
Abstract
Horizontal gene transfer mechanisms help in the transfer of distant genes generating pathogens with different types of virulence. Genomic Islands (GI) are the evidence representing microbial genome evolution. These genes are acquired by HGT process. It determines their adaptation to the environment, compatibility, and other gene expression mechanisms. The pathogenicity islands which are a subclass of Genomic Islands contribute to the virulent nature of the pathogen. Helicobacter pylori known for its colonization in the stomach and the intestinal regions poses the most genetic diversity among the pathogenic species of the bacterial community. H. pylori has the ability to adapt to the host system by changing its genetic features. Chronic infections show the maximum genetic differential ability of H. pylori. The strains are categorized into type 1 and type 2 in which each secretes certain antigens and cytotoxin enhancing its virulence. Numerous computational tools have been advanced to recognize and categorize these genomic islands. One such tool is Island viewer which enables the user to access published and even unpublished GIs. It is also linked with external databases like NCBI. GIs can be predicted based on single or multiple genomes. GIs do not frequently replicate like plasmids and they serve as markers for identifying evolutionary pathways. However, real-time tracking is still under research and needs to be developed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Horizontal gene transfer
- Genomic Islands
- Pathogenicity islands
- Helicobacter pylori
- Antigens
- Cytotoxins
- Genomes
- NCBI
- Plasmids
10.1 Introduction
Genomic Islands (GIs) are a group of bacterial or archeal genes, which are acquired via horizontal gene transfer (HGT) and contributes to microbial genome evolution. These are a pool of gene sequences ranging from 10–100 kb which encode various disease causing factors, antimicrobial resistant factors, metabolic pathway regulating factors and microbial adaptations. Ambulatory genetic elements that fall under GIs include integrons, transposons, integrative and conjugative elements (ICEs), and non-segregative phage elements. These gene sequences can be detected based on the methods of acquisition of genomic island genes. Some of the known methods are via conjugation, transformation or transduction, and adaptable elements such as enzymes that catalyzes insertion of viral DNA, replicative transposition mechanism, and insertional sequences that promote relocation of GIs. Once these elements get integrated into the genome they start their evolution process through mutations, rearrangements of genomic sequences, or by insertion and deletion of ambulant genetic elements. These GI sequences differ from the genomic DNA with the percentage of GC content and the usage of codons. These genes are acquired by the process of horizontal gene transfer mechanism. HGT involves the migration of genetic materials between genetically distant organisms which is also determined as lateral gene transfer. The microbes undergo transformation process or acquire the genetic material via transduction or conjugation process. The transfer mechanisms can be vertical or horizontal in which the former one is time consuming from parent to daughter cells where as the latter is between different species or genera which further contributes to evolutionary processes. Instead of representing evolution as tree, HGT has helped to represent natural selection as a existence of interdependent organisms (Khan et al. 2000). Genes of medicinal interest favor environmental selection and they share relationships with GIs (Hacker 2002). The extent of homogeneity amid the exchanged DNA and the bacterial host, the metabolic affinity, alterations to the environment, the shift of genes and their expression, the mismatch repair, and restriction modification systems affect the movement of genes.
10.2 Genomic Islands and its Contribution to Pathogenesis
Pathogenicity islands (PAI) are a subset of genomic islands which contributes to pathogenesis and other virulent characteristics. Certain adherence factors such as fimbrial extensions in E. coli are encoded by the GIs which do not cause any infections in gut flora but they adhere in the urinary tract and thus become true pathogenic islands.
Virulent genes are mostly present in local movable, genetic elements which include the PAIs (Boyd and Brüssow 2002; Shankar et al. 2002). The presence of PAIs is a recent discovery after studying the mechanisms of phages and plasmids. The PAIs have unfolded from lysogenic bacteriophages and plasmids, and they form part of substantial genomic regions that are present in different types of pathogenic bacteria but this does not apply to nonpathogenic bacteria. Pathogenic bacteria consist of more than one virulent gene, which has Guanine and Cytosine contents different from other chromosomal regions.
