
Abstract
Penta-Graphene (PG), proposed in 2014 on the basis of theoretical simulations, is a two dimensional (2D) metastable carbon allotrope composed of carbon pentagons. PG is proposed to be synthesized by exfoliation from T12-carbon by hydrogen intercalation. Theoretical calculations confirm that PG is appreciably stable and can sustain the temperature as high as 1000 K. Unlike graphene which needs to be functionalized for opening a band gap, PG is itself a quasi-direct band gap semiconductor having a band gap of ~3.25 eV. PG offers an unusual negative Poisson’s ratio and very high mechanical strength exceeding than that of graphene. According to prediction, successful synthesis of penta-graphene may bring forth considerable makeover in future nano-electronics arena. However, synthesis of PG is still a challenge. It is not only difficult to isolate PG from the plethora of alternative isomers; it may rapidly restructure itself towards graphene in the presence of even a trace amount of specific catalytic impurities. Though unstable in its pure form, theoretical simulation shows suitable chemical functionalization stabilizes the penta-graphene structure by partially releasing the strain, which paves the path towards successful synthesis of penta-graphene considering its functionalized state as the precursor. In this book chapter, the strikingly interesting features/properties, procedure for predicting the characteristics through theoretical simulations, synthesis process, potential applications and critical bottlenecks of functionalized penta-graphene have been comprehensively discussed with special emphasis on its future prospects for next generation electronic device applications. Mainly three types of chemical functionalizations have been considered employing (i) hydrogen, (ii) fluorine and (iii) oxygen. Improvement of stability of functionalized PG, in terms of energy, lattice dynamics, mechanical and thermal in comparison to that of pristine PG have been discussed. Underlying mechanism for the corresponding dramatic improvement in electronic, mechanical and thermal properties of PG for the above mentioned functionalization(s) have also been discussed. Hydrogenation and fluorination of PG changes the Poisson’s ratio from negative to positive and turns the PG from a semiconductor to an insulator. Thermal conductivity of PG enhances dramatically on full hydrogenation. The three types of functionalizations mentioned above, improve mechanical strength of PG by enhancing the value of failure stress and failure strain. Chemical functionalizations widen the application potentiality of PG. Improvement in thermal conductivity of PG by partial functionalization facilitates its application in thermoelectric devices. Band gap tailoring by judicious chemical functionalization lays the foundation stone for future application of PG in the field of optoelectronic and photovoltaic devices. Hydrogen adsorption remarkably enhances the magnetic moment of PG which is useful for potential application in magnetic storage technology and next generation spintronic nano-devices. However, the practical implementation (synthesis and subsequent technology development to couple it successfully with device fabrication) is still in nascent stage and the future research endeavor of the researchers is inclined towards mitigating that bottleneck.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Penta-graphene
- Chemical functionalization
- Electronic property
- Mechanical property
- Magnetic property
- Optical property
- Thermal property
1 Introduction
Carbon, the most important and major constituent of the cycles on the Earth, is also an interesting and versatile element in the periodic table due to its enormous flexibility in bonding characteristics. The vast and ever expanding family of carbon allotropes, constructed with sp3, sp2 and sp hybridized orbitals, offer a wide range of hierarchical nano-structures ranging from three dimensional (3D) to zero dimensional (0D) variety (such as, diamond (3D), graphene (2D), nanotubes (1D) and bucky balls (0D)) [1]. Such carbon allotropes are also be characterized with diversified material properties (viz., the electronic property of Carbon allotropes can vary drastically over the range; from metal to insulator). Graphene, constructed with sp2 hybridized carbon atoms arranged in a hexagonal honeycomb lattice (2D), has initiated a new era in nano-electronics due to its unprecedentedly advantageous electronic properties. The great success of graphene has triggered the interest of the researchers across the globe in exploring other 2D nanomaterials specially based on carbon [2]. Since then, a number of low-dimensional carbon allotropes have been proposed from theoretical consideration [3,4,5], among which Penta-Graphene (PG) is one of the most remarkable one. Zhang et al. first identified penta-graphene by simulation in 2014 [5]. It is a two dimensional metastable carbon allotrope and is constructed from a mixture of sp2- and sp3 hybridized carbon atoms arranged in pentagonal lattice which closely resembles to the well-known Cairo pentagonal tiling. The key difference between PG and graphene is that graphene is zero band gap material whereas penta-graphene is a semiconductor with a finite band gap of ~3.25 eV [5]. Although experimental synthesis of PG is still a crucial challenge, some of its unique electronic, thermal, mechanical, magnetic and optical features and their tunability through suitable functionalization (as predicted from theoretical studies) projected this material as a potential candidate for applications in nano-electronics and nano-mechanics for the coming era. Like graphene, PG can also form nanoribbon and nanotube structure which are beyond the scope of this chapter [6,7,8,9].
Prediction of either the existence of a material or the properties of a material (theoretically proposed or already existing) can be decided from “Density functional theory” (DFT) and “Molecular Dynamics” (MD) based calculations. DFT is the most successful ab initio/ first principle calculation which is based on the quantum mechanical description of the material and governed by the laws of condensed matter physics [10,11,12]. The underlying principle is that “the total energy of the system is a unique function of the electron density”. DFT calculations are based on ‘Kohn–Sham (KS)’ method which is an independent particle method for many-body electron problem. The framework of DFT is based on computing the ground state energy and charge density of many body system of electron by solving a set of Schrödinger like equations representing a fictitious but simplified system of non-interacting electrons moving in an effective potential [11, 12]. “Effective potential consists of external potential, Coulomb potential and Exchange-correlation (XC) potential. The XC potential is a measure of the quantum mechanical exchange and correlation of the particles” [13]. XC potential plays critical role in DFT calculation. The ground state energy of the material is identified from DFT calculation. “Ab initio Molecular Dynamics” (AIMD) simulation calculate the dynamics for systems for which the interatomic forces are derived from DFT based calculations.
The possibility of existence of penta-graphene and its properties were first predicted by Zhang et al. with the help of DFT based AIMD simulations performed using Vienna Ab initio Simulation Package (VASP) [14]. Interactions between ion cores and valance electrons were treated with “Projector augmented wave” (PAW) basis set [15]. Proper choice of “exchange-correlation” (XC) potential function is important for predicting material properties using DFT based calculations. Zhang et al. [5] used “generalized gradient approximation” (GGA) with ‘Perdew–Burke–Ernzerhof’ XC potential for most of the calculations. Since, electronic band structure calculations demand high precision, Zhang et al. [5] used ‘hybrid Heyd–Scuseria–Ernzerhof’ (HSE06) XC potential [17] only for calculation of electronic band structure of penta-graphene. ‘Phonopy package’ was used to determine phonon properties of PG using finite displacement method.
It is well established that electronic, magnetic, optical and mechanical properties of graphene can be tuned effectively by chemical functionalization [18, 19]. This triggered the curiosity among the researchers to find a way of tuning of various properties of PG through chemical functionalization. Although, penta-graphene is proposed very recently and its research is in very nascent stage, it is found that chemical functionalization will significantly widen the application of PG in electronic devices. Bonding characteristics (transformation from sp2 to sp3, change of bond length, angle etc.), electronic and phonon band structure of PG change upon chemical functionalization which is reflected in electronic, thermal, mechanical, optical and magnetic properties of the same. Thus, the properties of PG can be tuned for suitable electronic device application employing judicious chemical functionalizations. The property of PG can also be changed by doping which is out of scope of this book chapter [20,21,22].
2 Structure and Properties of PG
Penta-graphene was first theoretically proposed by Zhang et al. in 2014 by exfoliation of single layer from T12-carbon, whose existence is also recently predicted from DFT based calculations [5]. PG shows tetragonal lattice structure with six carbon atoms per unit cell having optimized lattice parameters; a = b = 3.64 Å. The carbon atoms in PG shows two types of hybridization viz. sp3 and sp2 which are customarily distinguished as C1 and C2, respectively [5].
Figure 1 shows the top and side view of the optimized structure of penta-graphene along with its the first Brillouin zone in reciprocal lattice space identifying the high symmetry path Γ–M–X–Γ where Γ = (0, 0, 0)2π/a, M = (1/2, 1/2, 0)2π/a and X = (0, 1/2, 0)2π/a. From the top view, PG looks like Cairo pentagonal tiling. Carbon-carbon bond lengths in PG are 1.55 Å (C1–C2) and 1.34 Å (C2–C2) which show pronounced character of single and double bonds, respectively. However, these bond lengths differ from the carbon-carbon bond length in diamond and graphite/graphene which are 1.54 and 1.42 Å, respectively. Thus, the bonding characteristics in PG are somewhat different than well-known diamond and graphite/graphene structure. The bond angle (θC2–C1–C2) in PG is 134.2° which also indicate the distorted sp3 character of C1 atoms. Since, sp3 carbon bonds are tetrahedral in nature; penta-graphene is not truly a planner structure [23]. A buckling of 0.6 Å is observed from the side view of PG. Thus, PG is a two dimensional sheet having a thickness of 1.2 Å. The structure of PG can be viewed as a “multidecker sandwich”, where the sp3 hybridized C atoms (C1) are sandwiched between the sp2 hybridized C atoms (C2). It is interesting to note that the structure of PG is similar to experimentally identified layered silver azide (AgN3) [5].

