
Abstract
This chapter reviews the fluorescence lifetime techniques currently applied in biomedical diagnostics. Specifically the chapter focuses on time-resolved fluorescence spectroscopy (TRFS) and fluorescence lifetime imaging (FLIM) technologies for in vivo tissue characterization, with special emphasis on the translational potential of these techniques and the prospects of autofluorescence to provide intrinsic contrast for the assessment and diagnosis of human diseases. The use of these techniques in a number of medical applications, including cancer (gastrointestinal tract, lung, head and neck, brain and breast), skin and eye diseases, and atherosclerotic cardiovascular disease, are discussed and their recent developments towards translational medicine are highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Fluorescence Lifetime
- Fluorescence Decay
- Pulse Nitrogen Laser
- Tissue Fluorescence
- Intrinsic Fluorophores
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Certain molecules in biological tissues have the ability to emit light for a short duration (typically <10−8 s), following the absorption of photons. This process is called fluorescence. The measurement of fluorescence emission is an established method for resolving the molecular composition of biological samples, using a variety of instruments, i.e. spectrophotometers, microarrays, microscopes, and endoscopes. Processing of fluorescence signal can reveal not only information on the specific molecular constituents but also on the local environment surrounding a specific fluorescence molecule or fluorophore. Fluorescence contrast and dynamics are often characterized by studying the excitation and emission spectra, quantum efficiency, polarization and fluorescence lifetime [1–6]. Among the most common tissue fluorophores are the aromatic amino acids (tyrosine, tryptophane, and phenylalanine), structural proteins (elastin, collagens, and collagen cross-links), enzyme metabolic co-factors [nicotin-amide adenine (phosphate) dinucleotide (NAD(P)H and flavin adenine dinucleotide (FAD)], lipid components and porphyrins. The optical properties of these fluorophores have been extensively studied, reviewed, and reported in literature [2–5, 7–9]. Detection of fluorescence emitted from intrinsic tissue fluorophores strongly depends on the surrounding micro-environment, making their in vivo characterization a challenging procedure [4].
The fluorescence emission of exogenous molecular probes has been also used for tissue diagnosis. With well-defined optical properties, exogenous probes are primarily used in near-infrared applications [10–14], where the background signal (autofluorescence) is minimal and excitation light penetrates deeper in tissue. However, among the numerous molecular probes under investigation only a few have been approved for human use by the U.S. Food and Drug Administration (FDA). These include fluorescein and indocyanine green (ICG) [7, 14], as well as porphyrin-based photosensitizers used in photodynamic therapy [15–21].
This chapter focuses on applications assessing the sources of endogenous fluorescence lifetime contrast and their prospects for in vivo tissue characterization and diagnosis. An in depth discussion of such applications is provided in the following sections.
2 Fluorescence Contrast for Tissue Characterization
Fluorescence measurements are commonly used for quantification of the optical properties of biological cells and tissues. Their interpretation can provide biochemical, functional and structural information of the sources of fluorescence contrast within these biological entities. Changes in tissue fluorescence properties can result from pathological transformations, therapeutic interventions, or during disease development, and thus intrinsic fluorescence has been frequently exploited as a diagnostic tool. Applications are diverse, ranging from cancer and intravascular diseases diagnostics to assessment of bioengineered tissues [2–5, 7–9, 15, 17, 20, 22–32]. Improvement in light delivery and collection systems via fiber optics is one of the key factors that lead to increased scientific and clinical interest towards tissue fluorescence measurements. Fiberoptic based systems have allowed for the development of non- or minimally invasive modules even for remote tissue fluorescence acquisition by using endoscopes or catheters [33–35]. Numerous published studies have clearly demonstrated the applicability of fluorescence spectroscopy and imaging for the assessment of various tissue pathologies.
Both steady-state (intensity and/or spectrum) and time-resolved (time- or frequency-domain) fluorescence measurements (Fig. 1) have been investigated for quantitative and qualitative compositional and pathophysiological analysis of biological tissues. Due to their rather simple and low-cost hardware implementation, the steady-state methods have been mostly exploited for human tissue diagnostics in vivo. Over the past decade commercial systems have emerged. One of the first fluorescence systems commercially available was the Light-Induced Fluorescence Endoscopy (LIFE) device from Xillix Technologies Corp. More recently Novadaq Technologies Inc. released the updated version of LIFE system, known as PINPOINT Endoscopic Fluorescence Imaging System, previously known as Onco-LIFE Autofluorescence Endoscopic Technology [36]. Karl Storz has also released an endoscopic system known as Video Bronchoscope for Autofluorescence Diagnostics, which was previously known as D-Light® System [37]. Both these systems have been developed to analyze fluorescence signals and visualize, through contrasting color assignment, bronchi and gastrointestinal (GI) tract diagnostic outcomes. The fluorescence excitation light for both of them is in the blue region of spectrum (400–450 nm for PINTPOINT and 380–440 nm for Video Bronchoscope for Autofluorescence Diagnostics), while the fluorescence detection is performed in the green region (470–560 nm) for the first system and in the green and red regions (475–800 nm) for the second. Other endoscopic fluorescence systems available nowadays are the SAFE-3000 system (408 nm excitation/430–700 nm detection) from Pentax [38], the AFI-Lucera® system (395–445 nm excitation/490–700 nm detection) from Olympus [39], DAFE (390–470 nm excitation/475–650 detection) from Richard Wolf [40], and Cellvisio (488/600 nm excitation/505–700/680–900 nm detection) from Mauna Kea Technologies [41] that can provide real-time microscopic images from endogenous or exogenous fluorophores.

a Spectro-temporal profile of fluorescence emission induced by sequential excitation light stimuli; b Fluorescence emission spectrum depicting integrated intensity values per wavelength; c Fluorescence transient signal as a result of a single excitation light stimulus
Commercial systems based on combined fluorescence and reflectance spectroscopy were also developed for the diagnosis of cervix cancer. Namely two such systems are Luma Cervical Imaging System (337 nm excitation/360–720 nm detection) from MediSpectra Inc. (now acquired from SprectraScience Inc. [42]) and LuViva® Advanced Cervical Scan (multiple excitation and detection wavelength combinations through filter wheel) from Guided Therapeutics [43].
Otolaryngology is another medical specialty for which commercial fluorescence visualization systems exist. Specifically, LED Medical Diagnostics, Inc. [44] has developed a device called VELscope Vx (400–460 nm excitation) for the direct visualization of fluorescence from oral abnormalities. This device does not provide any classification output; instead it assists the clinician to visually distinguish oral tissue differentiations, including oral pre-cancer and cancer, based on fluorescence contrast. Oral soft tissue exam is usually following to assess the lesion type.
Fluorescence intensity measurements, however, are affected by numerous experimental conditions. These include changes in excitation-collection coupling due to tissue motion and/or surface profile, the presence of intrinsic absorbers (e.g. hemoglobin presents high absorbance in both the excitation and emission spectral bands), and the photobleaching effect that can influence the fluorescence intensity over time. As a result, although many of these systems report high sensitivity in detecting tissue abnormalities, their specificity is relatively low [4, 45, 46]. In order to increase their overall diagnostic accuracy, either diffuse reflectance spectroscopy has been introduced or rigorous calibration procedures have been designed to correct for potential factors that non-linearly affect the measured fluorescence intensity values [6, 23, 28, 45, 47–63].
Time-resolved (lifetime) technique can address the limitations of steady-state fluorescence measurements. By resolving the dynamics of fluorescence decay such technique can improve the specificity of the fluorescence measurements [2, 5, 7, 8]. Fluorescence lifetime measures the average time a fluorophore remains in the excited states after a photon absorption event takes place. This metric can provide an additional source of contrast (lifetime values), which in the case of tissues can be of critical interest. Many of the intrinsic fluorophores present strongly overlapping fluorescence emission spectra [4, 8, 9], with distinct, however, lifetime values. Moreover, fluorescence lifetime is usually independent of the fluorophore’s concentration and quantum yield [2, 3, 8], property which can compensate for the variation in intensity values due to their dependence on changes in excitation/emission geometry and excitation light penetration depth.
In contrast to intensity measurements, the time-resolved fluorescence measurement can: (i) provide a means of discrimination between tissue constituents with overlapping fluorescence emission spectra but different fluorescence decay times; (ii) provide information regarding the physiochemical environment of the intrinsic fluorophores, as this figure is sensitive to various parameters of the surrounding biological microenvironment (pH, ion concentration and binding, enzymatic activity, temperature, redox state); (iii) provide robust in vivo quantitative measurements, as this metric is independent of all these parameters that affect intensity measurements (motion, absorption, concentration).
Nevertheless, fluorescence lifetime measurements have not yet been fully exploited in clinical environments. This is due to a few barriers that are still to be overcome. This includes the complexity of the instrumental setup combined with a high cost when compared with the steady-state methods. Recent advances in more compact and affordable light sources (i.e. solid state pulsed UV lasers) and fast electronics are expected to address this barrier. A second limiting factor is the lengthy data acquisition and analysis, which until recently has prevented real-time applications of lifetime techniques. The advance of computational units, with multicore CPUs and GPUs, and the continuously developing parallel computing algorithms, for both data acquisition and analysis, are expected to impact significantly this limiting factor and lead to real-time time-resolved fluorescence imaging applications within the next few years [33, 64–66].
Most time-resolved fluorescence studies reported have been realized in ex vivo tissue specimens [2, 4, 8, 26, 67]. In part, this is due to restrictions introduced by the lengthy data acquisition and analysis. Results from such studies, however, cannot be fully extrapolated to in vivo cases [26, 68, 69] since the tissue optical properties (including lifetime) change their values upon excision from the living organism. Nevertheless, recent advances in real-time fluorescence lifetime techniques have the potential to be applied in vivo, and combined with various established intra- and/or post-operative assessment techniques, could assist towards the development of new optical tissue diagnostic techniques.
3 Fluorescence Lifetime Spectroscopy and Imaging
Fluorescence intensity decay can be measured using either time-domain or frequency domain techniques. Although both techniques provide equivalent information about fluorescence decay dynamics, they are considerably different in their implementation and application. Furthermore, the time-domain techniques can be implemented using either time-correlated single-photon counting (TCSPC) or pulse sampling and gated detection techniques. The basic principles behind these techniques are shown in Fig. 2. Instrumentation based on these principles has been developed for both point spectroscopy (single channel) and imaging spectroscopy (multiple channels), also known as fluorescence lifetime imaging (FLIM) [2, 3, 7, 67, 69].

a Principle of time-correlated single-photon counting; b Principle of gated detection (e.g. by using a gated ICCD camera); c Principle of pulse sampling detection (e.g. by using a high sampling rate and analog bandwidth digitizer); d Principle of detection in frequency domain. The ratios B/A and b/a represent the modulation of emission and excitation, respectively, and their ratio represents the demodulation factor. All schematics were adapted from Lakowicz [3]
In addition, several signal processing techniques have been developed to approximate the fluorescence intensity decay. The multi-exponential model, and its more recent stretched exponential model, is the most widely applied approach to extract the fluorescence decay from the measured transient signals. Other models, such as the Laguerre expansion model or the emerging phasor approach, have also been described in literature [3, 70–72].
3.1 Time-Resolved Techniques Used in Tissue Characterization
The time-resolved fluorescence techniques are based on the measurement of fluorescence intensity as a function of time-delay following pulsed excitation [2, 3, 67, 73]. The most commonly used technique for such measurement is the TCSPC, which has been thoroughly described in literature for both spectroscopic and imaging applications [2, 3, 7, 67, 73, 74]. The extensive application of this technique has led to the development of general purpose TCSPC commercial systems such as FluoTime 300 from PicoQuant GmbH [75] and Simple-Tau 830 from Becker and Hickl GmbH [76].
The basic principle of TCSPC technique is depicted in Fig. 2a. A high repetition rate excitation pulsed light source is used to induce fluorescence emission. The time coincidence in the pulse period for each detected photon is measured and thus accumulation of photon detection events is realized by means of histogram memory. Repeating the process for a large number of excitation pulses, the photon distribution for the entire acquisition time is possible. This technique has been generally preferred for lifetime measurements due to its high sensitivity, near-ideal signal-to-noise ratio (all photons appear in the result and the standard deviation of each measurement can be estimated via Poisson statistics), and a very good time resolution (time resolution is not limited by the width of the single-electron response, but by the transit-time jitter in the detector). Moreover, by incorporating sample scanning modules in the system, spatial information can also be attributed to the measurements and thus formation of fluorescence lifetime images is possible. This represents a direct way to implement FLIM on conventional scanning microscopes [2, 74, 77, 78].
Two alternative techniques are the gated detection and pulse sampling techniques [3]. Implementation of the former requires the use of a time-gated detector, with the gating functionality usually provided by optical intensifiers, whose gain can be modulated through application of pulsed voltage. The basic principle of this technique is shown in Fig. 2b. The sample is illuminated with finite excitation pulses and the emitted transient signal is recorded at different relative delays with respect to the excitation pulse. Integration of the acquired intensities at each delay time reconstructs the entire fluorescence decay curve. Wide-field time-gated fluorescence lifetime imaging microscopy is an application that has exploited this technique [2, 79, 80], especially after the introduction of the intensified charge-coupled devices (ICCD) in the early 1990s.
