
Abstract
Discoveries of pattern recognition receptors (PRRs) and their functions are one of the most important advances in medicine and immunology, which has promoted developments of basic science and clinical diagnosis and therapeutics. Human bodies and inflammatory cells can sensitively sense, recognize and defeat infected microbes, such as bacteria, fungus, virus and others, and also some body’s own materials from damaged cardiac tissue via PRRs for fulminant myocarditis. We raise the proposal for fulminant myocarditis: exogenous pathogens invade the body, as antigens or ligands susceptible to cardiomyocytes, bind to PRRs on cardiomyocytes and inflammatory cells, sequentially triggering downstream signaling, especially NF-kB pathway, and inducing form of cytokine storm, resulting in fulminant myocarditis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


3.1 Brief Introduction
Human bodies as well as living beings can sensitively sense, recognize and defeat infected microbes, such as bacteria, fungus, virus and others, and also some materials from apoptosis and necrosis. It is a unique and important, and daunting task, which is essential for the survival of human bodies. This is an ability so called innate immune system of vertebrates by innate immune cells mainly including neutrophils, monocytes, macrophages, dendritic cells, natural killer cells, mast cells, eosinophils, and basophils. Furthermore, monocytes and macrophages swallow and manage antigens and transform to lymphocytes to have the adaptive or specific immunity.
Humans and organisms recognize self and non-self (exogenous microorganisms and materials) through pattern recognition receptors (PRRs). PRRs are distributed on both immune cells and non-immune cells, including endothelial cells, fibroblasts, and cardiomyocytes. Pathogen-associated molecular patterns (PAMPs) refer to PRR recognition of exogenous microorganisms and other pathogens, and corresponding damage-associated molecular patterns (DAMPs) refer to the recognition of endogenous molecules by PRRs [1]. Although some controversies exist and many action mechanisms need to be figured out it is widely accepted that both PAMP and DAMP trigger immune responses via the activation of classical and non-classical PRRs, so that PRRs signal to corresponding cells and initiate a series of cascade effects and inflammatory response. Importantly, activating immune cells and tissue cells not only stimulate innate immune and adaptive immune response, but also involve human disorders, even severe disease, such as fulminant myocarditis. In this chapter, we briefly introduce PRRS and pattern associated molecules and pathogen associated molecules, distribution and possible roles of PRRs to help understand pathogeny and pathogenesis of fulminant myocarditis and further is able to handle treatments patients with fulminant myocarditis.
3.2 Pattern Recognition Receptors (PRRs)
PRRs include five main classes, membrane-bound Toll-like receptors (TLRs) and NOD-like receptors (NLRs), retinoic acid inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs) and multiple intracellular DNA sensors [1]. In addition, several ion channels, G-protein-coupled receptors (GPCRs), and triggering receptors expressed on myeloid cells (TREMs) can sense DAMPs stimulation and have response although they are put under classical PRRs. Here we list all the PRRs and their roles in Table 3.1 [2].
3.2.1 Membrane-Bound TLRs
As one of the earliest PRRs discovered in immune system, Toll-like receptors (TLRs) have numerous family members and are essential in inflammatory responses. Up to date, at least 15 TLRs have been found and identified in mammalian and constitute a big family, of them 12 being mouse TLRs (TLR1–TLR9, TLR11–TLR13), 10 being human TLRs (TLR1–TLR10), and additional 2 discovered recently, respectively, TLR14 in fish species and chicken specific TLR15 [3,4,5]. PRRs are widely expressed on the cell membrane (TLR1–2, TLR4–6 and TLR10), intracellular compartment membranes (TLR3, TLR7–9) and distributed in the cytoplasm [6].