Pathogenicity islands are mobile genes which contain sequences of transposases or integrases and are also quite unstable (Dobrindt et al. 2002). The genes which are virulent in nature and make the PAIs and they are classified into groups based on the functions they perform, they are:
-
The factors responsible for adherence, enables the bacteria to adhere to host surface.
-
The uptake of Fe3+ ions and solubilization is controlled by siderophores.
-
Capsules help in preventing phagocytosis of the bacterial cells.
-
The endotoxins released by Gram-negative bacteria are capable to induce the host complement mechanism and dynamic signs of inflammation are also seen.
-
The exotoxins released can cause permanent impairment of eukaryotic cells by the modulation of signal transduction pathway.
-
Invasins are types III and IV restriction complexes that promote the entry of bacteria into eukaryotic cells which intervene bacterial entry into eukaryotic cells and mostly interfere with the apoptotic pathways of the host and also gain entry into the non-phagocytic cells (Dobrindt et al. 2002).
10.2.1 Evolution of PAIs
After the events of HGT, chromosomal incorporation by position-specified recombination takes place which further promotes the integration of the chromosomes. These integration processes cause genetic rearrangements inducing mobile genetic elements to become GIs. Genomic islands can also be formed due to gene loss or acquisition. Immobilization of GIs can be due to inactivation or excision of genes accountable for mobility along with replication of plasmids. The genetic factors from non-virulent bacteria can be identified on extrachromosomal replicons, i.e., phages or plasmids. The autonomously transmittable sizeable plasmid pHG1 of Ralstoniaeutropha H16 is composed of a group of functional genes which are compassed next to movable genetic elements. These group of genes consists of sequences that are necessary for utilization of inorganic substrates, reduction of nitrate and nitrite, degradation of biomasses which consists of aromatic compounds and uptake of iron, as well as for type IV and RP4-like sex pili (Schwartz et al. 2003). The substantial number of pHG1 genes that specify integrases and transposases specify the peak of recombinational duty of plasmids, which has further promoted the collection of diverse genotypic characteristics, in that way expanding the metabolic role of the recipients (Schwartz et al. 2003).
10.2.2 Antibiotic Resistance Islands
Antibiotic resistance factors are frequently correlated with ambulatory genetic elements which are responsible for producing virulence (Paulsen et al. 2003).
10.2.3 Secondary Metabolism
Operons that encrypt enzymes which are necessary for the production of secondary metabolites are key elements for HGT because they provide an advantage of different morphological features and are not necessary to the bacterial cell. Authentication of genes that take part in the synthesis of chief products of natural medicines is becoming an integral part of microbial studies. For example, the POLYKETIDE genes are exchanged horizontally among Streptomycetes (Egan et al. 1998).
10.3 Identification of GIs in Helicobacter Species
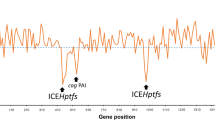
Helicobacter pylori, which is harmful to the Human gastric tract, is studied as a distinct genetically diverse species. In an experiment with duodenal ulcer, they contrasted the genome sequence of the duodenal ulcer strain and other H. pylori genomes to throw light on the structural arrangement of genes and genome selection mechanisms of these H. pylori species (Fig. 10.1). With these experimental evidence as described in Fig. 10.2, it is subjected that H. pylori possess a stretched out pan-genome. Different zones of plasticity are specific for different strains which suggests different pathways of evolution (Cover and Blaser 2009). H. pylori forms the majority of genetically variable pathogenic bacteria since it has high mutation rates (Fernandez-Gonzalez and Backert 2014). Most of the H. pylori strains consist of ambiguous plasmids, and the shuffled gene concatenation of plasmids which normally take part during evolution (Ilver et al. 1998).

