
Abstract
In recent times, agriculturally important plants face increasing challenges in maintaining productivity, disease control, and welfare of farmers with changing climatic conditions. To accomplish this, the generation and analysis of large volumes of data, especially in the emerging “OMICS” areas of genomics, proteomics, and bioinformatics, is imperative for decision-making over large volumes of data with respect to various crops. Analysis of this large amount of diverged data needs specific tools and techniques. There are various tools and techniques available for the analysis of such data. In this chapter, a detailed discussion on omics data analysis related tools and techniques have been made. This chapter provides a single platform to help the various researchers working in different domains of omics research for analyzing the data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
11.1 Introduction
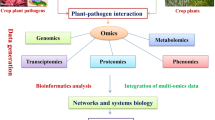
Various environmental factors like heat, cold, salinity, and drought severely affects plants growth and development that affects its production and productivity significantly. To address the abiotic stresses, defense mechanisms are often triggered by the plant to mitigate these unfavorable conditions. Understanding the mechanisms of plant defense systems at molecular level, there is a need to conduct a comprehensive study to decode the molecular mechanisms using various bioinformatics tools and techniques. Advanced DNA sequencing technology has accelerated the pace of genomics and transcriptomic studies in plants and animals to understand the molecular mechanisms. With the progress in omics approaches (viz. genomics, transcriptomics, proteomics, metabolomics, and phonemics) and its use in agriculture, a huge amount of data has been generated in molecular and biotechnology labs which can be used to identify novel genetic and chemical elements controlling various physiological processes and pathways of plant defense system. However, using only one approach is not sufficient to understand the complexity of stress response in plants. Recent development in the field of next generation sequencing technology (i.e., high-throughput data generation with reduced cost) in OMICS era generated a huge volume of molecular data. The major omics approaches are composed of genomics, transcriptomics, proteomics, metabolomics, and phenomics. These approaches provide a holistic view of molecular pathways at the cellular, tissue, or organism level. The integration of different omics-based approaches provides many folds of biological information which resulted in the development of a new branch of life science known as system biology (Hong et al. 2016; Chaudhary et al. 2019). However, analysis of high-throughput data from various omics-based approaches is one of the biggest challenges to interpret the plant defense mechanism(s). There are several tools, techniques, and databases available in public domain for various omics-based analyses independently. To handle this challenge due to generation and availability of multi-omics data, one has to use these tools in a more judicial and integrated way for deeper and novel biological insights. This chapter discusses various omics techniques such as genomics, transcriptomics, proteomics, metabolomics, and phenomics which are used to explore and understand the defense mechanism of plants at the molecular level to address abiotic stresses. Moreover, this chapter also provides a list of some important and widely used tools which can further be used to integrate the results of these omics approaches to draw a meaningful inferential conclusion.
11.2 OMICS Approaches to Study Plant Defense Mechanism
11.2.1 Genomics
-
(i)
Whole genome sequencing and resequencing: Genomics deals with the study of the complete genetic makeup of organisms or individuals. The field of genomics has grown exponentially in the past 20 years since the announcement of the first draft human genome in 2001. Further, the reduced sequencing costs and time accelerated the pace of whole genome sequencing due to the advancement of Next Generation Sequencing (NGS) technologies that have resulted in flooding of sequencing data (Fig. 11.1). This led to the development of advanced and efficient bioinformatics tools and techniques to handle such large-scale sequencing data for deeper and novel biological insights. We can consider mainly two groups of genomics, i.e., structural genomics and functional genomics. Structural genomics deals with locating the mapped genes and markers to individual chromosomes which results in producing physical map of the genome whereas functional genomics focuses on relating genome sequences with its transcriptome and proteome (encoded proteins) to describe gene functions and interactions. The most efficient way to study molecular mechanisms in plants is to decode the whole genome sequence. In plants, Arabidopsis was the first genome to be sequenced by an international consortium (Berardini et al. 2015). Plant genome sequence helps to explain the organization, regulation, and evolution of studied genomes. The advent of next generation sequencing (NGS) technologies allows millions of molecules to be sequenced simultaneously and whole genome sequencing has become substantially cheaper and faster than traditional sequencing methods (Goodwin et al. 2016). Availability of high-quality whole genome sequence data and a well-annotated reference genome is very crucial for genomics and transcriptomic-based research. The catalogue of annotated gene models, genome organization, and synteny-based knowledge, repeats, and most notably the basis for distinguishing genetic variants are more apparent advantages acquired from genome sequencing. The reference genome is also used as the basis for the annotation of other genomes of closely related species. However, sequencing of the whole genome (i.e., resequencing) is faster and cost-effective for the species which have already sequenced high-quality reference genomes. There are several Bioinformatics tools for assembling the reads of sequenced genome like Bowtie, Soap2, MIRA, Abyss, SOAPdenovo, and velvet (Wee et al. 2019).