All the figures are reprinted with permission from the Ref. [23]
a Top view, unit cell and b front view of the optimized structure of penta-graphene, Yellow, blue sphere represent C1 and C2 respectively, c first Brillouin zone and high symmetry points for 2-dimensional tetragonal lattice Γ = (0, 0, 0)2π/a, X = (0, 1/2, 0)2π/a and M = (1/2, 1/2, 0)2π/a
2.1 Stability Issues
Whenever a material is proposed theoretically, the first question arise regarding the stability of the material which can also be viewed as the feasibility of practical existence/synthesis of the material. Study of stability includes identifying ground state energy from the calculation of total energy by DFT as well as the behavior of the system in other perturbation like thermal, mechanical etc. Ab initio MD simulation within the framework of DFT is essential in studying the behavior of the system in perturbation.
Energetic Stability
Thermodynamic stability of penta-graphene has been examined by means of total energy calculation. PG is identified as metastable in comparison to graphene and other two dimensional carbon allotropes due to its violation of the IPR (isolated pentagon rule) [24]. However, it is more stable than some other nanostructures of carbon, like, 2D α-graphyne, 3D T-carbon and (3, 12)-carbon sheet [5].
Figure 2a shows the area dependence of total energy of penta-graphene along with graphene, graphyne, C20, (3, 12)-carbon sheet, a-graphyne [5]. It can be envisaged that penta-graphene is energetically stable than some experimentally synthesized carbon nanostructures such as, the smallest carbon nanotube and C20 (smallest fullerene). This study, therefore, affirmatively, indicates towards possibility of synthesis of the 2D penta-graphene sheet. The structural unit of both of the carbon allotropes, C20 and penta-graphene is fused pentagons. Since C20 is a highly curved structure, all carbon atoms in it are in strained condition with distorted sp2 hybridization. On the contrary, in penta-graphene, the curvature is reduced due to the presence of sp3 hybridized carbon atoms along with sp2 hybridized carbon atoms and thus the strain is released partially [5].

All the figures are reprinted with permission from the Ref. [5]
a Area dependence of total energy per atom for some 2D carbon allotropes. The total energy of the experimentally identified dodecahedral C20 cage is also calculated and plotted here for comparison. b Phonon band structures and PhDOS of penta-graphene. (Inset) High-symmetric q-point paths: Γ (0, 0) → X (1/2, 0) → M (1/2, 1/2) → Γ (0, 0)
Dynamic Stability
Calculation of lattice vibration gives the phonon dispersion curve which determines whether a crystalline structure is dynamically stable or not. For a dynamically stable crystal structure, there should not have any imaginary frequency in entire Brillouin zone [25]. Phonon band structure and phonon DOS of PG are shown in Fig. 2b. The dynamic stability of penta-graphene is confirmed from the absence of imaginary frequencies in entire Brillouin zone i.e. in all high symmetry directions. Like graphene, three distinct acoustic modes are observed in phonon band structure of PG [5].
Thermal Stability
To ensure the thermal stability, Zhang et al. performed “Ab initio molecular dynamics” (AIMD) simulation (using canonical ensemble) of 2D supercell of penta-graphene [5]. No structural reconstruction is observed for 6 ps with a time step of 1 fs, at room temperature (300 K). It is revealed, penta-graphene can sustain temperatures as high as 1000 K. PG is found to be dynamically stable even in presence of different types of defects like Stone–Wales-like defect, mono and di-vacancies, edge atoms and adatoms [5].
Mechanical Stability
To ensure the feasibility of practical existence of a theoretically proposed material, it is necessary to know the effect of lattice distortion on structural stability. The linear elastic constants of a mechanically stable crystal should follow the “Born–Huang criteria” to guarantee the positive-definiteness of strain energy caused by lattice distortion. Accordingly, the four nonzero 2D elastic constants viz., C11, C22, C12 and C66 should satisfy the criteria \( C_{11} C_{22} - C_{12}^{2} \) and C66 > 0 in a mechanically stable 2D sheet [26]. From the calculation of the change of energy due to the in-plane strain (uniaxial and biaxial), PG was found to satisfy the above criteria [5].
2.2 Properties of Pristine PG
Confirmation of stability of the material invokes the curiosity regarding the properties of penta-graphene. Researches are in progress in investigating the electronic, thermal, mechanical, magnetic, optical properties of penta-graphene [5, 20, 23, 27,28,29,30].
Electronic Properties
From the computation of band structure using—Scuseria–Ernzerhof (HSE06) potential functional, penta-graphene is found to be an indirect band-gap semiconductor with a band gap of ~3.25 eV (computed with HSE06 potential function) [5]. Figure 3 shows the electronic band structure of PG computed with hybrid HSE06 potential function (solid lines) and GW correction (dotted line). It is observed from the electronic band structure that the conduction band minimum lies on the M–Γ path whereas the valance band maximum (VBM) is located on the Γ–X path. However, there exists a sub-VBM on the M–Γ path whose energy value is very close to that of true VBM [5]. Thus, penta-graphene can also be considered as a “quasi-direct–band-gap” semiconductor. Analysis of partial DOS reveals that the major contribution of electronic states near the Fermi level is due to the sp2 hybridized C2 atoms. In penta-graphene, the pz orbitals of sp2-hybridized C2 atoms are spatially separated by the sp3-hybridized C1 atoms. Thus, full electron delocalization is hindered which gives rise to finite band gap. According to Zhang et al., there is possibility to achieve superconductivity in penta-graphene through hole doping since there are dispersion-less, partially degenerate valance bands which leads a high total DOS near the Fermi level [5, 31].