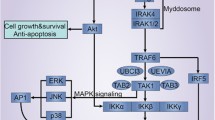
TLRs consist of three parts, a N-terminal domain (NTD), which located outside the membrane (ligand recognition domain), a middle single helix transmembrane domain, and a C-terminal domain (CTD) towards the cytoplasm (effector domain), respectively [7]. Ligand recognition domain is extracellular region which have leucine-rich repeats (LRRs) in charge of the pattern recognition of Toll/IL-1R (TIR) domain same as IL-1R and TLRs related with signal transduction (Fig. 3.1). Extracellular region specifically recognizes ligands. Once TLRs recognizing and binding with corresponding PAMPs or DAMPs ligands, TIR domain transform signal to cytoplasmic region and results in sequential inflammation in myeloid differentiation factor 88 (MyD88)-dependent and MyD88-independent pathways (including TIR domain-containing adaptor inducing IFN-β (TRIF), TRIF-related adaptor molecule (TRAM), B-cell adaptor for phosphoinositide (BCAP), and Sterile α- and armadillo-motif-containing protein (SARM)) (Fig. 3.2).

Similar structural features of TLR and IL-R

Distribution of TLRs in the cell membrane and endosomal compartments, and detection of various PAMPs by TLRs
3.2.2 TLR1–2 and TLR6
TLR2 recognizes various microbial components including Gram-positive bacteria’s peptidoglycan, lipoproteins/lipopeptides and lipoteichoic acid, mycobacteria’s lipoarabinomannan, Trypanosoma cruzi’s glycosylphosphatidylinositol anchors, Staphylococcus epidermis’s phenol-soluble modulin, fungi’s zymosan and Treponema maltophilum’s glycolipids and also LPS. This phenomenon may arise from the heterophilic dimerization of TLR2 with TLR1/6 and others, which are functionally linked to TLR2. In addition, TLR2 functionally cooperates with dectin-1 that recognize the fungal cell wall component b-glucan.
3.2.3 TLR4
It is well known that TLR4 is vital for LPS recognition, which is a classic example. TLR4 is also responsible for recognizing DAMPs, endogenous ligands, including the extra domain A of fibronectins, heat shock proteins (HSP60 and HSP70), heparan sulfate, oligosaccharides of hyaluronic acid and fibrinogen which, however, requires very high concentration [8].
3.2.4 TLR5
TLR5 is also located in cell membrane and it expresses in both immune cells and tissue cells (on the basolateral of intestinal epithelium, and subepithelial compartment of intestinal endothelial cells) [9, 10]. TLR5 can recognize flagellin by the interaction of TLR5 and flagellin. Because of its expression location, it can fight microbes via flagellin and dsRNA sensing and stimulation production of proinflammatory cytokines and interferons at the mucosal surface.
3.2.5 TLR3 and TLR7-9
TLR3 and TLR7-9 are known as nucleic acid-sensing TLRs. Because they are located in the endosomal compartment, and host-derived single strand RNAs are generally not delivered to endosomes, they are not aberrantly activated by self-nucleic acids. However, in some cases TLRs are nevertheless activated by internalized self-nucleic acids. TLR3 exists in cytoplasm and responds to double-stranded RNA and then induces cytokine production through a signaling pathway dependent on MyD88 [11]. TLR7/TLR8 recognize the same ligands in some cases, probably because they are highly conserved in structure [9]. Recent studies suggest that TLR7/TLR8 can recognize single-stranded RNA of viruses (including human immunodeficiency virus, and influenza virus). While TLR9 recognize CpG DNA no matter bacterial CpG DNA or viral-derived CpG DNA. A/D-type and B/K-type are the two major types of CpG DNA. B/K-type CpG DNA induces the release of IL-12 and TNF-α, and other inflammatory cytokines [9]. While A/D-type CpG DNA can effectively induce plasmacytoid dendritic cells to produce IFN-α.