-
(ii)
Identification of Molecular markers: The whole genome sequences can be extensively studied in discovering the molecular markers. One of the promising marker systems suitable for laboratories is microsatellites or simple sequence repeats (SSRs). A valuable resource for upcoming breeding programs are being developed for genome-wide identification of microsatellites and subsequently helps in markers development. MISA and GMATA are two most popular and widely used bioinformatics tools for identification of SSRs in the genomic data. But nowadays, SNP genotyping approaches are gaining mainstream acceptance with the introduction of cost-efficient and high-throughput genotyping techniques. SAM tools, GATK, Picard, etc. are some variant calling tools that are used to identify SNPs from the whole genome sequence assembly. The genotyping by sequencing (GBS) approach is an extremely multiplexed framework for building RRL (reduced representation libraries), finding molecular markers, and genotyping for crop improvement among the various other SNP-based genotyping approaches (Eltaher et al. 2018; Elbasyoni et al. 2018). GBS has been applied to many crop varieties as a result of low cost and innovative technology (Poland and Rife 2012; Kim et al. 2016). For example, a tomato GBS study led to the discovery of 8784 SNPs based on an approach to NGS and 88 percent of these SNPs are commonly found in tomato germplasm, (Sim et al. 2012). GBS is simple and cost-effective solution but use is still limited because it requires specialized skills in computational and data analysis. In the future, it can be a commonly used approach with the availability of easy-to-use computational packages and pipelines.
-
(iii)
QTL mapping and GWAS: Linkage mapping (LM) and association mapping (AM) by identifying marker–trait associations have contributed to the identification of QTL (Cockram and Mackay 2018). In many plant species, the importance has been given to mapping QTLs for many abiotic stresses, such as heat, salinity, drought, and cold. QTLs controlling seed germination under various stress conditions have been identified using QTL experiments. QTL mapping experiments are conducted to identify loci regulating stress resistance in particular, advancements in genomics have encouraged more complex approaches involving multi-parental populations such as nested association mapping (NAM) and Multi-parent advanced generation inter-cross (MAGIC). A Genome-wide association studies (GWAS) approach, on the other hand, has an advantage over linkage mapping (Linkage Disequilibrium, i.e., LD) as it examines the genetic variation and recombination events in germplasm collections and also offers higher precision mapping (Fukushima et al. 2009). This set is designed to capture the genetic variability for the trait of interest and represents the products of hundreds of historic recombination cycles, providing higher resolution during QTL mapping (Mackay et al. 2009). GWAS is systematically used to detect SNPs for agronomic characteristics in a germplasm collection (Pasam et al. 2012). However, associations detected in AM are often spurious because associations are based on LD, which not only depends on linkage but also on population stratification and relatedness among individuals. Nowadays, efforts have also been made to combine linkage-based QTL mapping with LD-based AM, and conduct joint linkage association mapping (JLAM) to overcome the limitations and exploit the benefits associated with each of the two approaches, i.e., linkage and LD.
