 0.43–8.04 μM. Compound 44c was found to be most effective with MIC values of 0.6 μM and 0.4 μM against D6 and W2 strains, respectively. Furthermore, none of the compounds were shown to be cytotoxic at the highest concentrations tested against Vero cell lines.

A series of 4-aminoquinoline-phthalimide hybrids were synthesized and tested for antimalarial activity against the chloroquine-resistant and mefloquine-sensitive strain (W2) of P. falciparum (Rani et al. 2018). To investigate SAR, the length of the alkyl chain linker between the phthalimide and 4-aminoquinoline, as well as the nature of substituents at the C3 or C4 position of phthalimide, was varied. And it was established that on increasing the number of carbon atoms in the linker, antimalarial activity increases. The optimum activity was achieved for the 6 and 8 carbons chain length. The compound 45v having a hexyl chain as a linker and a 3,4,5,6-tetrabromo substituent on the phthalimide ring was the most effective compound in the series, with an IC50 value of 0.10 μM. Some of the active compounds were tested for cytotoxicity using J774 murine macrophage cells and found to be noncytotoxic, with selectivity indexes ranging from 126 to 291. However, these analogues were not as effective as chloroquine (IC50 = 77 nM), prompting the research team to continue optimizing these compounds.

In continuation of previous research, a variable chain length amide linker was introduced between phthalimide and 4-aminoquinoline rings (Rani et al. 2019). Additionally, several cyclic tertiary amines were introduced at the fifth position of the phthalimide ring. All of the compounds were tested towards the chloroquine-resistant and mefloquine-sensitive strain (W2) of P. falciparum. Seven of the 36 compounds displayed better activity than chloroquine (IC50 = 0.23 μM). The SAR study revealed that compounds having m = 2 had improved antimalarial activity when compared to compounds with m = 1 or 3. In addition, compounds with tertiary amine substituents at the fifth position of the phthalimide ring demonstrated higher activity than their unsubstituted or 5-F-substituted analogues. Furthermore, it was found that none of the compounds were harmful to mammalian Vero cells. Among all compounds, 47j was shown to be the most active compound with an IC50 value of 0.097 μM.

Further, a new series of phthalimide-4-aminoquinoline hybrids (48a-48 t) with a variable length alkyl chain and a cyclic tertiary amine linked at the C4 or C5 position of the phthalimide ring was synthesized (Rani et al. 2020). The in vitro antimalarial activity of the synthesized compounds was tested against the chloroquine-resistant strain (W2) of P. falciparum, and most of them were found to be more effective than chloroquine (IC50 = 0.079 M). It was found that the type of the substituent at the C4 or C5 positions of the isoindoline-1,3-dione as well as the length of the alkyl chain linker had an impact on the antimalarial activity of the compounds. The author found that switching the substitution on the phthalimide ring from piperidine to morpholine and then to hydroxyethyl piperazine increased the antimalarial activity. The activity was also enhanced by moving the substituent from C4 to the C5 position. Furthermore, increasing the chain length of the linker resulted in improved antimalarial activity, except for the hydroxyethyl piperazine substituent, for which all the related compounds demonstrated good activities regardless of chain length. With an IC50 of 0.006 μM, compound 48r having hydroxyethyl-piperazine at the C5 position and a propyl chain as a linker was the most active compound among all. Heme binding studies revealed that heme could be the principal mechanism of action of these compounds. The heme binding constant of 48r was found to be comparable to that of chloroquine. Furthermore, none of the compounds showed cytotoxic effects on the mammalian Vero cells.

A series of 4-aminoquinoline-naphthalimide hybrids 49a-49y were synthesized and tested for in vitro antimalarial efficacy against the P. falciparum chloroquine-resistant strain (W2) (Shalini et al. 2020). Nineteen compounds out of 25 displayed better activity than chloroquine (IC50 = 232.7 nM). In general, compounds with shorter alkyl chain linkers outperformed compounds with longer alkyl chain linkers. The substitution by morpholine or piperidine on the naphthalimide ring displayed no noticeable impact on the antimalarial activity, whereas hydroxyethyl piperazine substituted analogues significantly increased antimalarial activity. Compound 49v, having propylene linker and hydroxyethyl piperazine substituent at the C-4 position of the naphthalimide ring, displayed the strongest activity (IC50 = 15.4 nM) and it was ~15 times more potent than chloroquine (IC50 = 232.6 nM). Furthermore, it was found that none of the compounds were cytotoxic to the mammalian Vero cell lines.

A series of 4-aminoquinoline-benzimidazole hybrids (50a-50e) and their ferrocenyl analogues (51a-51e) were synthesized and tested for in vitro antimalarial efficacy towards chloroquine-sensitive strain (NF54) and multidrug resistant strain (K1) of P. falciparum (Baartzes et al. 2019). The majority of the compounds were more effective against the K1 strain as compared to the NF54 strain of P. falciparum. Compounds 50c and 50e were discovered to be more active than chloroquine (IC50 = 0.205 μM) against the resistant strain, with IC50 values of 0.151 μM and 0.179 μM, respectively. The cytotoxic activities of the compounds were investigated against the non-tumorigenic Chinese hamster ovarian (CHO) cell line, and the majority of compounds proved to be nontoxic. The antimalarial effectiveness of the most potent phenyl (50c) and ferrocenyl (51c) aminoquinoline benzimidazole hybrids was also examined for in vivo activity in mice infected with P. berghei. At a dose level of 4 × 50 mg/kg, a considerable suppression of parasitaemia (58% for compound 50c and 92% for compound 51b) was observed. The β-hematin inhibitory activity of both compounds was also investigated which revealed more than four times higher potency of both compounds (IC50 = 16 μM) as compared to chloroquine (IC50 = 74 μM) to inhibit the formation of hemozoin.

A series of 4-aminoquinoline-ferrocene hybrids (52a-52g) with various linkers between the two pharmacophores were synthesized and tested for in vitro antimalarial activity towards chloroquine-sensitive (D10) and resistant (Dd2) strains of P. falciparum (N’Da and Smith 2014). Compounds having rigid linkers were shown to be ineffective, whereas those with flexible linkers were found to be active against both D10 and Dd2 strains of P. falciparum. Compound 52g with a 3-aminopropyl methylamine linker was the most active compound. Its antimalarial activity was found to be comparable to chloroquine against the susceptible strain D10 but significantly more active than chloroquine and the equimolar combination of chloroquine and ferrocene against the resistant strain D2d.