Flow chart of identification of GIs

General procedure of identification of mobile genes
10.4 Adaptation and Pathogenicity of GI in Helicobacter Species
Helicobacter pylori is a group of bacterial species which are specific for humans and are also known for colonizing the stomach (Hooi et al. 2017). Infectious symptoms of existence of H. pylori are diagnosed with gastric along with duodenal pathology which includes the chronic gastritis, peptic ulcers, and even gastric cancer in the population which also depends on the variation of virulence of bacteria, genetic attributes of the host, and environmental factors (Nr and Muller 2013; Cover and Blaser 2009). H. pylori possess more genetic diversity in the class of bacteria which pathogenic in nature (Fischer et al. 2010; Fernandez-Gonzalez and Backert 2014) and also more often takes part in the horizontal gene transfer (HGT) and adapts accordingly with the host environment via recombination processes (Covacci et al. 1993). The mutational abilities of DNA polymerase I help in recombination of genes according to the selected host (Fernandez-Gonzalez and Backert 2014). The combination of infections due to numerous H. pylori strains within one stomach shows the intensity of genetic variability (Covacci et al. 1993; Telford et al. 1994; Marchetti et al. 1995). The ability of H. pylori to alter its gene sequence is considered to be the key characteristic for its adaptation to different host systems, and also to the frequently changing gastric environment (Nr and Muller 2013). Current researches focus on the usage of H. pylori strains from patients who are infected in long terms (Covacci et al. 1993). Genes which code for outer membrane proteins change their genomic features and help in the production of diverse range of proteins. These proteins helps in the prolonged infection to the host organism (Chiapello et al. 2005).
H. pylori strains are classified into two different wide-ranging families temporarily addressed as types I and II, which are known as the bases of expression of vacuolating cytotoxin (VacA) and the CagA antigen (cytotoxin-associated gene A) (Fischer et al. 2010). Type I strain infections are seen in patients who are diagnosed with duodenal ulcers, tumors and duodenitis which with CagA and the cytotoxin which expresses together with other genes play a role in its virulence (Fischer et al. 2010; Fernandez-Gonzalez and Backert 2014; Covacci et al. 1993). The epidemiological studies are assisted by experiments in the mouse models. Type II strains are observed in patients who have gastric vandalization along with histological lesions and are similar to the biopsies from patients infected by H. pylori (Telford et al. 1994). The two isolates type I and type II of H. pylori can establish in mice and the type I strains are known to elevate gastric damages similar to the ones observed in humans (Marchetti et al. 1995).
10.5 Computational Tools Involved in GI Prediction
Distorted sequence configuration and occasional phylogenetic distribution are the two prominent attributes of horizontally transferred GIs. Nowadays, genomic islands prediction is done via computational methods which use either sequence of gene composition differences or comparative genetic approaches (Chiapello et al. 2005). Since more GIs are being predicted several databases related to GI have been developed. Islander (Mantri and Williams 2004), ICEBerg (Bi et al. 2012), and PAIDB (Yoon et al. 2007) are specific databases for GIs that have originated from tRNAs and pathogenicity islands (PAIs). Island viewer is one such database which precomputedly predicts the published GIs and unpublished genomic sequences can be submitted for analysis. If any GIs are predicted using two or more methods, then the annotations of the gene can be viewed just by clicking an image. Island viewer also links to external GI databases connected to NCBI and Joint Genome Institute (JGI). It also connects with the IslandPath which allows to probe of the features related to genomic islands for the users’ choice of the genome (Mantri and Williams 2004). Based on number of inputs of genomes prediction techniques can be broadly grouped into two different types rooted in one genome and multiple genomes (Lu and Leong 2016).
10.5.1 Prediction Methods Based on One Genome
Each species develop a specific gene composition due to mutational and selection events so the horizontally transferred genes show a different composition from other species. This assumption is used to discriminate species characteristics. The codons, amino acid usage, GC content, and k-mer frequencies (Lawrence and Ochman 1997) are used as criteria for score comparison. Computed threshold limits are fixed and any gene frequencies crossing the limit are classified as atypical genes. They further rely on gene sequence composition which is identified on the basis of Hidden Markov rule, DNA sequence composition which uses k-mer frequencies which window based or windowless methods and GI structures.