3.2.6 TLR10
As one of the least-known members of TLR family, the TLR10 gene was first cloned 20 years ago [12]. TLR10 is mainly distributed in immune cells including spleen, lymph nodes, thymus, tonsils and lungs, among which the highest expression is in B cells, followed by plasmacytoid dendritic cells. TLR10 also expresses in tissue cells, such as gastric mucosal tissues, cancer cells, TLR10 can recognize dsRNA and recruit MyD88 for signal transduction. In addition, it suppresses the production of type I interferon. Additionally, TLR10 can serve as co-receptor of many other TLRs, for example, it can work together with TLR2/TLR4 to recognize LPS and with TLR2/TLR6 to recognize synthetic diacylated lipoprotein (FSL-1).
3.2.7 NLRs
NLRs belong to intracellular PRRs, a large family of intracellular sensors that investigate the presence of PAMPs and DAMPs in the cytoplasm. NLRs are composed of three domains: (1) the central nucleotide-binding domain (NBD) that is critical in nucleic acid binding and oligomerization of NLRs; (2) the LRR of C-terminus for ligand recognition, and (3) an N-terminal effector domain comprising the caspase activation and recruitment domain and other protein interaction domain [3]. There are five subfamilies of NLRs: the NLRC subfamily, the NLRP subfamily, the NLRB subfamily; the NLRA subfamily; and NLRX subfamily [3, 7, 13]. After NLR oligomerization, pro-caspase-1 is recruited directly or via apoptosis-associated speck-like protein, and promoting the formation of inflammasome [13].
3.2.8 NLRP3 Inflammasome
The NLRP3 inflammasome is activated by a variety of molecules including microbial molecules, environmental stimuli, and many metabolites. The NLRs sense a common intracellular molecule. Many endogenous molecules have been linked mitochondrial damage to NLRP3 inflammasome activation, which recruit NLRP3 and trigger inflammasome. Ion fluctuations may involve NLRP3 inflammasome activation. Thus, the NLRP3 inflammasome is critical in various acute and chronic diseases.
3.2.9 RLRs
As a kind of intracellular PRRs, RLRs has three main members, melanoma differentiation-associated gene 5 (MDA5), RIG-I, and laboratory of genetics and physiology 2 (LGP2) [1, 3, 14]. Similar to TLR7 and TLR9, RLRs have innate antiviral immunity (Fig. 3.3).

RLRs’ structure and ligand recognition. In contrast to RIG-I, MDA5 has no self-inhibitory ability due to the lack of a repressor domain. LGP2 cannot transmit signal because there is no CARD
RIG-I induces the expression of IFN-β and therefore it has antiviral activity. RIG-I has three parts: the DexD/H helicase domain, RLR family domain with ATPase and helicase activities; The N-terminus with two caspase activation and recruitment domains (CARD) [15]. The C-terminus is composed of the C-terminal domain (CTD) for the recognition of viral RNA, and the repressor domain (RD) [3]. MDA5 is similar to RIG-I (Fig. 3.4). Very long double strand RNAs (>300 bp) can efficiently activate MDA5 regardless of the structure of the MDA5 terminus. LGP2 cannot recruit molecules due to the lack of CARD, but it plays a role in regulating the viral nucleic acid’s recognition by RIG-I and MDA5 [16]. LGP2 reduces the production of IFN and inflammatory factors by negatively regulating RIG-I-mediated viral dsRNA recognition, ultimately suppressing the antiviral innate immune response.

Development of innate and adaptive immunity
3.2.10 Cytoplasmic DNA Sensors
Cyclic GMP-AMP synthase (cGAS) and absent in melanoma 2 (AIM2) are the two main DNA sensors, which are critical in antimicrobial immunity. Activation of cGAS or AIM2 by self-DNA resulting from cell damage induces cellular senescence, and promotes cancers and inflammation.
3.2.11 CLRs
CLRs are phagocytic PRRs that are rarely studied and understood. Once CLRs recognizing and binding PAMPs through PRRs, pathogens are eliminated by phagocytosing into cytoplasmic vesicles for digestion.
3.2.12 Non-pattern Recognition Receptors
Non-pattern recognition receptors include TREMs, RAGE, ion channels and GPCRs. They sense DAMPs and PAMPs, and therefore promotes the activation and migration of immune cells after activated.