The figures are reprinted with permission from the Ref. [43]
Calculated HSE06 (solid lines) and GW0 (dotted lines) band structures of monolayer PG.
Mechanical Properties
The elastic strain per unit area in a 2D material can be expressed as [5];
where the constants, C11, C22, C12 and C66 are the components of the elastic modulus tensor and can be obtained from second order partial derivative of strain energy as a function of strain. \( \varepsilon_{xx} \), \( \varepsilon_{yy} \) and \( \varepsilon_{xy} \) are the components of strain tensor. C11, C22, C12 and C66 can be obtained by fitting the energy curves as a function of with uniaxial (\( \varepsilon_{yy} = 0 \)) and equi-biaxial strains (\( \varepsilon_{xx} = \varepsilon_{yy} \)).
Young’s modulus of a material is defined as \( E = \left( {C_{11}^{2} - C_{12}^{2} } \right)/C_{11} \) [5]. The value of Young modulus in penta-graphene is found to be 263.8 GPa nm whereas the corresponding value is 345 GPa nm in case of graphene [32]. The value of Young modulus in penta-graphene is comparable to that of h-BN monolayer [33].
Another important parameter which articulates the mechanical property of a material is Poisson’s ratio. It is defined as the negative ratio of the transverse strain to the corresponding axial strain. In case of most of the solids, this ratio is positive since under a uniaxial compression, solids expand in the transverse direction. Although the continuum mechanics theory does not discard the possibility of existence of negative Poisson’s ratio (NPR) in a stable linear elastic material, it is rarely found in nature. Very few artificial materials are characterized with NPR. Materials with NPR, known as “auxetic materials”, are of great scientific and technological interest [34, 35]. The major area of application of “auxetic materials” are in biomedical field as a material for structure/muscle/alignment anchors, surgical implant, prosthetic materials, dilator to open up blood vessels during heart surgery etc. Other application field of “auxetic materials” is piezoelectric sensors and actuators. Auxetic foams and fibers find application in filters and woven structures. Interestingly, penta-graphene exhibit negative Poisson’s ratio viz., ν12 = ν21 = C12/C11 = − 0.068 since C12 is negative for PG [5].
The NPR characteristics of PG has been testified comprehensively for both of the cases i.e. the lateral response in the x direction when the tensile strain is applied in the y direction and vice versa. Penta-graphene as a nano-auxetic material may have multiple applications in micro-mechanical and micro-electromechanical systems (MEMS).
In addition to in-plane stiffness, ideal strength (i.e. the maximum stress that a material can sustain) is also an important mechanical property of a nanomaterial. Figure 4a shows the stress–strain relationship of penta-graphene under equi-biaxial tensile strain [5]. It is observed from the curve that the strain at maximum stress is 21% (denoted by red arrow) beyond which the mechanical failure starts. However, the critical strain is less than 21% for penta-graphene since phonon instability i.e. phonon softening induced by Kohn anomaly occurs before the stress reaches its maximum [5, 36]. Figure 4b and c represent Phonon bands at the extreme of equi-biaxial strain of penta-graphene and graphene, respectively. It is found that in case of penta-graphene, phonon softening arise when the equi-biaxial strain reaches 17.2% whereas the corresponding value is 14.8% for graphene. Thus, the ideal strength of PG is higher than that of graphene. At the critical strained condition, the single bond lengths between C1 and C2 atoms are found to be ~1.77 Å, which is comparable with the experimentally and theoretically reported longest C–C bond length. Beyond critical strength, the structure fracture starts through breaking of the bonds between C1 and C2 atoms.

All the figures are reprinted with permission from the Ref. [5]
a Stress–strain relationship under equi-biaxial tensile strain. The red arrow denotes the maximum strain. b Phonon bands of penta-graphene at the extreme of equi-biaxial strain. c Phonon bands of graphene at the extreme of equi-biaxial strain. Blue lines and red circles represent phonons before and after the failure, respectively. (Insets) The high-symmetry q-point paths in the reciprocal space
Thermal Property
Thermal conductivity is an important physical parameter which asses heat dissipation ability of a material and high temperature application possibility. Joule heating is a major problem in electronic devices which affect performance, reliability and life time of the device. Moreover, lower heat capacity of nano-electronic devices is a critical issue. Hence, the device material should be thermally highly conductive so that the generated heat during device operation can dissipate fast. On the other hand, thermoelectric devices demands lower thermal conductivity. Although graphene is characterized with ultrahigh lattice thermal conductivity (3000–5800 W/mK), the absence of bandgap limits its application in most of the electronic devices including transistor. From the solution of linearized phonon “Boltzman transport equation” (BTE) with first principle calculation, lattice thermal conductivity of PG is found to be 645 W/mK at room temperature which is significantly lower in comparison to that of graphene. The presence of more scattering channels in buckled penta-graphene structure may be the possible reason behind significantly lower thermal conductivity of PG in comparison to that of graphene [27]. It is found that lattice thermal conductivity of stacked PG does not depend on the number of layers present. This characteristic of PG is also in sharp contrast to that of graphene. In metals, the thermal conductivity is mainly due to electrons while in semiconductors and insulators, phonons play a significant role in determining the resultant thermal conductivity.
From “classical equilibrium molecular dynamics simulations” using the original “Tersoff interatomic potential”, the intrinsic thermal conductivity of PG is found to be 167 W/mK [28]. In PG, the acoustic phonon lifetimes are 20 ps and 1–10 ps for lower (1–5 THz) and higher (5–15 THz) frequency range, respectively which are comparable to that of the graphene. However, the remarkably lower thermal conductivity of PG, in comparison to graphene, arises from the lower phonon group velocities and fewer collective phonon excitations in PG.
In penta-graphene, the phonon scattering rate is proportional to temperature and consequently, the thermal conductivity is inversely proportional to the temperature [28]. The variation of thermal conductivity of PG as a function of size of the simulating sample for constant temperature (300 K) is shown in Fig. 5a. Figure 5b shows the variation of thermal conductivity of PG as a function of temperature for a fixed size of the simulating sample (60 × 60 unit cell). From non-equilibrium molecular dynamics (NEMD) simulations using ReaxFF force field, thermal conductivity of infinitely long penta-graphene is found to be 112.35 W/mK [37]. The cause behind the difference in results is due to the method of simulation and the type of interatomic force field employed. Researchers are still in search of appropriate potential for penta-graphene [3].

All the figures are reprinted with permission from the Ref. [28]
a Thermal conductivity of penta-graphene at 300 K predicted by the Green-Kubo formula with respect to the size of simulating sample (Ns × Ns unit cells). b Thermal conductivity of penta-graphene as a function of temperature. The values are from simulations performed on the simulating sample of 60 × 60 unit cells. The red curve denotes the best-fitting
Optical Property
Although Kohn-Sham DFT estimate ground state energy most successfully, it cannot assess optical absorption accurately since “Kohn-Sham DFT eigenvalues” do not consider the electron addition and removal energies. It is necessary to include the calculation of electron-hole interaction to get information regarding the optical spectra. GW (G0W0) approximation/correction is a “many-body perturbation theory” which can successfully predict the optical properties of the materials. In this method, ground state energy and electronic density are computed by DFT Kohn-Sham framework and then, G0W0 i.e. the green function G and dynamically screened interaction W is computed from the obtained Kohn-Sham electronic structure. Applying G0W0 correction, the “quasi-direct band gap” of PG is found to lie in the range of 4.1–4.5 eV which corresponds to ultra-violet region (wavelength for optical absorption). Since visible light is not absorbed in PG, it is transparent in nature and photoelectron can’t be produced easily in PG [23].
3 Hydrogen Functionalization of Penta-Graphene
Hydrogen functionalization is the most widely studied functionalization method for penta-graphene. It is found that hydrogen functionalization not only stabilize the structure of penta-graphene but also tune its property flexibly.
Among sp3 (i.e. C1 atom) and sp2 (i.e. C2 atom) hybridized carbon atoms in penta-graphene, only sp2 hybridized carbon atoms are capable of adsorbing hydrogen. Figure 6 depicts the top and side views of the optimized structures of the fully hydrogenated penta-graphene. In fully hydrogenated penta-graphene, the ratio of C:H is 3:2, whereas this ratio is 1:1 in case of fully hydrogenated graphene. Li et al. [38] studied hydrogen functionalization of penta-graphene with the help of DFT based AIMD simulations using PAW method. Most of the calculations were performed using GGA-PBE exchange correlation (XC) potential functional whereas the hybrid HSE06 was considered for high precision electronic structure calculations. It is found that crystallographic symmetry of penta-graphene does not change even with full hydrogenation.