Recently, the pulse sampling technique (Fig. 2c) has gained popularity due to recent advances in fast digitizers. In this case the detector remains on until the entire fluorescence decay is recorded. The output signal from the detector is digitized through a high sampling frequency and analog bandwidth digitizers (e.g. digitizers with picoseconds resolution). For fluorophores with relative high quantum efficiency a single excitation pulse is enough to generate a measurable fluorescence pulse transient (decay). This technique has been employed in several point time-resolved fluorescence spectroscopy (TRFS) systems [25, 68, 81–86]. More recently a few groups have implemented scanning applications of this technique, including 1D-linear, 2D-raster or 2D-rotational scanning and reconstruction of fluorescence lifetime image [33, 66, 83, 87, 88]. A limitation of this technique, however, is that the unknown instrumental noise may influence the accurate recovery of the fluorescence decay. Recent studies try to investigate this issue and assess the accuracy levels of such systems [33].
In general, the time-domain techniques are based on complex and expensive opto-electronic equipment that remains a challenge for clinical translation. Both short pulsed (sub-nanosecond) laser and high speed detectors such as ICCDs and photomultipliers are required for the fluorescent transient signal detection. However, time-domain fluorescence measurements present some significant advantages [67]. The Fourier spectrum of the used pulsed sources is adequately broad to allow simultaneous acquisition of tissue complex fluorescence decay dynamics due to co-localization of the numerous endogenous fluorophores. In addition, measurements with low repetition rates (<10 kHz) of the excitation sources are not influenced by the ambient light of the surrounding environment, which is of great significance for the translation of such techniques to clinical applications [67].
Both single- and multi-photon [77, 89] techniques have been used for tissue fluorophores excitation. The latter approach is based on high repetition rate and tunable pulsed lasers in the near-infrared (NIR) region of spectrum, where numerous commercial options of such sources exist. Furthermore, these lasers reduce the global tissue phototoxicity and improve significantly the penetration depth, compared to the highly absorbed UV radiation. Nevertheless, the increased complexity of multi-photon based systems has limited their applicability only on tissues directly exposed to the excitation source, e.g. skin [89]. A commercial multi-photon excitation system (DermaInspect) for skin time-resolved fluorescence measurements has been developed by JenLab GmbH [77, 90].
The basic principle of time-resolved frequency domain technique is shown in Fig. 2d. This technique is based on an intensity-modulated excitation source and provides lifetime information through quantification of the phase shift between the sinusoidally modulated excitation and the corresponding emitted fluorescence. The repetition pattern of the intensity-modulated excitation light does not need to be purely sinusoidal, because any repetitive waveform can be decomposed into a sum of sinusoids through Fourier analysis [91]. Detection of frequency-domain fluorescence data is possible by wide-field excitation and a gain-modulated camera, or by scanning and detecting the fluorescence by gain-modulated point-detectors [73]. In addition, the finite response of the investigated sample results in demodulation of the emission as defined by the demodulation ratio, Fig. 2d [2, 3, 67]. Although frequency-domain technique requires relatively simpler instrumentation schemes than time-domain, acquisition of multiple fluorescence decay signals require repeated measurements with different modulation frequencies [3, 67]. This approach is common and accurate in cuvette-based measurements, but it is not practical for imaging applications because of the increased data acquisition and processing time. This limits the applicability of frequency-domain fluorescence measurements in clinical practice [91].
3.2 Analysis of Time-Resolved Fluorescence Measurements
Tissue endogenous fluorophores are characterized by a broad and overlapping absorption spectrum, resulting into simultaneous multiple fluorophore excitation. The measured fluorescence decay is consequently the superimposition of all individual decays and thus it cannot be modeled by a single exponential decay [3]. In addition, the shape of the measured fluorescent pulse transient is the result of the convolution between the actual fluorescence impulse response function (fIRF) and the instrumental impulse response function (iIRF). The broader the latter the higher impact on the measured decay [3, 71, 92]. Thus, in order to recover the fIRF from the measured data a deconvolution process is required to remove the instrumental response. In time-domain, lifetime measurements are made at N equally spaced sampling time points and thus it is usually expressed in the following discrete-time formula:
for k = 0, ···, N – 1. In this expression y(k) represents the kth measured decay, I(k) is the iIRF and h(k) is the desired fIRF. With ɛ k is represented the additive measurement noise. Thus recovery of the true fluorescence decay from the measured transient signals encompasses methods of rapid and accurate deconvolution of Eq. (1). Such methods are based on the mathematical models used to describe the fluorescence impulse response function. These models are generally categorized in two groups: parametric models, which require a priori knowledge about the dynamic behavior of the decay, and non-parametric models, which do not require such assumption.
For the first category of mathematical models the most commonly applied is the least-square iterative deconvolution, in conjunction with a multi-exponential approximation of the decay function [2, 3, 92]:
where τ i and A i correspond to the decay times and the amplitude coefficients, respectively, and M is the total number of existing fluorophores in the investigated sample. Equations (1) and (2) are used to formulate the physical model which, through the use of non-linear least square optimization methods (i.e. Gauss-Newton, Levenberg-Marquardt), is fitted to the measured decays and the multi-exponential parameters are approximated. The applied least-square optimization schemes are usually computationally expensive. Additionally, since these models do not form an orthonormal basis, the optimization function could converge to a minimum of the error function, which might be different with different initial values of the decay times and amplitude coefficients. Thus a unique solution of these models is not guaranteed [3].
However, the wide application of the models described in Eq. (2) is due to the fact that many physical phenomena can accurately be described through first-order differential equations [93]. Nevertheless this is true when considering known fluorescence species in controllable environments (i.e. cell imaging). In more complex systems, like the case of biological tissues, the type and/or the number of the intrinsic fluorophores is rarely known a priori. In such systems the multi-exponential approximation cannot be fully justified from a biological perspective. Moreover, the assumption of discrete fluorophores is not valid in most systems (i.e. a fluorophore in a mixture of solvents may be subject to various relaxation processes). Instead the system can be described by a range of exponential decays with a range of lifetime values [3, 67], known as stretched exponential models [71, 94].
In contrast, the application of non-parametric mathematical models does not require any a priori knowledge of the underlying decay characteristics or any information regarding the number of the fluorophores. By relying on expansion basis sets, these models are based on more simplified mathematical expressions and can be solved through more efficient numerical approaches. Such approaches include series based on Fourier [95], Laplace [96], exponential [97] or Laguerre [98] transforms. Without any information regarding the physical properties of the investigated fluorescence decays, the non-parametric models, however, are prone to converge to decays that do not bear any physical meaning, e.g. oscillations.
A robust non-parametric model which has been used to recover the fluorescence dynamics from biological tissues is based on a Laguerre basis of functions [69, 99, 100] (LBFs). By using this model the fIRF can be expressed as a series of orthonormal set of discrete time Laguerre basis functions b l (k):
where c l represent the Laguerre expansion coefficients and L is the order of the Laguerre functions. The discrete time Laguerre basis functions can be estimated through a recursive formula [101]:
with b l (– 1) = 0. This recursive formula derived from the analytical expression of the Laguerre basis functions [101]:
where the scale parameter a (0 < a < 1) determines the overall decay rate of the defined set of Laguerre basis functions.
In this approach the selection of the scale parameter is critical for the successful convergence of the applied optimization process. In addition, practical use of this model requires careful determination of the order of the Laguerre functions [71]. Recently this model had been revisited and a semi-parametric model was proposed by Liu et al. [102]. In the context of fluorescence decay deconvolution, the semi-parametric model provides constrain parameters to the applied optimization process, which are extracted by a priori knowledge of the underlying physical process. A robust technique for estimating the optimal scale factor of the Laguerre basis functions or their order has been reported [102]. The commonly used least-square deconvolution technique was also constrained by physical characteristics of the fluorescence decay signals. Specifically, these decays should always be positive, monotonically decreasing (not oscillating), strictly convex (exponential decays), and asymptotically should converge to zero [71, 102].
Another emerging non-parametric method for analysis and visualization of lifetime data is the phasor approach [72, 103, 104]. This technique, also known as AB-plot, does not require an iterative optimization process. It is rather a graphical representation of the lifetime values. To date it is mostly applied to the analysis of data from frequency-domain measurements [103]. Briefly, phase and modulation values of each measurement are projected in the two dimensional polar plot [72, 104]. The location of these projections, along with basic vector algebra, provides information regarding the fluorescence decays of the investigated specimens. Through application of the Fourier transform, the phasor approach is also applicable to time-domain fluorescence measurements [104]. Since phasor approach does not rely on fitting processes, it can provide rapid information about the measured fluorescence decays and is less sensitive to noise than the common optimization based procedures [104, 105]. Most of the phasor based fluorescence lifetime studies have been performed in vitro or cuvette measurements [72, 103, 104, 106, 107]. Recently, applications of this technique to fluorescence data from tissues measured in vivo have been reported in literature [108, 109].
4 In Vivo Assessment of Human Diseases Based on Time Resolved Fluorescence Measurements
Although numerous clinical studies have been reported, fluorescence lifetime techniques have not been fully exploited for in vivo tissue diagnosis. In this section we summarize the main fluorescence-based tissue diagnosis in patients and emphasize the TRFS and FLIM studies. In general, current reports demonstrate the application of these techniques to cancer diagnostics (GI tract, bronchi/lung, skin, head and neck, brain and breast), ophthalmic diseases, and atherosclerotic cardiovascular disease.
4.1 Cancer Diagnostics
4.1.1 Gastrointestinal Tract (GI) and Bronchi
Although cancers of esophagus, stomach, and colon are among the most common types of cancer worldwide, the current state-of-art in diagnosis techniques for early detection of the disease still rely on conventional white light inspection [110]. Moreover the availability of both commercial endoscopic systems with fluorescence imaging capability and conventional white-light endoscopic devises (i.e. endoscopes, bronchoscopes, and laryngoscopes) have enabled numerous studies of the GI and pulmonary tracts. Applications include cancer in the GI tract (e.g. colonic polyps and Barret’s esophagus), as well as cancer in the bronchi and larynx. Most of these studies are based on intensity measurements and the assessment of the detected contrast between normal and diseased tissues. In general this contrast, which is expressed as lower fluorescence intensity values and/or as spectral changes, is used to delineate the tumor boundaries. The results are overlaid as pseudo-color maps onto the conventional endoscopic images. Nevertheless, the sources of contrast are still unclear, and thus these tools present relatively limited specificity. For this reason an increasing number of clinical trials are currently conducted in an attempt to further evaluate the performance of fluorescence technique either as a standalone diagnostic tool or as a complementary method for other conventional endoscopic techniques [110].
Similarly to the steady-state studies, the in vivo human time-resolved fluorescence measurements were implemented using the existing clinical endoscopic systems. In order to access the tissue of interest, a fiber-optic was introduced in the accessory port of the commercial endoscope. Results from these studies are very promising for GI and bronchi early cancer detection [110]. However, in contrast to rather large number of steady-state in vivo human studies, the reports on time-resolved fluorescence are sparse (Table 1).
The first TRFS study was reported in 1998 by Mycek et al. [84] for the diagnosis of colonic polyps. The study employed a pulse-sampling TRFS instrument for the discrimination of adenomatous from hyperplastic colonic polyps. A pulsed nitrogen laser (337 nm, 4 ns, 20 Hz repetition rate) was used to excite the endogenous fluorophores, while the emitted fluorescence was detected at 550 ± 20 nm by an avalanche photodiode connected to a digital oscilloscope (500 MHz bandwidth, 2 GS/s sampling rate). A total of 17 patients were examined. The reported average lifetime values indicated that the emission decay from adenomatous (τavg = 9.3 ns) was faster than from non-adenomatous polyps (τavg = 10.5 ns). These values were approximated through the evaluation of the fluorescence pulse width, rather than the application of a model-based or model-free deconvolution approach. After analyzing these findings the authors claimed a sensitivity of 85 % and a specificity of 91 % in distinguishing adenomatous from non-adenomatous colon polyps. These statistical figures were revisited (73 % sensitivity; 70 % specificity) in a subsequent study from the same group and in a much larger data set (68 patients) [120].
More recently (2014) Coda et al. [121] reported comprehensive results from experiments conducted in colonic tissue using a fiberoptic-based TCSPC technique. The measurements were conducted in freshly biopsied/resected tissue specimens from patients undergoing endoscopy. Two pulsed diode lasers were used as excitation sources with central emission wavelengths of 375 and 435 nm. Both lasers emitted pulses of 50–150 ps duration and were configured to operate at 20 MHz. The emitted fluorescence was collected by 14 fluorescence detection fibers and imaged onto the input slit of a grating, which was attached to a 16 channel multi-anode photomultiplier tube (PMT) detector. The PMT was linked to a computer with a TCSPC card, allowing collection of fluorescence decays in 16 spectral channels spanning a wavelength range of 400–600 nm. A sketch of the developed system is shown in Fig. 3. The approximation of the lifetime values was based on the fitting onto a bi-exponential model. A total of 47 endoscopic samples from different regions of colon were collected from 17 patients undergoing colonoscopy. All measurements were implemented shortly after excision. By combining various spectroscopic parameters the neoplastic vs non-neoplastic polyps were discriminated with a sensitivity/specificity of 75 %/83 %. Although further work is required to investigate if the discrimination statistical figures of this study can be improved through the use of e.g. other excitation wavelengths, or combinations of fluorescence spectroscopy with other non-invasive optical techniques, this study provided thorough insight for the use of time-resolved fluorescence measurements to assess neoplastic colonic polyps. It also provided valuable information regarding the sources of any contrast resulted from the two excitation wavelengths.