3.2.13 RAGE
RAGE is expressed in various types of cells and tissues. RAGE can bind a series of endogenous ligands, including advanced glycation end products (AGEs), HMGB1, S100 proteins (S100s) [17]. In turn, these ligands upregulate RAGE expression, forming vicious circle. RAGE activation plays important roles in cardiovascular disease via inducing inflammation and activating NF-κB signaling.
3.2.14 PRMs
Both cellular PRRs and extracellular soluble PRMs belong to PRMs. Extracellular soluble PRMs play a key role in nonspecific humoral immunity [1, 3]. Extracellular soluble PRMs have various family members including pentraxin, collectin, and ficolin. Once the various pathogenic factors are identified, they are eliminated by complement, opsonization, and neutralization of inflammatory regulation activated by extracellular soluble PRMs; in addition, extracellular soluble PRMs interact with and regulate cell-associated PRRs to co-regulate the innate immune response. As typical representatives of pentraxin, liver-produced CRP and serum amyloid P components are nonspecific proteins in systemic inflammatory responses. They bind to phosphorylcholine and bacterial outer membrane protein A of pathogenic microorganisms. Mannose-binding lectin (MBL) is the main part of collectin, and is formed by homotrimer, of which consists of a CRD that recognize sugar structures (mannose, fucose and glucose, an alpha helix, and a backbone formed by collagen and surfactant protein (SP) helices) on the pathogen membranes. Although structurally similar to collectin, ficolin can recognize bacteria with fibrin-prototype carbohydrate recognition structures due to its binding with N-acetylglucosamine and LTA.
3.2.15 Myeloid Cells’s Triggering Receptors
There are two classes of TREM, TREM1 and TREM2, both of which are innate immune membrane receptors. TREM1 is mainly distributed in bone marrow cells and non-immune cells; TREM2 is highly expressed by myeloid cell types. Activating antibodies activate TREM1 on neutrophils and monocytes, promote the release of pro-inflammatory cytokine and chemokine, and enhance inflammatory responses. Intracellular proteins, including HMGB1, peptidoglycan recognition protein 1 (PGLYRP1), HSP70, and extracellular actin, are its ligands. A number of endogenous lipids and lipoproteins that bind to and activate TREM2 have been identified, although further identification is required.
3.2.16 G-Protein-Coupled Receptors
N-formyl peptide receptors (FPRs) and P2Y receptors (P2YRs) belonging to GPCRs, bind to various endogenous DAMPs and exogenous PAMPs to promote inflammation. FPR is widely distributed, and in addition to being expressed in leukocytes [18], it is also expressed in a variety of non-immune cells, including hepatocytes, and fibroblasts. Formylated peptides belong to both PAMPs and DAMPs. FPR has many ligands that are structurally and chemically unrelated. Activation of FPR by these ligands elicits distinct cellular responses. Serum amyloid A (SAA) and oxidized LDL participate in the pathogenesis of chronic inflammatory diseases by binding to FPR. Nucleotides such as ATP and UTP released into the extracellular space through necrosis and apoptosis bind to P2 purinergic receptors to trigger pro-inflammatory immune responses. P2Y2R signaling promotes the activation of a variety of immune cells. UDP activates the stromal cell’s P2Y6R signaling, whereas ADP-mediated activation of P2Y12R is critically involved in platelet activation and aggregation. P2X7R with seven members promotes immune responses in a number of ways.
3.2.17 Actions of PRRs
PRRs bind their ligands and act through various ways. They can co-act and have crosstalk between receptors.
3.2.18 Phagocytosis and TLRs
The first step in defensing against microorganisms is phagocytosis [9]. Once pathogens are recognized by TLRs, the expression of inflammatory molecules are enhanced, thereby promoting the development of adaptive immunity (Fig. 3.4). After binding to TLRs, pathogens are recognized and devoured by innate immune cells, and then antigenic peptides are presented to naive T cells. Simultaneously binding of antigen to TLRs induces inflammatory cytokines and costimulatory molecules, Naı̈ve T-cells, including Th1 cells are guided to produce adaptive immunity.