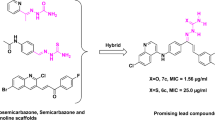
The chloroquine-quinoxaline 1,4-di-N-oxide hybrids were synthesized and tested for antimalarial efficacy against P. falciparum chloroquine-sensitive (3D7) and multidrug-resistant (FCR-3) strains (Bonilla-Ramirez et al. 2018). Furthermore, they tested in vitro and in vivo liver stage efficacy as well as the capacity to prevent transmission of the active compounds in the mosquito stage. Except for 53c, all of the compounds demonstrated antimalarial efficacy with IC50 values less than 1 μM towards the chloroquine susceptible strain; however, only two compounds displayed the same range of IC50 towards the multidrug-resistant strain. Compounds 53b and 53e were shown to be the most effective against both sensitive and resistant strains, with IC50 values ranging from 0.40 to 0.90 μM. Interestingly, compound 53b demonstrated moderate efficacy towards three liver stage parasites, most notably P. falciparum liver stage with an IC50 value of 6.39 μM.

Several quinoline-chalcone (54a-54k, 55a-55k) and quinoline-pyrazoline (56a-56g, 57a-57i) hybrids were synthesized and evaluated for in vitro antimalarial efficacy (Charris et al. 2019). As a preliminary screening for antimalarial activity, the in vitro β-hematin formation inhibitory activity was investigated. Ten compounds were found to inhibit more than 80% of heme crystallization, with chloroquine inhibiting 98.52%. These compounds were then chosen for in vivo testing in mice infected with P. berghei. The antimalarial effectiveness of the compounds was assessed by their ability to reduce parasitaemia and increase survival on the fourth day after infection as compared to the untreated control group. Mice were given either compound (20 mg/kg) or chloroquine (20 mg/kg) intraperitoneally once daily for 4 days. On the fourth day, the mean survival time and parasitaemia percentage were compared to control mice given only saline. Compounds 54e and 54h were found to be the most active as they extended the infected mice’s average survival period to 22.44 and 24.60 days, respectively, which is comparable to chloroquine (24.99 days) and threefold better than the control (6.8 days). They were, however, unable to reduce or postpone the progression of malaria in the same way that chloroquine did (1.40% and 1.81% parasitaemia for compounds 54e and 54h, respectively on day 4, compared to 0.32% for chloroquine).

3 Anticancer Activity
Cancer is a leading cause of death worldwide. The number of new cancer cases is increasing daily even though better medications and targeted cancer therapies are readily available. No single treatment of cancer is completely successful and patients frequently receive a combination of therapies such as surgery, radiation therapy, immunotherapy, chemotherapy, gene therapy, or hormonal therapy, depending on the type and stage of cancer, patient’s health status, age and personal characteristics (http://www.cancer.gov/) (Cancer. gov n.d.). Due to the genotoxicity, resistance and toxic effects, anticancer drugs have limited therapeutic applicability (Aydemir and Bilaloglu 2003). Thus, there is always a need for comparatively more effective and safer compounds for the treatment of cancer. Recently, quinoline has attracted much attention from researchers across the globe due to its presence in well-known anticancer drugs such as camptothecin and cabozantinib (Srivastava et al. 2005). The number of quinoline compounds having anticancer action has grown over the past few years, according to scientific research papers (Musiol 2017; Afzal et al. 2015; Marganakop et al. 2014; Aly 2010; Tseng et al. 2013; El-Gamal et al. 2014; Solomona et al. 2019; Ahadi and Emami 2020; Akkachairin et al. 2020).
A series of N′-substituted methylene-4-(quinoline-4-amino) benzoyl hydrazides 58a-58p were synthesized and tested against HepG2 cells (Li et al. 2020). Compounds 58h and 58j were found to have significant antiproliferative activity, with IC50 values of 12.6 μM and 27.3 μM, respectively. With IC50 values of 9.6 μM and 6.3 μM, respectively, against SMMC-7721 and Huh7 cells (human hepatic cancer cell lines), compound 58h also demonstrated strong cytotoxicity. Inspiringly, both 58h and 58j were less cytotoxic in healthy cells as compared to hepatocellular cancer cells. Additional bioassays revealed that 58h and 58j can reduce the mRNA level and expression level of c-Myc. Furthermore, both compounds were found to induce the pro-apoptotic protein Bax to be up-regulated and the anti-apoptotic protein Bcl-2 to be down-regulated. The anticancer activity of 58h and 58j in HepG2 cells was discovered to be due to their anti-survival effects, induction of apoptotic cell death, cell cycle arrest and inhibition of cell migration. According to SAR studies, alkyl substitutions (58a and 58b) at the nitrogen atom (N′) of N-acylhydrazone do not affect the cytotoxic activity significantly. On the other hand, different substituents on the benzene ring attached to the N′-methylene group showed a substantial correlation with cytotoxic activity. Compounds 58d, 58e and 58k having carboxyl (COOH) or nitro (NO2) groups on the benzene ring were found to be inactive.

The hydrazone derivatives of quinoline were synthesized and their anticancer activity was determined towards the full NCI 60 human cancer cell line panel (Katariya et al. 2020). Nine compounds (59c, 59d, 59e, 60a, 60b, 60c, 60d, 60e and 60f) exhibited significant anticancer activity at 10 μM concentration and were subsequently examined at ten-fold dilutions of five different concentrations (0.01, 0.1, 1, 10 and 100 μM). All of the compounds displayed antiproliferative action with GI50 values in the range of 0.33–4.87 μM and LC50 values in the range of 4.67–100 μM. Compound 60e had the highest activity of all, with GI50 values from 0.33 to 2.05 μM and LC50 values from 5.15 to 24.4 μM. Furthermore, a molecular docking analysis showed that compounds exhibited strong interaction with DNA and GTP nucleotides and also in the active site of the topoisomerase enzyme via hydrogen bonding.