-
(iv)
Genomic Selection: The declining cost of SNP assays has made it possible to genotype vast numbers of experimental lines in stress-tolerant crop breeding programs to introduce the Genomic Selection (GS) method. The GS method is successful in simultaneously controlling all the loci that lead to the growth of the trait, regardless of the magnitude of their individual impact. The GS solution overcomes the disadvantage of QTL mapping-based breeding where it is difficult to track/identify small-effect QTLs. Importantly, the small effects of QTLs can collectively have greater effects on abiotic traits of economic significance. Due to epistatic interactions, the most economically significant traits are complex and influenced by unexpected trait expressions (Deshmukh et al. 2014). Therefore, by using all available molecular markers in conjunction with the phenotypic data of a training population, GS is the best way to predict genetic values for selection. A model has been developed to classify and analyze genotypic and phenotypic data to evaluate the phenotypic variation based on their genotypes of their whole genomes (genetic composition) (Yan et al. 2009). To estimate breeding values, different GS models like nonlinear regressions (RKHS and RF), Bayesian approaches (Bayes A and B), and penalized regressions (RR, LASSO, and EN) have been used in many studies.

Workflow diagram of omics approaches for study of plant defense mechanism
11.2.2 Transcriptomics
For the efficient management of abiotic stress, understanding the gene regulatory cascades for stress responses is very important. The best strategy for investigating plant response regulation and identifying genes involved in mechanisms of stress tolerance is to collect and compare the transcriptome of different tissue types at various developmental stages. Thus, understanding the transcriptome of different tissues at developmental stages will lead to better understand the associated phenotypic variation. Several tools and techniques are available to obtain expression profiling for assessment of transcriptomic results both gene-by-gene and collectively for several genes at a time.
-
(i)
Microarray
Microarray technology is based on hybridization between the target DNA and probe DNA designed with known sequences. It is capable of covering tens of thousands of genes at a time, it has made a significant contribution to research. It is well developed and is still being used as a major platform for transcriptome analysis of sequenced species, despite its shortcomings in the variety of target transcripts in the dynamic spectrum of quantification compared with NGS-based RNA-seq technology (Wang et al. 2009; Jazayeri et al. 2014). Microarray is used to identify the differentially expressed genes in response to abiotic stresses, including salinity, heat, cold, drought, and oxidative stress. Numerous studies have been conducted in several plant species using microarray approaches to identify genes having significant roles in stress tolerance mechanisms as well as for the understanding of diverse molecular mechanisms (Kumar et al. 2018, 2019; Nagaraju et al. 2019, 2020).
-
(ii)
RNAseq
This approach is based on high-throughput Next Generation Sequencing. RNAseq relies on high-speed sequencing of short cDNA fragments (typically 30–400 bp) reverse-transcribed from mRNAs. Further, number of cDNA fragments aligned to the reference sequence indicates the abundance of the mRNA. RNA sequencing (RNAseq) has become the most cost-effective, reliable, and high-throughput transcriptomic technology with the quick advancement of next generation sequencing. In contrary to microarray, the RNAseq approach is not only confined to comparing the transcripts levels, but also it is useful in discovery of novel genes and spliced forms, especially in non-model plants. Numerous reports on the application of RNAseq technology in case of plants are available (Ye et al. 2017; Xiong et al. 2017; Guan et al. 2019). RNAseq technology has also been applied to unsequenced organisms (Ekblom and Galindo 2010) as several computational tools enable de novo assembly of the reads without the availability of a reference genome (Oshlack et al. 2010; Grabherr et al. 2011). Although management of the huge data sets generated poses many challenges and this technology is becoming a mainstream of transcriptome analysis.
-
(iii)
HiCEP
High-coverage gene expression profiling (HiCEP) is based on the amplified-fragment-length polymorphism technique. This approach can detect changes in transcript expression with high coverage (Fukumura et al. 2003). Amplified DNA fragments are first derived from mRNA followed by capillary electrophoresis. Their abundances are estimated by the peak observed through electropherogram. The relevant peaks are then fractionated and sequenced.