Optical configuration of the fiber-optic probe-based fluorescence lifetime spectroscopy system. Insets show the arrangement of the optical fibers in the custom fiber bundle at the distal end of the probe and at the output of the fluorescence detection branch of the proximal end of the probe. Cores shown in green are fluorescence detection fibers; fibers colored blue are used to deliver the laser excitation; the orange and red cores were unused in this study. Adapted from Coda et al. [121]
Another application of TRFS in the GI tract was reported by Pfefer et al. [85]. It concerns the diagnosis of high grade dysplasia in Barrett’s esophagus in 37 patients. The instrumental setup used for this study is a modified version of the TRFS system used by Mycek et al. [84] and allows for simultaneous acquisition of both wavelength- and time-resolved fluorescence data from tissue upon 337 and 400 nm excitation. The fluorescence decays in this study were approximated through an iterative optimization process and by modeling the decays as a bi-exponential model. It was found that for 337 nm excitation the dysplastic tissues emit fluorescence with faster decay than the non-dysplastic tissues, while 400 nm did not provide effective means of tissue diagnosis. Moreover, this study revealed that changes in TRFS data were not significant enough to support the development of diagnostic algorithms. A retrospective statistical analysis indicated that none of the parameters calculated from TRFS could achieve sensitivity and specificity values above 60 %.
TRFS measurements have also been applied to detection of cancer in the bronchi. One such study was implemented by Uehlinger et al. [46]. This study was conducted in 11 high-risk lung cancer patients undergoing conventional white-light bronchoscopy. The excitation source of the apparatus used was a nitrogen laser-pumped dye laser emitting at 405 nm (500 ps pulse width, 1–50 Hz repetition rate), while a streak camera positioned at the imaging plane of a spectrograph spectrally (640 channels, 15 nm resolution) and temporally (480 channels, 280 ps resolution) resolved the transient fluorescence signals. The fluorescence decays were analyzed using the Levenberg-Marquardt iterative nonlinear least-squares deconvolution fitting algorithm, assuming an a priori bi-exponential model. This study concluded that the decay parameters obtained from healthy bronchial mucosa and preneoplastic or neoplastic lesions show no statistically significant differentiation. In fact, the quantified differences are smaller than inter- and intra-patient variations of the healthy mucosa. These results suggest that the heterogeneities in the healthy mucosa affect the fluorescence decay measurements and that make the discrimination of diseased vs. healthy tissue difficult. Nevertheless, this study provided useful insights into the mechanisms underlying the fluorescence spectral and intensity contrasts in bronchial tissue.
Overall these studies do not conclude on statistically significant results for the detection of cancer in the GI tract and bronchi, they have significantly contributed to the better understanding of the fluorescence sources in these tissue types. Further systematic investigations are required to correlate other spectroscopic parameters, or combinations of such parameters, with histopathological findings, in an attempt to increase the diagnostic power of fluorescence lifetime technique.
4.1.2 Head and Neck Cancer
The relative accessibility of the oral cavity to flexible (fiber optic), semi-flexible (light guide), or rigid (direct illumination) light guides led to the development of a large number of both point spectroscopy and wide-field imaging devices to assess various lesion types. Table 2 summarizes the main fluorescence studies of oral cancer in patients.
As in the case of GI tract or bronchi, most of the studies in head and neck cancer (see Table 2) are based on either steady-state spectroscopy or intensity imaging. In order to improve specificity of the fluorescence measurements most of these studies included complementary diagnostic techniques, such as diffuse reflectance spectroscopy [57, 60], light scattering spectroscopy [57], depth sensitive probes [60], and digital image processing [127]. Only a limited number of studies demonstrate the applications of fluorescence lifetime techniques for the diagnosis of oral carcinoma in patients. These are described in brief below.
Chen et al. [128] employed a point spectroscopy lifetime technique consisting of a handheld optical fiber-based probe connected to a commercial TCSPC system (HORIBA Jobin Yvon IBH, UK). A pulsed laser diode (wavelength 408 nm, pulse width 70 ps) was used for tissue excitation. The fluorescence emission was resolved in 632-nm band pass filter (bandwidth 20 nm) corresponding to porphyrin fluorescence. The acquired signals from 55 patients were fitted to a bi-exponential model and an ANOVA test applied to the resulting average lifetime values was used to determine their ability to resolve normal oral mucosa (NOM), epithelial hyperplasia (EH), verrucous hyperplasia (VH), and epithelial dysplasia (ED). In addition a Fisher discriminate analysis was used to find optimal thresholds for different types of samples and to develop the classification algorithm. This study resulted in an accuracy rate of 75 % for VH and EH, of 93 % for ED, and of 100 % for NOM samples.
Meier et al. [83] reported the use of a point spectroscopy TRFS technique based on pulse sampling for diagnosis of head and neck squamous cell carcinoma (HNSCC). The study was conducted in 9 patients. The measurements were taken from 53 sites including buccal mucosa, tongue, vocal cord and floor of the mouth. The fluorescence was induced with a pulsed nitrogen laser (337 nm, 700 ps pulse width), spectrally resolved with a monochromator (360–610 nm spectral range, 5 nm interval), detected with a gated multichannel plate photo-multiplier tube, and time-resolved with a digital oscilloscope (2.5 GHz bandwidth, 40 GS/s sampling rate). A Laguerre basis function deconvolution scheme was employed to approximate the fluorescence decays. The lifetime values were computed at 1/e of the intensity’s maximum value. With this apparatus both spectrally and temporally resolved fluorescence characteristics were recorded and comparison between the malignant and normal tissue was performed using 2-way ANOVA. Significant spectral intensity peaks of normal and malignant tissue were noted at two spectral bands: 370–400 nm (corresponding to the collagen emission) and 440–470 nm (corresponding to the NAD(P)H emission). In general, this study revealed that across all wavelengths between 360–610 nm, the absolute fluorescence intensity was significantly decreased in carcinoma compared to the normal mucosa. These findings agree with other fluorescence steady-state studies [57, 60, 125–127]. In addition, when considering all anatomical sites, the average lifetime at the 370–400 nm spectral band did not present any statistically significant differentiation, while at the 440–470 nm spectral band, the average lifetime was 1.50 ± 0.05 ns for normal areas and 1.38 ± 0.05 ns for malignant tissue (P < 0.001). These differences are most probably attributed to different ratios of free and bounded NAD(P)H in the two tissue types. When bound to proteins, the NAD(P)H fluorescence lifetime is known to increase multifold [3, 8].
Sun et al. [124] recently reported on the findings of a FLIM based study from 26 sites in the oral cavity (Fig. 4). This study was conducted in a subset of patients involved in Meier’s TRFS study [83] described above and used the same pulsed nitrogen laser (337 nm, 700 ps pulse width) for fluorescence excitation. However, the emitted fluorescence was recorded by a gated optically intensified CCD camera (minimum gating time of 200 ps; repetition rate up to 200 kHz) and a band-pass filter (460 ± 25 nm). Both excitation and emission lights were delivered via a customized imaging probe with a high numerical aperture (NA = 0.48) excitation fiber and a collection fiber image bundle (10,000 fibers, ~0.6 mm total diameter) and a GRIN lens objective (FOV = 4 mm). The Laguerre basis function based deconvolution scheme was also employed to approximate the fluorescence decays. The findings in this study were equivalent to the ones in the TRFS study. Specifically, the average fluorescence lifetime of HNSCC presented smaller values when compared with that of normal tissue.

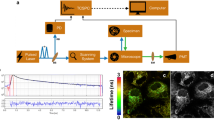
Fluorescence lifetime imaging (FLIM) system for intraoperative diagnosis of oral carcinoma. a Endoscopic FLIM schematic. b Picture of the semiflexible integrated endoscope used in patients studies. c Picture of the FLIM probe applied to oral cavity. Adapted from Sun et al. [124]
The results from these two independent studies (Meier et al. [83] and Sun et al. [124]) demonstrate that upon 337 nm excitation the approximation of the fluorescence decay dynamics in the 460 nm centered band can provide statistically significant discrimination of oral carcinoma tumors (regardless of their location) from the surrounding normal tissue. Further measurements are required to sufficiently account for intra- and inter-patient variability and to construct a dense database for the development of efficient diagnostic tools.
4.1.3 Brain Tumors
The highly infiltrative nature of brain tumors makes the delineation of positive margins a challenging procedure. The development of an intra-operative tool that would assist neurosurgeons in assessing the infiltration of tumor cells in normal brain tissue is still an unmet clinical need. As shown in Table 3, overall only a few groups have explored the application of fluorescence spectroscopy techniques to diagnosis of brain tumors. Moreover, the clinical application of TRFS and FLIM techniques is even less explored [81, 82, 129]. These studies are briefly reviewed in the following.
Fluorescence decay characteristics for both high and low grade glioma were primarily assessed in three studies [81, 82, 129]. These characteristics were compared to the corresponding quantities from normal cortex and white matter. All of them were conducted in patients undergoing surgery for brain tumor removal. Measurements were conducted through customized fiber-optic probes that were used to remotely access the brain tissues during conventional craniotomy procedures.
In the two TRFS studies [81, 82] fluorescence was induced with a pulsed nitrogen laser (337 nm, 700 ps pulse width), spectrally resolved with a monochromator (360–550 nm spectral range, 10 nm interval), detected with a gated multichannel plate photomultiplier tube, and time-resolved with a digital oscilloscope (1 GHz bandwidth, 5 GS/s sampling rate). A Laguerre based deconvolution scheme was used to approximate the fluorescence decays. Univariate statistical analysis (one-way ANOVA) was used to identify the set of spectroscopic parameters that best discriminate between distinct tissue types, as defined by the histopathologic analysis. The first of these studies was conducted on 17 patients [81], while fluorescence from 42 patients was acquired for the second study [82]. Results demonstrate that intensity ratio between 390 ± 10 nm (collagen emission peak) and 460 ± 10 nm (NAD(P)H emission peak) bands, as well as the corresponding fluorescence decay dynamics, have the potential to discriminate between normal brain tissues and low and high grade gliomas. Moreover, by using combined spectral and temporal parameters of the TRFS measurements it was possible to classify low-grade gliomas with high sensitivity and specificity (100 and 98 %, respectively) [82]. However, when high-grade gliomas were assessed, the sensitivity was limited (47 %), but specificity remained high (94 %). A possible source for the low sensitivity was the heterogeneous nature of the investigated tumors, as well as phenotypic diversity.
FLIM measurements [129] were conducted in 13 sites from 3 patients. A 337 nm pulsed nitrogen laser (337 nm, 700 ps pulse width) was used to excite fluorescence, while detection was achieved through a fast-gated ICCD equipped with a bandpass filter (460 ± 25 nm). The schematic of the FLIM system is shown in Fig. 5, along with some characteristic measurements. To distinguish between tissue types (tumor vs. normal), FLIM decay characteristics were assessed using ANOVA. Findings were complementary to the two TRFS studies. Specifically, fluorescence decay parameters at 460 nm have the potential to successfully discriminate between gliomas and normal brain tissues.

a Schematic of the FLIM instrument setup, including a picture of the tip of the imaging bundle probe positioned on the interrogated area of the cortex. Representative fluorescence intensity and lifetime images. b, c, and d are intensity images. e, f, and g are fluorescence average lifetime images. h, i, and j show fluorescence lifetime histograms. j Depicts lifetime histograms of two ROIs identified in (g), where the arrow indicates the tumor area. For each image, the average lifetime value was retrieved from the 2 × 2 binning pixel of four original pixels (2 × 2 square). All average lifetime values for each binning pixel in the ROI were plotted together to show a histogram of average lifetime distribution. Adapted from Sun et al. [129]
A common conclusion in all three studies is that further investigation is required in order to account for gliomas genetic sub-classification associated with prognostics. Fluorescence emission of the diverse brain tumor phenotype (e.g. malignant astrocytomas, mixed gliomas and oligodendroglioma) has not yet been fully investigated in order to conclude on the ability of fluorescence signatures in analyzing and distinguishing gliomas.
4.1.4 Other Cancers
Additional clinical applications reported include the detection of cervical cancer and assessment of breast cancer margins. Representative studies are summarized in Table 4.
Several steady-state fluorescence studies of cervical cancer were reported. Nevertheless most of them have employed combined fluorescence and diffuse reflectance measurements to improve tissue classification accuracy. Recently Hariri Tabrizi et al. [23] published a review summarizing all optical spectroscopy techniques used for this clinical problem. Other pertinent reviews were also previously reported [45]. In addition, commercial devices based on fluorescence and reflectance spectroscopy have been developed for the diagnosis of cervix cancer, some of which have been described previously [42, 43]. In general, optical spectroscopy has been applied as a complementary technique to conventional colposcopy. It has been shown, however, that both optical spectroscopy and conventional colposcopy show equivalent performance when applied as stand-alone diagnostic tools [45]. Promising results were reported by many studies; however, further investigations with larger samples are required [23]. Nevertheless, despite the increased scientific interest in fluorescence measurements for cervical cancer diagnosis, no time-resolved spectroscopic or imaging techniques were reported. The inherent nature of the fluorescence lifetime measurements can potentially improve the specificity of the fluorescent measurements conducted in cervix and eventually can initialize a totally new clinical application area for time-resolved techniques.