Twenty-six new 4-aminoquinoline derivatives were synthesized and evaluated for their ability to inhibit cell proliferation in A549, A2780/T, A2780S, HCT116, HCT-8, HCT-8/T, HCT-8/V, MCF-7, MCF-7/ADR and MDA-MB-231 cells (Zhou et al. 2017). Among all, compound 63h demonstrated the most effective cytotoxic activity with IC50 values in the range of 1.5–3.9 nM towards all the examined cancer cell lines and displayed significant activity against the multidrug-resistant cancer cells also. The immunofluorescence tests, EBI examination, microtubule dynamics test and molecular modelling investigations identified compound 63h as a novel depolymerization agent binding to the colchicine active site. Moreover, compound 63h displayed significant in vivo anticancer activity in the HCT116 xenograft model without appreciable body weight loss.

A series of new 4-aminoquinoline derivatives 65a-65s and 66a-66y were synthesized and evaluated for their anticancer action against cervical carcinoma (Hela), gastric carcinoma (BGC-823), human colorectal carcinoma (HCT-116 and RKO), non-small-cell lung carcinoma (A549 and NCI-H1650) and ovarian carcinoma (A2780) cells (Su et al. 2019). At a concentration of 10 μM, compounds 65a-65s exhibited no cytotoxic activity against any of the cancer cell lines. Compounds 66a-66n and 66w demonstrated anticancer effects selectively towards Hela cell lines with IC50 values <10 μM. Compounds 66b, 66d-66j, 66l-66n and 66w demonstrated selectivity for A2780 cancer cell lines, with IC50 values smaller than 10 μM. Furthermore, compounds 66a, 66e, 66f, 66h, 66j, 66m and 66n demonstrated antiproliferative action specifically against BGC-823 cell lines with IC50 values less than 10 μM. Surprisingly, five compounds, 66p, 66s, 66v, 66x and 66y, demonstrated strong anticancer activity against all the examined cancer cell lines with IC50 values <10 μM. Compound 66x was discovered to be the most effective compound against HCT-116, Hela, A2780 and RKO cell lines, with IC50 values of 2.56, 2.71, 3.46 and 3.67 μM, respectively. SAR analysis revealed that the presence of a bulky group at the seventh position and 4-methoxyaniline at the fourth position of the quinoline nucleus is favourable for antiproliferative activity. The in vivo anticancer activity of the representative compound 66x was also determined, and the results revealed that compound 66x significantly slowed down tumour growth and reduced tumour size in animal models. According to mechanistic studies, compound 66x suppresses colorectal cancer growth through an ATG5-dependent autophagy mechanism.

A series of quinoline-based chalcones (67a-67f) and pyrazoles (68a-68f, 69a-69f, 70a-70f, 71a-71f and 72a-72f) were synthesized (Ramírez-Prada et al. 2017). Fourteen compounds (67a, 67c, 67d, 68a, 68c, 69a, 69c, 69d, 70a, 70c, 71a, 71c, 71d and 72a) were selected by NCI for a single dose (10 μM) testing against 60 different human cancer cell lines. Out of all, four compounds, 67c, 67d (chalcones) and 69a, 69c (pyrazolines), demonstrated significant activity (GI50 = 0.28–11.7 μM and LC50 = 2.6–100 μM) at 10 μM concentration and were chosen for five dose screening. Compounds 67c, 67d and 69a, 69c demonstrated high GI50 values in five-dose screening against a variety of cell lines, and some of the values were less than 1 μM. The GI50 and LC50 values of compound 67c ranged from 0.49 to 3.85 μM and 23.7 to >100 μM, respectively. The best cytostatic effect of 67c was demonstrated in the SR leukaemia cell line (GI50 = 0.49 μM), and the best cytotoxic effect was demonstrated in the U251 CNS cancer cell line (LC50 = 23.7 μM), both of which were superior to the reference drug adriamycin. Compound 67d was discovered to be the most efficient for which the GI50 values ranged from 0.31 to 2.49 μM and LC50 values from 2.6 to >100 μM. The best cytostatic effect of 67d was demonstrated towards the leukaemia cell line RPMI-8226 (GI50 = 0.31 μM), and the best cytotoxic effect was demonstrated towards the ovarian cancer cell line SK-OV-3 (LC50 = 2.6 μM), both of which were superior to adriamycin. The GI50 and LC50 values of compound 69a ranged from 0.28 to 9.42 μM and 5.1 to >100 μM, respectively. Compound 69a had the best cytostatic effect on the breast cancer cell line MDA-MB-468 (GI50 = 0.28 μM) and the greatest cytotoxic effect on the melanoma cell line SK-MEL-5 (LC50 = 5.1 μM). The GI50 and LC50 values of compound 69c ranged from 0.37 to 11.7 μM and 17.4 to >100 μM, respectively. The strongest cytostatic effect was determined towards the melanoma cell line MDA-MB-435 (GI50 = 0.37 μM) and the greatest cytotoxic effect towards the melanoma cell line SK-MEL-5 (LC50 = 17.4 μM).

Four different series of aminoquinoline derivatives, 73a-73f, 74a-74f, 75a-75f and 76a-76f, were synthesized and evaluated for their antiproliferative activity against MCF-7, H460, HCT 116 and SW620 cancer cell lines (Zorc et al. 2019). Compounds 73a-73f and 74a-74f demonstrated low to moderate activity against all four cancer cell lines. Compounds 75a-75f and 76a-76f, on the other hand, demonstrated good antiproliferative activity in micromolar concentrations. Compound 75f demonstrated potent anticancer activity in MCF-7 and HCT 116 cells, with IC50 values of 0.4 μmol/L and 0.3 μmol/L, respectively. Compounds 76d and 76f also demonstrated strong activity towards MCF-7 cell lines with IC50 values of 0.4 and 0.3 μmol/L, respectively.


4-Aminoquinoline and 1,3,5-triazine-based hybrid molecules were synthesized and evaluated for their anticancer activity against the four cancer cell lines, HeLa, MCF-7, HL-60 and HepG2, as well as MCF 12A (normal epithelial cell) (Bhat et al. 2020). Compound 77e was shown to be the most efficient compound against all the cell lines tested. When chloro (77e) was replaced with fluoro (77i), the activity against all cell lines was significantly reduced. The replacement of fluoro (77i) with nitro (77c) results in a further decrease in activity. Interestingly, none of the compounds displayed toxicity towards the normal cell lines MCF-12A.