10.5.2 Prediction Methods Focussed on Multiple Genomes
GIs are predicted based on irregular phylogenetic distribution. The comparing process involves the use of sequence alignment tools like BLAST for local alignment and MAUVE for global alignment (Darling et al. 2004). To promote genome selection, IslandPick builds a distance matrix of the whole genome and makes use of respective cut-offs to sort out adequate genomes to distinguish with the query genome. This method is completely automatic. The whole-genome pairwise positioning is done by MAUVE to get large distinct zones in the query genome. Finally, genome duplication is eliminated using BLAST and these zones are taken as assumed GIs. Different tools to predict genomic islands are given in Table 10.1.
10.6 Future Perspectives of GIs
GIs have various advantages over plasmid since they easily incorporate into the host’s chromosome. They do not need to be replicated often like plasmids and a single facsimile of GI can be conferred per genome (Gaillard et al. 2008). GIs render a key role in the development of pathogenic species among bacteria. Studies on the GIs of H. pylori has disclosed that the HopH gene helps in the colonization of mucosal surfaces in gastrointestinal linings (Yamaoka et al. 2002). Proinflammatory signaling events can be identified via transcriptional profiling in H. pylori-stimulated epithelia which further helps in studies on the regulation of gene expression in different strains (Yamaoka et al. 2004). The CT dinucleotide repeats differ in different strains from different countries. These characteristics can be used for the calculation of mutation rates in different species. However the non-bioinformatical part it is difficult to visualize the GIs. Alien hunter and GI hunter predictions can give circular representations of the GIs. MTGI can give dynamic simulations of GIs in circular manner but cannot give unique interpretations. Hence, there is a need to develop these technologies further.
10.7 Conclusion
The prediction and analysis of GIs are now becoming a key part of microbial examinations. Helicobacter species is one the most virulent strains and is also capable of rapid mutation. GI identification is critical for the study of genomic islands in Helicobacter species. Recently acquired gene sequences can be probed for studies of disease outbreaks, strain mutations among patients, resistance, etc. Different computational methods can identify different features of bacterial GIs. There are no accurate GI prediction tools to present, especially for the horizontally transferred genes. The development of more comprehensive methods would further help researchers in real-time tracking of GI studies.
References
Arvey AJ, Azad RK, Raval A, Lawrence JG (2009) Detection of genomic islands via segmental genome heterogeneity. Nucleic Acids Res 37(16):5255–5266
Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, Ou HY (2012) ICEberg: a web-based resource for integrative and conjugative elements found in bacteria. Nucleic Acids Res 40(D1):D621–D626
Boyd EF, Brüssow H (2002) Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol 10(11):521–529
Che D, Wang H, Fazekas J, Chen B (2014) An accurate genomic island prediction method for sequenced bacterial and archaeal genomes. J Proteom Bioinform 7(8):214
Chiapello H, Bourgait I, Sourivong F, Heuclin G, Gendrault-Jacquemard A, Petit MA, El Karoui M (2005) Systematic determination of the mosaic structure of bacterial genomes: species backbone versus strain-specific loops. BMC Bioinformatics 6(1):1–10
Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N (1993) Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci 90(12):5791–5795
Cover TL, Blaser MJ (2009) Helicobacter pylori in health and disease. Gastroenterology 136(6):1863–1873
Darling AC, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14(7):1394–1403
Dobrindt U, Hentschel U, Kaper JB, Hacker J (2002) Genome plasticity in pathogenic and nonpathogenic enterobacteria. Pathogenicity Islands and the Evolution of Pathogenic Microbes:157–175
Egan S, Wiener P, Kallifidas D, Wellington EM (1998) Transfer of streptomycin biosynthesis gene clusters within streptomycetes isolated from soil. Appl Environ Microbiol 64(12):5061–5063
