
Abstract
Apoptosis is programmed cell death, which sustains the equilibrium between survival and death in eukaryotic cells. It is a tightly regulated cell death program that aims at eliminating harmful, damaged, or unwanted cells. This wisely programmed cell death is central in the development of all multicellular organisms, which is highlighted by the prevalence of diseases associated with abnormal apoptosis. For example, defect in apoptosis is a hallmark of cancer, whereas excessive cell death occurs in several neurodegenerative disorders. The cell death signals are responsible for maintenance of the genomic integrity, while defective cell death may stimulate carcinogenesis. These signals are convoluted and are controlled at various points. Tumor cells survive by taking help of several different molecular mechanisms to inhibit apoptosis and acquire resistance to apoptotic agents, for example, by the expression of anti-apoptotic proteins such as Bcl-2 or by the downregulation or mutation of pro-apoptotic proteins such as BAX. This chapter includes recent developments in the field and reviews new evidences of the interconnection between apoptosis and cancer. Various molecules that can be regulated to facilitate apoptosis in myriad of cancers are also enlisted. Overall, the chapter discusses about the development of various treatments and approaches to combat cancer by targeting anti-apoptotic proteins belonging to Bcl-2 and IAP families.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
5.1.1 Apoptosis: A Programmed Cell Death
The average adult human body makes around 50–70 billion new cells every day by somatic cell division also called mitosis. Therefore, to maintain a constant number of cells in the body, they must also die a natural death. In humans, about a million of cells are made every second through mitosis, whereas a similar number is destroyed by the process of apoptosis – this is known as cellular homeostasis (Vaux and Korsmeyer 1999). The word “apoptosis” (Greek origin) is divided into two parts, apo (from) and ptosis (falling), that describes falling of leaves from a tree (Wong 2011). Including apoptosis, there are various types and subtypes of cell death. The primary ones are as follows:
-
1.
Necrosis: Uncontrolled cell death. It is generally the outcome of cellular injury and/or infection. Phenotypically, the cells burst, emitting their contents into the surrounding tissue fluid; this stimulates inflammation, pain, and swelling.
-
2.
Apoptosis (programmed cell death): It is a programmed, multistep pathway that leads to cell death for the normal functioning of cellular growth and tissue homeostasis. It is developed as an irreversible practice to proficiently remove nonfunctional and old cells (Letai 2008). There are stepwise events, such as enzyme activation, required for this (unlike necrosis). In apoptosis, genetic control is preserved till the end (Vo and Letai 2010).
-
3.
Autophagy: It is a naturally occurring controlled mechanism by which a cell can dismantle its redundant and defective components in a systematic manner (Yu et al. 2004).
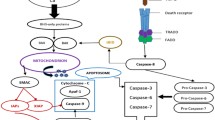
Generally, the morphological features of a dying cell decide the mode of cell death. Consequently, apoptosis has been designated as type I programmed cell death, autophagy as type II, and necrosis as a type of death that combines characteristics of both type I and type II. With the advent of new biochemical methods over the past few decades, a more conclusive categorization of cell death types has become possible. Cell death classifications have yielded categories which render to biochemical characteristics, morphological features, immunological facets, or functional references. Normally, cell cleaning process or apoptosis starts with breaking down of the genetic materials across the nuclear margins, followed by cell fragmentation and generation of cell debris, which in turn are consumed by phagocytes (Figs. 5.1 and 5.2). A series of biochemical events leading to apoptosis includes activation of proteases like caspases, cleavage of DNA and proteins, and exposure of phosphatidylserine on the surface of the cell. Along with elimination of unwanted cells and tissues, apoptosis also plays a major role in development of the embryo and neurologic pruning. Categorically, phases of apoptosis include the initiation phase and execution phase (Kroemer et al. 2007).

Ultrastructural examination of typical apoptotic cell using transmission electron microscopy (TEM). Untreated control HEK293T cell. (a) exhibit normal cell morphology including normal cell nuclei, cytoplasm, and cell organelles. (b) Drug-treated cell indicates the appearance characteristics of apoptotic cell such as membrane blebbing, nuclear condensation, and damage to cell organelles. Magnification, x8,000. Image is under publication (Bose Lab, unpublished results)

Hallmarks of apoptotic cell death. Cartoon illustrating the basic stages of the apoptotic pathway. Apoptosis includes cellular shrinking, chromatin condensation, and margination at the nuclear periphery with the eventual formation of membrane-bound apoptotic bodies that contain organelles, cytosol, and nuclear fragments
5.1.2 Initiation Phase
This phase of apoptosis mostly involves a complex entanglement of a number of proteins. It depends on the cell type, apoptotic stimulus, and activation of proteases, nucleases, and other effector molecules. Normally, it gets activated by several intracellular and extracellular stresses (Bender and Martinou 2013). Extracellular signals that induce apoptosis include dysfunctioning growth factors, hypoxia, and environmental radiation. Intracellular signals include DNA damage, damage caused by chemotherapeutic drugs, telomere malfunction, and viral infections. The two different important pathways that mediate initiation phase are discussed in the following sections.