A series of molecular hybrids were synthesized by conjugating C5-curcuminoids and 4-aminoquinoline moieties through the 1,2,3-triazole ring (Kandi et al. 2015). Some of the molecules were selected by NCI for a single dose (10 μM) testing against 60 different human cancer cell lines. Only two compounds 78d and 78f demonstrated significant inhibition of several cell lines and were chosen for five-dose screening. Compound 78d was shown to be more efficient (GI50 = 0.189–2.17 μM and LC50 = 0.77–100 μM) than 78f against all the cancer cell lines. The strongest cytostatic effect of 78d was demonstrated against the melanoma cancer cell line LOX IMVI (GI50 = 0.189 μM), and the strongest cytotoxic effect was demonstrated against the colon cancer cell line HCT-116 (LC50 = 0.77 μM). Mechanistic studies like Annexin V screening, DNA fragmentation and caspase activation showed apoptotic induction as a cause for their anticancer activity.

4 Antitubercular Activity
According to WHO, approximately ten million new cases and 1.5 million deaths were reported in 2020 due to tuberculosis (https://www.who.int/news-room/fact-sheets/detail/tuberculosis WHO n.d.-b; Kumar et al. 2015a, b). The drug-sensitive variant of tuberculosis can be treated with a conventional 6-month course of four antimycobacterial drugs (isoniazid, ethambutol, pyrazinamide and rifampicin). However, the treatment of multidrug-resistant (MDR) and extensively drug-resistant (XDR) tuberculosis remains a big challenge worldwide (Bahuguna and Rawat 2020). There is an urgent need to find new and better therapeutic options for the therapy due to the resistance developed by M. tuberculosis against several first-line medications. Quinoline has always drawn the attention of researchers due to the presence of this nucleus in various already established antimicrobial drugs such as ciprofloxacin, moxifloxacin mefloquine and gatifloxacin. Bedaquiline, a new quinoline-based compound, received FDA approval in 2012 for its use in the treatment of MDR-TB. Unfortunately, resistance to bedaquiline was also observed shortly after it was used in clinical settings (Veziris et al. 2017). Therefore, it is necessary to continuously discover antitubercular agents with new mechanisms of action.

4-Quinolinyl hydrazone derivatives 79a-79u were synthesized and tested for in vitro antimycobacterial activity against M. tuberculosis H37Rv (Candéa et al. 2009). Some of the compounds showed reasonably good activity (MIC = 2.5–6.25 μg/mL). Three compounds 79f, 79i and 79o exhibited very good activity with a MIC value of 2.5 μg/mL. The activity of these three compounds was found to be better than the standard antitubercular drugs ethambutol (MIC = 3.12 μg/mL). According to the SAR analyses, compounds containing chloro, fluoro, hydroxy and methoxy groups showed good activity, whereas bromo, cyano and nitro group-containing compounds were shown to be less effective. All of the compounds were also examined for their cytotoxic activities using the BCG-infected and non-infected macrophage cell line J774s and the majority of the compounds were found to be noncytotoxic.

4-Aminoquinoline derivatives were synthesized and assessed for in vitro antimycobacterial efficacy against H37Rv strain of M. tuberculosis (De Souza et al. 2009). The compounds containing 2–4 carbon atoms in the diamine part displayed weak activity (MIC = 100 μg/mL), whereas compounds with 6, 8 and 10 carbon atoms demonstrated significantly increased activity. For example, in the series (81a-81f), the MIC value varies from 100 μg/mL (81a, 81b, 81c, where n = 2, 3, 4, respectively) to 25 μg/mL (81d, n = 6) to 6.25 μg/mL (81e, n = 8) to 3.12 μg/mL (81f, n = 10). The chlorine atom at the seventh position in the quinoline nucleus was found to be essential for the antitubercular activity, which can be demonstrated by the substitution of the chlorine atom by hydrogen in the compound 82f (MIC = 12.5 μg/mL), which was responsible for the decrease in the antitubercular activity of the compound 83d (MIC = 50.0 μg/mL).

A series of novel 4-aminoquinoline-chalcone and 4-aminoquinoline-pyrimidine conjugates were synthesized and examined for in vitro antitubercular activity against H37RV M. tuberculosis (Sharma et al. 2009). Quinolinyl-chalcones (84a-84p) outperformed quinolinyl-pyrimidine hybrids in terms of antitubercular activity (85a-85p). With MIC values of 3.12 μg/mL and being noncytotoxic to Vero and MBMDM cell lines, the 2,3-dimethoxyphenyl (84g) and 2,5-dimethoxyphenyl (84h) substituted derivatives of chalcones were found to be the most effective against M. tuberculosis, while their pyrimidine analogues (85g, 85h) were discovered to be ineffective against the same strain.

Four different series of 4-aminoquinolines derivatives 86a-86 t, 87a-87e, 88a-88c and 89a-89d were synthesized (Eswaran et al. 2010). Except for 88a-88c, all the tested compounds exhibited good to moderate inhibitory activity against M. tuberculosis and multidrug-resistant tuberculosis (MDR-TB). Compounds 86k, 86s and 89a demonstrated very good activity and they exhibited 99% growth inhibition of M. tuberculosis at 3.12 μg/mL concentration, while compounds 86d, 86g, 86i, 86l, 86q, 87b, 89b and 89d showed significant activity and inhibited 95% growth of M. tuberculosis at 6.25 μg/mL concentration. Additionally, all the compounds were tested against MDR-TB (M. tuberculosis is resistant to three antitubercular drugs, namely, isoniazid, rifampicin and ethambutol). The majority of compounds exhibited decent activity (MIC = 6.25–25 μg/mL) against MDR-TB strains, and many of them were found to be more effective than isoniazid and rifampicin. Interestingly, nine compounds, namely, 86d, 86g, 86i, 86k, 86l, 86s, 87d, 89a and 89b, were discovered to be more efficient than isoniazid (MIC = 12.5 μg/mL) with a MIC value of 6.25 μg/mL, whereas compounds 86c, 86h, 86m, 86o, 86q, 87a, 87c, 89c and 89d were discovered to be more efficient than rifampicin (MIC = 25 μg/mL) with MIC value of 12.5 μg/mL. Further, compounds 86b, 86n, 86p and 88b were shown to be as potent as rifampicin. Surprisingly, compound 89a unexpectedly exhibited strong antimycobacterial action against sensitive and MDR strains of tuberculosis with MIC values of 3.12 and 6.25 μg/mL, respectively.


