
Abstract
Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of myeloid cells with immature phenotypes and immunosuppressive functions. This population of cells has been extensively studied over the past decade owing to an increasing recognition of their pivotal role in pathological conditions including cancers, infectious diseases, sepsis, and autoimmune diseases. Various treatments targeting MDSCs are currently under development or in clinical trials with the aim to restore functional immunity against tumors or pathogens. Recent advances in immune metabolism demonstrate the role of metabolic pathways, especially lipid metabolism, in the differentiation and function of MDSCs in tumor environments. Therefore, a comprehensive understanding of lipid metabolism in MDSCs would facilitate the development of novel therapies against tumors through metabolic reprograming of MDSCs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
7.1 Lipid Metabolism and the Origin of MDSCs
Lipid metabolism involves lipid catabolism and anabolism, providing energy and substrate to sustain cellular activities. Lipid metabolism begins with food digestion to breakdown triglycerides into fatty acids, glycerol, and cholesterol by lipases. Small intestinal epithelial cells absorb fatty acids and monoglycerides to reassemble into triglycerides. Subsequently, triglyceride and cholesterol are packed with lipoproteins to form lipid transport complexes like chylomicrons, which travel into the blood circulation [1]. When lipid transport complexes move across tissues, capillary endothelial cells release and breakdown triglycerides into fatty acids and glycerol for consumption in metabolizing cells [2]. Glycerol participates in glycolysis or gluconeogenesis mostly in the cytosol of liver cells whereas fatty acids are stored mainly in the form of triglycerides or used through fatty acid beta-oxidation (FAO) for energy [3]. Long-chain fatty acids are transported via carnitine palmitoyltransferase system from cytosol to mitochondrial where the FAO takes place. The sequential removal of two carbon units from a beta position of fatty acyl-CoA molecules produces acetyl-CoA, a key intermediate for carbohydrate, protein, and lipid metabolism. Acetyl-CoA sustains tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS) [4]. If fatty acids are completely oxidized to CO2 and water, they yield the highest ATP on an energy per gram basis compared to carbohydrates and proteins. On the contrary to FAO, fatty acid synthesis commences with carboxylation of acetyl-CoA derived from citrate in cytosol into malonyl CoA by acetyl CoA carboxylase 1 (ACC-1). Malonyl CoA in turn potently inhibits the carnitine palmitoyltransferase 1 (CPT-1) activity, the rate-limiting enzyme in FAO. Condensation of malonyl CoA to acetyl CoA and another seven cycles of reaction to produce a saturated long-chain fatty acid palmitate by fatty acid synthase. De novo synthesized fatty acids and intermediates of other metabolic pathways participate in the generation of other lipids or incorporated into triglycerides for lipid droplet formation.
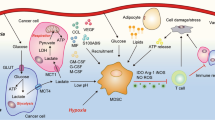
Tissue-specific metabolic environment has a great influence on the hematopoiesis and peripheral immune homeostasis. Take bone marrow (BM) as an example (Fig. 7.1), adipocytes occupy up to 70% of adult BM cavity and thus make BM the third largest fat depot in the body behind subcutaneous and visceral fat [5]. These cells are not just “space fillers” [6], but rather metabolically active and release fatty acids from triacylglycerol droplet into hematopoietic milieu, therefore creating a lipid rich microenvironment for hematopoietic cells [7]. While long-term hematopoietic stem cell (LT-HSC) relies on anaerobic glycolysis for energy [8], short-term HSC (ST-HSC) and committed progenitors live on FAO for self-renew and differentiation [9]. When LT-HSCs differentiate, the expression of genes related to lipid metabolism became increasingly vigorous. HSC numbers in BM of mice and human increase with age, partially due to loss of quiescence for LT-HSCs and enhanced proliferation of ST-HSCs [10, 11]. A similar observation was reported in diet-induced obesity mice [12]. Moreover, aged and obese mice manifest biased hematopoiesis toward myeloid lineages and reduced lymphoid progenitors [13, 14]. Excessive accumulation of fat content in the marrow with aging and obesity is thought to contribute to this altered hematopoiesis through lipolysis of triglycerides in adipocytes. Indeed, fatty acids released from BM adipocytes are utilized by leukemic blasts via fatty acid-binding protein (FABP)-4, supporting the survival and proliferation of acute myeloid leukemia blast cells [15]. As such, lipid-rich BM microenvironment may favor the hematopoiesis toward myelopoiesis through metabolic reprogramming of HSC for FAO.