5.1.2.1 Extrinsic Pathway Mediated by Receptor
It is induced by external events of cells mediated by the death receptors. In this pathway, members of the tumor necrosis factor (TNF) receptor superfamily of transmembrane proteins are mainly involved. The most notable feature of TNF receptors is a region called “death domain” which roughly consists of 80 amino acids (Bradley and Pober 2001). Transferring death signals across cellular membrane is the most significant role of these death domain regions. Two most well-known death receptors are the type 1 TNF receptor (TNFR-1) and a related protein called Fas (CD95). Upon receiving apoptotic stimuli, receptors trimerize after binding to their respective extracellular ligands, TNF and Fas ligand (FasL) (Hengartner 2001). Post trimerization, these death receptors recruit adapter proteins such as TNF receptor-associated death domain (TRADD) and Fas-associated death domain (FADD) through homotypic protein-protein interactions. Subsequently, the adaptor molecules bind to initiator cysteine proteases such as caspase 8/10 through similar homotypic interactions and that forms a complex called death inducing signaling complex (DISC) (Singh et al. 2016). DISC formation is a prerequisite of autoactivation of upstream or initiator caspases 8/10 with the subsequent activation of downstream caspases 3/7 (Wong 2011) that lead to initiation of the caspase cascade, hence apoptosis (Schneider and Tschopp 2000). Extrinsic cell death pathway is strongly controlled by relative expression of both pro-apoptotic and anti-apoptotic proteins, and a disproportion in that regulation leads to several diseases.
5.1.2.1.1 Defects in Extrinsic Pathway Signaling and Carcinogenesis
Genetic studies on mice in which certain genes encode death ligands or their receptors have given sufficient proof of role in tumor suppression by cellular immune mechanisms. Fas ligand (FasL) is essential when there is cytotoxic T lymphocytes (CTL)-mediated killing of tumor cells, and TRAIL/Apo2L (TNF-related apoptosis-inducing ligands) is important for natural killer (NK)-cells-mediated tumor suppression. However, a number of cancer cells escape immune annihilation by resisting the response of the death receptor pathway to FasL. Although numerous available cancer cell lines express various cell surface receptors, they show an inherent resistance to TRAIL. It somehow indicates that during the course of evolution of tumors in vivo, an enforced selection of malignant clones takes place, which are capable of defying immune attack. Some of the examples are downregulation of the Fas receptor, abundance of nonfunctioning or precursor of Fas receptors, and generation of large amount of soluble Fas receptor. Moreover, some cancer cells can execute a “counterattack” facilitated by Fas ligand that results in apoptotic reduction of activated tumor-sensitive lymphocytes (Koyama et al. 2001). Thus, it is very important to have a promising therapeutic approach that can rely on making extrinsic pathway to act on nonessential cells.
5.1.2.2 Intrinsic Pathway or In-House (Mitochondria Dependent)
This pathway is initiated by internal cell death gestures, mainly severe nuclear DNA damage, low oxygen content in the cells, viral infection, etc. It is majorly controlled by proteins in the Bcl-2 (B-cell lymphoma 2) family, comprising a total of 25 known proteins. They are involved to either excite (pro-apoptotic) or inhibit apoptosis (anti-apoptotic) (Hengartner 2000).Under normal conditions, there is an equilibrium among pro-apoptotic and anti-apoptotic proteins present in the cell.
The BH3-only (Bcl-2 Homology-3) proteins when respond to signs, such as loss of genetic material to undergo apoptosis, position themselves to the mitochondrial outer membrane and trigger the two important proteins Bax or Bak. When stimulated, Bax and Bak heterodimer cause mitochondrial outer membrane permeabilization (MOMP) (Bender and Martinou 2013). This heterodimer penetrates the membrane and creates pores all over the membrane that help other apoptotic factors like cytochrome-c to come out into the cytosol. Later on, it binds to caspase 3 forming a crucial compound/complex known as apoptosome comprising cytochrome-c, Apaf-1 (actin filament-associated protein 1), and nonfunctional initiator caspase-9 (Kroemer et al. 2007). The newly formed composite structure works by hydrolyzing adenosine triphosphate (ATP) and form a functional caspase 9. The active/functional caspase 9 further reacts with the executioner caspases 3, 6, and 7 to carry out their respective functions that ultimately help in cellular apoptosis. This pathway is distinct from the one emanating from extracellular signals. Apart from them, some apoptotic factors like Apoptosis-inducing factor (AIF), second mitochondria-derived activator of caspase (SMAC), and Omi/high-temperature requirement protein A-2 (HtrA2) are also exuded out from the intermembrane space of mitochondria into the cytoplasm (Kroemer et al. 2007).