4-Aminoquinoline-ferrocene hybrids were synthesized and tested for their in vitro antimycobacterial activity against the Mtb mc2 7000 strain of M. tuberculosis (Mahajan et al. 2011). Only one compound 90a exhibited good activity (MIC = 2.5–5 μg/mL) comparable to the standard drug ethambutol (MIC = 1–2.5 μg/mL), while all other compounds demonstrated no activity even at very high dosages (100 μg/mL). Unexpectedly, ferroquine (FQ) also exhibited potent activity (MIC = 10–15 μg/mL), although its activity was found to be less than that of compound 90a.

A series of novel 4-aminoquinoline derivatives comprising semicarbazone and thiosemicarbazone were synthesized and screened for their in vitro antimycobacterial activity against H37Rv M. tuberculosis (Alegaon et al. 2020). All of the compounds showed significant antitubercular activity (MIC = 1.56–50 μM). The majority of the compounds were discovered to be more effective than pyrazinamide (MIC = 50 μM). Compounds, 91l and 91p were the most potent of all the compounds examined, with a MIC value of 1.56 μM, and were equally effective as the standard antitubercular drug isoniazid. Three compounds 91j, 91r and 92j exhibited superior activity than ciprofloxacin (MIC = 12.5 μM) and pyrazinamide (MIC = 50 μM) with a MIC value of 6.25 μM. Nine compounds 91a, 91b, 91d, 91e, 91m, 91n, 92a, 92b, 92k and 92l were shown to be just as effective as ciprofloxacin (12.5 μM). Cytotoxicity of the compounds having MIC values less than 12.5 μM was determined against normal HDF cell line using MTT cell proliferation assay. None of the active compounds showed toxic effects on HDF cell lines.

5 Anticoronavirus Activity
Coronavirus disease (COVID-19) is a viral infection caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The first coronavirus case was discovered in December 2019 in the Chinese city of Wuhan, and it quickly spread to almost every country in the world (Gorbalenya et al. 2020). As a result, in March 2020, WHO declared this a worldwide emergency. It has infected over 500 million people and killed over six million people worldwide, as well as causing long-term health problems in those who survived (https://covid19.who.int/) (WHO n.d.-a). COVID-19 causes mild to severe respiratory diseases (Clark et al. 2020). So far, seven human coronaviruses (HCoV) have been discovered (Wang et al. 2020a, b). SARS-CoV-1, Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2, belonging to the beta coronavirus family, are extremely deadly viruses. The HCoV-229E and HCoV-NL63 (both alpha coronaviruses) and HCoV-OC43 and HCoV-HKU1 (both beta coronaviruses), on the other hand, cause minor upper respiratory tract infections. Treatment of COVID-19 depends upon the infection’s severity. Proper rest and antipyretic medication are often enough for milder symptoms. More severe cases may require hospitalization, supplementary oxygen, ventilation and other supportive measures. Even though several vaccines have already been developed, no efficient antiviral treatment for COVID-19 has been established. As a result, it is highly desirable to discover new and effective drugs for the treatment of SARS-CoV-2. The most common approach to providing new drugs, especially in a short period, is the repurposing of drugs which have already received approval and license for use in treating other diseases (Bazotte et al. 2021; Pushpakom et al. 2019).
A commonly used antiviral drug called remdesivir was repurposed and discovered to exhibit potential action against SARS-CoV-2 in vitro as well as in preclinical research and human clinical trials (Wang et al. 2020a, b). It was the first SARS-CoV-2 treatment that the FDA had approved. Despite initial activity data that looked promising, there were not enough clinical investigations, and recently, due to limited efficacy, its usage for treating COVID-19 is no longer advised (Young et al. 2021; Jiang et al. 2021; Kokic et al. 2021). The other two antimalarial drugs chloroquine and hydroxychloroquine have also been reported to be effective against SARS-CoV-2 (Cortegiani et al. 2020). Although both drugs were highly effective against SARS-CoV-2, neither in vitro testing nor clinical trial studies produced findings that were persuasive enough to be approved for either single use or use in combination with other antiviral medications (Giacomelli et al. 2021). Due to the promising activity of quinoline-based compounds against SARS-CoV-2, researchers have been working on this moiety to generate more effective therapeutic molecules.
The anticoronavirus activity of ten quinoline analogues was investigated against HCoV-229E (alpha coronavirus), HCoVOC43, SARS-CoV-1 and SARS-CoV-2 (all are beta coronaviruses) (Persoons et al. 2021). Except for piperaquine, all analogues demonstrated antiviral activity against one or more coronaviruses tested. The most potent analogues against the various coronaviruses were chloroquine and hydroxychloroquine, with EC50 values in the range of 0.12–12 μM. With an EC50 value of 0.12 μM and a selectivity index of 165, chloroquine has particularly remarkable action against HCoV-OC43 in HEL cells. Overall, the reduced cytotoxicity of hydroxychloroquine in the different cell lines, which led to more favourable selectivity indices, made the profile of the drug more appealing. Although mefloquine demonstrated very good antiviral action against the various coronaviruses, it was associated with significant toxicity in Huh7, HEL and Vero E6 cell lines. In addition to showing strong antiviral activity, amodiaquine and ferroquine also had lower cytotoxicity and higher selectivity indices. Furthermore, primaquine, quinidine, quinine and tafenoquine exhibited antiviral action against a particular coronavirus only at higher concentrations (EC50 = >13 μM), which suggests a lack of selective antiviral action.
The 4-aminoquinoline derivatives were synthesized and tested for antiviral activity against SARS-CoV-2 (Herrmann et al. 2022). All the tested compounds had anti-SARS-CoV-2 activity comparable to or greater than the reference compound chloroquine. The EC50 values in Caco-2 cells ranged from 5.9 μM to 18.9 μM (EC50 of chloroquine = 12.7 μM) and from 1.5 μM to 2.9 μM in Vero76 cells (EC50 of chloroquine = 3.1 μM). Compound 93 was shown to be the most efficient against Vero 76 cells, with an EC50 value of 1.5 μM, while compound 94 was the most effective against Caco-2 cells, with an EC50 value of 5.9 μM.