Lipid metabolism and the origin of MDSCs. (i) LT-HSC relies on anaerobic glycolysis for energy, while ST-HSC and committed progenitors live on FAO for energy. (ii) During LT-HSC differentiation, the expression of lipid metabolism-related genes increases, accompanied by a decrease in the expression of glycolysis-related genes. (iii) Aging or obesity would lead to an increase in adipocytes and fatty acids in BM, forming a lipid-rich microenvironment. In the BM of aged and obese human or mice: (iv) the quiescence of LT-HSC is disrupted and ST-HSCs prefer to differentiate into myeloid progenitors, accompanied by a metabolic switch from glycolysis to FAO and OXPHOS for energy supply. (v) Immature myeloid cells accumulate and generate immunosuppressive MDSCs. MDSC Myeloid-derived suppressor cells, LT-HSCs Long-term hematopoietic stem cells, ST-HSCs Short-term hematopoietic stem cells, FAO Fatty acid beta oxidation, BM Bone marrow, OXPHOS Oxidative phosphorylation
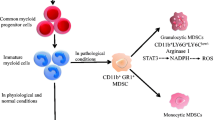
Myeloid-derived suppressor cells (MDSCs) (mouse: CD11b+ Gr1+; human: CD11b+ CD33+ HLA−DR−/lo) are immunosuppressive myeloid cell populations first described in tumors, subsequently identified in chronic inflammation, and recently in neonates [16,17,18,19]. Inhibition of MDSC mediated suppression of innate and adaptive immunity is crucial for effective immunotherapy against tumor and pathogens. Based on surface markers, MDSCs resemble neutrophils and monocytes with immature phenotypes, thus can be further divided into PMN-MDSCs (mouse: Ly6G+Ly6C−/lo; human: CD15+CD14−) and M-MDSCs (mouse: Ly6C+/hiLy6G−; human: CD14+CD15−) subpopulations, respectively [20]. The origin of MDSCs is still under debate. Initially, MDSCs are proposed as myeloid precursors blocked from differentiation in BM and recruited by chemokines secreted from tumor cells. This hypothesis is supported by in vivo experiments that all-trans retinoic acid and vitamin D3 drive the immature myeloid cells in tumors into functional macrophages, granulocytes, and DCs [21, 22]. However, recent studies challenge this theory by demonstrating that monocytes and neutrophils are plastic and acquire immunosuppressive function within tumor microenvironment (TME) [23]. In any case, both hypotheses acknowledge the role of TME in the development of MDSCs. Although cytokines secreted by tumor cells have been indicated in the manipulation of myelopoiesis in BM or TME [24, 25], accumulating evidence points that metabolic reprogramming of MDSCs from glycolysis to FAO and OXPHOS by tumor cells play a pivotal role in the generation and function of MDSCs. Besides tumor tissues, MDSCs also accumulate in adipose tissue and bone marrow, two other lipid-rich environments. It has been reported that MDSCs are elevated in obese individuals, as well as in spleen and adipose tissue in obese mice. The level of MDSCs in adipose tissue of mice on a high-fat diet is 1.5–3 times higher than their lean counterparts [26]. As discussed above, bone marrow of obese and aged individuals also harbors a large reservoir of immature myeloid cells. These evidence together suggest that tissue-specific lipid microenvironment greatly impacts MDSC generation.
7.2 Influence of TME on MDSC Energy Metabolism
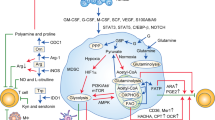
Four distinct mechanisms are employed by MDSCs in TME to promote tumor growth and metastases: (a) suppression of tumor-specific immunity, (b) establishing a TME to benefit tumor growth, (c) facilitating tumor metastasis, (d) induction of tumor stem cell and promotion of epithelial-to-mesenchymal transition [27]. In TME, factors determining MDSC expansion, differentiation, migration, and immunosuppression, include cytokines and growth hormones (e.g., PGE2, TGF-β, GM-CSF, IL-6, and IFN-γ), low PH, hypoxia, and nutrient availability [28]. These triggers participate in shaping MDSC functions by exploiting their plasticity and reprogramming their metabolic fate [29]. Below, we discuss the key metabolic features in TME and its influence on the MDSC energy metabolism (Fig. 7.2).