5.1.2.2.1 Defects in Intrinsic Pathway Signaling and Carcinogenesis
One of the key proteins found to be associated with intrinsic pathway of apoptosis is caspase 9, whose impairment leads to detrimental consequences. For example, caspase-9-knockout mice show juvenile death due to abnormalities in the brain structure/function due to accumulation of increased number of unnecessary cells (Hakem et al. 1998; Kuida et al. 1998). Furthermore, lymphocytes within the thymus glands from the knockout mice display susceptibility toward common variety of anticancer drugs and chemo-radiations (Kuida et al. 1998).
There are reports suggesting reduced expression of caspase 9 in the epithelial cells from colorectal cancer patient’s cells related to cells from healthy individual (Hector et al. 2012). Apart from that, anti-apoptotic proteins (e.g., Bcl-2, Bcl-XL (B-cell lymphoma 2-Xetra large), Bcl-2 L2 (B-cell lymphoma 2 like protein 2), and Mcl-1 (induced myeloid leukemia cell differentiation protein)), which have conserved BH1–BH4 domains are known to promote cell survival, whereas pro-apoptotic proteins are associated with stress-dependent apoptosis. There is a second group of multidomain BH3-only proteins such as Bax and Bak, which are essential for apoptosis (Reed 2006). Anti-apoptotic proteins block apoptosis by counteracting the actions of Bax/Bak complex through a known mechanism (Green 2006). Overexpression of anti-apoptotic Bcl-2 or Bcl-XL occurs in more than half of all cancers (Amundson et al. 2000) by rendering cancer cells resistant to apoptotic stimuli that also include majority of cytotoxic anticancer drugs.
5.1.2.3 Perforin/Granzymes
As a part of human cellular immunity, in response to several stress conditions, cells such as cytotoxic T lymphocytes can cause apoptosis. The procedure involves activation of such cells followed by secretion of special proteins called perforin and its associated enzymes. Their main job is to form pores onto the cell membrane of the target cell. The additional particles use these pores to enter the cell. After entering the cell, they release their enzymes (granzymes A and B) that start execution of apoptosis by causing destruction of cellular structure and function (Trapani and Smyth 2002).
5.1.3 Execution Phase
The initiation phase activates the execution phase. The execution phase involves the activation of a group of cysteine proteases named as caspases which plays cell execution (Wong 2011; Bender and Martinou 2013). These proteins are ubiquitously present in all cell types, and their expression is enough to carry out its function. Among all, caspase 3 is a well-known executioner caspase. Once this protein takes charge as an executioner, apoptosis is definite. It is activated in major functions mainly DNA and chromatin damage, cell division, as well as signal transduction.
5.2 Resistance to Apoptosis in Cancer
Uncontrolled proliferation of cells give birth to variety of cancers (King and Cidlowski 1998). Moreover, the role of malfunctioning apoptotic machineries during cancer metastasis is well documented in myriad of cancers (Kerr and Searle 1972). Normally, tumor cells use various molecular mechanisms to acquire resistance to apoptosis. These processes mostly include upregulation of anti-apoptotic proteins like Bcl-2 and downregulation of pro-apoptotic proteins such as BAX. As regulation of both proteins is controlled/regulated by the p53 gene (Miyashita et al. 1994), some forms of B-cell lymphoma show Bcl-2 overexpression (Fig. 5.3). Therefore, many similar examples represent the first line of evidences that failure of apoptosis contributes to cancer (Vaux 1998).