Breast cancer detection and margins assessment is another challenging and still unmet clinical need that fluorescence measurements can address. As summarized in Table 4, a few SSFS studies were conducted in the attempt to intra-operatively assess breast cancer margins on fresh excised tissue samples. However, all of them conclude that steady-state fluorescence measurements cannot discriminate between fibrotic and malignant tissues and thus many proposed approaches are combined with diffused reflectance measurements [28, 52]. Up to date only one paper reports a time-resolved fluorescence study focused on breast cancer margins assessment [135]. The customized system included a supercontinuum pulsed laser source with a 5 ps pulse width and a broadband spectrum (~440 nm to 2 μm) at a repetition rate of 20 MHz. The excitation wavelength was selected using a 447 ± 30 nm bandpass filter. The emitted fluorescence was detected at four spectral bands (532 ± 5, 562 ± 20, 632 ± 11, and 684 ± 12 nm) via a cooled PMT and a PC-based single photon counting card (Fig. 6). All measurements were conducted on a total of 93 locations across 6 breast cancer cases (34 from invasive ductal carcinoma regions, 31 from fibrotic tissue regions and 28 from adipose tissue regions) immediately after their resection through mastectomies or lumpectomies. Fluorescence decay was approximated via a bi-exponential model, and a multinomial logistic regression was used as the classification tool. This study concluded that the mean fluorescence lifetime values were significantly different between invasive ductal carcinoma and the two other types of benign breast tissue, predominantly at 532 and 562 nm. At these wavelengths the lifetime contrast was attributed mainly to flavins and lipopigments. Also this study concluded that DRS technique is more robust in distinguishing the malignant tissues from the other two types, than lifetime measurements.

a A simplified block diagram of the dual-modality data acquisition system; b an illustration of the probe tip geometry; c A picture showing breast specimen being measured by the optical fiber. The colors on the edges of the specimen are due to ink labeling done by the pathologist as a conventional procedure to mark anatomical/structural sides of the breast sample. AFLM Autofluorescence lifetime measurements; LRS Light reflectance spectroscopy. Adapted from Sharma et al. [135]
Based on these results, the same group expected that the methodology described in [135] is highly likely to work for detection and classification in other types of cancer as well. Thus, recently the system in Fig. 6 was also used for the detection of prostate cancer of fresh ex vivo radical prostatectomy specimens from 37 patients with high volume, intermediate-to-high-grade PCa (Gleason score, GS ≥ 7) [136]. Findings were equivalent to the breast cancer margins assessment study [135]. However, since there is limited understanding of the origin of fluorescence sources at these wavelengths, it is expected that further investigation of the molecular contrast sources will assist towards the identification of fluorescence decay parameters with best discrimination power.
4.2 Human Skin Diagnostics
Because of the direct accessibility of skin, fluorescence techniques have been broadly applied for the analysis of skin physiology, optical biopsy of skin, and detection of dermatological disorders. The latter includes fungal infections, skin age, hair pigment, and cancer. Table 5 summarizes the most representative studies. The fluorescence contrast sources commonly investigated in these studies are the endogenous bio-molecules such as flavins, NAD(P)H coenzymes, metal-free porphyrins, components of lipofuscin, melanin, elastin, collagen and keratin. Moreover, a broad range of customized fluorescence-based devices have been developed, such as multi-photon excitation and high resolution scanning systems, or free-optics based systems. The direct accessibility of the skin has permitted the extensive exploitation of both steady-state [47, 48, 58, 137–141] and time-resolved techniques [77, 86, 89, 142–144]. For example SSFS measurements have been shown to present the potential to discriminate non-melanoma skin cancer (NMSC) [86, 142]. Nevertheless, the wide intra-patient variability and the heterogeneity of skin fluorescence strongly influence the development of any classification architecture. This lead to limited specificity in skin tissue diagnosis, even in studies where fluorescence measurements are complemented with DRS measurements [58].
One of the most commonly applied techniques for human skin diagnostics is the multi-photon scanning approach, including FLIM. This approach offers the opportunity for non-invasive optical sectioning of skin and disease stage in vivo. Many of these studies are summarized in recent reviews [6, 22, 89, 140]. Studies conducted on excised human skin [89] have provided valuable information on the sources of fluorescence lifetime contrast and their potential to distinguish between various skin conditions. Nevertheless, measurements on excised tissues are not suitable to assess the applicability, as well as the accuracy metrics, of this technique for skin diagnostics. For example, skin freezing is prone to reduce NAD(P)H, the dominant fluorophore in the viable epidermis, and most likely affects skin fluorescent measurements ex vivo.
The development of a commercial multi-photon excitation system (DermaInspect) for skin time-resolved fluorescence measurements has contributed to the increased number of in vivo skin fluorescence studies in humans. The developed FLIM module includes a tunable NIR light source (titanium sapphire laser: repetition rate of 80/90 MHz, pulse width 75–120 fs FWHM), a x-y galvano-scanner module, and a TCSPS unit. This technique is not only compatible with fast scanning but also capable of recording FLIM images in several wavelength channels simultaneously. Moreover, multi-exponential fit is commonly used to analyze and approximate fluorescence decay, with the bi-exponential model being the most frequent. One detailed study using DermaInspect device was reported by Dimitrow et al. [137]. This was the first clinical in vivo study of benign and malign melanocytic skin lesions using this imaging device. A total of six distinctive malignant melanoma features were measured and statistically assessed from 83 melanocytic skin lesions. Sensitivity values up to 95 % (range: 71–95 %) and specificity values up to 97 % (range: 69–97 %) were achieved for diagnostic classification. In addition, this study showed that architectural disarray of the epidermis, poorly defined keratinocyte cell borders, as well as the presence of pleomorphic or dendritic cells were of prime importance. By means of this procedure accuracy values up to 97 % were reached.
The TRFS applications for skin diagnostics are sparse [86, 142]. One representative study was reported by Blackwell et al. [142]. Within the framework of this study TCSPC measurements were conducted on 75 patients (37 healthy subjects and 38 diabetic subjects) and on various locations of a 31-year-old healthy Caucasian male. The excitation source was a LED (375 ± 5 nm, 700 ps FWHM), while the emitted fluorescence was recorded via a photomultiplier attached to a monochromator at four distinct wavelengths (442, 460, 478, and 496 nm). The intrinsic fluorescence decays were approximated via a tri-exponential model and fitted to the acquired data with application of the Levenberg-Marquardt method. In this study fluorescence characteristics were assessed as a function of location, gender, skin complexion and age and results indicated that specific decay components can be associated to free and bound NADH, while others to advanced glycation end product (AGE) crosslinks.
Subsequent studies in this area, however, have not been reported despite the promising results reported in current studies. The increased heterogeneity of human skin is the most significant limiting factor. Thorough histological investigations, in conjunction with further lifetime measurements are required to assess (a) the intrinsic fluorophores of human skin and (b) any possible correlation between fluorescence decay parameters and the intrinsic fluorophores as a function of different wavelengths and of different skin types (location, gender, etc.).
4.3 Eye Diseases Diagnostics
Intrinsic fluorescence has been also investigated as a diagnostic tool for human eye diseases. Applications include the detection of metabolic alternations in early stages of age-related macular degeneration and diabetic retinopathy. Similar to the human skin, the human eye can be directly accessed. In addition, the availability of commercial fluorescence imaging systems has led to numerous in vivo fluorescence case studies. Table 6 depicts the most representative reports.
Time-resolved fluorescence measurements of the eye are significantly challenging due to numerous reasons. For example the eye is sensitive to direct illumination and special considerations must be adopted regarding the applied wavelengths and exposure levels. Other reasons include the efficiency of light propagation through the ocular media and the ability to define a single imaging plane as the eye anatomy and its multilayer structure present variations between individuals. An additional challenge is the compensation of the motion blur, which is especially present during ocular imaging applications.
In an attempt to address such problems, Schweitzer et al. [150] developed a FLIM-based scanning laser ophthalmoscope (SLO) that was used in several other studies of this group [148, 151, 152]. The excitation source of this system was a picosecond laser (448 nm, 70 ps FWHM), while the emitted fluorescence was detected by a TCSPC module at two consecutive spectral bands (490–560 and 560–700 nm). A tri-exponential model was used to fit the recorded fluorescence decays. Results from these studies demonstrate that a good correspondence can be found between the first component of the tri-exponential model and the retinal pigment epithelium in the 490–560 nm band. Lifetime values from the second component were attributed to the neuronal retina, while the third component was correlated to the connective tissue in the choroid and was covered by the fluorescence emitted from the crystalline lens. Moreover, by investigating healthy subjects, this group reported highest repeatability at the papillo-macular bundle, followed by the fovea and the optic disc. Repeatability was higher in the 490–560 nm band (mean coefficient of variation 7.9 %) than in the 560–700 nm band (mean coefficient of variation 17.7 %) [148].
The contribution of multiple fluorophores distributed among the different fundus layers cannot be efficiently approximated via the tri-exponential model used in these studies. Nevertheless, these studies demonstrate a very unique and challenging application of FLIM. The development of real-time imaging applications could account for many of the existing challenges permitting the implementation of further studies.
4.4 Atherosclerotic Cardiovascular Diseases
One of the most significant, but still unmet, clinical needs is the development of an imaging technology for in vivo analysis of atherosclerotic cardiovascular diseases. Such tool should be capable of discriminating the constituents of atherosclerotic plaque, as its composition plays an important role on plaque instability and susceptibility to rupture [153–156]. Fluorescence techniques, both steady-state and time-resolved, have been extensively investigated as prospect tools for research and diagnosis of atherosclerotic cardiovascular diseases, mostly because of their ability to detect the emitted fluorescence from elastin, collagen, lipid components and other constituents in normal and diseased arterial wall. Current studies have shown the ability of fluorescence measurements to detect plaque disruption [157], plaques with thin fibrous caps [158], and lipid-rich lesions [159]. A detailed review of the most recent fluorescence lifetime studies has been recently reported by Marcu [26].
Nevertheless, most of time-resolved fluorescence studies, TRFS or FLIM, were realized either on excised specimens or on in vivo animal models [33, 70, 153, 156–163]. These are summarized in Table 7. However, only very few in vivo studies in humans were reported.
One of the very few applications of point TRFS measurements for in vivo assessment of plaque fluorescence in humans was reported by Marcu et al. [68]. Within the framework of this study tissue fluorescence was induced with a pulsed nitrogen laser (337 nm, 700 ps pulse width), while the emitted fluorescence was dispersed by an imaging spectrograph/monochromator (360–550 nm; 10 nm spectral increments) and detected with a gated microchannel plate PMT connected to a digital oscilloscope (bandwidth 1 GHz, sampling rate 5 Gsamples/s). A schematic of the developed system is shown in Fig. 7 (Module A). A Laguerre-based deconvolution method was used to approximate the fluorescence decays. In order to identify a set of spectroscopic parameters that best discriminate between different tissue types, a univariate statistical analysis (one-way ANOVA) was used. The developed classification architecture was based on a stepwise linear discriminant analysis (SLDA) approach. This study was applied on 65 patients undergoing carotid endartectomy (CEA) procedure. However, due to limitations imposed by clinical protocols, only 28 areas were measured. Moreover, out of these only 10 measurements were correlated to histopathologic analysis of the plaques. Some representative results from this study are shown in Fig. 7 (Module B). This small number of measurements underscores the challenge to validate the fluorescence results against gold standard methods in histopathology. Results from this study indicated that the spectral intensities and time-dependent parameters, at discrete emission wavelengths, allow for discrimination (sensitivity > 81 %, specificity > 94 %) of various compositional and pathological features associated with plaque vulnerability, including infiltration of macrophages into intima and necrotic/lipid core under a thin fibrous cap. Moreover the results revealed a linear correlation with plaque biochemical content, i.e. elastin, collagen, inflammatory cells, and necrosis.

Schematic representation of the TR-LIFS system used to induce, collect, record and analyze the fluorescence emission of the carotid plaques and sample results. Adapted from Marcu et al. [68]
To date no TRFS or FLIM studies in vivo during catheterization have been reported. Such attempts would require advanced engineering of specialized catheters and blood flushing systems. Recent reports demonstrate a progress in the development of such catheters capable for in vivo measurements [33, 70, 165]. Another limiting factor for using these techniques in vivo is the required time for data acquisition. Spectroscopic measurements are in general relatively slow procedures (a few seconds per point for the acquisition of the entire spectrum). Recently (2014) a scanning TRFS device has been reported that confronts this challenge, as it is capable of acquiring fluorescence decays in multiple spectral bands in a few microseconds [33]. Moreover, [33] reports, for the first time, the development and validation of a hybrid intravascular diagnostic system combining multispectral fluorescence lifetime imaging (FLIM) and intravascular ultrasound (IVUS) for cardiovascular imaging applications. The developed system allowed for helical scanning of narrow arterial vessels and sequential acquisition of time-resolved fluorescence data and ultrasonic data. The guide-wire artifact, present in data from both modalities, allowed for fluorescence lifetime data and ultrasonic images co-registration at the lumen/intima interface. The functionality of this hybrid system was tested ex vivo in explanted intact swine hearts, experiments that demonstrated the ability of this bimodal system to operate reliably in coronary arteries and to provide registered biochemical and structural information. Some representative results from this study are shown in Fig. 8.