TME shapes the energy metabolism of MDSCs. (i) To meet the demand for rapid proliferation, tumor cells initiate a metabolic shift toward glycolysis, which is known as Warburg Effect. The high glycolytic rate in tumor cells results in massive lactate release, forming an acidulated microenvironment. Besides, uncontrolled proliferation of tumor cells and abnormal blood vessel formation cause hypoxia in TME. (ii) MDSCs reprogram their metabolic pathways toward glycolysis to compete with tumor cells for limited oxygen and glucose. (iii) Tumor cells enhance fatty acid uptake and synthesis and recruit adipocytes in TME to produce excessive exogenous fatty acids, which makes a lipid-rich environment for infiltrating MDSCs. MDSCs are then driven to adopt FAO as their primary energy source. (iv) The acidic and lipid-rich TME promotes the development of MDSCs and enhances their immunosuppressive function. MDSCs consume amino acids in the TME through enzymes (Arg-1, IDO, iNOS, etc.) and transporters like Cat-2 and Xc, contributing to amino acid starvation in the TME, which in turn leads to inhibition of tumor antigen-specific proliferation of T cells. (v) Toxic ROS/RNS and amino acid metabolites (polyamine and kynurenine) released by MDSCs induce apoptosis of T cells and promote the generation of Treg from naïve CD4 T cells. TME Tumor microenvironment, MDSC Myeloid-derived suppressor cells, FAO Fatty acid beta oxidation, Arg-1 arginase-1, iNOS induced nitric oxide synthase, IDO indoleamine 2,3 dioxygenase, Cat-2 amino acid transporter 2B, Xc cysteine-glutamine antiporter, ROS reactive oxygen species, RNS reactive nitrogen species
Rapid proliferation of tumor cells requires large amounts of ATPs and substrates to fulfill energetic and biosynthetic demands. As such, tumor cells rewire their metabolic pathway toward glycolysis even under aerobic condition, also known as Warburg Effect [30]. Although aerobic glycolysis is insufficient to generate ATP per unit of glucose compared to mitochondrial respiration, it generates a comparable amount of ATP within a given period of time [31]. Massive lactate secretion from tumor glycolysis acidifies TME and altered the myelopoiesis to promote the generation of MDSCs [32]. Uncontrolled proliferation of tumor cells and abnormal blood vessel formation cause hypoxia in TME. Hypoxia stimulates the expression of HIF-1α in tumor-infiltrating immune cells, which is the downstream of PI3K-AKT-mTOR pathway [33]. Activation of HIF-1α favors the glycolysis over OXPHOS by upregulation of glucose and lactate transporters, along with glycolytic enzymes [34, 35]. This results in dampened lymphocyte activation and blocked maturation of myeloid subsets, which requires sufficient glucose for effector functions. Dynamic metabolic flux analysis (dMFA) study shows that MDSC maturation in TME correlates with a high glycolytic flux contributing to up to 95% of the global ATP turnover rate, demonstrating that developing MDSCs obtain energy primarily through glycolysis [36]. In addition, glycolysis activation by metformin or providing lactate promotes the proliferation of MDSCs in TME [37]. Hence, MDSCs would compete with tumor cells for limited glucose and oxygen, but eventually acclimate its metabolic pathway to the available nutrients and hypoxia in TME.
Nitrogen metabolism of MDSCs has been extensively studied in the context of immunosuppression of T cell function in TME. Amino acids such as l-arginine, tryptophan, and cysteine are either depleted from TME by MDSCs expressed enzymes like Arginase-1 (Arg-1), iNOS, and indoleamine 2,3 dioxygenase (IDO), or sequestered by transporters like cationic amino acid transporter 2B (Cat-2) or cysteine-glutamine antiporter (Xc) in MDSCs [38,39,40]. Amino acid starvation leads to inhibition of tumor antigen-specific proliferation of T cells through downregulation of CD3ζ and induction of cell cycle arrest at G0–G1 phase [41,42,43]. Moreover, products from amino acid metabolism such as polyamine, kynurenine, and toxic ROS/RNS (NO, O2−, H2O2, and PNT) can further induce apoptosis of cytotoxic CD8 T cells and promote conversion of naïve CD4 T cells into Tregs [43,44,45,46].
Aerobic glycolysis provides tumor cells with excessive carbon for de novo synthesis of nucleotides, lipids, and proteins to support their uncontrolled proliferation. Of note, enhanced uptake of glucose promotes synthesis of reducing agent NADPH from oxidative branch of pentose phosphate pathway to participate in lipid synthesis [47, 48]. As a result, reactivation of lipid biosynthesis and storage have been frequently reported in cancer tissues, especially when tumor cells outgrew tissue blood supply of nutrients and oxygen. Despite that tumor cells synthesize most fatty acids de novo [49], aggressive tumor cells often intermingle with adipocytes in TME to obtain exogenous fatty acids [50, 51]. Therefore, similar to BM and adipose tissue microenvironment, TME is also a lipid-rich environment for infiltrating immune cells. We will discuss further on how this lipid-rich TME influences immunosuppressive function of MDSCs.
7.3 Lipid Metabolism and Immunosuppressive Function of MDSC
Recent studies on mouse models conclude that tumor-associated MDSCs upregulate FAO as a primary energy source as opposed to glycolysis to exert immunosuppressive functions, in comparison to peripheral myeloid cells and spleen MDSCs. This conclusion is supported by increased mitochondrial mass, fatty acid uptake via CD36, expression of key enzymes in FAO (CPT-1, acyl-CoA dehydrogenase, 3-hydroxyacyl-CoA dehydrogenase), and oxygen consumption rate (OCR)/extracellular acidification rate (ECAR) ratio [52]. Of note, consistent with what has been observed in the mouse study, enhanced fatty acid uptake and increased level of FAO-related enzymes in MDSCs were also detected in the blood and tumor from cancer patients. Along this line, lipid overload in MDSCs correlates with their suppressive function. Interestingly, most lipids in MDSCs are found to be oxidized via MPO and ROS, and the polyunsaturated fatty acids, which are more susceptible to oxidation, could promote the suppressive function of MDSCs [29]. Importantly, pharmacologic inhibition of FAO in mouse significantly alleviated tumor progression and improved antitumor outcome of adoptive T-cell therapy. Mechanically, this is because pharmacologic inhibition of FAO decreased the production of inhibitory cytokines and prevented the immune inhibitory capability of MDSCs. Moreover, combining chemotherapy and pharmacologic FAO inhibition eliminated the immunosuppressive effects of MDSCs and consequently enhanced the therapeutic outcome [52]. Lysosomal acid lipase (LAL) is required for hydrolysis of cholesteryl and triglycerides in lysosomes to generate fatty acid for FAO. MDSCs in LAL-deficient mice switched from FAO to glucose-dependent oxidative pathway, with enhanced proliferation but compromised immunosuppression [53]. Together, these results indicate that modulating FAO activity may provide an interesting option to inhibit the immunosuppressive function of MDSCs and improve the outcome of clinical therapy [54].