11.2.3 Proteomics
Proteomics is the large-scale study of proteins in a studied organism or system. The proteome represents a complete set of proteins that are produced by an underlying organism or system. Proteomics has enabled us to identify and validate the ever-increasing numbers of proteins. Proteins are important for living organisms as they produce a variety of functions. Modern proteomic technologies have made it possible to detect vast number of proteins in plant samples easily and simultaneously (Vanderschuren et al. 2013). Over the last few years in plant science, high-throughput quantitative proteomics studies gained considerable significance in characterizing proteomes and their differential regulation during plant growth, biotic and abiotic stresses. Proteomics experiments often found that many insect attack-responsive proteins were associated with the cycle of tricarboxylic acid (TCA) and also involved in carbon metabolism, which suggested that carbon metabolism was altered during insect attack for defense. High abundance of proteins such as ribulose-1,5-bisphosphate carboxylase oxygenase (Rubisco) creates considerable difficulties using shotgun plant proteomics for the whole proteome characterization. To understand the defense mechanisms during plant–insect interactions, an enhanced proteomic system, called Polyethyleneimine Assisted Rubisco Cleanup (PARC) was used (Zhang et al. 2013). George et al. (2011) reported the differential protein expression in maize (Zea mays L.) in response to infestation of a chewing (Spodoptera littoralis) and a boring insect (Busseola fusca).
-
(i)
Gel-Based Electrophoresis:
In the first dimension, proteins are isolated either by an immobilized pH gradient strip or by an isoelectric focusing tube, and then followed by SDS polyacrylamide gel electrophoresis in the second dimension (Komatsu et al. 2007, 2012, 2013a, b, 2015). Protein spots are extracted from the gel after staining, reduced by dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. A form of Mass Spectrometry (MS), such as nano-liquid chromatography (LC) tandem MS or nano-LC MS/MS, will then analyze the peptide mixtures. While 2D gel-based methods offer a visual description of proteins including intact protein profiles and they are not sufficient for the detection and identification of proteins with low abundance or with extreme molecular weights, isoelectric points, or hydrophobicity.
-
(ii)
Gel-free proteomics:
Gel-free proteomics includes both label-free and labeling methods. In case of label-free method, protein samples are purified by chloroform-methanol extraction and reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin and lysyl endopeptidase. They are analyzed by nano-LC MS/MS (Komatsu et al. 2013b). Differentially expressed proteins are identified from the spectrum obtained by scanning with MASCOT Daemon client software against a peptide database. For identification and annotation of homologous proteins, positive matches are searched against protein databases available at NCBI (www.ncbi.nlm.nih.gov) through BLASTP. It is now a commonly used technology in proteomics, since its protocol is simple and helpful in identifying proteins in a large scale.
11.2.4 Metabolomics
Metabolomics is a promising approach that provides a biochemical snapshot of phenotype of an organism. Metabolomics makes it possible to systematically classify and measure low-molecular weight molecules which are closely related to essential toxicological and nutritional features. Information on genes, proteins, and transcriptomes are not adequate to thoroughly classify a cell but the broad variety of primary and secondary metabolites found in a cell must also be examined. Numerous studies have been done to explain the function of metabolites in plants under conditions of biotic and abiotic stresses. Plant chemical compounds that are not active in photosynthetic and core metabolic processes are linked to the evolution of the chemical defense mechanism against stress in plants (Mithöfer and Boland 2012; Gjindali et al. 2021). These compounds are classified as secondary metabolites that do not play any significant role in the plant’s growth, development, or reproduction rather these compounds serve as signaling molecules or direct defense chemicals and include alkaloids, terpenoids, cyanogenic glycosides, glucosinolates, and phenolics (Bennett and Wallsgrove 1994; Zebelo and Maffei 2012). To study the chemicals involved in the interactions of living organisms, including the chemical defense system during plant–insect contact, a special area called “chemical ecology” is developed (Mithöfer and Boland 2012). Plants have to sacrifice some of the central metabolism by allocating energy to this defense while activating the defense response mechanism controlled by the secondary metabolites against insect attack. Along with secondary metabolism, during an insect or pathogen attack, the primary metabolism of a plant is often differentially influenced (Barah et al. 2013). It has been in use for the past decades to study the selective control of primary or secondary metabolites during plant–insect activity (Salem et al. 2020).