Disrupted balance between Bcl-2 family of proteins: Many proteins have been reported to exert pro- or anti-apoptotic activity in the cell. It is not the absolute quantity but rather the ratio of these pro- and anti-apoptotic proteins that plays an important role in the regulation of cell death. Besides, over- and underexpression of certain genes have been found to contribute to carcinogenesis by reducing apoptosis in the cancer cells
5.3 Disrupted Balance of Pro-apoptotic and Anti-apoptotic Proteins
5.3.1 Role of Bcl-2 Family of Proteins in Apoptosis
This family of proteins is considered as a chief doorkeeper to the apoptotic response. Gene involving Bcl-2 family of proteins was first identified as one of the promoters of cell death whose functioning revolved around activation of pro-apoptotic proteins or inhibition of anti-apoptotic proteins. Anti- and pro-apoptotic members belonging to this group of structurally related proteins interact with each other. Among them, one of the subgroups include Bid, Bad, Bim, Bmf, Puma, and Noxa proteins that contain a single Bcl-2 homology 3 domain (BH3-only proteins) and have pro-apoptotic activity. Two other protein subcategories have multiple BH domains. The first subcategory, including Bcl-2-associated X protein (Bax), Bcl-2 homologous antagonist/killer (Bak), and Bcl-2 family apoptosis regulator (Bok), is pro-apoptotic; the other subcategory, including Bcl-2, Bcl-XL, and Mcl-1, is anti-apoptotic (Fig. 5.4). At least one BH domain is confined in each of the Bcl-2 family members and contributes to the functioning of the members (Strasser et al. 2011).

Bcl-2 family: organization, functions, and characteristics. Schematic shows the important Bcl-2 family proteins in humans along with the main conserved structural motifs (BH domains), grouped according to function. TM, transmembrane domain. BH, Bcl-2 homology
The Bcl-2 family members can be classified into 3 functional groups: anti-apoptotic, pro-apoptotic effectors, and pro-apoptotic activators. According to a number of literature reports, pro-apoptotic activators, which comprise BH3 motifs, mostly function as important mediators in stress-related cellular responses like DNA damage (Certo et al. 2006). However, effectors group of Bcl-2 proteins initiates pore formation in the mitochondrial membrane after being activated by BH3 containing activators (Garcia-Saez 2012; Wei et al. 2001).
However, in preclinical models, it has been established that Bcl-2 binding to BH3-only activators disrupts the interactions between activators and pore-forming effectors, which in turn refrain the effectors from forming mitochondrial pores. The dynamic balance occurring between anti-apoptotic and pro-apoptotic members may involve combinations of under- or overexpression of one or more anti-apoptotic as well as pro-apoptotic proteins. This further helps to determine whether the cell initiates apoptosis or not (Strasser et al. 2011; Garcia-Saez 2012). For example, there are reports on Bcl-2 overexpression resulting survival of prostate tumor cells from apoptosis (Raffo et al. 1995). Moreover, in breast carcinoma and neuroblastoma cancer cells, overexpression of Bcl-2 is observed which leads to reduced TRAIL-induced apoptosis (Fulda et al. 2002). Similarly, various in vivo studies show that in c hronic lymphocytic leukemia (CLL) cells, Bcl-2/Bax ratio increases with the decrease in pro-apoptotic Bax protein. Moreover, it is also seen that apoptosis induced by drug application decreases the ratio and rate of drug induction is inversely proportional to the ratio (Goolsby et al. 2005).
5.3.2 Tumor Cells May Become Dependent on Bcl-2 for Survival
Recently, a phenomenon known as “oncogene addiction” has been identified. It is the dependence of a cancer cell on one signaling pathway or hyperactive gene for the cell’s survival and growth. Similar oncogene addiction is also seen in various cancer cells whose survival become directly dependent on pathways involving Bcl-2 proteins (Certo et al. 2006). In case of these tumor cells, the pro-apoptotic activators get sequestered due to Bcl-2 binding. However, with a sufficient increase in the activators quantity, Bcl-2 sequestering by those activators can be invoked resulting apoptosis. Hence, this sensitivity toward Bcl-2 modulation can be used as a therapeutic tool for the treatment of Bcl-2-dependent cancers (Deng et al. 2007).
5.4 Expression Levels in Various Cancers
Expressions of functional Bcl-2 (increased ratio of anti-apoptotic to pro-apoptotic proteins) alone can regulate the extent of developing cancer. Following are the different cancer types and its association with aberrant expression of Bcl-2 blood cancer/leukemia.
CLL patients have always shown a noticeable Bcl-2 gene rearrangement with upregulated expression (Hanada et al. 1993). Increased expression is also found frequently in a cute m yeloid l eukemia (AML) (Wei et al. 2001) and in almost all patients with a cute l ymphocytic l eukemia (ALL) (Gala et al. 1994).
5.4.1 Non-Hodgkin lymphoma
In follicular lymphoma, most of the patients shows a t(14;18) chromosomal translocation in abnormal cells (Tsujimoto et al. 1984), which is believed to cause overexpression of Bcl-2 protein (Chen-Levy et al. 1989). This is true with diffused large B-cell lymphoma patients, in whom relatively high Bcl-2 expression is found (Hermine et al. 1996).