Besides FAO for ATP supply, fatty acid synthesis, lipogenesis, and lipid accumulation also link to suppressive function of MDSCs [55, 56]. Gabrilovich et al. demonstrated that targeting key enzymes of lipid synthesis ACC-1 with 5-tetradecycloxy-2 furoic acid (TOFA) to block fatty acid synthesis in MDSCs revert the suppression of T cell activation. Recently, Veglia, et al. identified MDSCs utilize fatty acid transport protein 2 (FATP-2) for transporting arachidonic acid and synthesizing prostaglandin E2 (PGE-2) to suppress T cell-mediated antitumor immunity. Selective inhibition of FATP2 abolished the suppressive activity of PMN-MDSCs and substantially delayed tumor progression in mice [57]. Interestingly, PGE2, a lipid mediator, generated by COX-2 from fatty acid arachidonic acid [58] was proved to promote MDSC generation in tumor-bearing mice [59]. And further research indicated that inhibiting PEG2 or COX2 could decrease MDSC generation and delay tumor progression [59, 60]. Prevention of lipid droplet formation in MDSC like cell line MSC-2 by diacylglycerol acyltransferases impairs the immunosuppression [61]. Therefore, although fatty acid synthesis and oxidation cannot occur under classical view, it appears that MDSCs adapt to hypoxic and nutrient-deprived TME to activate both metabolic pathways simultaneously to exert its suppressive function (Fig. 7.3).

lipid metabolism in MDSCs correlate with their suppressive function. (i) Fatty acid uptake, fatty acid beta oxidation (FAO), fatty acid synthesis, and lipogenesis increase in MDSCs and result in upregulation of immunosuppressive function. (ii) Lipid accumulation in MDSC leads to lipid droplet formation and associate with their immunosuppressive functions. (iii) Pharmacologic inhibition of fatty acid transport, FAO, and fatty acid synthesis in MDSCs blocks immunosuppressive functions. MDSC Myeloid-derived suppressor cells, FAO Fatty acid beta oxidation, FATP2 fatty acid transport protein 2, COX2 cyclooxygenase 2, PGE2 prostaglandin E2, CPT-1 Carnitine palmitoyl transferase 1, ACAD acyl-CoA dehydrogenase, ACC-1 Acetyl-CoA carboxylase, TOFA 5-tetradecycloxy-2 furoic acid
7.4 Genetic and Epigenetic Pathways Involved in Lipid Metabolisms of MDSC
Although how lipid metabolism in MDSCs influences their immunosuppressive function is not fully understood, several signaling pathways have been implicated to play a role in this process. For example, liver X receptors (LXR) are members of nuclear hormone receptor family that regulate lipid homeostasis. Their physiological activators are oxysterols and intermediates in the cholesterol biosynthetic pathway. Under the administration of LXR agonist, PMN-MDSCs and M-MDSCs were effectively decreased in mouse models and cancer patients [62]. In addition, the LXR target gene ApoE triggers activation of cytotoxic T cells to enhance antitumor immune defenses [62]. Another known “lipid sensing” receptor is peroxisome proliferator-activator receptors (PPARs), which are activated by free fatty acids, prostaglandins, and sterols. Several PPARs subtypes elicit the expression of FAO genes and coordinate cell fate and inflammation. Cardiolipin, the main phospholipid in the inner mitochondrial membrane, promotes IL-10 expression in MDSCs of tumor-bearing mice by activating PPARγ [63, 64], and the effect can be reversed by PPARγ inhibitor [65]. Moreover, PPARα agonist activated CPT and thereby enhanced fatty acid catabolism in melanoma-specific CD8+ TILs, ultimately leading to delayed tumor progression [66]. These studies indicate targeting PPAR pathways as a promising strategy for rebalancing lipid metabolism in MDSCs. Further research supported cross-regulation between PPARs and LXRs pathways to increase atherosclerosis susceptibility and elucidated that activation of this regulatory network has additive effects in controlling ATP binding cassette transporter A1 (ABCA1) expression, thereby reversing cholesterol transport in macrophages [67, 68].