Fifteen hybrid compounds containing artemisinin and 4-aminoquinoline were synthesized and tested for their antiviral activity against SARS-CoV-2. All hybrids containing artesunic acid (97–103) demonstrated very good anti-SARS-CoV-2 activity, with EC50 values in the range of 7.8–46 μM. With an EC50 value of 7.8 μM, compound 103 having a morpholine ring was discovered to be the most active. It was also substantially more active than its parent compound, artesunic acid (EC50 > 50 μM). From the quinoline-based hybrids, compound 111 containing adamantane ring (EC50 = 1.5 μM) was the most effective, outperforming both the reference compounds chloroquine (EC50 = 3.8 μM) and remdesivir (EC50 = 4.0 μM). Notably, synthetic peroxide-based hybrids (104–109) were also extremely effective against SARS-CoV-2, with the compound 106 being the most active (EC50 = 11.5 μM). Remarkably, none of the hybrid molecules had cytotoxic effects on Vero E6 cells.

6 Conclusion
The synthesis of new derivatives of 4-aminoquinolines has proven to be an effective chemotherapeutic approach. In this article, the effectiveness of compounds, having an aminoquinoline scaffold, against malaria parasites, tuberculosis bacteria, cancer-causing agents, and coronavirus has been discussed. Some of the compounds discussed in this article may serve as a starting lead for the development of novel medications to treat a variety of diseases brought on by parasites and other pathogens.
References
Afzal O, Kumar S, Haider MR et al (2015) A review on anticancer potential of bioactive heterocycles quinoline. Eur J Med Chem 97:871–910
Ahadi H, Emami S (2020) modification of 7-piperazinylquinolone antibacterials to promising anticancer lead compounds: synthesis and in vitro studies. Eur J Med Chem 187:11
Akkachairin B, Rodphon W, Reamtong O et al (2020) Synthesis of neocryptolepines and carbocycle fused quinolines and evaluation of their anticancer and antiplasmodial activities. Bioorg Chem 98:103732
Alegaon S, Kashniyal K, Kuncolienkar S et al (2020) Synthesis and biological evaluation of some 4-aminoquinoline derivatives as potential antitubercular agents. Futur J Pharm Sc 6:2
Al-Ghorbani M, Begum B, Zabiulla MS et al (2015) Piperazine and morpholine: synthetic preview and pharmaceutical application. J Chem Pharm Res 7:281–301
Aly EI (2010) Design, synthesis and in vitro cytotoxic activity of new 4-anilino-7-chloro quinoline derivatives targeting EGFR tyrosine kinase. J Am Sci 6:73–83
Ambatkar MP, Khedekar PB (2019) Quinoline as TRPV1 antagonists: a new approach against inflammation. J Drug Deliv Ther 9:782–788
Aydemir N, Bilaloglu R (2003) Genotoxicity of two anticancer drugs, gemcitabine and topotecan, in mouse bone marrow in vivo. Mutat Res 537:43–51
Baartzes N, Stringer T, Seldon R et al (2019) Bioisosteric ferrocenyl aminoquinoline-benzimidazole hybrids: antimicrobial evaluation and mechanistic insights. Eur J Med Chem 180:121–133
Bahuguna A, Rawat DS (2020) Recent trends and strategies for the anti-tubercular drug development. Med Res Rev 40:263–292
Bazotte RB, Hirabara SM, Serdan TAD et al (2021) 4-Aminoquinoline compounds from the Spanish flu to COVID-19. Biomed Pharmacother 135:111138
Bhagat S, Arfeen M, Das G et al (2019) Design, synthesis and biological evaluation of 4-aminoquinolineguanylthiourea derivatives as antimalarial agents. Bioorg Chem 91:103094
Bhat HR, Masih A, Shakya A et al (2020) Design, synthesis, anticancer, antibacterial, and antifungal evaluation of 4-aminoquinoline-1,3,5-triazine derivatives. J Heterocycl Chem 57:390–399
Bonilla-Ramirez L, Rios A, Quiliano M et al (2018) Novel antimalarial chloroquine- and primaquine-quinoxaline 1,4-di-N-oxide hybrids: design, synthesis, plasmodium life cycle stage profile, and preliminary toxicity studies. Eur J Med Chem 158:68–81
Cancer.gov (n.d.). http://www.cancer.gov/. Accessed 10 July 2022
Candéa ALP, Ferreira ML, Pais KC et al (2009) Synthesis and antitubercular activity of 7-chloro-4 quinolinylhydrazones derivatives. Bioorg Med Chem Lett 19:6272–6274
Çapci A, Lorion MM, Wang H et al (2019) Artemisinine(iso)quinoline hybrids by CH activation and click chemistry: combating multidrug-resistant malaria. Angew Chem Int Ed 58:13066–13079
Charris JE, Monasterios MC, Acosta ME et al (2019) Antimalarial, antiproliferative, and apoptotic activity of quinoline-chalcone and quinolinepyrazoline hybrids. A dual action. Med Chem Res 28:2050–2066
Chokkar N, Kalra S, Chauhan M et al (2019) A review on quinoline derived scaffolds as anti-HIV agent. Mini Rev Med Chem 19:510–526
Chopra R, Chibale K, Singh K (2018) Pyrimidine-chloroquinoline hybrids: synthesis and antiplasmodial activity. Eur J Med Chem 148:39–53
Clark A, Jit M, Warren-Gash C et al (2020) Global, regional, and national estimates of the population at increased risk of severe COVID-19 due to underlying health conditions in 2020: a modelling study. Lancet Glob Health 8:1003–1017
Cortegiani A, Ingoglia G, Ippolito M et al (2020) A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J Crit Care 57:279–283
de Souza MVN, Pais KC, Kaiser CR et al (2009) Synthesis and in vitro antitubercular activity of a series of quinoline derivatives. Bioorg Med Chem 17:1474–1480
Deshpande S, Kuppast B (2016) 4-aminoquinolines: an overview of antimalarial chemotherapy. Med Chem 6:1–11
Dondorp AM, Nosten F, Yi P et al (2009) Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467