5.4.2 Cancerous Solid Tumors
Involvement of Bcl-2 in non-hematologic cancers like small cell carcinoma, breast carcinoma, and prostate and lung cancers has been observed where its unregulated expressions lead to aforementioned malignancies (Karnak and Xu 2010; Hellemans et al. 1995; Jiang et al. 1995; Anagnostou et al. 2010). In case of lung cancer, overexpression in >85% of patients has been reported (Jiang et al. 1995). Moreover, in neuroblastoma, colorectal, ovarian, and urinary bladder cancers, substantial levels of Bcl-2 expression have been observed (Henriksen et al. 1995; Lamers et al. 2012; Swellam et al. 2004; Zhao et al. 2005; Pena et al. 1999) (Table 5.1).
5.5 The p53 Protein
Tumor protein 53 (or TP 53) is a tumor-suppressor protein encoded by the TP53 gene, due to its specific molecular weight, that is, 53 kDa. The association of p53 with cancer surfaced in the late 1990s, which showed that gain of its oncogenic function was the direct outcome of its mutation (Levine et al. 1991). p53 is widely known as the “guardian of the genome” due to its significant role in a large number of cellular processes like chromosomal segregation, cell cycle regulation, recombination of genetic material, gene amplification, as well as apoptosis (Oren and Rotter 1999; Lane 1992). More than 50% of different types of human cancers are associated with malfunctioning of p53 gene (Levine et al. 1991). Recently, abnormal expression of p53 has also been reported in melanoma cells where its reduced activity leads to melanoma cells proliferation (Avery-Kiejda et al. 2011) Similarly, one of the research groups observed decreased survival, reduced apoptosis, and enhanced level of proliferative markers in a mouse model expressing truncated version of p53(Δ122p53) (Slatter et al. 2011). Moreover, p53 knockdown also decreases the growth of cellular colonies in malignant tumor cells, which is due to apoptotic induction (Vikhanskaya et al. 2007).
5.6 Inhibitors of Apoptosis Proteins (IAPs)
Regulation of apoptosis involves a group of structurally related and functionally analogous group of proteins called the IAPs. The family is characterized by having one or two Baculovirus IAP repeat (BIR) protein domain (LaCasse et al. 2008). Since then, eight IAPs have been discovered, namely, Apollon (BRUCE, BIRC6), c-IAP1 (BIRC2), c-IAP2 (BIRC3), IAP-like protein 2 (BIRC8), Livin/ML-IAP (BIRC7), NAIP (BIRC1), survivin (BIRC5), and X-linked IAP (XIAP, BIRC4) (Vucic and Fairbrother 2007). Generally, they are involved in inhibition of caspase activity by either degrading the active forms of caspases or holistically interacting with them to prevent substrate-caspase binding (Wei et al. 2008).
It has been reported that in many cancers, there is a versatile expression of IAP proteins. For example, unusual levels of IAP were identified in the pancreatic cancer cells due to its link with chemoresistance. A study has further reported that there is a significant increase in the levels of cIAP-2 in many cancers (Lopes et al. 2007). According to recent reports, in cancers such as melanoma and lymphoma, significantly high levels of Livin/ML-IAP (BIRC7) has been reported (Vucic et al. 2000; Ashhab et al. 2001).While in gliomas, Apollon (BRUCE, BIRC6) upregulation exhibited chemoresistance for drugs cisplatin and camptothecin (Chen et al. 1999).Overexpression of survivin in various cancers has also been described. Together with XIAP, overexpression of survivin has been observed in non-small-cell lung carcinomas (NSCLCs). According to their study, tumors that upregulate both the proteins are capable of chemoresistance in all the apoptosis-inducing conditions (Krepela et al. 2009).
5.7 Potential Treatment Strategies Against Cancer
Role of dysregulated apoptosis pathways not only is limited to initiation of malignancy and cancer development but also leads to chemo- and radiation-resistant tumor cells. Initial responses of the cancer cells toward different therapies subside gradually as the cells start resisting apoptosis by increasing anti-apoptotic protein expressions which in turn block the pro-apoptotic pathways. Therefore, modern cancer research and drug development predominantly involve in finding the fundamental mechanisms governing apoptosis which may help in constructing therapeutic strategies to combat with cancer resistance and enhance patient survival.
5.7.1 Bcl-2 Antagonists
Bcl-2 family of proteins can be therapeutically targeted by developing Bcl-2 antagonists which may inhibit their activities as well as silence the upregulated anti-apoptotic genes or proteins. Significant role of Bcl-2 proteins in disrupting apoptosis has led to several drug-based advances in twenty-first century, which focus mainly on developing molecules that antagonize Bcl-2 family of proteins and quantitatively reduce their levels in cells. For example, a drug has been developed on the basis of an antisense nucleotide to bcl-2 and has been successfully tested in a wide range of human cancers (Frankel 2003). Moreover, Bcl-2 antagonists have been designed by a number of research groups to primarily mimic BH3-only peptides (Baell and Huang 2002; Kutzki et al. 2002; Becattini et al. 2004; Qian et al. 2004). These agents are useful in treating cancers such as follicular lymphoma in which elevated levels of Bcl-2 occur.