Changes in lipid metabolism can also influence cell differentiation and functions by epigenetic modifications. For example, the phospholipid derivative lysophosphatidic acid (LPA) activates a family of GPCRs named LPAR1–6 and leads to recruitment of HDAC1 coincides with decreased histone acetylation in the TRAIL death receptors promoter, which ultimately promotes survival of cancer cells [69]. Moreover, many DNA/histone-modifying enzymes often require cofactors and substrates that are also critical intermediate metabolites. For example, increased FAO leads to elevation of acetyl-CoA, which served as a carrier of acyl groups by histone acetyltransferases to promote open chromatin state and activate transcription. Compared with other immune cells, evidence to support the direct link between metabolites or cofactors and epigenetic regulation in MDSCs is still lacking [70, 71].
Besides providing most of the acetyl groups on histones, fatty acids are also involved in another histone modification: histone acylation. Up till now, a variety of short and long-chain acyl groups have been identified covalently attached to a histone lysine residue, including propyl, butyryl, crotonyl, myristoyl, and palmitoyl. Each fatty acid modifying group confers distinct biochemical properties that influence subcellular localization, intracellular trafficking, and protein–lipid interactions. A compelling study to link lysine fatty acylation to immune response shows the role of HDAC11 in the regulation of serine hydroxymethyltransferase 2 (SHMT2) defatty-acylation in type I IFN-mediated signaling [72]. HDAC11, the only class IV HDAC member, has been rediscovered as a highly potent and efficient defatty-acylase of lysine as compared to its deacetylase activity [72]. Of note, HDAC11 acts as a negative regulator of MDSCs expansion and function [73]. A better understanding of the mechanisms and functional consequences of reversible lysine fatty acylation may provide new insights into regulating lipid metabolic homeostasis. In the future, we expect to see the signaling pathway inhibitors, epigenetic inhibitors, and metabolite antagonists that specifically interfere with the above mentioned regulatory process being applied in combination to manipulate the fate of MDSCs and its function.
7.5 Issues of Translating MDSC Knowledge from Bench to Bed
Although several key discoveries made in mouse studies have been validated on human, there are still differences between human and murine MDSCs. For example, LOX-1, a low-density lipoprotein receptor, has been proposed as a candidate marker for human immunosuppressive PMN-MDSCs and numbers of LOX-1+ PMN-MDSCs correlate with cancer progression [74]. On the contrary, PMN-MDSCs from LOX-1 knockout mice did not exhibit differences in migration toward tumor tissue, nor in suppressive function against T cells compared to wild type PMN-MDSCs. Moreover, Arg-1 expression in M-MDSCs and TAMs are well documented in murine studies, whereas Arg-1 is constitutively expressed in human neutrophils and inducible in other monocytic cells under disease setting [75]. Another example of these differences could be the fact that mice lacks cholesteryl ester transfer protein (CETP), which transferring CE from HDL toward ApoB containing lipoproteins [76]. Consequently, mice and human display a substantially different plasma lipoprotein profile. These species barriers might hinder the translation process from mouse studies to effective therapies targeting metabolisms of MDSCs.
Despite the success achieved in past years on studying lipid metabolism and mouse MDSCs, research on human MDSCs is still scarce and key questions regarding human MDSC biology remain largely unexplored. How chronic tumor microenvironment influence the generation process of human MDSCs? Through which mechanism are they recruited to the tumor microenvironments? Would it be possible to manipulate human MDSC generation or migration toward tumor microenvironment through altering lipid metabolism? To address these questions, novel research models and tools are required. For instance, severe immune-deficient mice engrafted with human CD34+ HSC develop multi-lineage human immune cells, which can serve as a unique platform to study human MDSC biology in vivo [77, 78]. These humanized mouse models can be further transplanted with tumor cells or tissues to dissect the factors of TME on both the generation and the recruitment of MDSCs [79, 80]. Moreover, liver humanization protocol has succeeded in addition to human immune system reconstitution to create a double humanized mouse. These complex yet highly clinically relevant models would provide an in vivo platform to investigate the metabolic and immune cross-talk among human MDSCs, tumor, and liver (Fig. 7.4). On average, 95% of new cancer drug candidates failed during clinical tests due to the inconsistency between murine and human immune systems [81] and lack of reliable preclinical animal models, which could accurately recapitulate patient tumor microenvironment [82]. Hence, it is critical to appreciate the differences between human and murine MDSCs in order to accelerate therapeutic development. With the advance of humanized mouse models, screening, and validation of MDSC targeted therapeutics would be easier and faster in the future.

Liver/Tumor/Immune humanized mouse models. To further investigate lipid metabolism in human tumor-associated MDSCs in vivo, we propose a liver, tumor, and immune system triple humanized mouse model. Human MDSC and immune system can be generated from human CD34+ HSC engrafted to severe immune-deficient mice. And the liver can be humanized by injecting human liver cells into spleen of a genetic engineered mouse strain with spontaneous or induced liver damage. This humanized mouse model can be further transplanted with primary tumor cells or tissues from patients or tumor cell lines to create a triple humanized mouse model. With the availability of this triple humanized mouse model, we can decipher the factors involved in the generation and recruitment of MDSCs to tumor microenvironment and calibrate the balance between lipid catabolism and anabolism in MDSC function and tumor progression. MDSC Myeloid-derived suppressor cell, HSC hematopoietic stem cells
References
Jo Y, Okazaki H, Moon YA, Zhao T. Regulation of lipid metabolism and beyond. Int J Endocrinol. 2016;2016:5415767. https://doi.org/10.1155/2016/5415767.