Fernandez-Gonzalez E, Backert S (2014) DNA transfer in the gastric pathogen helicobacter pylori. J Gastroenterol 49(4):594–604
Fischer W, Windhager L, Rohrer S, Zeiller M, Karnholz A, Hoffmann R, Zimmer R, Haas R (2010) Strain-specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res 38(18):6089–6101
Gaillard M, Pernet N, Vogne C, Hagenbüchle O, van der Meer JR (2008) Host and invader impact of transfer of the clc genomic island into Pseudomonas aeruginosa PAO1. Proc Natl Acad Sci 105(19):7058–7063
Hacker J (2002) Urinary tract infection: From basic science to clinical application. In Genes and Proteins Underlying Microbial Urinary Tract Virulence (pp. 1–8). Springer, Boston, MA
Hooi JK, Lai WY, Ng WK, Suen MM, Underwood FE, Tanyingoh D et al (2017) Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 153(2):420–429
Hsiao W, Wan I, Jones SJ, Brinkman FS (2003) IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19(3):418–420
Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T (1998) Helicobacter pylori adhesin binding fucosylatedhisto-blood group antigens revealed by retagging. Science 279(5349):373–377
Khan AS, Kniep B, Oelschlaeger TA, Van Die I, Korhonen T, Hacker J (2000) Receptor structure for F1C fimbriae of uropathogenic Escherichia coli. Infect Immun 68(6):3541–3547
Langille MG, Hsiao WW, Brinkman FS (2008) Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9(1):1–10
Lawrence JG, Ochman H (1997) Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol 44(4):383–397
Lu B, Leong HW (2016) Computational methods for predicting genomic islands in microbial genomes. Comput Struct Biotechnol J 14:200–206
Mantri Y, Williams KP (2004) Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res 32(suppl_1):D55–D58
Marchetti M, Arico B, Burroni D, Figura N, Rappuoli R, Ghiara P (1995) Development of a mouse model of helicobacter pylori infection that mimics human disease. Science 267(5204):1655–1658
Nr S, Muller A (2013) Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Natl Rev Microbiol 11(6):385–399
Paulsen IT, Banerjei L, Myers GSA, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H (2003) Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299(5615):2071–2074
Schwartz E, Henne A, Cramm R, Eitinger T, Friedrich B, Gottschalk G (2003) Complete nucleotide sequence of pHG1: a Ralstoniaeutropha H16 megaplasmid encoding key enzymes of H2-based lithoautotrophy and anaerobiosis. J Mol Biol 332(2):369–383
Shankar N, Baghdayan AS, Gilmore MS (2002) Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417(6890):746–750
Telford JL, Ghiara P, Dell'Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce MF, Censini S, Covacci A, Xiang Z (1994) Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J Exp Med 179(5):1653–1658
Tu Q, Ding D (2003) Detecting pathogenicity islands and anomalous gene clusters by iterative discriminant analysis. FEMS Microbiol Lett 221(2):269–275
Vernikos GS, Parkhill J (2006) Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22(18):2196–2203
Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF, Surovcik K, Meinicke P, Merkl R (2006) Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7(1):1–12
Yamaoka Y, Kikuchi S, El-Zimaity HM, Gutierrez O, Osato MS, Graham DY (2002) Importance of helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 123(2):414–424
Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY (2004) Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology 126(4):1030–1043
Yoon SH, Park YK, Lee S, Choi D, Oh TK, Hur CG, Kim JF (2007) Towards pathogenomics: a web-based resource for pathogenicity islands. Nucleic Acids Res 35(suppl_1):D395–D400
Zhang R, Ou HY, Gao F, Luo H (2014) Identification of horizontally-transferred genomic islands and genome segmentation points by using the GC profile method. Curr Genomics 15(2):113–121
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sudharsan, M.S., K, V., Hari, S., Punniavan, S. (2023). Genomic Islands in Helicobacter Species. In: Mani, I., Singh, V., Alzahrani, K.J., Chu, DT. (eds) Microbial Genomic Islands in Adaptation and Pathogenicity. Springer, Singapore. https://doi.org/10.1007/978-981-19-9342-8_10
Download citation
DOI: https://doi.org/10.1007/978-981-19-9342-8_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-9341-1
Online ISBN: 978-981-19-9342-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)