5.7.2 SMAC (Second Mitochondria-Derived Activator of Caspase)/Diablo Mimetics
Development of small molecule and peptide mimetics that mimic SMAC can be another novel approach as it is a pro-apoptotic mitochondrial protein that is also an endogenous inhibitor of the IAP family of cellular proteins.
IAPs represent the last line of defense for the cancer cells against apoptosis. Clinically, IAPs have been proven to be a significant factor in cancer cell survival, development, and poor prognosis. Their association with tumor resistance to therapies is considered as one of the important therapeutic targets to selectively induce apoptosis in tumor cells.
The main objective behind designing SMAC mimetics (also called IAP antagonists) is to downregulate cellular IAPs that ultimately induce cancer cell death.
One of the SMAC mimetics, TL32711, in its clinical studies on patient-derived tumor xenograft models has successfully led to tumor regression.
Moreover, it also helps in preserving cancer cell sensitivity toward stimulation generated by pro-apoptotic elements like TNF-α or TRAIL, as shown in an in vitro human-derived cancer cell lines.
Collectively, these mimetics has been a great deal in cancer research and therapeutics with great potential to overcome the limitations of current anticancer therapies.
5.7.3 MDM2-p53 Complex
Novel agents that bind MDM2 (mouse double minute 2 homolog) have been developed that displace p53 from the complex, thereby activating the p53 pathway. p53 activation subsequently leads to cell cycle arrest and apoptosis (Vassilev et al. 2004).Development of such agents as drugs would have the advantage of specifically targeting tumors overexpressing MDM2, leaving the normal cells aside.
5.7.4 Death Receptor Ligands (TNF and TRAIL)
Tumor necrosis factor (TNF) is a well-known anticancer agent in animal models, and has been found to cause death of tumor cells. Interestingly, it acts on the surrounding cells that are associated with feeding the tumor (Stoelcker et al. 2000).
Another important molecule, TRAIL (LeBlanc and Ashkenazi 2003), acts directly on cancer cells and causes apoptosis. It has been well established that TRAIL knockout mice works hand in hand with natural killer (NK) cells in order to keep metastasis from happening (Takeda et al. 2001; Cretney et al. 2002). Moreover, induction of cell death has also been observed in tumor cells from various cancer patients due to the activation of TRAIL-producing macrophages which further release biological molecules that consequently result in upregulation of particular markers on tumor cells (Herbeuval et al. 2003).
5.7.5 Monoclonal Abs (Rituximab) and Apoptosis in Cancer
One of the most potent agents for treatment of lymphocytic cancer such as non-Hodgkin’s lymphoma is a monoclonal antibody (Rituximab) against the antigen B220 present in B-cells. This monoclonal antibody mainly acts by inducing apoptosis that ultimately leads to cell death (Cartron et al. 2004).
5.8 Conclusion
Cancer Is a Failure of Apoptosis
Apoptosis or programmed cell death is one of the ways through which the cell cycle is maintained and kept under check. This basic defense mechanism has always been the main reason why cancer cells do not thrive and lead to abnormal cell growth or cancer. For example, cells from the epidermal layer of the skin when exposed to harmful ultraviolet radiations in this case as a part of defense mechanism, programmed cell death is normally activated.
This helps in eliminating those injured cells that otherwise would survive and develop into cancerous growths.
In metastasis or malignant cancer, in order be alive, cells always travel from one organ to the other through the blood system.
This is usually prevented by apoptosis as cells typically “self-destroy” when they lose contact with neighboring cells or the extracellular matrix (Hanahan and Weinberg 2000).
One of the hallmarks of cancer is to escape apoptosis (Hanahan and Weinberg 2011). Cancer cells display many characteristics; for example, they interfere into cell cycle checkpoints and withstand exposure to cytotoxic agents (Letai 2008) that subsequently lead to their survival. Since apoptosis is an important impediment toward developing cancer, avoiding apoptosis is integral to tumor development and resistance to therapy (Cory et al. 2003; Plati et al. 2011). Cancer cells are capable of escaping apoptosis and continuously dividing. One of the important reasons for this behavior is loss of tumor-suppressor p53.