El-Gamal MI, Khan MA, Abdel-Maksoud MS et al (2014) A new series of diarylamides possessing quinoline nucleus: synthesis, in vitro anticancer activities, and kinase inhibitory effect. Eur J Med Chem 87:484–492
Eswaran S, Adhikari AV, Chowdhury IH et al (2010) New quinoline derivatives: synthesis and investigation of antibacterial and antituberculosis properties. Eur J Med Chem 45:3374–3383
Giacomelli A, Pagani G, Ridolfo AL et al (2021) Early administration of lopinavir/ritonavir plus hydroxychloroquine does not alter the clinical course of SARS-CoV-2 infection: a retrospective cohort study. J Med Virol 93:1421–1427
Gorbalenya AE, Baker SC, Baric RS et al (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5:536–544
Heravi MM, Zadsirjan V (2020) Prescribed drugs containing nitrogen heterocycles: an overview. RSC Adv 10:44247–44311
Herrmann L, Hahn F, Wangen C et al (2022) Anti-SARS-CoV-2 inhibitory profile of new quinoline compounds in cell culture-based infection models. Chem Eur J 28:e202103861
Hu YQ, Gao C, Zhang S et al (2017) Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur J Med Chem 139:22–47
Jiang Y, Chen D, Cai D et al (2021) Effectiveness of remdesivir for the treatment of hospitalized COVID-19 persons: a network meta-analysis. J Med Virol 93:1171–1174
Kandi SK, Manohar S, Ve’lez Gerena CE et al (2015) C5-curcuminoid-4-aminoquinoline based molecular hybrids: design, synthesis and mechanistic investigation of anticancer activity. New J Chem 39:224–234
Katariya KD, Shah SR, Reddy D (2020) Anticancer, antimicrobial activities of quinoline based hydrazone analogues: synthesis, characterization and molecular docking. Bioorg Chem 94:103406
Kaur R, Kumar K (2021) Synthetic and medicinal perspective of quinolines as antiviral agents. Eur J Med Chem 215:113220
Kerru N, Gummidi L, Maddila S et al (2020) A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 25:1909
Kokic G, Hillen HS, Tegunov D et al (2021) Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun 12:279
Kondaparla S, Soni A, Manhas A et al (2017) Antimalarial activity of novel 4-aminoquinolines active against drug resistant strains. Bioorg Chem 70:74–85
Kondaparla S, Manhas A, Dola VR et al (2018) Design, synthesis and antiplasmodial activity of novel imidazole derivatives based on 7-chloro-4-aminoquinoline. Bioorg Chem 80:204–211
Krafts K, Hempelmann E, Skorska-Stania A (2012) From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy. Parasitol Res 11:1–6
Kremsner PG, Krishna S (2004) Antimalarial combinations. Lancet 364:285–294
Kumar D, Khan SI, Tekwani BL et al (2015a) 4-Aminoquinoline pyrimidine hybrids: synthesis, antimalarial activity, heme binding and docking studies. Eur J Med Chem 89:490–502
Kumar D, Negi B, Rawat DS (2015b) The anti-tuberculosis agents under development and the challenges ahead. Future Med Chem 7:1981–2003
Li B, Zhu F, He F et al (2020) Synthesis and biological evaluations of N′-substituted methylene-4-(quinoline-4-amino) benzoylhydrazides as potential anti-hepatoma agents. Bioorg Chem 96:103592
Mahajan A, Kremer L, Louw S et al (2011) Synthesis and in vitro antitubercular activity of ferrocene-based hydrazons. Bioorg Med Chem Lett 21:2866–2868
Manohar S, Rajesh UC, Khan SI et al (2012) Novel 4-aminoquinoline-pyrimidine based hybrids with improved in vitro and in vivo antimalarial activity. ACS Med Chem Lett 3:555–559
Manohar S, Tripathi M, Rawat DS (2014) 4-Aminoquinoline based molecular hybrids as antimalarials: an overview. Curr Top Med Chem 14:1706–1733
Manohar S, Pavan VS, Taylor D et al (2015) Highly active 4-aminoquinoline–pyrimidine based molecular hybrids as potential next generation antialarial agents. RSC Adv 5:28171
Marganakop SB, Kamble RR, Hoskeri J et al (2014) Facile synthesis of novel quinoline derivatives as anticancer agents. Med Chem Res 23(6):2727–2735
Matada BS, Pattanashettar R, Yernale NG (2021) A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg Med Chem 32:115973
Maurya SS, Khan SI, Bahuguna A et al (2017) Eur J Med Chem 129:175–185
Maurya SS, Bahuguna A, Khan SI et al (2019) N-substituted aminoquinoline-pyrimidine hybrids: synthesis, in vitro antimalarial activity evaluation and docking studies. Eur J Med Chem 162:277–289
Meshnick S, Dobson M (2001) The history of antimalarial drugs. In: Rosenthal P (ed) Antimalarial chemotherapy. Humana Press, Totowa, NJ, pp 15–25
Meunier B (2008) Hybrid molecules with a dual mode of action: dream or reality. Acc Chem Res 41:69–77
Mukherjee S, Pal M (2013) Quinolines: a new hope against inflammation. Drug Discov Today 18:389–398
Muregi FW, Ishih A (2010) Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design. Drug Dev Res 71:20–32
Musiol R (2017) An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin Drug Discov 12:583–597
Musiol R, Serda M, Hensel-Bielowka S et al (2010) Quinoline-based antifungals. Curr Med Chem 17:1960–1973
Musiol R, Malarz K, Mularski J (2017) Quinoline alkaloids against neglected tropical diseases. Curr Org Chem 21:1896–1906
N’Da DD, Smith PJ (2014) Synthesis, in vitro antiplasmodial and antiproliferative activities of a series of quinoline-ferrocene hybrids. Med Chem Res 23:1214–1224
Narula AK, Azad CS, Nainwal LM (2019) New dimensions in the field of antimalarial research against malaria resurgence. Eur J Med Chem 181:111353
Noedl H, Se Y, Schaecher K et al (2008) Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 359:2619–2620
Parashar RK, Negi B (2015) Chemistry of heterocyclic compounds. Ane Books Pvt Ltd, New Delhi. ISBN 978–93–8546-292-4