Bimodal imaging of ex vivo coronary artery with fluorescence marked drug-eluting stent (DES) deployed in the lumen. a Overview of the pig heart with catheter inserted in the left anterior descending artery. b Close-up of the drug-eluting stent showing the painted sections. c Fluorescence intensity spectra of fluorescent paints, arterial tissue, and the spectral bands of the wavelength selection module. d Fluorescence intensity images, average lifetime images, and intensity weighted lifetime images of four spectral channels showing the stent structure (green paint marked struts are pointed by arrows and the guide wire (vertical line). e IVUS cross-sectional image of the vessel with fluorescence lifetime registered over the arterial wall. Stent struts and guide wire can be identified with both FLIM and IVUS contrast. Adapted from Ma et al. [33]
The system in [33] has been also evaluated in vivo in a pig artery, demonstrating for the first time the ability of a TRFS system to acquire data in a blood vessel in vivo and under a continuous pull-back motion [70]. To address the increased blood optical absorption the distal end of the catheter was enclosed into 1.5 mm inner diameter silicon tubing. During the imaging sequence, the tube was inflated with saline until it sealed the vessel (standard balloon occlusion procedure). Fluorescence signal from the wall was then acquired through the balloon without obstruction from blood. This group is currently working towards the development and evaluation of a co-axial catheter configuration, enabling the FLIM and IVUS components to rotate and acquire data simultaneously. This will maintain the co-registration of the two modalities even during vessel wall motion. In addition, as the developed system is capable of acquiring data in timeframes equivalent to the conventional bolus injection approaches, this research group is investigating the possibility to address the blood optical absorption without the use of the silicon tubing via injection of a flushing agent with appropriate optical property.
5 Challenges in Clinical Translation for Time-Resolved Fluorescence Techniques
Current studies summarized above have demonstrated that steady-state fluorescence measurements can provide good sensitivity for various tissue diagnostic applications, but they present limited specificity. The latter is usually improved when complementary techniques are used, like diffuse reflectance spectroscopy. Nevertheless, due to the heterogeneity of the various tissues and an increased intra-patient variability the overall specificity remains relatively low when compared with the standard histopathological diagnostics.
The measurement of lifetime can provide an alternative means to evaluate tissue fluorescence without the need of any exogenous contrast and has the potential to improve specificity of the diagnostic methods. However, with a few exceptions, most of time-resolved studies have enrolled a limited number of patients, and thus the diagnostic potential of these techniques has not been fully evaluated yet. Overall, two critical challenges remain to be addressed in order to facilitate practical time-resolved fluorescence measurements in vivo in patients.
First, fluorescence lifetime techniques require rather complex and expensive instrumentation. This, in addition to the limited number of available excitation sources and sensitive detectors, has prevented the development of practical clinical time-resolved devices. Moreover, the majority of current TRFS and FLIM systems are characterized by a slow data acquisition speed, which either makes the measurement subject to motion artifacts or results in a limited number of measurements. Moreover, most of TRFS or FLIM studies conducted in vivo employed pulsed nitrogen lasers as an excitation light source. Although the wavelength and output power of these lasers is suitable for most studies, their low repetition rate has precluded the development of high-speed spectroscopic or imaging modalities. On the other hand, most ultraviolet and blue range emission pulsed diode lasers, operating at high repetition rates do not provide adequate output power levels. Recent advances on pulsed solid-state lasers in the ultraviolet spectral region, including compact fiber lasers, can address this need and could become an important component of future time-resolved fluorescence systems. Similarly to the excitation sources, there is a limited choice of fast and sensitive detectors. Most applications employ fast MCP-PMTs or avalanche photodiodes, streak cameras, gated ICCDs and TCSPC modules. These detectors are suitable for clinical applications; however, the limited number of manufacturers has still retained their prices at relatively increased levels. Overall, a broader availability of both excitation sources and detectors could boost the TRFS and FLIM research in clinical environments.
Second, the lack of robust and fast analytical methods for accurate approximation of the fluorescence decays from time-resolved measurements represents a challenge in the development of real-time tissue diagnostic systems. There is also a need for accurate interpretation of the decay components in relation to tissue pathophysiology. A major drawback of parametric models is that they require a priori knowledge about the fluorescence decay model (e.g. multi-exponential approximation function). Knowledge of such model would increase both accuracy and time efficacy of the deconvolution problem. However, an optimal model does not exist, mainly because tissue fluorescence is emitted by several biological molecules with overlapping emission spectra. In addition the characteristics of fluorescence decay strongly depend on the molecular micro-environment, thus no unique solution can be associated with one molecular species. Moreover, accurate estimation of the fluorescence decays would require encountering for scattering and absorption within the investigated tissue, as well as accurate modeling of the tissue surface profile. This can be accomplished through the analytical model of radiative transfer equation, or any of its deterministic or stochastic approximations (i.e. diffusion approximation, Monte Carlo). However, finding a solution for such models besides being time consuming is also based on a priori knowledge of the fluorophores distribution within the tissues, as well as the distribution of any other molecule that absorbs or scatters the wavelengths of interest. In order to simplify the solution of this optimization problem, most fluorescence lifetime studies apply an exponential approximation (mostly bi- or tri-exponential) to evaluate the fluorescence decay dynamics. Although this approach is adequate when a dominant fluorescence source is present, it cannot accurately approximate the true number and their relative contribution of the existing tissue fluorophores. In addition, the solution of the optimization problem with this model strongly depends on the applied initial values of the time constant and fractional contribution of the exponential function. Such problems are prone to converge to different local minima of the error function. Thus a unique solution of these models is not always guaranteed [3].
Several studies have shown that non-parametric models (model-free) are very promising alternatives for evaluating the fluorescence decay without a priori knowledge of the decay function. Although these models do not bear any physical meaning, they succeed in fitting the fluorescence decay with increased time efficacy. One such example is the Laguerre functions-based deconvolution. Also, more recently, the phasor approach has gained a significant attention mainly because it does not require any iterative optimization process to approximate the measured fluorescence decays. Overall, these techniques can provide rapid quantification of the fluorescence decays and are less sensitive to the inherited noise than most of the other deconvolution methods [104, 105]. These non-parametric studies enable further development of real-time TRFS or FLIM diagnostic systems.
An additional clinical translation challenge for fluorescence lifetime based techniques is the ability to correlate the fluorescence decay parameters measured in vivo with the numerous pathophysiological conditions in tissue. Fluorescence data from in vivo studies are typically validated with results from conventional histopathological analysis of tissue removed from optically interrogated regions. This approach, however, is subject to both sampling/registration errors as well as to changes in tissue conditions post-excision (physiology, metabolism, morphology, etc.). As a result, a quantitative interpretation of the fluorescence decay data is practically impossible, especially for metabolically active tissues such as tumors.
Finally, one major drawback of fluorescence measurements is the shallow penetration depth of the excitation light, typically in the ultraviolet-visible wavelength range. For example, single-photon measurements are based on ultraviolet excitation sources, which can provide a tissue interrogation depth bellow 0.5 mm. Multi-photon applications are based on near-infrared excitation sources, which enable higher penetration (~1.5 mm), however it remains mainly a shallow interrogation. As a result TRFS and FLIM measurements are limited to the surface of the investigated tissues/organs. Nevertheless, because early pathological molecular changes usually occur at the surface of the tissue/organ, these techniques are very sensitive to biochemical changes within small tissue volume (high spatial resolution). Future development of hybrid multimodal systems combining techniques capable of providing complementary information (i.e. structural—OCT, ultrasound; molecular—near infrared molecular fluorescence; biochemical—TRFS, FLIM, Raman) could expand the diagnostic ability of current fluorescence-based techniques.
The use of exogenous fluorescence probes can address several of these challenges. Most of exogenous probes are designed for visible and near-infrared excitation sources, which increases the tissue sampling depth when compared with fluorescence measurements that takes advantage of endogenous contrast that are typically excited at the ultraviolet wavelength range. In addition, the high quantum efficiency of the exogenous fluorophores increases the sensitivity of these techniques, while efforts are made to increase also their specificity by improved targeting properties of the fluorophores. A number of clinical studies using exogenous probes or photosensitizers for tissue diagnosis or fluorescence guided surgery are at the edge of clinical translation. A few extensive reviews focused on fluorescence based intraoperative guidance and decision making were recently published by de Boer et al. [166] and Hsu et al. [167]. Fluorescence-guided tumor resection (FGR) during neurosurgery, in particular, has gained significant traction during the last few years [19, 21]. Nevertheless, as noted above, among the numerous molecular probes under investigation only a few have been approved for human use by the FDA (i.e. fluorescein and indocyanine green [7, 14]; porphyrin-based photosensitizers [15–21]). Extensive studies are required to demonstrate the pharmacodynamics/pharmacokinetics along with the binding specificity of such molecular probes. Potential toxicity is an additional factor that hampers the testing and use of new fluorescent probes in humans.
6 Conclusion
Current TRFS and FLIM studies reported by several research groups have demonstrated the potential of fluorescence lifetime measurements as a diagnostic tool to discriminate between normal and diseased tissue in clinical environments. However, the relative limited number of these studies, as well as the small sample population enrolled to date, do not allow for full assessment of the diagnostic potential of TRFS and/or FLIM techniques. Further investigation on both the approximation of the fluorescence decay dynamics and their interpretation in relation to the histopathological analysis is required. Inter- and intra-patient variability studies are also important for the in vivo applicability of these techniques.
Recent advances in excitation sources and detectors, as well as in data analysis and management (i.e. GPU programming, solid-state drives) can influence the future development of compact TRFS and FLIM systems with fast data acquisition and analysis capabilities suitable for practical studies in clinical environments. Extrapolating on the current clinical successes of the fluorescence systems based on intensity and spectral information only, it is anticipated that the addition of time-resolved fluorescence measurements capabilities to current steady-state devices can improve their diagnostic accuracy.
Finally, TRFS and FLIM modules could easily be adapted to work in conjunction with existing SSFS devices and thus take advantage of the extensive evaluation of these devices in clinical settings. Such approach could boost further investigation of TRFS and FLIM as diagnostic tools and whether such technologies can be used to improve the specificity of fluorescence measurements. An alternative approach would be the development of multimodal devices, where structural and biochemical information would be fused to provide direct characterization of various tissue pathologies. Either approach can enable the investigation of TRFS and FLIM potential to impact the clinical management of critical human diseases and the intraoperative decision-making.