All the figures are reprinted with permission from the Ref. [38]
a Top view of optimized fully hydrogenated penta-graphene. Unit cell is identified by square marked by red dashed lines and b side views of the optimized fully hydrogenated penta-graphene. Δ represent thickness of PG. Yellow, gray and white spheres represent C1, C2 and Hydrogen, atoms respectively
In fully hydrogenated PG, both of the bond lenghts (C1–C2 and C2–C2) are 1.55Å which resembles the single bond character. The bond angles θC2–C1–C2 and θC1–C2–C1 are 116.9° and 105.9°, respectively, indicating the distorted sp3 hybridization of the carbon atoms [38]. Change of bond length of C2–C2 from 1.34 to 1.55 Å, indicates that the C2–C2 bond transforms from double bond to single bond due to hydrogenation. After hydrogenation of penta-graphene, the thickness buckling increases. Vertical distance between topmost and bottommost carbon atom in fully hydrogenated penta-graphene is 1.62 Å whereas the value is 1.20 Å for pristine penta-graphene.
3.1 Stability Issues
The binding energy of each hydrogen atom in penta-graphene is defined as [38];
where, \( E_{total} ,E_{PG} , and E_{adatom} \) are total energy of the ground state configuration of the unit cell of fully hydrogenated penta-graphene, pristine penta-graphene, and isolated hydrogen atom, respectively. There are four adsorbed hydrogen atoms per unit cell. The calculated binding energy is 3.65 eV whereas the corresponding value is 2.47 eV for graphene. Thus, adsorption of hydrogen in penta-graphene is energetically favorable over that of graphene. In fully hydrogenated PG, all sp2 hybridized carbon atoms are transformed into sp3 hybridized atoms. C2–C2 bonds get elongated and bonding characteristic changes from double bond to single bond. The strain stored in pristine PG is released partially due to such hydrogenation leading towards more stability of functionalized PG than its pristine counterpart.
Figure 7 shows the variations of total potential energy of fully hydrogenated penta-graphene during the AIMD simulations for 300 K and 1000 K. It is found that the structures remain integrated and the average value of the total potential energies remains nearly constant throughout the simulation time for both the temperature 300 and 1000 K [38]. Thus, it can be inferred that hydrogenated PG sheets are not only thermally stable at room temperature, but it can also sustain at a temperatures as high as 1000 K.

All the figures are reprinted with permission from the Ref. [38]
The variations of total potential energy of full hydrogenated PG during the AIMD simulations at a 300 K and b 1000 K. The insets are snapshots of the top and side view of the structures at the end of simulations
Figure 8 represents phonon band structure of fully hydrogenated PG along the high symmetric paths (Γ–X–M–Γ) in the first Brillouin zone. The dynamic stability of the hydrogenated penta-graphene is confirmed by the absence of imaginary frequencies in the entire Brillouin zone. Like penta- graphene, there exist three distinct acoustic modes and the two-fold degeneracy on the X–M path in hydrogenated penta-graphene also [38]. A large phonon gap is observed in phonon band structure of penta-graphene. From the vibrational activity at the Γ point in the Brillouin zone, Raman and Infrared (IR) spectra of hydrogenated penta-graphene can be studied. In hydrogenated penta-graphene, Raman-activity was found at 2945.89, 2911.14, 1267.57, 1179.84, 1115.23, 990.23 and 780.64 cm−1 and IR-activity was found at 2943.53 and 2911.14 cm−1, respectively.

All the figures are reprinted with permission from the Ref. [38]
a1 Phonon band structures and corresponding atom-resolved PhDOS of fully hydrogenated PG. The inset represents the high-symmetric q-point paths: Γ (0, 0)–X (1/2, 0)–M (1/2, 1/2)–G (0, 0). a2 Phonon band structures of fully hydrogenated PG in the low frequency region
3.2 Properties of Hydrogenated Penta-Graphene
Electronic Property
Like penta-graphene, fully hydrogenated PG is also characterized with indirect band gap since its conduction band minimum (CBM) and valence band maximum (VBM) are located at different k points in the momentum space. Figure 9a shows the electronic band structure and atom-decomposed partial DOS of fully hydrogenated penta-graphene computed with PBE and HSE06 functional. The computed band gap of fully hydrogenated penta-graphene with PBE and hybrid HSE06 functional are 4.29 and 5.35 eV, respectively. It is well known that calculation with PBE functional underestimate the bandgap of a material.

Hydrogen functionalization changes penta-graphene from a semiconductor to an insulator [38]. Due to hydrogenation, the CBM/VBM of penta-graphene shifts from the M–Γ/Γ–X path to the M–Γ path. From the analysis of partial DOS of fully hydrogenated penta graphene, it is found that the electronic states near the Fermi level are mainly contributed from C1 and C2 atoms.
Mechanical Property
Molecular Dynamics simulation confirms that hydrogenation significantly enhance the mechanical strength of penta-graphene [39]. Figure 9b shows stress–strain curves of pristine and hydrogenated PG with four different hydrogenation coverage of 25, 50, 75, and 100% at 300 K [39]. Stress increases linearly with the increase of tensile strain and attain a maximum after which stress drops sharply indicating a material degradation. The strain for which the stress is the maximum is identified as the failure strain and the corresponding strain is the failure stress. The simulated failure stress and failure strain value for pristine penta-graphene are 50.2 GPa and 0.127, respectively which are significantly lower than that of graphene (130 GPa and 0.25, found experimentally and 125.2 GPa and 0.191, found by MD simulations) [39]. It is revealed from Fig. 9 that fully hydrogenated PG is characterized with the highest value of failure stress and strain. This phenomenon is strikingly in contrast with graphene where hydrogenation (up to 30%) significantly reduces the value of failure stress and failure strain. For a hydrogenation range of 30–100%, mechanical properties of graphene are almost insensitive to functionalization [40].
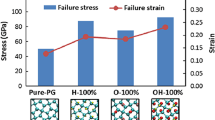
The failure stress and failure strain of pristine and hydrogenated PGs are represented in the form of a bar diagram summarized in Fig. 10a and b, respectively. When PG is hydrogenated to 25%, the failure stress and failure strain decrease by 16.9 and 22.8%, respectively than that of pristine PG. With further increase of hydrogenation, failure stress failure and strain increase monotonically. For 100% hydrogenated PG, failure stress and failure strain increase by 74.9 and 52.8% with respect to that of pristine PG.