p53 knockout cells are incapable of sensing DNA damage that drives apoptosis (Hanahan and Weinberg 2000). Anti-apoptotic Bcl-2 family members and IAPs also promote cell proliferation when upregulated (Vo and Letai 2010). Although there has been a huge advancement in understanding the apoptotic pathway and targeting it for therapeutic intervention, the challenges in this area are still manifold. Complex machineries of cancer cells enable them to develop resistance to apoptosis by acquiring new mutations which nullify the drug-induced targeted therapies. For example, apoptosis is initiated in cancer cells when drug inhibits the activity of Bcl-2 family proteins. However, a new mutation generated in the cancer cells may upregulate the caspase inhibitors, and the drug will not be effective anymore (Wong 2011). Therefore, a comprehensive understanding of the apoptotic pathway along with the cross talks among different pro- and anti-apoptotic proteins might open new avenues in devising new strategies to effectively combat cancer.
References
Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96(2):245–254
Wong RS (2011) Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30:87
Letai AG (2008) Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer 8(2):121–132
Vo TT, Letai A (2010) BH3-only proteins and their effects on cancer. Adv Exp Med Biol 687:49–63
Yu L et al (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304(5676):1500–1502
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1):99–163
Bender T, Martinou JC (2013) Where killers meet--permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb Perspect Biol 5(1):a011106
Bradley JR, Pober JS (2001) Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20(44):6482–6491
Hengartner MO (2001) Apoptosis: corralling the corpses. Cell 104(3):325–328
Singh N, Hassan A, Bose K (2016) Molecular basis of death effector domain chain assembly and its role in caspase-8 activation. FASEB J 30(1):186–200
Schneider P, Tschopp J (2000) Apoptosis induced by death receptors. Pharm Acta Helv 74(2–3):281–286
Koyama S, Koike N, Adachi S (2001) Fas receptor counterattack against tumor-infiltrating lymphocytes in vivo as a mechanism of immune escape in gastric carcinoma. J Cancer Res Clin Oncol 127(1):20–26
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407(6805):770–776
Hakem R et al (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94(3):339–352
Kuida K et al (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94(3):325–337
Hector S et al (2012) Clinical application of a systems model of apoptosis execution for the prediction of colorectal cancer therapy responses and personalisation of therapy. Gut 61(5):725–733
Reed JC (2006) Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ 13(8):1378–1386
Green DR (2006) At the gates of death. Cancer Cell 9(5):328–330
Amundson SA et al (2000) An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res 60(21):6101–6110
Trapani JA, Smyth MJ (2002) Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2(10):735–747
King KL, Cidlowski JA (1998) Cell cycle regulation and apoptosis. Annu Rev Physiol 60:601–617
Kerr JF, Searle J (1972) A mode of cell loss in malignant neoplasms. J Pathol 106(1):xi
Miyashita T et al (1994) Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9(6):1799–1805
Vaux DL (1998) Immunopathology of apoptosis--introduction and overview. Springer Semin Immunopathol 19(3):271–278
Strasser A, Cory S, Adams JM (2011) Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J 30(18):3667–3683
Certo M et al (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9(5):351–365
Garcia-Saez AJ (2012) The secrets of the Bcl-2 family. Cell Death Differ 19(11):1733–1740
Wei MC et al (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292(5517):727–730
Raffo AJ et al (1995) Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res 55(19):4438–4445
Fulda S, Meyer E, Debatin KM (2002) Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 21(15):2283–2294
Goolsby C et al (2005) Bcl-2 regulatory pathway is functional in chronic lymphocytic leukemia. Cytometry B Clin Cytom 63(1):36–46
Deng J et al (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12(2):171–185
Hanada M et al (1993) bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 82(6):1820–1828
Gala JL et al (1994) High expression of bcl-2 is the rule in acute lymphoblastic leukemia, except in Burkitt subtype at presentation, and is not correlated with the prognosis. Ann Hematol 69(1):17–24
Tsujimoto Y et al (1984) Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226(4678):1097–1099
Chen-Levy Z, Nourse J, Cleary ML (1989) The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol Cell Biol 9(2):701–710
Hermine O et al (1996) Prognostic significance of bcl-2 protein expression in aggressive non-Hodgkin's lymphoma. Groupe d’Etude des Lymphomes de l’Adulte (GELA). Blood 87(1):265–272
Karnak D, Xu L (2010) Chemosensitization of prostate cancer by modulating Bcl-2 family proteins. Curr Drug Targets 11(6):699–707
Hellemans P et al (1995) Prognostic value of bcl-2 expression in invasive breast cancer. Br J Cancer 72(2):354–360