Persoons L, Vanderlinden E, Vangeel L et al (2021) Broad spectrum anti-coronavirus activity of a series of anti-malaria quinoline analogues. Antivir Res 193:105127
Pushpakom S, Iorio F, Eyers PA et al (2019) Drug repurposing: Progress, challenges and recommendations. Nat Rev Drug Discov 18:41–58
Raj R, Land KM, Kumar V (2015) 4-Aminoquinoline-hybridization en route towards the development of rationally designed antimalarial agents. RSC Adv 5:82676
Ramírez-Prada J, Robledo SM, V’elez ID et al (2017) Synthesis of novel quinoline–based 4,5-dihydro–1H–pyrazoles as potential anticancer, antifungal, antibacterial and antiprotozoal agents. Eur J Med Chem 131:237–254
Rangappa SK, Siddappa AP (2014) Quinoline: a promising antitubercular target. Biomed Pharmacother 68:1161–1175
Rani A, Singh A, Gut J et al (2018) Microwave-promoted facile access to 4-aminoquinoline-phthalimides: synthesis and anti-plasmodial evaluation. Eur J Med Chem 143:150–156
Rani A, Legac J, Rosenthal PJ et al (2019) Substituted 1,3-dioxoisoindoline-4-aminoquinolines coupled via amide linkers: synthesis, antiplasmodial and cytotoxic evaluation. Bioorg Chem 88:102912
Rani A, Kumar S, Legac J et al (2020) Design, synthesis, heme binding and density functional theory studies of isoindoline-dione-4-aminoquinolines as potential antiplasmodials. Future Med Chem 12:193–205
Rawat DS, Beena (2013) Antituberculosis drug research: a critical overview. Med Res Rev 33:693–764
Reddy PL, Khan SI, Ponnan P et al (2017) Design, synthesis and evaluation of 4-aminoquinoline-purine hybrids as potential antiplasmodial agents. Eur J Med Chem 126:675–686
Shalini J, Legac AA, Adeniyi P et al (2020) Functionalized naphthalimide-4-aminoquinoline conjugates as promising antiplasmodials, with mechanistic insights. ACS Med Chem Lett 11:154–161
Shamsuddin MA, Ali AH, Zakaria NH et al (2021) Synthesis, molecular docking, and antimalarial activity of hybrid 4-aminoquinoline-pyrano[2,3-c]pyrazole derivatives. Pharmaceuticals 14:1174
Sharma M, Chaturvedi V, Manju YK et al (2009) Substituted quinolinylchalcones and quinolinylpyrimidines as a new class of antiinfective agents. Eur J Med Chem 44:2081–2091
Sharma B, Kaur S, Legac J et al (2020) Synthesis, antiplasmodial and cytotoxic evaluation of 1H-1,2,3-triazole/acyl hydrazide integrated tetrahydro-b-carboline-4-aminoquinoline conjugates. Bioorg Med Chem Lett 30:126810
Singh K, Kaur T (2016) Pyrimidine-based antimalarials: design strategies and antiplasmodial effects. Med Chem Commun 7:749–768
Singh S, Kaur G, Mangla V et al (2015) Quinoline and quinolones: promising scaffolds for future antimycobacterial agents. J Enzyme Inhib Med Chem 30:492–504
Singh B, Chetia D, Kumawat MK (2021) Synthesis and in vitro antimalarial activity evaluation of some new 1,2-diaminopropane side-chain-modified 4-aminoquinoline Mannich bases. Pharm Chem J 55:7
Solomona VR, Pundira S, Lee H (2019) Design and synthesis of 4-piperazinyl quinoline derived urea/thioureas for anti-breast cancer activity by a hybrid pharmacophore approach. J Enzyme Inhib Med Chem 34(1):620–630
Srivastava V, Negi AS, Kumar JK et al (2005) Plant-based anticancer molecules: a chemical and biological profile of some important leads. Bioorg Med Chem 13:5892–5908
Su T, Zhu J, Sun R et al (2019) Design, synthesis and biological evaluation of new quinoline derivatives as potential antitumor agents. Eur J Med Chem 178:154–167
Tseng CH, Chen YL, Hsu CY et al (2013) Synthesis and antiproliferative evaluation of 3-phenylquinolinylchalcone derivatives against non-small cell lung cancers and breast cancers. Eur J Med Chem 59:274–282
Vandekerckhove S, D’hooghe M (2015) Quinoline-based antimalarial hybrid compounds. Bioorg Med Chem 23:5098–5119
Veziris N, Bernard C, Guglielmetti L et al (2017) Rapid emergence of mycobacterium tuberculosis bedaquiline resistance: lessons to avoid repeating past errors. Eur Respir J 49:1601719
Wang J, Xu C, Wong YK et al (2019) Artemisinin, the magic drug discovered from traditional Chinese medicine. Engineering 5:9–32
Wang M, Cao R, Zhang L et al (2020a) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271
Wang Y, Grunewald M, Perlman S (2020b) Coronaviruses: an updated overview of their replication and pathogenesis. Methods Mol Biol 2203:1–29
WHO (n.d.-a). https://covid19.who.int/. Accessed 20 July 2022
WHO (n.d.-b). https://www.who.int/news-room/fact-sheets/detail/tuberculosis. Accessed 15 July 2022
Woodward R, Doering W (1944) The total synthesis of quinine. J Am Chem Soc 66:849
Yadav P, Shah K (2021) Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry. Bioorg Chem 109:104639
Young B, Tan TT, Leo YS (2021) The place for remdesivir in COVID-19 treatment. Lancet Infect Dis 21:20–21
Zhou Y, Yan W, Cao D et al (2017) Design, synthesis and biological evaluation of 4-anilinoquinoline derivatives as novel potent tubulin depolymerization agents. Eur J Med Chem 138:1114–1125
Zorc B, Rajic Z, Perkovic I (2019) Antiproliferative evaluation of various aminoquinoline derivatives. Acta Pharma 69:661–672
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kumar, D., Negi, B., Rawat, D.S. (2023). Diverse Pharmacological Activities of 4-Aminoquinoline and its Derivatives. In: Singh, P.P. (eds) Recent Advances in Pharmaceutical Innovation and Research. Springer, Singapore. https://doi.org/10.1007/978-981-99-2302-1_10
Download citation
DOI: https://doi.org/10.1007/978-981-99-2302-1_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-2301-4
Online ISBN: 978-981-99-2302-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)