All the figures are reprinted with permission from the Ref. [39]
a Failure stress and b failure strain of pristine and hydrogenated PGs with different hydrogenation functionalities
Structural transformation from pentagon to polygon (6-, 7-, 8-, or more atom rings) is the cause behind the failure of pristine as well as partially hydrogenated PGs beyond failure strain [39]. For 100% hydrogenated PG, the failure mechanism is entirely different. Any structural transition induced failure is not observed in fully hydrogenated PG. In the 100% hydrogenated PG, all the sp2-hybridized carbon atoms transforms to sp3-hybridized. Thus, fully hydrogenated PG is more stable structure and is characterized with higher value of failure stress and failure strain (87.8 GPa and 0.194). In fully hydrogenated PG, the failure mechanism is initiated (for a strain 0.1944) with C–C bond breaking and a visible crack is formed in the structure [39]. With further increase of strain, the crack rapidly spreads in the direction perpendicular to the loading direction and causes fully hydrogenated PG to rupture finally [39].
These improvements in the mechanical properties of hydrogenated PG remain almost unaltered for the temperature range of 200–600 K. Figures 5a and b represent the failure stress and failure strain as a function of temperature for pristine and fully hydrogenated PG. It can be observed that with the increase in temperature from 200 to 600 K, the value of failure stress and failure strain decrease slightly for pristine as well as fully hydrogenated PGs (Fig. 11).

All the figures are reprinted with permission from the Ref. [39]
Effect of temperature on a the failure stress and b the failure strain of pristine PG and fully hydrogenated PG
Decrease of failure stress and failure strain with temperature can be caused by the stronger thermal vibration of atoms at higher temperature which renders the atomic bonds more likely to reach the critical bond length and subsequently to break. In spite of the degradation of the mechanical properties with increasing temperature, the failure stress and failure strain of fully hydrogenated PG are still much higher (about 71–88% higher) than that of pristine PG [39].
In Edge Hydroxyl, OP: The “in-plane Young’s modulus” reduces to 205.50 GPa from 263.8 GPa upon full hydrogenation of PG [38]. In contrast to the pristine penta-graphene (−0.068), Poisson’s ratio of fully hydrogenated PG is found to be positive (0.243) [38].
Thermal Property
From first-principles lattice dynamics and iterative solution of the phonon “Boltzmann transport equation” (BTE), it is found that thermal conductivity of fully hydrogenated PG is increased by 76% compared to its pristine counterpart. This is in contrast to hydrogenated graphene which shows a dramatic decrease in thermal conductivity in comparison to pristine graphene. Figure 12a shows Variation of thermal conductivity as a function of sample length of penta-graphene and fully hydrogenated penta-graphene [41].

Another study based on non-equilibrium molecular dynamics simulations using ‘ReaxFF force field’ on hydrogenated penta-graphene at different levels of coverage shows that initially with increasing coverage (up to 25% hydrogen), thermal conductivity decreases (nearly 57%), but with further increase in the coverage, thermal conductivity increases [37]. Interestingly, the fully functionalized (100%) PG shows nearly the same thermal conductivity as that of the pristine one. The result is depicted in Fig. 20a. The difference in results may arise from the considered size of the sample for simulation since the results of MD simulations is sensitive to the model size due to the boundary scattering [42]. For reduced sample length, the associated phonon-boundary scattering is more which may reduce the simulated value of thermal conductivity of hydrogenated PG. However, researches are in progress to optimize the simulation environment to obtain more realistic results.
Figure 12b shows the vibration power spectra (VPS) of carbon atoms in pristine and hydrogenated (25 and 100%) PG. In pristine graphene, peaks appear in VPS at low frequency domain. In case of 25% hydrogenated penta-graphene, peaks are significantly suppressed. Change in VPS leads to a local acoustic mismatch which leads to phonon scattering [37]. Consequently, thermal conductivity decreases. Although peaks are suppressed at low frequency in 100% hydrogenated graphene, there exist peaks at high frequencies (50 and 100 THz). Thus, higher thermal conductivity in 100% hydrogenated graphene is attributed to high frequency vibration [37].
Optical Property
Like pristine PG, fully hydrogenated PG is also characterized by indirect band gap with CBM and VBM at M and Γ point respectively [43]. The computed band gap of with GW approximation is ~5.78 eV. New bands are found at conduction band edge of fully hydrogenated PG which eventually leads to possible optical absorption (Fig. 13).

The figures are reprinted with permission from the Ref. [43]
Electronic band structure and partial densities of states of fully hydrogenated PG computed with GW approximation
Magnetic Property
From spin polarized DFT calculations, it is found that hydrogen adsorption induce magnetic moment in PG [44]. The magnetic moment of PG increases 137 times upon hydrogenation. This result indicates the potential application of PG in magnetic storage and next generation spintronic devices.
4 Fluorine Functionalization of Penta-Graphene
Like hydrogen functionalization, fluorine functionalization was also found to be very effective in tuning different physio-chemical and electronic properties of penta-graphene.
Figure 14 depicts the top and side views of the optimized structures of fluorinated penta-graphene. The structure of fluorinated penta-graphene is similar to that of hydrogenated PG, where C2-C2 bond changes from double bond to single bond and thus stabilizes the structure by releasing strain. In fluorinated penta-graphene, the bond lengths of C1–C2 and C2–C2 are 1.57Å and 1.58Å, respectively. Like hydrogen, fluorine also gets adsorb on sp2 hybridized carbon atom. The bond angles θC2–C1–C2 and θC1–C2–C1 are 117.8° and 106.7°, respectively. The vertical distance between the topmost and the bottommost carbon atom in fully fluorinated PG is 1.6 Å [38].

All the figures are reprinted with permission from the Ref. [38]
a Top and b side views of the optimized structures of fluorinated penta-graphene. The square marked by red dashed lines denotes the unit cell. The yellow, gray and cyan spheres represent the C1, C2 and fluorine C atoms
4.1 Stability Issues
The binding energy of fluorine in fluorinated graphene is 4.22 eV which is higher than hydrogen functionalized PG [38].
Figure 15 shows the variations of total potential energy of 100% fluorinated penta-graphene during the AIMD simulations for 300 K and 1000 K, respectively. The insets are the top and the side view of the structure at the end of the simulation. The fully fluorinated PG sheet remains integrated and the average value of total potential energy remains almost constant during the MD simulations for both of the temperatures (300 and 1000 K). Thus, like 100% hydrogenated PG, fully fluorinated PG is also a stable structure and can withstand a temperature as high as 1000 K.

All the figures are reprinted with permission from the Ref. [38]
The fluctuations of total potential energy of fully fluorinated penta-graphene during the AIMD simulations at a 300 K and b 1000 K. The insets are snapshots of the structures at the end of simulations
Figure 16a shows the phonon band structures and corresponding atom-resolved PhDOS of fully fluorinated penta-graphene [38]. Absence of imaginary frequency in the entire Brillouin zone confirms the dynamical stability of fluorinated penta-graphene. The phonon gap in fluorinated PG is less in magnitude than that of hydrogenated PG. It is observed that high frequency (> 40THz) phonon modes are absent in fluorinated PG which is in contrast to the hydrogenated PG. The frequencies of pronounced Raman active modes of fluorinated PG are 1219.33, 1077.20, 1012.18 and 491.26 cm−1 while the IR-active modes are 1261.45, 1130.71 and 545.58 cm−1.

All the figures are reprinted with permission from the Ref. [38]
a Phonon band structures and corresponding atom-resolved PhDOS of fully fluorinated penta-graphene. b Electronic band structure and atom-decomposed partial DOS of fluorinated penta-graphene. Blue dashed lines and red solid lines correspond to the PBE and HSE06 results, respectively
4.2 Properties of Fluorinated Penta-Graphene
Electronic Property
Figure 16b represents the electronic band structure and atom-decomposed partial DOS of fluorinated penta-graphene [38]. Fluorinated PG is also characterized with indirect band gap. The computed band gap of fully fluorinated penta-graphene with PBE and hybrid HSE06 functional are 3.36 and 4.78 eV, respectively. From the analysis of partial DOS, it is found that, the states near Fermi level is more contributed by fluorine than C1 or C2.
Mechanical Property
Young’s modulus of fluorinated PG is found to be 239.12 GPa which is lower than that of pristine PG but higher than that of hydrogenated PG. Poisson’s ration of fluorinated PG is positive (0.236) [38]. Figure 17 represents strain–energy relationship of the fluorinated and hydrogenated penta-graphene sheets under uniaxial strain and equi-biaxial strain.