Feingold KR, Grunfeld C. Introduction to lipids and lipoproteins. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext. South Dartmouth, MA: MDText.com; 2000.
Berg JM, Tymoczko J L, Stryer L. Section 16.3. Glucose can be synthesized from noncarbohydrate precursors. In: Biochemistry. 5th ed. New York: W H Freeman; 2002.
Abo Alrob O, Lopaschuk GD. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem Soc Trans. 2014;42(4):1043–51. https://doi.org/10.1042/BST20140094.
Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, Klibanski A. Marrow fat and bone—new perspectives. J Clin Endocrinol Metab. 2013;98(3):935–45. https://doi.org/10.1210/jc.2012-3634.
Guerra DAP, Paiva AE, Sena IFG, Azevedo PO, Batista ML Jr, Mintz A, Birbrair A. Adipocytes role in the bone marrow niche. Cytometry A. 2018;93(2):167–71. https://doi.org/10.1002/cyto.a.23301.
Robles H, Park S, Joens MS, Fitzpatrick JAJ, Craft CS, Scheller EL. Characterization of the bone marrow adipocyte niche with three-dimensional electron microscopy. Bone. 2019;118:89–98. https://doi.org/10.1016/j.bone.2018.01.020.
Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–611. https://doi.org/10.1084/jem.20050967.
Lee MKS, Al-Sharea A, Dragoljevic D, Murphy AJ. Hand of FATe: lipid metabolism in hematopoietic stem cells. Curr Opin Lipidol. 2018;29(3):240–5. https://doi.org/10.1097/MOL.0000000000000500.
Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108(50):20012–7. https://doi.org/10.1073/pnas.1116110108.
Pearce DJ, Anjos-Afonso F, Ridler CM, Eddaoudi A, Bonnet D. Age-dependent increase in side population distribution within hematopoiesis: implications for our understanding of the mechanism of aging. Stem Cells. 2007;25(4):828–35. https://doi.org/10.1634/stemcells.2006-0405.
Singer K, DelProposto J, Morris DL, Zamarron B, Mergian T, Maley N, Cho KW, Geletka L, Subbaiah P, Muir L, Martinez-Santibanez G, Lumeng CN. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol Metab. 2014;3(6):664–75. https://doi.org/10.1016/j.molmet.2014.06.005.
Adler BJ, Green DE, Pagnotti GM, Chan ME, Rubin CT. High fat diet rapidly suppresses B lymphopoiesis by disrupting the supportive capacity of the bone marrow niche. PLoS One. 2014;9(3):e90639. https://doi.org/10.1371/journal.pone.0090639.
Kuranda K, Vargaftig J, de la Rochere P, Dosquet C, Charron D, Bardin F, Tonnelle C, Bonnet D, Goodhardt M. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell. 2011;10(3):542–6. https://doi.org/10.1111/j.1474-9726.2011.00675.x.
Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, Piddock RE, Fenech M, Zaitseva L, Abdul-Aziz A, Turner J, Watkins JA, Lawes M, Bowles KM, Rushworth SA. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129(10):1320–32. https://doi.org/10.1182/blood-2016-08-734798.
He YM, Li X, Perego M, Nefedova Y, Kossenkov AV, Jensen EA, Kagan V, Liu YF, Fu SY, Ye QJ, Zhou YH, Wei L, Gabrilovich DI, Zhou J. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat Med. 2018;24(2):224–31. https://doi.org/10.1038/nm.4467.
Li M, Zhu D, Wang T, Xia X, Tian J, Wang S. Roles of myeloid-derived suppressor cell subpopulations in autoimmune arthritis. Front Immunol. 2018;9:2849. https://doi.org/10.3389/fimmu.2018.02849.
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–44. https://doi.org/10.1182/blood-2007-07-099226.
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–802. https://doi.org/10.4049/jimmunol.181.8.5791.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150.
Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441–9.
Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–307. https://doi.org/10.1158/0008-5472.CAN-06-1690.
Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. 2017;8(2):3649–65. https://doi.org/10.18632/oncotarget.12278.
Sonda N, Simonato F, Peranzoni E, Cali B, Bortoluzzi S, Bisognin A, Wang E, Marincola FM, Naldini L, Gentner B, Trautwein C, Sackett SD, Zanovello P, Molon B, Bronte V. miR-142-3p prevents macrophage differentiation during cancer-induced myelopoiesis. Immunity. 2013;38(6):1236–49. https://doi.org/10.1016/j.immuni.2013.06.004.
Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, Bogner PN, Farren MR, Lee KP, Liu K, Abrams SI. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123(10):4464–78. https://doi.org/10.1172/JCI68189.
Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. 2011;286(26):23591–9. https://doi.org/10.1074/jbc.M111.237123.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Investig. 2015;125(9):3356–64. https://doi.org/10.1172/jci80005.
Porta C, Marino A, Consonni FM, Bleve A, Mola S, Storto M, Riboldi E, Sica A. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis. 2018;39(9):1095–104. https://doi.org/10.1093/carcin/bgy088.
Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, Wan X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol. 2019;10:1399. https://doi.org/10.3389/fimmu.2019.01399.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–8. https://doi.org/10.1016/j.tibs.2015.12.001.
Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, Locasale JW. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. elife. 2014;3:e03342. https://doi.org/10.7554/eLife.03342.
Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–95. https://doi.org/10.4049/jimmunol.1202702.
Yu Q, Dong L, Li Y, Liu G. SIRT1 and HIF1alpha signaling in metabolism and immune responses. Cancer Lett. 2018;418:20–6. https://doi.org/10.1016/j.canlet.2017.12.035.
LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18(4):356–65. https://doi.org/10.1038/ncb3330.
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496(7444):238–42. https://doi.org/10.1038/nature11986.
Goffaux G, Hammami I, Jolicoeur M. A dynamic metabolic flux analysis of myeloid-derived suppressor cells confirms immunosuppression-related metabolic plasticity. Sci Rep. 2017;7(1):9850. https://doi.org/10.1038/s41598-017-10464-1.
Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, Lu J, Yang Z, Wang J, Zhao Y. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. 2016;6:20250. https://doi.org/10.1038/srep20250.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42. https://doi.org/10.1016/j.immuni.2005.03.013.
Sica A, Strauss L, Consonni FM, Travelli C, Genazzani A, Porta C. Metabolic regulation of suppressive myeloid cells in cancer. Cytokine Growth Factor Rev. 2017;35:27–35. https://doi.org/10.1016/j.cytogfr.2017.05.002.
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77. https://doi.org/10.1158/0008-5472.CAN-09-2587.
Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43(3):435–49. https://doi.org/10.1016/j.immuni.2015.09.001.
Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, Ochoa AC. L-arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J Immunol. 2010;185(9):5198–204. https://doi.org/10.4049/jimmunol.1001224.
Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, Park HJ, Zabaleta J, Ochoa AC. L-arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. 2004;232(1–2):21–31. https://doi.org/10.1016/j.cellimm.2005.01.004.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. https://doi.org/10.1038/nri3175.
Grohmann U, Puccetti P. The coevolution of IDO1 and AhR in the emergence of regulatory T-cells in mammals. Front Immunol. 2015;6:58. https://doi.org/10.3389/fimmu.2015.00058.
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, Savino B, Colombo P, Jonjic N, Pecanic S, Lazzarato L, Fruttero R, Gasco A, Bronte V, Viola A. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208(10):1949–62. https://doi.org/10.1084/jem.20101956.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. https://doi.org/10.1126/science.1160809.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. https://doi.org/10.1016/j.ccr.2012.02.014.
Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16(11):732–49. https://doi.org/10.1038/nrc.2016.89.
Balaban S, Lee LS, Schreuder M, Hoy AJ. Obesity and cancer progression: is there a role of fatty acid metabolism? Biomed Res Int. 2015;2015:274585. https://doi.org/10.1155/2015/274585.
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, Klemm DJ, Woolthuis CM, Stranahan AW, Park CY, Jordan CT. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19(1):23–37. https://doi.org/10.1016/j.stem.2016.06.001.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236–47. https://doi.org/10.1158/2326-6066.CIR-15-0036.
Du H, Ding X, Yan C. Metabolic reprogramming of myeloid-derived suppressive cells. Onco Targets Ther. 2017;4(3–4):29–30. https://doi.org/10.18632/oncoscience.349.
Sica A, Strauss L. Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J Leukoc Biol. 2017;102(2):325–34. https://doi.org/10.1189/jlb.4MR1116-476R.
Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline science: high fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. 2018;103(3):395–407. https://doi.org/10.1002/JLB.4HI0517-210R.
Ostrand-Rosenberg S. Myeloid derived-suppressor cells: their role in cancer and obesity. Curr Opin Immunol. 2018;51:68–75. https://doi.org/10.1016/j.coi.2018.03.007.
Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, Ricciotti E, DiRusso C, Murphy ME, Vonderheide RH, Lieberman PM, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Lin C, Nefedova Y, Black P, Kagan VE, Gabrilovich DI. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–8. https://doi.org/10.1038/s41586-019-1118-2.
Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–70. https://doi.org/10.1016/j.cell.2015.08.015.
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507–13. https://doi.org/10.1158/0008-5472.CAN-06-4174.
Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR, Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71(7):2664–74. https://doi.org/10.1158/0008-5472.CAN-10-3055.
Wu H, Weidinger C, Schmidt F, Keye J, Friedrich M, Yerinde C, Willimsky G, Qin Z, Siegmund B, Glauben R. Oleate but not stearate induces the regulatory phenotype of myeloid suppressor cells. Sci Rep. 2017;7(1):7498. https://doi.org/10.1038/s41598-017-07685-9.
Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, Takeda S, Tafreshian KN, Rowinsky E, Szarek M, Waltzman RJ, McMillan EA, Zhao C, Mita M, Mita A, Chmielowski B, Postow MA, Ribas A, Mucida D, Tavazoie SF. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172(4):825–840.e818. https://doi.org/10.1016/j.cell.2017.12.026.
Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23(6):631–9. https://doi.org/10.1016/j.semcdb.2012.01.003.
Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23(7):351–63. https://doi.org/10.1016/j.tem.2012.05.001.
Chakraborty K, Raundhal M, Chen BB, Morse C, Tyurina YY, Khare A, Oriss TB, Huff R, Lee JS, St Croix CM, Watkins S, Mallampalli RK, Kagan VE, Ray A, Ray P. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat Commun. 2017;8:13944. https://doi.org/10.1038/ncomms13944.
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–391.e379. https://doi.org/10.1016/j.ccell.2017.08.004.
Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, Brewer HB, Fruchart JC, Clavey V, Staels B. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7(1):53–8. https://doi.org/10.1038/83348.
Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7(1):161–71. https://doi.org/10.1016/s1097-2765(01)00164-2.
Ishdorj G, Graham BA, Hu X, Chen J, Johnston JB, Fang X, Gibson SB. Lysophosphatidic acid protects cancer cells from histone deacetylase (HDAC) inhibitor-induced apoptosis through activation of HDAC. J Biol Chem. 2008;283(24):16818–29. https://doi.org/10.1074/jbc.M710177200.
Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354(6311):481–4. https://doi.org/10.1126/science.aaf6284.
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–80. https://doi.org/10.1126/science.1164097.
Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang X, Huang W, Seto E, Lin H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci U S A. 2019;116(12):5487–92. https://doi.org/10.1073/pnas.1815365116.
Sahakian E, Powers JJ, Chen J, Deng SL, Cheng F, Distler A, Woods DM, Rock-Klotz J, Sodre AL, Youn JI, Woan KV, Villagra A, Gabrilovich D, Sotomayor EM, Pinilla-Ibarz J. Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63(2):579–85. https://doi.org/10.1016/j.molimm.2014.08.002.
Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, Partlova S, Garfall A, Vogl DT, Xu X, Knight SC, Malietzis G, Lee GH, Eruslanov E, Albelda SM, Wang X, Mehta JL, Bewtra M, Rustgi A, Hockstein N, Witt R, Masters G, Nam B, Smirnov D, Sepulveda MA, Gabrilovich DI. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2):aaf8943. https://doi.org/10.1126/sciimmunol.aaf8943.
Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, Luckner C, Doschko G, Soler G, Eichmann K, Muller FM, Ho AD, Goerner M, Modolell M. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105(6):2549–56. https://doi.org/10.1182/blood-2004-07-2521.
Masson D, Jiang XC, Lagrost L, Tall AR. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J Lipid Res. 2009;50(Suppl):S201–6. https://doi.org/10.1194/jlr.R800061-JLR200.
Ding S, Li S, Zhang S, Li Y. Genetic alterations and checkpoint expression: mechanisms and models for drug discovery. Adv Exp Med Biol. 2020;1248:227–50. https://doi.org/10.1007/978-981-15-3266-5_10.
Li Y, Di Santo JP. Modeling infectious diseases in mice with a “humanized” immune system. Microbiol Spectr. 2019;7(2) https://doi.org/10.1128/microbiolspec.BAI-0019-2019.
Meraz IM, Majidi M, Meng F, Shao R, Ha MJ, Neri S, Fang B, Lin SH, Tinkey PT, Shpall EJ, Morris J, Roth JA. An improved patient-derived xenograft humanized mouse model for evaluation of lung cancer immune responses. Cancer Immunol Res. 2019;7(8):1267–79. https://doi.org/10.1158/2326-6066.CIR-18-0874.
Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, Fan Y, Yang H, Lyer SG, Bonney GK, Loh E, Chang KTE, Tan TC, Zhai W, Chan JKY, Chow EK, Chee CE, Lee GH, Dan YY, Chow PK, Toh HC, Lim SG, Chen Q. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67(10):1845–54. https://doi.org/10.1136/gutjnl-2017-315201.
Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8. https://doi.org/10.4049/jimmunol.172.5.2731.
Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163(1):39–53. https://doi.org/10.1016/j.cell.2015.08.068.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Liu, W., Song, H., Li, X., Ren, D., Ding, S., Li, Y. (2021). Lipid Metabolism in Tumor-Associated Myeloid-Derived Suppressor Cells. In: Li, Y. (eds) Lipid Metabolism in Tumor Immunity. Advances in Experimental Medicine and Biology, vol 1316. Springer, Singapore. https://doi.org/10.1007/978-981-33-6785-2_7
Download citation
DOI: https://doi.org/10.1007/978-981-33-6785-2_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-33-6784-5
Online ISBN: 978-981-33-6785-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)