Recent developments in high-throughput metabolite profiling methods and advanced combinatorial protocols available in plant metabolomics are liquid chromatography–mass spectrometry (LC-MS), gas chromatography–mass spectrometry (GC-MS), Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS), ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS), flow-injection electrospray ionization mass spectrometry (FI-ESI-MS) and nuclear magnetic resonance (NMR). However, it is computationally difficult to analyze the enormously diversified plant metabolites produced using these methods (Allwood et al. 2008; Ernst et al. 2014). Hence in analyzing and processing highly complex biological data, the role of bioinformatics is very crucial.
11.2.5 Phenomics
Phenomics is the study of high-throughput phenotypic variation analysis, which is a complex web of genotype, phenotype, and environment interactions. Phenome represents a set of phenotypes. Studies of the genome and phenome with individuals or large populations are complementary to each other (Yasunori and Sinha 2014). Plants with stable phenotypes are strong genomic tools and are also a target to identify the alleles by high-throughput sequencing. Advances in sequencing technology have increased genotyping efficiencies, while phenotypic characterization has progressed more slowly over the past decade, restricting the characterization of quantitative characteristics, especially those associated with stress tolerance (White et al. 2012). There are recent developments in phenotyping methods which allow the identification of specific characteristics. Phenomics technology requires advanced imaging systems, sensors, automations, and computational resources for the phenotyping in plants. These make phenomics a high-throughput approach that is capable of handling thousands of genotypes for the evaluation of hundreds of phenotypic parameters simultaneously (White et al. 2012; Ubbens and Stavness 2017; Tardieu et al. 2017). There are various phenomics platforms available to investigate physiological parameters in plants under different stress conditions, e.g., one such tool is scan analyzer 3D. As phenomic data collection is an expensive and time-consuming method, integrated technological developments would help to minimize the associated costs and increase phenomic throughput.
11.3 Bioinformatics Tools and Techniques for Integration of Multi-OMICS Data
Due to availability of large-scale multi-omics data and their availability in public domain, e.g., in the form of various databases and repositories, poses a major challenge for bioinformatics community for integrating different tools and techniques so that one can draw biologically useful inferences because the use of only one approach at a time cannot lead to understand the defense mechanism robustly. Even after having lots of development in this area, integration of heterogeneous omics data to draw meaningful biological inferences is a major challenge (Keurentjes et al. 2011). However, to develop ultimate products like climate-smart cultivars, efficient integration of different tools, techniques, and approaches appears to be a promising strategy. For example, GWAS and QTL mapping both identify a genomic region or marker that is associated with underlying trait of interest and further in discovering the candidate genes. As the use of RNAseq data with gene expression profiles gives an idea about the functions of unknown genes. So, relating GWAS and QTL with their respective transcriptome will give the clue to identify differentially expressed candidate genes.
There are large number of user-friendly computational platforms are developed for the integration of multi-omics data (Table 11.1). Details of such few tools and software are given in Table 11.1.