All the figures are reprinted with permission from the Ref. [38]
Strain–energy relationship of the hydrogenated and fluorinated penta-graphene sheets under a uniaxial strain and b equi-biaxial strain
5 Oxygen Functionalization
Two type of oxygen functionalization is studied in penta-graphene viz. functionalization with epoxy group (–O–) and with hydroxyl group (–OH). Figure 18a and 18b represent top and side view of atomistic configurations of PG fully functionalized with epoxy (–O–) group and hydroxyl group (–OH), respectively. Hydroxyl group creates bond with single sp2 hybridized C2 atom(s) whereas epoxy group creates bond with two C2 atom(s).

All the figures are reprinted with permission from the Ref. [39]
Top and side view of atomistic configurations of PG fully functionalized with a epoxide group and b hydroxyl group. Cyan, red and yellow balls are carbon, oxygen and hydrogen atoms respectively
5.1 Properties of Oxygenated Penta-Graphene
Mechanical property
Figure 19a depicts the stress-strain curves of fully functionalized PG with epoxy (–O–) and hydroxyl (–OH) groups along with that of pristine and fully hydrogenated PG for comparison. In terms of failure stress and failure strain, all functionalized PG structures outperform the pristine PG [39]. Among the three different functional groups, the fully functionalized PG with hydroxyl groups possesses the highest failure stress and failure strain (86.6 and 82.4%), followed by the PG having hydrogen (74.9 and 52.8%) and epoxy groups (49.3 and 45.2%) [39].

All the figures are reprinted with permission from the Ref. [39]
a Stress-strain curves of pristine and fully functionalized PGs with different functional groups. b Failure stress and failure strain of pristine and fully functionalized PGs with different functional groups
The lower failure stress and failure strain value of functionalized PG with epoxy group may be caused by the bonding characteristics of epoxy group with PG. Epoxy group is bonded with two carbon atoms in PG. As a result, the local sp3 bonds of PG are distorted and stress is generated. It can be observed from Fig. 18a that there exists a non-zero initial stress in the stress-strain curve of epoxide-functionalized PG. It is also notable that beyond the failure stress, in epoxide-functionalized (100%) and hydroxyl-functionalized PG, the failure mechanism starts with bond breaking as in case of 100% hydrogenated PG.
Thermal property
Computation of thermal conductivity of oxygen functionalized PG for different percentage of coverage (of oxygen) in PG shows that, initially thermal conductivity decreases with increasing percentage of coverage (up to 25%). Beyond 25%, thermal conductivity increases with increasing percentage of coverage. Figure 20a shows the normalized thermal conductivity of functionalized PG for different coverage of hydrogen and oxygen atoms [37]. It is observed that the trends of variation of thermal conductivity with percentage of coverage are similar for oxygen and hydrogen functionalization of PG. Thus, fully oxygenated PG is also characterized with nearly equal thermal conductivity of pristine PG [37].