References
J. McGinty, C. Dunsby, E. Auksorius, R.K.P. Benninger, P. De Beule, D.S. Elson, N. Galletly, D. Grant, O. Hofmann, G. Kennedy, S. Kumar, P.M.P. Lanigan, H. Manning, I. Munro, B. Önfelt, D. Owen, J. Requejo‐Isidro, K. Suhling, C.B. Talbot, P. Soutter, M.J. Lever, A.J. deMello, G.S. Stamp, M.A.A. Neil, P.M.W. French, in Chapter 4 Multidimensional fluorescence imaging, ed. by T.W.J. Gadella. Laboratory Techniques in Biochemistry and Molecular Biology (Elsevier, 2009)
D. Elson, N. Galletly, C. Talbot, J. Requejo-Isidro, J. McGinty, C. Dunsby, P.P. Lanigan, I. Munro, R.P. Benninger, P. de Beule, E. Auksorius, L. Hegyi, A. Sandison, A. Wallace, P. Soutter, M.A. Neil, J. Lever, G. Stamp, P.W. French, in Multidimensional fluorescence imaging applied to biological tissue, ed. by C. Geddes, J. Lakowicz. Reviews in Fluorescence (Springer, US, 2006)
J.R. Lakowicz, Principles of Fluorescence Spectroscopy (Springer, New York, 2006)
M.A. Mycek, B.W. Pogue, Handbook of Biomedical Fluorescence (Marcel Dekker, New York, 2003)
R. Richards-Kortum, E. Sevick-Muraca, Quantitative optical spectroscopy for tissue diagnosis. Annu. Rev. Phys. Chem. 47, 555–606 (1996)
E.G. Borisova, L.P. Angelova, E.P. Pavlova, Endogenous and exogenous fluorescence skin cancer diagnostics for clinical applications. IEEE J. Sel. Top. Quantum Electron. 20, 211–222 (2014)
M.Y. Berezin, S. Achilefu, Fluorescence lifetime measurements and biological imaging. Chem. Rev. 110, 2641–2684 (2010)
D. Chorvat Jr, A. Chorvatova, Multi-wavelength fluorescence lifetime spectroscopy: a new approach to the study of endogenous fluorescence in living cells and tissues. Laser Phys. Lett. 6, 175–193 (2009)
G.A. Wagnieres, W.M. Star, B.C. Wilson, In vivo fluorescence spectroscopy and imaging for oncological applications. Photochem. Photobiol. 68, 603–632 (1998)
X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu, Y. Wei, Aggregation induced emission-based fluorescent nanoparticles: fabrication methodologies and biomedical applications. J. Mat. Chem. B 2, 4398–4414 (2014)
X.M. Yi, F.L. Wang, W.J. Qin, X.J. Yang, J.L. Yuan, Near-infrared fluorescent probes in cancer imaging and therapy: an emerging field. Int. J. Nanomed. 9, 1347–1365 (2014)
K. Umezawa, D. Citterio, K. Suzuki, New trends in near-infrared fluorophores for bioimaging. Anal. Sci. 30, 327–349 (2014)
C. Darne, Y. Lu, E.M. Sevick-Muraca, Small animal fluorescence and bioluminescence tomography: a review of approaches, algorithms and technology update. Phys. Med. Biol. 59, R1–R64 (2014)
H. Kobayashi, M. Ogawa, R. Alford, P.L. Choyke, Y. Urano, New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 110, 2620–2640 (2010)
S. Andersson-Engels, C. Klinteberg, K. Svanberg, S. Svanberg, In vivo fluorescence imaging for tissue diagnostics. Phys. Med. Biol. 42, 815–824 (1997)
N. Dognitz, D. Salomon, M. Zellweger, J.P. Ballini, T. Gabrecht, N. Lange, H. van den Bergh, G. Wagnieres, Comparison of ala- and ala hexyl-ester-induced ppix depth distribution in human skin carcinoma. J. Photochem. Photobiol., B 93, 140–148 (2008)
N. Haj-Hosseini, J. Richter, S. Andersson-Engels, K. Wardell, Optical touch pointer for fluorescence guided glioblastoma resection using 5-aminolevulinic acid. Lasers Surg. Med. 42, 9–14 (2010)
H. Xie, P. Svenmarker, J. Axelsson, S. Grafe, M. Kyriazi, N. Bendsoe, S. Andersson-Engels, K. Svanberg, Pharmacokinetic and biodistribution study following systemic administration of fospeg(r)—a pegylated liposomal mthpc formulation in a murine model. J. Biophotonics 8(1–2), 142–152 (2013)
B.W. Pogue, S. Gibbs-Strauss, P.A. Valdes, K. Samkoe, D.W. Roberts, K.D. Paulsen, Review of neurosurgical fluorescence imaging methodologies. IEEE J. Sel. Top. Quantum Electron. 16, 493–505 (2010)
R. Rydell, C. Eker, S. Andersson-Engels, A. Krogdahl, P. Wahlberg, K. Svanberg, Fluorescence investigations to classify malignant laryngeal lesions in vivo. Head Neck 30, 419–426 (2008)
D. Bechet, S.R. Mordon, F. Guillemin, M.A. Barberi-Heyob, Photodynamic therapy of malignant brain tumours: a complementary approach to conventional therapies. Cancer Treat. Rev. 40, 229–241 (2014)
E. Drakaki, C. Dessinioti, A.J. Stratigos, C. Salavastru, C. Antoniou, Laser-induced fluorescence made simple: implications for the diagnosis and follow-up monitoring of basal cell carcinoma. J. Biomed. Opt. 19, 030901 (2014)
S. Hariri Tabrizi, S.M. Aghamiri, F. Farzaneh, H.C.M. Sterenborg, The use of optical spectroscopy for in vivo detection of cervical pre-cancer. Lasers Med. Sci. 29, 831–845 (2014)
D.F. Boerwinkel, M.K. Shariff, M. di Pietro, J.A. Holz, M.C. Aalders, W.L. Curvers, R.C. Fitzgerald, J.J. Bergman, Fluorescence imaging for the detection of early neoplasia in barrett’s esophagus: old looks or new vision? Eur. J. Gastroenterol. Hepatol. 26, 691–698 (2014)
L. Marcu, B.A. Hartl, Fluorescence lifetime spectroscopy and imaging in neurosurgery. IEEE J. Sel. Top. Quantum Electron. 18, 1465–1477 (2012)
L. Marcu, Fluorescence lifetime in cardiovascular diagnostics. J. Biomed. Opt. 15, 011106-011106-10 (2010)
R.R. Alfano, Advances in ultrafast time resolved fluorescence physics for cancer detection in optical biopsy. AIP Adv. 2, 011103 (2012)
C. Zhu, G.M. Palmer, T.M. Breslin, J. Harter, N. Ramanujam, Diagnosis of breast cancer using fluorescence and diffuse reflectance spectroscopy: a monte-carlo-model-based approach. J. Biomed. Opt. 13, 034015 (2008)
N. Ramanujam, Fluorescence spectroscopy of neoplastic and non-neoplastic tissues. Neoplasia 2, 89–117 (2000)
I. Georgakoudi, W.L. Rice, M. Hronik-Tupaj, D.L. Kaplan, Optical spectroscopy and imaging for the noninvasive evaluation of engineered tissues. Tissue Eng. Part B Rev. 14, 321–340 (2008)
B.Z. Fite, M. Decaris, Y. Sun, Y. Sun, A. Lam, C.K. Ho, J.K. Leach, L. Marcu, Noninvasive multimodal evaluation of bioengineered cartilage constructs combining time-resolved fluorescence and ultrasound imaging. Tissue Eng. Part. C Methods 17, 495–504 (2011)
P. Ashjian, A. Elbarbary, P. Zuk, D.A. DeUgarte, P. Benhaim, L. Marcu, M.H. Hedrick, Noninvasive in situ evaluation of osteogenic differentiation by time-resolved laser-induced fluorescence spectroscopy. Tissue Eng. 10, 411–420 (2004)
D. Ma, J. Bec, D.R. Yankelevich, D. Gorpas, H. Fatakdawala, L. Marcu, Rotational multispectral fluorescence lifetime imaging and intravascular ultrasound: bimodal system for intravascular applications. J. Biomed. Opt. 19, 066004 (2014)
U. Utzinger, R.R. Richards-Kortum, Fiber optic probes for biomedical optical spectroscopy. J. Biomed. Opt. 8, 121–147 (2003)
J.A. Udovich, N.D. Kirkpatrick, A. Kano, A. Tanbakuchi, U. Utzinger, A.F. Gmitro, Spectral background and transmission characteristics of fiber optic imaging bundles. Appl. Opt. 47, 4560–4568 (2008)
http://novadaq.com/. Accessed 2014
https://www.karlstorz.com/. Accessed 2014
http://www.pentaxmedical.de/. Accessed 2014
http://medical.olympusamerica.com/. Accessed 2014
http://www.richard-wolf.com/. Accessed 2014
http://www.maunakeatech.com/. Accessed 2015
http://www.spectrascience.com/. Accessed 2014
http://www.guidedinc.com/. Accessed 2014
http://www.leddental.com/. Accessed 2014
M. Cardenas-Turanzas, J.A. Freeberg, J.L. Benedet, E.N. Atkinson, D.D. Cox, R. Richards-Kortum, C. MacAulay, M. Follen, S.B. Cantor, The clinical effectiveness of optical spectroscopy for the in vivo diagnosis of cervical intraepithelial neoplasia: where are we? Gynecol. Oncol. 107, S138–S146 (2007)
P. Uehlinger, T. Gabrecht, T. Glanzmann, J.P. Ballini, A. Radu, S. Andrejevic, P. Monnier, G. Wagnieres, In vivo time-resolved spectroscopy of the human bronchial early cancer autofluorescence. J. Biomed. Opt. 14, 024011 (2009)
N. Rajaram, T.J. Aramil, K. Lee, J.S. Reichenberg, T.H. Nguyen, J.W. Tunnell, Design and validation of a clinical instrument for spectral diagnosis of cutaneous malignancy. Appl. Opt. 49, 142–152 (2010)
L. Brancaleon, A.J. Durkin, J.H. Tu, G. Menaker, J.D. Fallon, N. Kollias, In vivo fluorescence spectroscopy of nonmelanoma skin cancer. Photochem. Photobiol. 73, 178–183 (2001)
Y. Fawzy, H. Zeng, Intrinsic fluorescence spectroscopy for endoscopic detection and localization of the endobronchial cancerous lesions. J. Biomed. Opt. 13, 064022 (2008)
J.A. Freeberg, D.M. Serachitopol, N. McKinnon, R. Price, E.N. Atkinson, D.D. Cox, C. MacAulay, R. Richards-Kortum, M. Follen, B. Pikkula, Fluorescence and reflectance device variability throughout the progression of a phase ii clinical trial to detect and screen for cervical neoplasia using a fiber optic probe. J. Biomed. Opt. 12, 034015 (2007)
A. Vaitkuviene, V. Gegzna, R. Kurtinaitiene, J.V. Vaitkus, Cervical smear photodiagnosis by fluorescence. Photomed. Laser Surg. 30, 268–274 (2012)
M.D. Keller, S.K. Majumder, M.C. Kelley, I.M. Meszoely, F.I. Boulos, G.M. Olivares, A. Mahadevan-Jansen, Autofluorescence and diffuse reflectance spectroscopy and spectral imaging for breast surgical margin analysis. Lasers Surg. Med. 42, 15–23 (2010)
S.S. Nazeer, A. Saraswathy, A.K. Gupta, R.S. Jayasree, Fluorescence spectroscopy to discriminate neoplastic human brain lesions: a study using the spectral intensity ratio and multivariate linear discriminant analysis. Laser Phys. 24(2), 025602 (2014)
H. Xie, Z. Xie, M. Mousavi, N. Bendsoe, M. Brydegaard, J. Axelsson, S. Andersson-Engels, Design and validation of a fiber optic point probe instrument for therapy guidance and monitoring. J. Biomed. Opt. 19, 071408 (2014)
W.C. Lin, A. Mahadevan-Jansen, M.D. Johnson, R.J. Weil, S.A. Toms, In vivo optical spectroscopy detects radiation damage in brain tissue. Neurosurgery 57, 518–25; discussion 518–525 (2005)
C. Piazza, D.B.F.G. Peretti, P. Nicolai, Biologic endoscopy: optimization of upper aerodigestive tract cancer evaluation. Curr. Opin. Otolaryngol. Head Neck Surg. 19, 67–76 (2011)
M.G. Muller, T.A. Valdez, I. Georgakoudi, V. Backman, C. Fuentes, S. Kabani, N. Laver, Z. Wang, C.W. Boone, R.R. Dasari, S.M. Shapshay, M.S. Feld, Spectroscopic detection and evaluation of morphologic and biochemical changes in early human oral carcinoma. Cancer 97, 1681–1692 (2003)
N. Rajaram, J.S. Reichenberg, M.R. Migden, T.H. Nguyen, J.W. Tunnell, Pilot clinical study for quantitative spectral diagnosis of non-melanoma skin cancer. Lasers Surg. Med. 42, 716–727 (2010)
C. Redden Weber, R.A. Schwarz, E.N. Atkinson, D.D. Cox, C. Macaulay, M. Follen, R. Richards-Kortum, Model-based analysis of reflectance and fluorescence spectra for in vivo detection of cervical dysplasia and cancer. J. Biomed. Opt. 13, 064016 (2008)
R.A. Schwarz, W. Gao, C. Redden Weber, C. Kurachi, J.J. Lee, A.K. El-Naggar, R. Richards-Kortum, A.M. Gillenwater, Noninvasive evaluation of oral lesions using depth-sensitive optical spectroscopy. Cancer 115, 1669–1679 (2009)
S.A. Toms, W.C. Lin, R.J. Weil, M.D. Johnson, E.D. Jansen, A. Mahadevan-Jansen, Intraoperative optical spectroscopy identifies infiltrating glioma margins with high sensitivity. Neurosurgery 61, 327–35; discussion 335–336 (2007)
G.M. Palmer, R.J. Viola, T. Schroeder, P.S. Yarmolenko, M.W. Dewhirst, N. Ramanujam, Quantitative diffuse reflectance and fluorescence spectroscopy: tool to monitor tumor physiology in vivo. J. Biomed. Opt. 14, 024010 (2009)