Jiang SX et al (1995) Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. J Pathol 177(2):135–138
Anagnostou VK et al (2010) High expression of BCL-2 predicts favorable outcome in non-small cell lung cancer patients with non squamous histology. BMC Cancer 10:186
Henriksen R, Wilander E, Oberg K (1995) Expression and prognostic significance of Bcl-2 in ovarian tumours. Br J Cancer 72(5):1324–1329
Lamers F et al (2012) Targeted BCL2 inhibition effectively inhibits neuroblastoma tumour growth. Eur J Cancer 48(16):3093–3103
Swellam M et al (2004) Incidence of Bcl-2 expression in bladder cancer: relation to schistosomiasis. Clin Biochem 37(9):798–802
Zhao DP et al (2005) Prognostic significance of bcl-2 and p53 expression in colorectal carcinoma. J Zhejiang Univ Sci B 6(12):1163–1169
Pena JC et al (1999) Bcl-xL and Bcl-2 expression in squamous cell carcinoma of the head and neck. Cancer 85(1):164–170
Campos L et al (1993) High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81(11):3091–3096
Huang JZ et al (2002) The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood 99(7):2285–2290
Iqbal J et al (2006) BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol 24(6):961–968
Levine AJ, Momand J, Finlay CA (1991) The p53 tumour suppressor gene. Nature 351(6326):453–456
Oren M, Rotter V (1999) Introduction: p53 – the first twenty years. Cell Mol Life Sci 55(1):9–11
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358(6381):15–16
Avery-Kiejda KA et al (2011) P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 11:203
Slatter TL et al (2011) Hyperproliferation, cancer, and inflammation in mice expressing a Delta133p53-like isoform. Blood 117(19):5166–5177
Vikhanskaya F et al (2007) Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53. Nucleic Acids Res 35(6):2093–2104
LaCasse EC et al (2008) IAP-targeted therapies for cancer. Oncogene 27(48):6252–6275
Vucic D, Fairbrother WJ (2007) The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res 13(20):5995–6000
Wei Y, Fan T, Yu M (2008) Inhibitor of apoptosis proteins and apoptosis. Acta Biochim Biophys Sin Shanghai 40(4):278–288
Lopes RB et al (2007) Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer 120(11):2344–2352
Vucic D et al (2000) ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 10(21):1359–1366
Ashhab Y et al (2001) Two splicing variants of a new inhibitor of apoptosis gene with different biological properties and tissue distribution pattern. FEBS Lett 495(1–2):56–60
Chen Z et al (1999) A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 264(3):847–854
Krepela E et al (2009) Increased expression of inhibitor of apoptosis proteins, survivin and XIAP, in non-small cell lung carcinoma. Int J Oncol 35(6):1449–1462
Frankel SR (2003) Oblimersen sodium (G3139 Bcl-2 antisense oligonucleotide) therapy in Waldenstrom's macroglobulinemia: a targeted approach to enhance apoptosis. Semin Oncol 30(2):300–304
Baell JB, Huang DC (2002) Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem Pharmacol 64(5–6):851–863
Kutzki O et al (2002) Development of a potent Bcl-x(L) antagonist based on alpha-helix mimicry. J Am Chem Soc 124(40):11838–11839
Becattini B et al (2004) Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem Biol 11(3):389–395
Qian J et al (2004) Discovery of novel inhibitors of Bcl-xL using multiple high-throughput screening platforms. Anal Biochem 328(2):131–138
Vassilev LT et al (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303(5659):844–848
Stoelcker B et al (2000) Tumor necrosis factor induces tumor necrosis via tumor necrosis factor receptor type 1-expressing endothelial cells of the tumor vasculature. Am J Pathol 156(4):1171–1176
LeBlanc HN, Ashkenazi A (2003) Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 10(1):66–75
Takeda K et al (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 7(1):94–100
Cretney E et al (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168(3):1356–1361
Herbeuval JP et al (2003) Macrophages from cancer patients: analysis of TRAIL, TRAIL receptors, and colon tumor cell apoptosis. J Natl Cancer Inst 95(8):611–621
Cartron G et al (2004) From the bench to the bedside: ways to improve rituximab efficacy. Blood 104(9):2635–2642
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Cory S, Huang DC, Adams JM (2003) The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22(53):8590–8607
Plati J, Bucur O, Khosravi-Far R (2011) Apoptotic cell signaling in cancer progression and therapy. Integr Biol (Camb) 3(4):279–296
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wagh, A.R., Bose, K. (2019). Apoptosis in Cancer Cell Signaling and Current Therapeutic Possibilities. In: Bose, K., Chaudhari, P. (eds) Unravelling Cancer Signaling Pathways: A Multidisciplinary Approach. Springer, Singapore. https://doi.org/10.1007/978-981-32-9816-3_5
Download citation
DOI: https://doi.org/10.1007/978-981-32-9816-3_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9815-6
Online ISBN: 978-981-32-9816-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)