All the figures are reprinted with permission from the Ref. [37]
a Normalized thermal conductivity of functionalized PG at different coverage of hydrogen and oxygen atoms. b VPS of carbon atoms in pristine and oxidized PG
Figure 20b shows the vibration power spectra (VPS) of carbon atoms in pristine and oxygenated (25 and 100%) PG. In 25% oxygenated PG, VPS peaks are also significantly suppressed as in case of hydrogenated PG. In case of 100% oxygenated PG, although peaks are suppressed in the lower frequencies (where pristine PG shows pronounced peaks), the same at higher frequencies is observed to be dominant [37].
6 Challenges in Implementation and Future Direction of Research
A set of criteria has been put forward by Ewels et al. [45] whether or not the theoretically proposed penta-graphene can experimentally be synthesized. The experimental feasibility of synthesis of a theoretically proposed material not only demands that the energy of the structure should be at a minima on the local potential energy surface but also its energy should be isolated by high potential barrier from its related isomers [45]. Accordingly, after synthesis also, PG may rapidly restructure itself towards graphene in the presence of even a trace amount of specific catalytic impurities. Some of the researchers considered the growth of large-area aligned pentagonal graphene domains on Cu foils by chemical vapor deposition as the first step towards the experimental fabrication of PG [37, 46]. However, there exists lots of confusion/controversy regarding the effectiveness of this approach. However, theoretical simulation shows suitable chemical functionalization stabilizes penta-graphene. In most of the functionalizations i.e. hydrogen, fluorine and hydroxyl, sp2 bonded carbon atoms of penta-graphene transforms to sp3 bonded ones. Thus, these functionalizations stabilize the penta-graphene structure by partially releasing strain. The binding energy of hydrogen and fluorine in PG are also found to be higher than that of graphene. It suggests possible synthesis of penta-graphene considering its functionalized state as a precursor just like the case of experimentally synthesized dodecahedral C20 fullerene where hydrogenated or brominated C20 is considered as the precursor [38].
Guo et al. studied the characteristics of the vdW heterojunction composed of penta-graphene and graphene to explore the future device applications of PG [47]. For potential device application Hydrogen adsorption capability of PG steers the application of PG as a substrate for hydrogen spillover or hydrogen storage [48] where Pt4, Pd4 Ni4, and Ti4 can play the role of a catalyst. The oxygen adsorption capability of PG indicate the application of it as a potential, metal-free, and low-cost catalyst for low-temperature CO oxidation where CO transforms to CO2 by the pre-adsorbed O2 on the PG sheet [49]. This property can further be extended to gas sensor device development as the basic principle of operation is in line with the conventional chemi-resistive gas sensors [50–52]. Hydrogenation and fluorination transforms penta-graphene from semiconductor to insulator. High band gap of hydrogenated and fluorinated penta-graphene can find their possible application as dielectric layers in field effect transistors (FETs), relinquishing the requirement of conventional silicon oxides [38]. Hydrogen adsorbed magnetic penta-graphene (M–PG) and weakly magnetic penta-graphene (WM–PG) are predicted to be the most stable state among other hydrogen adsorbed PG. On hydrogen adsorption, the magnetic moment of the PG increases by 137 times which can be utilized in magnetic storage technology [44].
Band gap tailoring as well as appearance of new bands due to hydrogen passivation paves the path for future application of PG in the field of optoelectronic and photovoltaic devices also [43]. It is observed that CBM and VBM of PG shift along momentum axis upon chemical functionalization. If indirect band gap PG can be transformed to direct band gap semiconductor by band tailoring through suitable chemical functionalization, then it would open new opportunities in the field of optoelectronic devices.
7 Conclusion
Penta-graphene is a two dimensional carbon allotrope which has been proposed theoretically very recently. Although, its practical synthesis/implementation is yet a crucial challenge, some of its unique electrical, thermal, mechanical, magnetic and optical properties highlighting promising application in nano-electronic devices have motivated researchers to study the material thoroughly. Zero bandgap characteristic of graphene limits its application in various cases whereas PG is a quasi-direct band gap semiconductor with a bandgap of ~3.25 eV. It can withstand a temperature as high as 1000 K. Thermal conductivity of PG is significantly lower in comparison to graphene which enhances the possibility of using PG in thermoelectric devices. Mechanical strength of PG also outperforms graphene. There would have enormous application of PG in biomedical and sensor field as nano auxetic materials (having negative Poisson’s ratio). However, in spite of such a wide spectra of interesting properties, practical realization of pristine PG is a crucial challenge. Judicious chemical functionalization could be possible solutions for achieving stable PG structure. Functionalized PG (hydrogenated and fluoridated) is shown to be (theoretically) more stable than its pristine counterpart suggesting the possibility of synthesis of PG functionalized state as a precursor. Interestingly, PG was proposed to be synthesized by hydrogen intercalation from T12-carbon in the pioneer work on PG. Moreover, electronic, mechanical, thermal, optical and magnetic property of PG can be significantly tailored by judicious chemical functionalization. Mechanical strength of PG was found to improve significantly upon chemical functionalization especially for hydrogen and hydroxyl groups. Thermal conductivity of PG dramatically increases upon hydrogenation which is in contrast to the graphene, where hydrogenation reduces thermal conductivity. Possibility of tuning thermal conductivity by varying the percentage of coverage of functional groups widens the application of PG in thermoelectric devices. Band gap tailoring of PG is also possible by suitable chemical functionalization which is of huge interest for nano-electronic devices. Magnetization can also be induced in PG by hydrogen functionalization which finds application in next generation spintronic devices. Mainly the effects of three types of functionalizations (hydrogen, fluorine and oxygen) of PG have been studied so far. There are many others in the queue (which are found to be effective in other 2D materials) to be studied. However, successful synthesis of PG, with desired stability and predicted properties, is still a crucial challenge.
References
Zhang, X., Wei, L., Tan, J., Zhao, M.: Prediction of an ultrasoft graphene allotrope with Dirac cones. Carbon 105, 323–329 (2016). https://doi.org/10.1016/j.carbon.2016.04.058
Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Grigorieva, I.V., Firsov, A.A.: Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004). https://doi.org/10.1126/science.1102896
Winczewski, S., Shaheen, M.Y., Rybicki, J.: Interatomic potential suitable for the modeling of penta-graphene: molecular statics/molecular dynamics studies. Carbon 126, 165–175 (2018). https://doi.org/10.1016/j.carbon.2017.10.002
Su, C., Jiang, H., Feng, J.: Two-dimensional carbon allotrope with strong electronic anisotropy. Phys. Rev. B 87, 075453 (2013). https://doi.org/10.1103/PhysRevB.87.075453
Zhang, S., Zhou, J., Wanga, Q., Chend, X., Kawazoef, Y., Jenac, P.: Penta-graphene: a new carbon allotrope. PNAS 112, 2372–2377 (2015). https://doi.org/10.1073/pnas.1416591112
He, C., Wanga, X.F., Zhang, W.X.: Coupling effects of the electric field and bending on the electronic and magnetic properties of penta-graphene nanoribbons. Phys. Chem. Chem. Phys. 19, 18426–18433 (2017). https://doi.org/10.1039/c7cp03404k
Rajbanshi, B., Sarkar, S., Mandal, B., Sarkar, P.: Energetic and electronic structure of penta-graphene nanoribbons. Carbon 100, 118–125 (2016). https://doi.org/10.1016/j.carbon.2016.01.014
Yuan, P.F., Zhang, Z.H., Fan, Z.Q., Qiu, M.: Electronic structure and magnetic properties of penta-graphene nanoribbons. Phys. Chem. Chem. Phys. 19, 9528–9536 (2017). https://doi.org/10.1039/c7cp00029d
Quijano-Briones, J.J, Fernández-Escamilla, H.N, Tlahuice-Flores, A.: Chiral penta-graphene nanotubes: structure, bonding and electronic properties. Comput. Theor. Chem. 1108, 70–75 (2017). http://dx.doi.org/10.1016/j.comptc.2017.03.019
Hasnip, P.J, Refson, K., Probert, M.I.J., Yates, J.R., Clark, S.J., Pickard, C.J.: Density functional theory in the solid state. Philos. Trans. Royal Soc. Math. Phys. Eng. Sci. vol. 372 (2014). https://doi.org/10.1098/rsta.2013.0270
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136, B864–B87 (1964). https://doi.org/10.1103/Phys-Rev.136.B864
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). https://doi.org/10.1103/PhysRev.140.A1133