C. Zhu, E.S. Burnside, G.A. Sisney, L.R. Salkowski, J.M. Harter, B. Yu, N. Ramanujam, Fluorescence spectroscopy: an adjunct diagnostic tool to image-guided core needle biopsy of the breast. IEEE Trans. Biomed. Eng. 56, 2518–2528 (2009)
D.-U. Li, B. Rae, R. Andrews, J. Arlt, R. Henderson, Hardware implementation algorithm and error analysis of high-speed fluorescence lifetime sensing systems using center-of-mass method. J. Biomed. Opt. 15, 017006 (2010)
X. Zhu, D. Zhang, Efficient parallel levenberg-marquardt model fitting towards real-time automated parametric imaging microscopy. PLoS ONE 8, e76665 (2013)
D.R. Yankelevich, D. Ma, J. Liu, Y. Sun, Y. Sun, J. Bec, D.S. Elson, L. Marcu, Design and evaluation of a device for fast multispectral time-resolved fluorescence spectroscopy and imaging. Rev. Sci. Instrum. 85, 034303 (2014)
R. Cubeddu, D. Comelli, C. D’Andrea, P. Taroni, G. Valentini, Time-resolved fluorescence imaging in biology and medicine. J. Phys. D-Appl. Phys. 35, R61–R76 (2002)
L. Marcu, J.A. Jo, Q.Y. Fang, T. Papaioannou, T. Reil, J.H. Qiao, J.D. Baker, J.A. Freischlag, M.C. Fishbein, Detection of rupture-prone atherosclerotic plaques by time-resolved laser-induced fluorescence spectroscopy. Atherosclerosis 204, 156–164 (2009)
L. Marcu, J.A. Jo, P. Butte, Fluorescence lifetime spectroscopy in cardio and neuroimaging, ed. by N. Iftimia, W.R. Brugge D.X. Hammer. Advances in optical imaging for clinical medicine (Wiley, New Jersey, 2011)
J. Bec, D.M. Ma, D.R. Yankelevich, J. Liu, W.T. Ferrier, J. Southard, L. Marcu, Multispectral fluorescence lifetime imaging system for intravascular diagnostics with ultrasound guidance: in vivo validation in swine arteries. J. Biophoton. 7, 281–285 (2013)
J. Liu, D.S. Elson, L. Marcu, in Analysis of time-domain fluorescence measurements using least-squares deconvolution. Fluorescence lifetime Spectroscopy and Imaging (CRC Press, 2014)
M.A. Digman, E. Gratton, in The phasor approach to fluorescence lifetime imaging: exploiting phasor linear properties. Fluorescence Lifetime Spectroscopy and Imaging (CRC Press, 2014)
W. Becker, Fluorescence lifetime imaging–techniques and applications. J. Microsc. 247, 119–136 (2012)
W. Becker, in Fluorescence lifetime imaging techniques: time-correlated single-photon counting. Fluorescence Lifetime Spectroscopy and Imaging (CRC Press, 2014)
http://www.picoquant.com/. Accessed 2014
http://www.becker-hickl.com/. Accessed 2014
K. Konig, Clinical multiphoton tomography. J. Biophotonics 1, 13–23 (2008)
S. Kumar, C. Dunsby, P.A. De Beule, D.M. Owen, U. Anand, P.M. Lanigan, R.K. Benninger, D.M. Davis, M.A. Neil, P. Anand, C. Benham, A. Naylor, P.M. French, Multifocal multiphoton excitation and time correlated single photon counting detection for 3-d fluorescence lifetime imaging. Opt. Express 15, 12548–12561 (2007)
J. McGinty, C. Dunsby, P.M.W. French, in Fluorescence lifetime imaging techniques: time-gated fluorescence lifetime imaging. Fluorescence Lifetime Spectroscopy and Imaging (CRC Press, 2014)
J. McGinty, N.P. Galletly, C. Dunsby, I. Munro, D.S. Elson, J. Requejo-Isidro, P. Cohen, R. Ahmad, A. Forsyth, A.V. Thillainayagam, M.A. Neil, P.M. French, G.W. Stamp, Wide-field fluorescence lifetime imaging of cancer. Biomed. Opt. Express 1, 627–640 (2010)
P.V. Butte, Q. Fang, J.A. Jo, W.H. Yong, B.K. Pikul, K.L. Black, L. Marcu, Intraoperative delineation of primary brain tumors using time-resolved fluorescence spectroscopy. J. Biomed. Opt. 15, 027008 (2010)
P.V. Butte, A.N. Mamelak, M. Nuno, S.I. Bannykh, K.L. Black, L. Marcu, Fluorescence lifetime spectroscopy for guided therapy of brain tumors. Neuroimage 54, S125–S135 (2011)
J.D. Meier, H. Xie, Y. Sun, Y. Sun, N. Hatami, B. Poirier, L. Marcu, D.G. Farwell, Time-resolved laser-induced fluorescence spectroscopy as a diagnostic instrument in head and neck carcinoma. Otolaryngol. Head Neck Surg. 142, 838–844 (2010)
M.A. Mycek, K.T. Schomacker, N.S. Nishioka, Colonic polyp differentiation using time-resolved autofluorescence spectroscopy. Gastrointest. Endosc. 48, 390–394 (1998)
T.J. Pfefer, D.Y. Paithankar, J.M. Poneros, K.T. Schomacker, N.S. Nishioka, Temporally and spectrally resolved fluorescence spectroscopy for the detection of high grade dysplasia in barrett’s esophagus. Lasers Surg. Med. 32, 10–16 (2003)
J.D. Pitts, M.-A. Mycek, Design and development of a rapid acquisition laser-based fluorometer with simultaneous spectral and temporal resolution. Rev. Sci. Instrum. 72, 3061–3072 (2001)
Y. Sun, A.J. Chaudhari, M. Lam, H.T. Xie, D.R. Yankelevich, J. Phipps, J. Liu, M.C. Fishbein, J.M. Cannata, K.K. Shung, L. Marcu, Multimodal characterization of compositional, structural and functional features of human atherosclerotic plaques. Biomed. Opt. Express 2, 2288–2298 (2011)
S. Cheng, R.M. Cuenca, B. Liu, B.H. Malik, J.M. Jabbour, K.C. Maitland, J. Wright, Y.S.L. Cheng, J.A. Jo, Handheld multispectral fluorescence lifetime imaging system for in vivo applications. Biomed. Opt. Express 5, 921–931 (2014)
M.S. Roberts, Y. Dancik, T.W. Prow, C.A. Thorling, L.L. Lin, J.E. Grice, T.A. Robertson, K. Konig, W. Becker, Non-invasive imaging of skin physiology and percutaneous penetration using fluorescence spectral and lifetime imaging with multiphoton and confocal microscopy. Eur. J. Pharm. Biopharm. 77, 469–488 (2011)
http://www.jenlab.de/. Accessed 2014
J. Eichorst, K. Teng, R.M. Clegg, in Fluorescence lifetime imaging techniques: Frequency-domain FLIM. Fluorescence Lifetime Spectroscopy and Imaging (CRC Press, 2014)
D.V. O’Connor, W.R. Ware, J.C. Andre, Deconvolution of fluorescence decay curves. A critical comparison of techniques. J. Phys. Chem. 83, 1333–1342 (1979)
A.A. Istratov, O.F. Vyvenko, Exponential analysis in physical phenomena. Rev. Sci. Instrum. 70, 1233–1257 (1999)
M.N. Berberan-Santos, E.N. Bodunov, B. Valeur, Mathematical functions for the analysis of luminescence decays with underlying distributions 1. Kohlrausch decay function (stretched exponential). Chem. Phys. 315, 171–182 (2005)
J.C. Andre, L.M. Vincent, D. O’Connor, W.R. Ware, Applications of fast fourier transform to deconvolution in single photon counting. J. Phys. Chem. 83, 2285–2294 (1979)
K. Ishii, T. Tahara, Two-dimensional fluorescence lifetime correlation spectroscopy. 1. Principle. J. Phys. Chem. B 117, 11414–11422 (2013)
W.R. Ware, L.J. Doemeny, T.L. Nemzek, Deconvolution of fluorescence and phosphorescence decay curves. A least-squares method. J. Phys. Chem. 77, 2038–2048 (1973)
J.M.I. Maarek, L. Marcu, W.J. Snyder, W.S. Grundfest, Time-resolved fluorescence spectra of arterial fluorescent compounds: reconstruction with the laguerre expansion technique. Photochem. Photobiol. 71, 178–187 (2000)
J.A. Jo, Q. Fang, T. Papaioannou, J.D. Baker, A.H. Dorafshar, T. Reil, J.H. Qiao, M.C. Fishbein, J.A. Freischlag, L. Marcu, Laguerre-based method for analysis of time-resolved fluorescence data: application to in-vivo characterization and diagnosis of atherosclerotic lesions. J. Biomed. Opt. 11, 021004 (2006)
J.A. Jo, Q. Fang, T. Papaioannou, L. Marcu, Fast model-free deconvolution of fluorescence decay for analysis of biological systems. J. Biomed. Opt. 9, 743–752 (2004)
V.Z. Marmarelis, Identification of nonlinear biological systems using laguerre expansions of kernels. Ann. Biomed. Eng. 21, 573–589 (1993)
J. Liu, Y. Sun, J. Qi, L. Marcu, A novel method for fast and robust estimation of fluorescence decay dynamics using constrained least-squares deconvolution with laguerre expansion. Phys. Med. Biol. 57, 843–865 (2012)
G. Redford, R. Clegg, Polar plot representation for frequency-domain analysis of fluorescence lifetimes. J. Fluoresc. 15, 805–815 (2005)
M.D. Digman, E. Gratton, in Fluorescence lifetime microscopy: the phasor approach, ed. by. E.H. Egelman. Comprehensive Biophysics (Elsevier, Amsterdam, 2012)
A. Leray, C. Spriet, D. Trinel, R. Blossey, Y. Usson, L. Héliot, Quantitative comparison of polar approach versus fitting method in time domain FLIM image analysis. Cytom. Part A 79A, 149–158 (2011)
M. Štefl, N.G. James, J.A. Ross, D.M. Jameson, Applications of phasors to in vitro time-resolved fluorescence measurements. Anal. Biochem. 410, 62–69 (2011)
F. Fereidouni, A. Esposito, G.A. Blab, H.C. Gerritsen, A modified phasor approach for analyzing time-gated fluorescence lifetime images. J. Microsc. 244, 248–258 (2011)
F. Fereidouni, A.N. Bader, A. Colonna, H.C. Gerritsen, Phasor analysis of multiphoton spectral images distinguishes autofluorescence components of in vivo human skin. J. Biophotonics 7, 589–596 (2014)
T.B. Krasieva, C. Stringari, F. Liu, C.H. Sun, Y. Kong, M. Balu, F.L. Meyskens, E. Gratton, B.J. Tromberg, Two-photon excited fluorescence lifetime imaging and spectroscopy of melanins in vitro and in vivo. J. Biomed. Opt. 18, 031107 (2013)
S. Coda, A.V. Thillainayagam, State of the art in advanced endoscopic imaging for the detection and evaluation of dysplasia and early cancer of the gastrointestinal tract. Clin. Exp. Gastroenterol. 7, 133–150 (2014)
P. Von Breitenbuch, T. Jeiter, S. Schreml, G. Glockzin, A. Agha, P. Piso, H.J. Schlitt, Autofluorescent imaging in patients with peritoneal carcinomatosis. Surg. Innov. 21, 187–193 (2014)
D.F. Boerwinkel, J.A. Holz, M.C.G. Aalders, M. Visser, S.L. Meijer, M.I. Van Berge Henegouwen, B.L.A.M. Weusten, J.J.G.H.M. Bergman, Third-generation autofluorescence endoscopy for the detection of early neoplasia in barrett’s esophagus: a pilot study. Dis. Esophagus 27, 276–284 (2014)
D.F. Boerwinkel, J.A. Holz, D.M. Hawkins, W.L. Curvers, M.C. Aalders, B.L. Weusten, M. Visser, S.L. Meijer, J.J. Bergman, Fluorescence spectroscopy incorporated in an optical biopsy system for the detection of early neoplasia in barrett’s esophagus. Dis. Esophagus (2014). doi:10.1111/dote.12193
B. Mayinger, M. Jordan, P. Horner, C. Gerlach, S. Muehldorfer, B.R. Bittorf, K.E. Matzel, W. Hohenberger, E.G. Hahn, K. Guenther, Endoscopic light-induced autofluorescence spectroscopy for the diagnosis of colorectal cancer and adenoma. J. Photochem. Photobiol. B-Biol. 70, 13–20 (2003)
B. Zaric, B. Perin, H.D. Becker, F.J.H. Felix, R. Eberhardt, S. Jovanovic, T. Orlic, M. Panjkovic, B. Zvezdin, A. Jovelic, M. Bijelovic, V. Jurisic, M. Antonic, Combination of narrow band imaging (nbi) and autofluorescence imaging (afi) videobronchoscopy in endoscopic assessment of lung cancer extension. Med. Oncol. 29, 1638–1642 (2012)
M. Hanibuchi, S. Yano, Y. Nishioka, T. Miyoshi, K. Kondo, H. Uehara, S. Sone, Autofluorescence bronchoscopy, a novel modality for the early detection of bronchial premalignant and malignant lesions. J. Med. Invest. 54, 261–266 (2007)
D. Hüttenberger, T. Gabrecht, G. Wagnières, B. Weber, A. Linder, H.J. Foth, L. Freitag, Autofluorescence detection of tumors in the human lung-spectroscopical measurements in situ, in an in vivo model and in vitro. Photodiagnosis Photodyn. Ther. 5, 139–147 (2008)
T. Gabrecht, A. Radu, P. Grosjean, B. Weber, G. Reichle, L. Freitag, P. Monnier, H. van den Bergh, G. Wagnières, Improvement of the specificity of cancer detection by autofluorescence imaging in the tracheo-bronchial tree using backscattered violet light. Photodiagnosis Photodyn. Ther. 5, 2–9 (2008)