3.2.19 TLR Signaling Pathways
The TLR signaling pathway includes a variety of molecules (protein kinases, transcription factors, etc.), all of which converge on canonical signaling pathways.
The NF-kB Signaling: NF-κB, which is composed of p50 and p65, is critically involved in cellular inflammation and immune responses. After TLRs recognize and receive stimulation of antigens, they trigger the expression of proteins in TLR-mediated signaling pathways [9]. The dimerization of TLRs which is formed via TLR1 or TLR6, as well as homodimers is necessary for the recognition of microorganisms. The cytoplasmic TIR domain of dimerized TLRs recruit MyD88, which then induces inflammatory cytokines, such as TNF-α and IL-12 [9]. MyD88 is essential for all TLRs. Different activation of TLRs may results in different profiles. Activation of TLR3-4 and TLR7-9 signaling pathways lead to induction of type I interferon (IFN) in distinct mechanisms, but not TLR2 and TLR5 mediated pathways.
MyD88-dependent signaling pathway plays a key role in TLR1-2 and TLR4, TLR6 signaling. Upon stimulation, IRAK-4, TRIF, TIRAP, and TRAM are recruited to TLRs by MyD88, which subsequently promote nuclear factor κB (NF-κB) and MAPK activation through the IRAK complex and two non-catalytic subunits, and ultimately lead to the production of tumor necrosis factor (TNF), IL-6, IL-1, chemokines and other pro-inflammatory cytokines [9, 19].
MAPKs are a class of serine-threonine protein kinases that respond to a variety of extracellular stimuli. MAPKs are activated by IRAK-1 in a MyD88-dependent pathway of TLRs, activated p38 MAPK leads to the secretion of pro-inflammatory molecules [3].
The TBK1–IRF-3 signaling: IRF-3 is a transcription factor and is critically involved in the synthesis of type I IFN. TRIF is the adaptor protein in the MyD88-independent pathway, and promotes the expression of type I interferon, and therefore exerts antiviral effect.
In addition, there is an inflammasome signaling. Inflammasome is a multi-protein complex in the cytoplasm assembled by PRRs. NLRP1, NLRP3, NLRC4, IPAF inflammasome, and AIM2 are inflammasome. After recognition of PAMPs or DAMPs by inflammasome, Caspase-1 is recruited and activated. Then proIL-1β/proIL-18 is spliced into mature cytokines by activated Caspase-1 (Fig. 3.5).

PRR-mediated inflammasome signaling and signal transduction
3.3 Pyroptosis and its Pathway
Pyroptosis, Pyroptotic cell death, is controlled by the inflammatory caspases. Caspase 1 is pro-inflammatory and is linked with the secretion of mature IL-1β and IL-18 [20]. Pyroptosis was originally defined as “caspase 1-dependent necrosis” due to its strict requirements for caspase 1 [21]. Further, the inflammasome complex and the non-canonical inflammasome pathway were discovered in 2002 and 2011, respectively. They were identified in pyroptosis in inflammasome-dependent form. After those, there are many advances and importantly it was found that various members of gasdermin family play key roles in pyroptosis processing. Therefore, pyroptosis need redefinition. More recently, it was defined as “gasdermin-induced cell necrosis,” a new definition that applies to all gasdermin members that may lead to cell death.