11.4 Concluding Remarks
The recent developments in modern high-throughput sequencing technologies have flooded the web with the availability of biological data from various platforms. Recent efforts for development in integrating omics data are not sufficient in understanding such vast biological data. However, the integrative system-based approach, i.e., integrating multi-omics data generated from heterogeneous platforms, using various bioinformatics tools, techniques, and approaches, is the only solution to this problem of understanding and finding meaningful biological conclusions. Although efficient adaptation of bioinformatics tools and techniques depends on their availability and user-friendly manner. So, there is a need to develop more user-friendly and easy-to-use bioinformatics tools and pipelines for end users, such as accessibility, easy to use tutorials and manuals, and interactive options to analyze multi-platform data. This will help the researchers to understand the biological system in a more realistic way, and will definitely help to translate this understanding to develop better crop varieties with improved defense mechanisms.
References
Allwood JW, Ellis DI, Goodacre R (2008) Metabolomic technologies and their application to the study of plants and plant–host interactions. Physiol Plant 132:117–135. https://doi.org/10.1111/J.1399-3054.2007.01001.X
Barah P, Winge P, Kusnierczyk A et al (2013) Molecular signatures in Arabidopsis thaliana in response to insect attack and bacterial infection. PLoS One 8. https://doi.org/10.1371/JOURNAL.PONE.0058987
Bennett RN, Wallsgrove RM (1994) Secondary metabolites in plant defence mechanisms. New Phytol 127:617–633. https://doi.org/10.1111/J.1469-8137.1994.TB02968.X
Berardini TZ, Reiser L, Li D et al (2015) The Arabidopsis information resource: making and mining the ‘gold standard’ annotated reference plant genome. Genesis 53:474. https://doi.org/10.1002/DVG.22877
Brown TB, Cheng R, Sirault XRR et al (2014) Trait capture: genomic and environment modelling of plant phenomic data. Curr Opin Plant Biol 18:73–79. https://doi.org/10.1016/J.PBI.2014.02.002
Chaudhary J, Khatri P, Singla P et al (2019) Advances in omics approaches for abiotic stress tolerance in tomato. Biol 8:90. https://doi.org/10.3390/BIOLOGY8040090
Cockram J, Mackay I (2018) Genetic mapping populations for conducting high-resolution trait mapping in plants. Adv Biochem Eng Biotechnol 164:109–138. https://doi.org/10.1007/10_2017_48
Deshmukh R, Sonah H, Patil G et al (2014) Integrating omic approaches for abiotic stress tolerance in soybean. Front Plant Sci 5. https://doi.org/10.3389/FPLS.2014.00244
Ekblom R, Galindo J (2010) Applications of next generation sequencing in molecular ecology of non-model organisms. Hered 1071(107):1–15. https://doi.org/10.1038/hdy.2010.152
Elbasyoni IS, Lorenz AJ, Guttieri M et al (2018) A comparison between genotyping-by-sequencing and array-based scoring of SNPs for genomic prediction accuracy in winter wheat. Plant Sci 270:123–130. https://doi.org/10.1016/J.PLANTSCI.2018.02.019
Eltaher S, Sallam A, Belamkar V et al (2018) Genetic diversity and population structure of F3:6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front Genet 0:76. https://doi.org/10.3389/FGENE.2018.00076
Ernst M, Silva DB, Silva RR et al (2014) Mass spectrometry in plant metabolomics strategies: from analytical platforms to data acquisition and processing. Nat Prod Rep 31:784–806. https://doi.org/10.1039/C3NP70086K
Fukumura R, Takahashi H, Saito T, Tsutsumi Y, Fujimori A, Sato S, Tatsumi K, Araki R, Abe M (2003) A sensitive transcriptome analysis method that can detect unknown transcripts. Nucleic Acids Res 31(16):e94. https://doi.org/10.1093/nar/gng094