L. Thiberville, M. Saiaiin, S. Lachkar, S. Dominique, S. Moreno-Swirc, C. Vever-Bizet, G. Bourg-Heckly, Human in vivo fluorescence microimaging of the alveolar ducts and sacs during bronchoscopy. Eur. Resp. J. 33, 974–985 (2009)
M.-A. Mycek, K. Vishwanath, K.T. Schomacker, N.S. Nishioka, Fluorescence spectroscopy for in vivo discrimination of pre-malignant colonic lesions. Biomed. Optical Spectrosc. Diagn. OSA Trends Opt. Photonics 38, SuB2 (2000)
S. Coda, A.J. Thompson, G.T. Kennedy, K.L. Roche, L. Ayaru, D.S. Bansi, G.W. Stamp, A.V. Thillainayagam, P.M.W. French, C. Dunsby, Fluorescence lifetime spectroscopy of tissue autofluorescence in normal and diseased colon measured ex vivo using a fiber-optic probe. Biomed. Opt. Express 5, 515–538 (2014)
C. Venugopal, S.S. Nazeer, A. Balan, R.S. Jayasree, Autofluorescence spectroscopy augmented by multivariate analysis as a potential noninvasive tool for early diagnosis of oral cavity disorders. Photomed. Laser Surg. 31, 605–612 (2013)
H. Hanken, J. Kraatz, R. Smeets, M. Heiland, M. Blessmann, W. Eichhorn, T.S. Clauditz, A. Gröbe, A. Kolk, M. Rana, The detection of oral pre- malignant lesions with an autofluorescence based imaging system (velscopetm)—a single blinded clinical evaluation. Head Face Med. 9, (2013)
Y. Sun, J.E. Phipps, J. Meier, N. Hatami, B. Poirier, D.S. Elson, D.G. Farwell, L. Marcu, Endoscopic fluorescence lifetime imaging for in vivo intraoperative diagnosis of oral carcinoma. Microsc. Microanal. 19, 791–798 (2013)
I. Pavlova, C.R. Weber, R.A. Schwarz, M.D. Williams, A.M. Gillenwater, R. Richards-Kortum, Fluorescence spectroscopy of oral tissue: Monte carlo modeling with site-specific tissue properties. J. Biomed. Opt. 14, 014009 (2009)
P. Chaturvedi, S.K. Majumder, H. Krishna, S. Muttagi, P.K. Gupta, Fluorescence spectroscopy for noninvasive early diagnosis of oral mucosal malignant and potentially malignant lesions. J. Canc. Res. Ther. 6, 497–502 (2010)
D. Roblyer, C. Kurachi, V. Stepanek, M.D. Williams, A.K. El-Naggar, J.J. Lee, A.M. Gillenwater, R. Richards-Kortum, Objective detection and delineation of oral neoplasia using autofluorescence imaging. Cancer Prev. Res. 2, 423–431 (2009)
H.-M. Chen, C.-P. Chiang, C. You, T.-C. Hsiao, C.-Y. Wang, Time-resolved autofluorescence spectroscopy for classifying normal and premalignant oral tissues. Laser Surg. Med. 37, 37–45 (2005)
Y. Sun, N. Hatami, M. Yee, J. Phipps, D.S. Elson, F. Gorin, R.J. Schrot, L. Marcu, Fluorescence lifetime imaging microscopy for brain tumor image-guided surgery. J. Biomed. Opt. 15, 056022 (2010)
A.C. Croce, S. Fiorani, D. Locatelli, R. Nano, M. Ceroni, F. Tancioni, E. Giombelli, E. Benericetti, G. Bottiroli, Diagnostic potential of autofluorescence for an assisted intraoperative delineation of glioblastoma resection margins. Photochem. Photobiol. 77, 309–318 (2003)
W.C. Lin, S.A. Toms, M. Johnson, E.D. Jansen, A. Mahadevan-Jansen, In vivo brain tumor demarcation using optical spectroscopy. Photochem. Photobiol. 73, 396–402 (2001)
W.K. Huh, R.M. Cestero, F.A. Garcia, M.A. Gold, R.S. Guido, K. McIntyre-Seltman, D.M. Harper, L. Burke, S.T. Sum, R.F. Flewelling, R.D. Alvarez, Optical detection of high-grade cervical intraepithelial neoplasia in vivo: Results of a 604-patient study. Am. J. Obstet. Gynecol. 190, 1249–1257 (2004)
R.J. Nordstrom, L. Burke, J.M. Niloff, J.F. Myrtle, Identification of cervical intraepithelial neoplasia (cin) using uv-excited fluorescence and diffuse-reflectance tissue spectroscopy. Lasers Surg. Med. 29, 118–127 (2001)
Y. Pu, W. Wang, Y. Yang, R.R. Alfano, Native fluorescence spectra of human cancerous and normal breast tissues analyzed with non-negative constraint methods. Appl. Opt. 52, 1293–1301 (2013)
V. Sharma, S. Shivalingaiah, Y. Peng, D. Euhus, Z. Gryczynski, H. Liu, Auto-fluorescence lifetime and light reflectance spectroscopy for breast cancer diagnosis: potential tools for intraoperative margin detection. Biomed. Opt. Express 3, 1825–1840 (2012)
V. Sharma, E.O. Olweny, P. Kapur, J.A. Cadeddu, C.G. Roehrborn, H. Liu, Prostate cancer detection using combined auto-fluorescence and light reflectance spectroscopy: ex vivo study of human prostates. Biomed. Opt. Express 5, 1512–1529 (2014)
E. Dimitrow, M. Ziemer, M.J. Koehler, J. Norgauer, K. König, P. Elsner, M. Kaatz, Sensitivity and specificity of multiphoton laser tomography for in vivo and ex vivo diagnosis of malignant melanoma. J. Invest. Dermatol. 129, 1752–1758 (2009)
J. Hegyi, V. Hegyi, T. Ruzicka, P. Arenberger, C. Berking, New developments in fluorescence diagnostics. J. Dtsch. Dermatol. Ges. 9, 368–373 (2011)
M.J. Koehler, K. König, P. Elsner, R. Bückle, M. Kaatz, In vivo assessment of human skin aging by multiphoton laser scanning tomography. Opt. Lett. 31, 2879–2881 (2006)
K. König, A. Ehlers, I. Riemann, S. Schenkl, R. Bückle, M. Kaatz, Clinical two-photon microendoscopy. Microsc. Res. Tech. 70, 398–402 (2007)
B.R. Masters, P.T.C. So, Confocal microscopy and multi-photon excitation microscopy of human skin in vivo. Opt. Express 8, 2–10 (2001)
J. Blackwell, K.M. Katika, L. Pilon, K.M. Dipple, S.R. Levin, A. Nouvong, In vivo time-resolved autofluorescence measurements to test for glycation of human skin. J. Biomed. Opt. 13, 014004 (2008)
E. Dimitrow, I. Riemann, A. Ehlers, M.J. Koehler, J. Norgauer, P. Elsner, K. König, M. Kaatz, Spectral fluorescence lifetime detection and selective melanin imaging by multiphoton laser tomography for melanoma diagnosis. Exp. Dermatol. 18, 509–515 (2009)
B.R. Masters, P.T.C. So, E. Gratton, Multiphoton excitation fluorescence microscopy and spectroscopy of in vivo human skin. Biophys. J. 72, 2405–2412 (1997)
M. Oishi, A. Oishi, K. Ogino, Y. Makiyama, N. Gotoh, M. Kurimoto, N. Yoshimura, Wide-field fundus autofluorescence abnormalities and visual function in patients with cone and cone-rod dystrophies. Invest. Ophthalmol. Vis. Sci. 55, 3572–3577 (2014)
T.R. Burke, T. Duncker, R.L. Woods, J.P. Greenberg, J. Zernant, S.H. Tsang, R. Theodore Smith, R. Allikmets, J.R. Sparrow, F.C. Delori, Quantitative fundus autofluorescence in recessive stargardt disease. Invest. Ophthalmol. Vis. Sci. 55, 2841–2852 (2014)
T. Duncker, J.P. Greenberg, R. Ramachandran, D.C. Hood, R. Theodore Smith, T. Hirose, R.L. Woods, S.H. Tsang, F.C. Delori, J.R. Sparrow, Quantitative fundus autofluorescence and optical coherence tomography in best vitelliform macular dystrophy. Invest. Ophthalmol. Vis. Sci. 55, 1471–1482 (2014)
M. Klemm, A. Dietzel, J. Haueisen, E. Nagel, M. Hammer, D. Schweitzer, Repeatability of autofluorescence lifetime imaging at the human fundus in healthy volunteers. Curr. Eye Res. 38, 793–801 (2013)
M. Hammer, E. Königsdörffer, C. Liebermann, C. Framme, G. Schuch, D. Schweitzer, J. Strobel, Ocular fundus auto-fluorescence observations at different wavelengths in patients with age-related macular degeneration and diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 246, 105–114 (2008)
D. Schweitzer, M. Hammer, F. Schweitzer, R. Anders, T. Doebbecke, S. Schenke, E.R. Gaillard, In vivo measurement of time-resolved autofluorescence at the human fundus. J. Biomed. Opt. 9, 1214–1222 (2004)
D. Schweitzer, S. Quick, S. Schenke, M. Klemm, S. Gehlert, M. Hammer, S. Jentsch, S.J. Fischer, Comparison of parameters of time-resolved autofluorescence between healthy subjects and patients suffering from early amd. Ophthalmologe 106, 714–722 (2009)
D. Schweitzer, S. Quick, M. Klemm, M. Hammer, S. Jentsch, J. Dawczynski, Time-resolved autofluorescence in retinal vascular occlusions. Ophthalmologe 107, 1145–1152 (2010)
Y. Honda, P.J. Fitzgerald, Frontiers in intravascular imaging technologies. Circulation 117, 2024–2037 (2008)
P. Libby, M. Aikawa, Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat. Med. 8, 1257–1262 (2002)
B.D. MacNeill, H.C. Lowe, M. Takano, V. Fuster, I.K. Jang, Intravascular modalities for detection of vulnerable plaque: current status. Arterioscler. Thromb. Vasc. Biol. 23, 1333–1342 (2003)
P.R. Moreno, J.E. Muller, Identification of high-risk atherosclerotic plaques: a survey of spectroscopic methods. Curr. Opin. Cardiol. 17, 638–647 (2002)
A. Christov, E. Dai, M. Drangova, L. Liu, G.S. Abela, P. Nash, G. McFadden, A. Lucas, Optical detection of triggered atherosclerotic plaque disruption by fluorescence emission analysis. Photochem. Photobiol. 72, 242–252 (2000)
K. Arakawa, K. Isoda, T. Ito, K. Nakajima, T. Shibuya, F. Ohsuzu, Fluorescence analysis of biochemical constituents identifies atherosclerotic plaque with a thin fibrous cap. Arterioscler. Thromb. Vasc. Biol. 22, 1002–1007 (2002)
L. Marcu, M.C. Fishbein, J.M.I. Maarek, W.S. Grundfest, Discrimination of human coronary artery atherosclerotic lipid-rich lesions by time-resolved laser-induced fluorescence spectroscopy. Arterioscler. Thromb. Vasc. Biol. 21, 1244–1250 (2001)
G.O. Angheloiu, J.T. Arendt, M.G. Müller, A.S. Haka, I. Georgakoudi, J.T. Motz, O.R. Scepanovic, B.D. Kuban, J. Myles, F. Miller, E.A. Podrez, M. Fitzmaurice, J.R. Kramer, M.S. Feld, Intrinsic fluorescence and diffuse reflectance spectroscopy identify superficial foam cells in coronary plaques prone to erosion. Arterioscler. Thromb. Vasc. Biol. 26, 1594–1600 (2006)
J.J. Baraga, R.P. Rava, P. Taroni, C. Kittrell, M. Fitzmaurice, M.S. Feld, Laser induced fluorescence spectroscopy of normal and atherosclerotic human aorta using 306–310 nm excitation. Lasers Surg. Med. 10, 245–261 (1990)
A.L. Bartorelli, M.B. Leon, Y. Almagor, L.G. Prevosti, J.A. Swain, C.L. McIntosh, R.F. Neville, M.D. House, R.F. Bonner, In vivo human atherosclerotic plaque recognition by laser-excited fluorescence spectroscopy. J. Am. Coll. Cardiol. 17, 160B–168B (1991)
J.E. Phipps, Y. Sun, M.C. Fishbein, L. Marcu, A fluorescence lifetime imaging classification method to investigate the collagen to lipid ratio in fibrous caps of atherosclerotic plaque. Lasers Surg. Med. 44, 564–571 (2012)
O.R. Šćepanovic, M. Fitzmaurice, A. Miller, C.R. Kong, Z. Volynskaya, R.R. Dasari, J.R. Kramer, M.S. Feld, Multimodal spectroscopy detects features of vulnerable atherosclerotic plaque. J. Biomed. Opt. 16, 011009 (2011)
J. Bec, H.T. Xie, D.R. Yankelevich, F.F. Zhou, Y. Sun, N. Ghata, R. Aldredge, L. Marcu, Design, construction, and validation of a rotary multifunctional intravascular diagnostic catheter combining multispectral fluorescence lifetime imaging and intravascular ultrasound. J. Biomed. Opt. 17, 106012 (2012)
E. de Boer, N.J. Harlaar, A. Taruttis, W.B. Nagengast, E.L. Rosenthal, V. Ntziachristos, G.M. van Dam, Optical innovations in surgery. Br. J. Surg. 102, e56–e72 (2015)
M. Hsu, M. Gupta, L.M. Su, J.C. Liao, Intraoperative optical imaging and tissue interrogation during urologic surgery. Curr. Opin. Urol. 24, 66–74 (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Gorpas, D., Marcu, L. (2016). Fluorescence Lifetime Spectroscopy and Imaging Techniques in Medical Applications. In: Olivo, M., Dinish, U. (eds) Frontiers in Biophotonics for Translational Medicine. Progress in Optical Science and Photonics, vol 3. Springer, Singapore. https://doi.org/10.1007/978-981-287-627-0_1
Download citation
DOI: https://doi.org/10.1007/978-981-287-627-0_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-287-626-3
Online ISBN: 978-981-287-627-0
eBook Packages: EngineeringEngineering (R0)