Gasdermin family at least include five classes of members, gasdermin A, B, C, D and E. These gasdermin proteins all involve pyroptosis through pore formation role in cell membrane. Each gasdermin protein has conserved amino- terminal domain (NTD) which has membrane-forming activity, and carboxy-terminal domains. In the quiescent state, the gasdermin protein maintains itself in an autoinhibitory state by binding of NTD with the carboxy-terminal domain (CTD) [22]. However, the aspartic acid residue of GSDMD in the linker between NTD and CTD can be recognized and cleaved by activated caspase-1, caspase-4, caspase-5 and caspase-11, and the cleaved NTD is in non-covalent binding state. Due to its high affinity for membrane phospholipids, the NTD of GSDMD translocate to the plasma membrane, induces pyroptotic bubble formation and membrane lysis. Therefore, membrane lysis induced by GSDMD is the final step in LPS-induced pyroptosis mediated by caspase-11, caspase-4 and caspase-5. In fact, NTD of other gasdermins has similar lipid-binding properties and induces intracellular pyroptosis. Following figure shows mechanisms of gasdermin activation and pore formation and pyroptosis occurs [21] (Fig. 3.6).

Gasdermin D mechanism to pyroptosis
Gasdermins express in different tissues including the organs in digestive, respiratory, urinary, reproductive, circulatory systems and other tissues. Especially, we note that the heart and artery have expression. Recently, some studies found that in mice with coxsackievirus B3 induced myocarditis, heart levels of GSDMD p30 and IL-1β and HMGB1 were elevated and the inhibition of pyroptosis signaling attenuated myocarditis, which suggest that pyroptosis involves in pathophysiology of myocarditis [23, 24]. Therefore, we have reason to believe, pyroptosis play a vital role in part in fulminant myocarditis.
3.3.1 Expression of PRRs and Responses to Ligands in Heart
Human and mammal heart express almost all PRRs and after the stimulation of ligands the expression of PRRS in whole heart and cardiomyocytes is dramatically upregulated. S. aureus peptidoglycan, E. coli LPS and S. typhimurium flagellin induced chemokine KC production by activating TLR2, TLR4 and TLR5. The IL-6 and MIP2 production mediated by LPS and lagellin, respectively, both significantly reduced by pyrrolidine dithiocarbamate (PDTC), a chemical inhibitor of NF-κB. These indicate that TLRs promotes the expression of proinflammatory cytokines, chemokines and cell surface adhesion molecules by NF-κB signaling [25].
Although the most types of PRRs express in different tissues and cell types in human, their expression patterns are different and different PAMPs and DAMPs or ligands have different target PRRs. On the other hand, same PRRs in different cells may have different response to same ligand because some cell may be refractory to some special ligand stimulation, such as platelet-activating factor receptor (PAFr), one of GPCRs, express in cardiomyocytes, endothelial cells and neurons. However, after exposure to phosphocholine-containing bacterial components, three types of cells uptake in a PAFr-dependent manner, but with different pathophysiological consequences. Non-inflammatory manifestations appear in endothelial cells and neurons, while cardiomyocytes rapidly lose contractility and die, because endothelial cells and neurons are refractory to the phosphorylcholine-containing bacterial components and there is no classic-type NF-B response [26]. Therefore, we can meet fulminant myocarditis, fulminant pancreatic, fulminant type I diabetes, fulminant hepatitis and others after exposed to different pathogens and injury in clinic, which may have same or similar pathogenesis and pathophysiological mechanisms.