Ghosh, K., Rahaman, H., Bhattacharyya, P.: Potentiality of density-functional theory in analyzing the devices containing graphene-crystalline solid interfaces: a review. IEEE Trans. Electron Dev 64, 4738–4745 (2017). https://doi.org/10.1109/TED.2017.2755688
Kresse, G., Furthmüller, J.: Efficient iterative schemes for abinitio total-energy calculations using a plane-wave basis set. Phys. Rev. B, Condens. Matt. 54, 11169–11186 (1996). https://doi.org/10.1103/Phys.RevB.54.11169
Blöchl, P.E.: Projector augmented-wave method. Phys. Rev. B: Condens. Matter 50, 17953–17979 (1994). https://doi.org/10.1103/PhysRevB.50.17953
Perdew, J.P, Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3866–3868 (1996). https://doi.org/10.1103/physrevlett.77.3865
Heyd, J., Scuseria, G.E., Ernzerhof, M.: Erratum: hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 124, 219906 (2006). https://doi.org/10.1063/1.1564060
Kuila, T., Bose, S., Mishra, A.K., Khanra, P., Kim, N.H., HeeLe, J.: Chemical functionalization of graphene and its application. Prog. Mater Sci. 57, 1061–1105 (2012). https://doi.org/10.1016/j.pmatsci.2012.03.002
Maity, I., Ghosh, K., Rahaman, H., Bhattacharyya, P.: Selectivity tuning of graphene oxide based reliable gas sensor devices by tailoring the oxygen functional groups: a DFT study based approach. IEEE Trans. Dev. Mater. Reliab. 17, 738–745 (2017). https://doi.org/10.1109/TDMR.2017.2766291
Berdiyorov, G.R., Dixit, G., Madjet, M.E.: Band gap engineering in penta-graphene by substitutional doping: first-principles Calculations. J. Phys.: Condens. Matter 28, 475001 (2016)
Enriquez, J.I.G., Villagracia, A.R.C.: Hydrogen adsorption on pristine, defected, and 3d-block transition metal-doped penta-graphene. Int. J. Hydrogen Energy 41, 12157–12166 (2016). https://doi.org/10.1016/j.ijhydene.2016.06.035
Quijano-Briones, J.J., Ferna´ndez-Escamilla, H.N., Tlahuice-Flores, A.: Doped penta-graphene and hydrogenation of its related structures: a structural and electronic DFT-D study. Phys. Chem. Chem. Phys. 18, 15505—15509 (2016). https://doi.org/10.1039/c6cp02781d
Einollahzadeh, H., Dariani, R.S., Fazeli, S.M.: Computing the band structure and energy gap of penta-graphene by using DFT and G0W0 approximations. Solid State Commun. 229, 1–4 (2016). https://doi.org/10.1016/j.ssc.2015.12.012
Tan, Y.Z., Xie, S.Y., Huang, R.B., Zheng, L.S.: The stabilization of fused-pentagon fullerene molecules. Nat. Chem. 1, 450–460 (2009). https://doi.org/10.1038/nchem.329
Togo, A., Tanaka, I.: First principles phonon calculations in materials science. Scripta Mater. 108, 1–5 (2015). https://doi.org/10.1016/j.scriptamat.2015.07.021
Ding, Y., Wang, Y.: Density functional theory study of the silicene-like SiX and XSi3 (X = B, C, N, Al, P) honeycomb lattices: The various buckled structures and versatile electronic properties. J. Phys. Chem. C 117, 18266–18278 (2013). https://doi.org/10.1021/jp407666m
Wang, F.Q., Yu, J., Wang, Q., Kawazoe, Y., Jena, P.: Lattice thermal conductivity of penta-graphene. Carbon 105, 424–429 (2016). https://doi.org/10.1016/j.carbon.2016.04.054
Xu, W., Zhang, G., Li, B.: Thermal conductivity of penta-graphene from molecular dynamics study. J. Chem. Phys. 143, 154703 (2015). https://doi.org/10.1063/1.4933311
Le, M.Q.: Mechanical properties of penta-graphene, hydrogenated penta-graphene, and penta-CN2 sheets. Comput. Mater. Sci. 136, 181–190 (2017). https://doi.org/10.1016/j.commatsci.2017.05.004
Sun, H., Mukherjee, S., Singh, C.V.: Mechanical properties of monolayer penta-graphene and phagraphene: a first-principles study. Phys. Chem. Chem. Phys. 18, 26736–26742 (2016). https://doi.org/10.1039/c6cp04595b
Savini, G., Ferrari, A.C., Giustino, F.: First-principles prediction of doped graphane as a high-temperature electron-phonon superconductor. Phys. Rev. Lett. 105, 037002 (2010). https://doi.org/10.1103/physrevlett.105.037002
Lee, C., Wei, X., Kysar, J.W., Hone, J.: Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321(5887), 385–388 (2008). https://doi.org/10.1126/science.1157996
Andrew, R.C., Mapasha, R.E., Ukpong, A.M., Chetty, N.: Mechanical properties of graphene and boronitrene. Phys. Rev. B 85, 125428 (2012). https://doi.org/10.1103/PhysRevB.85.125428
Jiang, J.W, Park, H.S.: Negative Poisson’s ratio in single-layer black phosphorus. Nat. Commun. 5, 4727 (2014). doi.org/10.1038/ncomms5727
Greaves, G.N., Greer, A.L., Lakes, R.S., Rouxel, T.: Poisson’s ratio and modern materials. Nat. Mater. 10, 823–837 (2011). https://doi.org/10.1038/nmat3134
Marianetti, C.A., Yevick, H.G.: Failure mechanisms of graphene under tension. Phys. Rev. Lett. 105, 245502 (2010). https://doi.org/10.1103/PhysRevLett.105.245502
Zhang, Y.Y., Pei, Q.X., Cheng, Y., Zhang, Y.W., Zhang, X.: Thermal conductivity of penta-graphene: the role of chemical functionalization. Comput. Mater. Sci. 137, 195–200 (2017). https://doi.org/10.1016/j.commatsci.2017.05.042
Li, X., Zhang, S., Wang, F.Q., Guo, Y., Liu, J., Wang, Q.: Tuning the electronic and mechanical properties of penta-graphene via hydrogenation and fluorination. Phys. Chem. Chem. Phys. 18, 14191 (2016). https://doi.org/10.1039/c6cp01092j
Zhang, Y., Pei, Q., Sha, Z., Zhang, Y., Gao, H.: Remarkable enhancement in failure stress and strain of penta-graphene via chemical functionalization. Nano Res. 10, 3865–3874 (2017). https://doi.org/10.1007/s12274-017-1600-9
Pei, Q.X., Zhang, Y.W., Shenoy, V.B.: A molecular dynamics study of the mechanical properties of hydrogen functionalized graphene. Carbon 48, 898–904 (2010). https://doi.org/10.1016/j.carbon.2009.11.014
Wu, X., Varshney, V., Lee, J., Zhang, T., Wohlwend, J.L., Roy, A.K., Luo, T.: Hydrogenation of penta-graphene leads to unexpected large improvement in thermal conductivity. Nano Lett. 16, 3925–3935 (2016). https://doi.org/10.1021/acs.nanolett.6b01536
Schelling, P.K., Phillpot, S.R., Keblinski, P.: Comparison of atomic-level simulation methods for computing thermal conductivity. Phys. Rev. B 65, 144306 (2002). https://doi.org/10.1103/PhysRevB.65.144306
Wang, Z., Dong, F., Shen, B., Zhang, R.J., Zheng, Y.X., Chen, L.Y., Wang, S.Y., Wang, C.Z., Ho, K.M., Fan, Y.J., Jin, B.Y., Su, W.S.: Electronic and optical properties of novel carbon allotropes. Carbon 101, 77–85 (2016b). https://doi.org/10.1016/j.carbon.2016.01.078
Liu, L.L., Wang, Y., Chen, C.P., Yu, H.X., Zhao, L.S., Wang, X.C.: Tuning the electronic and magnetic properties of penta-graphene using a hydrogen atom: a theoretical study. RSC Adv. 7, 40200 (2017). https://doi.org/10.1039/c7ra06956a
Ewels, C.P., Rocquefelte, X., Kroto, H.W., Rayson, M.J., Briddon, P.R., Heggie, M.I.: Predicting experimentally stable allotropes: Instability of penta-graphene. PNAS 112, 15609–15612 (2015). https://doi.org/10.1073/pnas.1520402112
Xia, K., Artyukhov, V.I., Sun, L., Zheng, J., Jiao, L., Yakobson, B.I., Zhang, Y.: Growth of large-area aligned pentagonal graphene domains on high-index copper surfaces. Nano Res. 9, 2182–2189 (2016). https://doi.org/10.1007/s12274-016-1107-9
Guo, Y., Wang, F.Q., Wang, Q.: An all-carbon vdW heterojunction composed of penta-graphene and graphene: tuning the Schottky barrier by electrostatic gating or nitrogen doping. Appl. Phys. Lett. 111, 073503 (2017). https://doi.org/10.1063/1.4986604
Guo, J.H., Liu, D.D., Li, X.D., Liu, H.Y., Chen, G.: Pt4, Pd4, Ni4, and Ti4 catalyzed hydrogen spillover on penta-graphene for hydrogen storage: the first-principles and kinetic Monte Carlo study. Int. J. Hydrogen Energy 43, 2247–2255 (2018). https://doi.org/10.1016/j.ijhydene.2017.11.169
Krishnan, R., Su, W.S., Chen, H.T.: A new carbon allotrope: penta-graphene as a metal-free catalyst for CO oxidation. Carbon 114, 465–472 (2017). https://doi.org/10.1016/j.carbon.2016.12.054
Bhowmik, B., Manjuladevi, V., Gupta, R.K., Bhattacharyya, P.: Highly selective low-temperature acetone sensor based on hierarchical 3-D TiO2 nanoflowers. IEEE Sensors J. 16, 3488–3495 (2018). https://doi.org/10.1109/JSEN.2016.2530827
Bhattacharyya, P., Basu, P.K., Mukherjee, N., Mondal, A., Saha, H., Basu, S.: Deposition of nanocrystalline ZnO thin films on p-Si by novel galvanic method and application of the heterojunction as methane sensor. J. Mater. Sci. Mater. Electr. 18, 823–829 (2007). https://doi.org/10.1007/s10854-006-9105-4
Ghosh, K., Hahapatra, N.S., Rahaman H., Bhattacharyya, P.: Prediction of adsorption probability of oxidizing and reducing species on 2-D hybrid junction of rGO-ZnO from first principle analysis. IEEE Trans. Nanotechnol. 18, 119–125 (2019). https://doi.org/10.1109/TNANO.2018.2884171
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ghosh, K., Rahaman, H., Bhattacharyya, P. (2019). Chemically Functionalized Penta-Graphene for Electronic Device Applications: Journey from Theoretical Prediction to Practical Implementation. In: Khan, A., Jawaid, M., Neppolian, B., Asiri, A. (eds) Graphene Functionalization Strategies. Carbon Nanostructures. Springer, Singapore. https://doi.org/10.1007/978-981-32-9057-0_14
Download citation
DOI: https://doi.org/10.1007/978-981-32-9057-0_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9056-3
Online ISBN: 978-981-32-9057-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)