Finally, we try to raise the proposals for fulminant myocarditis based on our some experiments and clinical practice (Fig. 3.7) [27, 28, 21]. (1) exogenous pathogens, either infection of virus, bacteria, fungus or other microbe, chemicals including proteins, nucleus acids, antibiotics, allopurinol and others such as seafood, or some vaccination, enter the body, as antigens or ligands susceptible to cardiomyocytes, bind to PRRs on cardiomyocytes, either TLRs or others, membrane PRRS or cytoplasm PRRs, sequentially triggering downstream signaling, especially NF-kB pathway. Thus, inflammatory storm form. Importantly, these PAMPs successively or simultaneously activate innate immune cells, monocytes, lymphocytes, neutrophils, eosinophils, and then together with cardiomyocytes produce inflammatory storm; (2) These reactions also induce adaptive immune response, including production of anti-myocardial antibodies by B-lymphocytes and direct attack to heart by toxic T-cells; (3) PAMPs and DAMPs induce pyroptosis and their consequence results in proinflamattory storm. These inflammatory factors can strongly stun the heart, inhibiting calcium handling and pump function; (4) Immune check-point inhibitors induced fulminant myocarditis: Programmed cell death protein 1 (PD-1)-mediated and cytotoxic T-lymphocyte antigen 4 (CTLA4)-mediated pathways, and immune checkpoint pathways, are activated by tumor cells to evade recognition by the immune system [23, 29, 30]. Immune check-point inhibitors activate the antitumour immune of T-cells via targeting the molecules of these pathways. However, T-cells targeted antigen may be shared with tumors, heart and muscles and thus, the activated T-cells will attack heart and induce fulminant myocarditis. Additionally, immune check-point inhibitors can activation of cardiac antigen-reactive T cells block antigen-presenting cells.

Development of fulminant myocarditis
References
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112. https://doi.org/10.1038/s41577-019-0215-7.
Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. https://doi.org/10.1016/j.smim.2003.10.003.
Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6:291. https://doi.org/10.1038/s41392-021-00687-0.
Wu M, Guo L, Zhu KC, Guo HY, Liu BS, Zhang N, Jiang SG, Zhang DC. Molecular characterization of toll-like receptor 14 from golden pompano Trachinotus ovatus (Linnaeus, 1758) and its expression response to three types of pathogen-associated molecular patterns. Comp Biochem Physiol B Biochem Mol Biol. 2019;232:1–10. https://doi.org/10.1016/j.cbpb.2019.02.010.
Oven I, Resman Rus K, Dusanic D, Bencina D, Keeler CL Jr, Narat M. Diacylated lipopeptide from mycoplasma synoviae mediates TLR15 induced innate immune responses. Vet Res. 2013;44:99. https://doi.org/10.1186/1297-9716-44-99.
O'Keeffe M. Conventional dendritic cells may be ideal targets for vaccine strategies in the aged. Immunol Cell Biol. 2012;90:665–6. https://doi.org/10.1038/icb.2012.16.
Sameer AS, Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. Biomed Res Int. 2021;2021:1157023. https://doi.org/10.1155/2021/1157023.
Ando K, Hasegawa K, Shindo K, Furusawa T, Fujino T, Kikugawa K, Nakano H, Takeuchi O, Akira S, Akiyama T, et al. Human lactoferrin activates NF-kappaB through the toll-like receptor 4 pathway while it interferes with the lipopolysaccharide-stimulated TLR4 signaling. FEBS J. 2010;277:2051–66. https://doi.org/10.1111/j.1742-4658.2010.07620.x.
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. https://doi.org/10.1093/intimm/dxh186.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. https://doi.org/10.1038/ni1112.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732–8. https://doi.org/10.1038/35099560.
Su SB, Tao L, Deng ZP, Chen W, Qin SY, Jiang HX. TLR10: insights, controversies and potential utility as a therapeutic target. Scand J Immunol. 2021;93:e12988. https://doi.org/10.1111/sji.12988.
Yang X, Lin G, Han Z, Chai J. Structural biology of NOD-like receptors. Adv Exp Med Biol. 2019;1172:119–41. https://doi.org/10.1007/978-981-13-9367-9_6.
Wei LM, Jiao PR, Song YF, Han F, Cao L, Yang F, Ren T, Liao M. Identification and expression profiling analysis of goose melanoma differentiation associated gene 5 (MDA5) gene. Poult Sci. 2013;92:2618–24. https://doi.org/10.3382/ps.2013-03064.
Buskiewicz IA, Koenig A, Huber SA, Budd RC. Caspase-8 and FLIP regulate RIG-I/MDA5-induced innate immune host responses to picornaviruses. Futur Virol. 2012;7:1221–36. https://doi.org/10.2217/fvl.12.115.
Sharma A, Kontodimas K, Bosmann M. The MAVS immune recognition pathway in viral infection and sepsis. Antioxid Redox Signal. 2021;35:1376–92. https://doi.org/10.1089/ars.2021.0167.
Wang H, Li W, Goldstein R, Tracey KJ, Sama AE. HMGB1 as a potential therapeutic target. Novartis Found Symp. 2007;280:73–85. discussion 85-91, 160-164
Chen K, Bao Z, Gong W, Tang P, Yoshimura T, Wang JM. Regulation of inflammation by members of the formyl-peptide receptor family. J Autoimmun. 2017;85:64–77. https://doi.org/10.1016/j.jaut.2017.06.012.
Xiong D, Song L, Geng S, Jiao Y, Zhou X, Song H, Kang X, Zhou Y, Xu X, Sun J, et al. Salmonella coiled-coil- and TIR-containing TcpS evades the innate immune system and subdues inflammation. Cell Rep. 2019;28:804-818.e807. https://doi.org/10.1016/j.celrep.2019.06.048.
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. https://doi.org/10.1016/s0966-842x(00)01936-3.
Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–57. https://doi.org/10.1038/s41577-019-0228-2.
Rathinam VAK, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol. 2019;20:527–33. https://doi.org/10.1038/s41590-019-0368-3.
Collen D, Lijnen HR, Todd PA, Goa KL. Tissue-type plasminogen activator. A review of its pharmacology and therapeutic use as a thrombolytic agent. Drugs. 1989;38:346–88. https://doi.org/10.2165/00003495-198938030-00003.
Liu N, Su H, Zhang Y, Liu Z, Kong J. Cholecalciterol cholesterol emulsion attenuates experimental autoimmune myocarditis in mice via inhibition of the pyroptosis signaling pathway. Biochem Biophys Res Commun. 2017;493:422–8. https://doi.org/10.1016/j.bbrc.2017.09.006.
Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93. https://doi.org/10.1016/j.cardiores.2006.09.011.
Fillon S, Soulis K, Rajasekaran S, Benedict-Hamilton H, Radin JN, Orihuela CJ, El Kasmi KC, Murti G, Kaushal D, Gaber MW, et al. Platelet-activating factor receptor and innate immunity: uptake of gram-positive bacterial cell wall into host cells and cell-specific pathophysiology. J Immunol. 2006;177:6182–91. https://doi.org/10.4049/jimmunol.177.9.6182.
Hang W, Chen C, Seubert JM, Wang DW. Fulminant myocarditis: a comprehensive review from etiology to treatments and outcomes. Signal Transduct Target Ther. 2020;5:287. https://doi.org/10.1038/s41392-020-00360-y.
Zhou N, Zhao Y, Jiang J, Shen L, Li J, Wan J, Ma X, Zhang J, Ammirati E, Wang DW. Impact of mechanical circulatory support and immunomodulation therapy on outcome of patients with fulminant myocarditis: Chinese registry of fulminant myocarditis. Signal Transduct Target Ther. 2021;6:350. https://doi.org/10.1038/s41392-021-00700-6.
Tajiri K, Aonuma K, Sekine I. Immune checkpoint inhibitor-related myocarditis. Jpn J Clin Oncol. 2018;48:7–12. https://doi.org/10.1093/jjco/hyx154.
Sury K, Perazella MA, Shirali AC. Cardiorenal complications of immune checkpoint inhibitors. Nat Rev Nephrol. 2018;14:571–88. https://doi.org/10.1038/s41581-018-0035-1.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wang, D.W., Zhou, R. (2022). Pattern Recognition Receptors and Fulminant Myocarditis. In: Wang, D.W. (eds) Fulminant Myocarditis. Springer, Singapore. https://doi.org/10.1007/978-981-19-5759-8_3
Download citation
DOI: https://doi.org/10.1007/978-981-19-5759-8_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-5758-1
Online ISBN: 978-981-19-5759-8
eBook Packages: MedicineMedicine (R0)