Fukushima A, Kusano M, Redestig H et al (2009) Integrated omics approaches in plant systems biology. Curr Opin Chem Biol 13:532–538. https://doi.org/10.1016/J.CBPA.2009.09.022
George D, Babalola OO, Gatehouse AMR (2011) Differential protein expression in maize (Zea mays) in response to insect attack. African J Biotechnol 10:7700–7709. https://doi.org/10.4314/ajb.v10i39
Gjindali A, Herrmann HA, Schwartz J-M et al (2021) A holistic approach to study photosynthetic acclimation responses of plants to fluctuating light. Front Plant Sci 0:651. https://doi.org/10.3389/FPLS.2021.668512
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 176(17):333–351. https://doi.org/10.1038/nrg.2016.49
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 297(29):644–652. https://doi.org/10.1038/nbt.1883
Guan Y, Li G, Chu Z et al (2019) Transcriptome analysis reveals important candidate genes involved in grain-size formation at the stage of grain enlargement in common wheat cultivar “Bainong 4199.”. PLoS One 14:e0214149. https://doi.org/10.1371/JOURNAL.PONE.0214149
Hong J, Yang L, Zhang D, Shi J (2016) Plant metabolomics: an indispensable system biology tool for plant science. Int J Mol Sci 17. https://doi.org/10.3390/IJMS17060767
Jazayeri SM, Melgarejo-Muñoz LM, Romero HM (2014) Rna-Seq: a glance at technologies and methodologies. Acta Biológica Colomb 20:23–35. https://doi.org/10.15446/abc.v20n2.43639
Katari MS, Nowicki SD, Aceituno FF et al (2010) Virtual plant: a software platform to support systems biology research. Plant Physiol 152:500–515. https://doi.org/10.1104/PP.109.147025
Keurentjes JJB, Angenent GC, Dicke M et al (2011) Redefining plant systems biology: from cell to ecosystem. Trends Plant Sci 16:183–190. https://doi.org/10.1016/J.TPLANTS.2010.12.002
Kim C, Guo H, Kong W et al (2016) Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci 242:14–22. https://doi.org/10.1016/j.plantsci.2015.04.016
Komatsu S, Han C, Nanjo Y et al (2013a) Label-free quantitative proteomic analysis of abscisic acid effect in early-stage soybean under flooding. J Proteome Res 12:4769–4784. https://doi.org/10.1021/PR4001898
Komatsu S, Kuji R, Nanjo Y et al (2012) Comprehensive analysis of endoplasmic reticulum-enriched fraction in root tips of soybean under flooding stress using proteomics techniques. J Proteome 77:531–560. https://doi.org/10.1016/J.JPROT.2012.09.032
Komatsu S, Sakata K, Nanjo Y (2015) ‘Omics’ techniques and their use to identify how soybean responds to flooding. J Anal Sci Technol 61(6):1–8. https://doi.org/10.1186/S40543-015-0052-7
Komatsu S, Shirasaka N, Sakata K (2013b) ‘Omics’ techniques for identifying flooding–response mechanisms in soybean. J Proteome 93:169–178. https://doi.org/10.1016/J.JPROT.2012.12.016
Komatsu M, Waguri S, Koike M et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163. https://doi.org/10.1016/J.CELL.2007.10.035
Kumar A, Batra R, Gahlaut V et al (2018) Genome-wide identification and characterization of gene family for RWP-RK transcription factors in wheat (Triticum aestivum L.). PLoS One 13:e0208409. https://doi.org/10.1371/JOURNAL.PONE.0208409
Kumar A, Sharma M, Gahlaut V et al (2019) Genome-wide identification, characterization, and expression profiling of SPX gene family in wheat. Int J Biol Macromol 140:17–32. https://doi.org/10.1016/J.IJBIOMAC.2019.08.105
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma 91(9):1–13. https://doi.org/10.1186/1471-2105-9-559
Lê Cao K-A, González I, Déjean S (2009) Integr omics: an R package to unravel relationships between two omics datasets. Bioinformatics 25:2855–2856. https://doi.org/10.1093/BIOINFORMATICS/BTP515
Mackay TFC, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 108(10):565–577. https://doi.org/10.1038/nrg2612
Medina I, Carbonell J, Pulido L et al (2010) Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res 38:W210–W213. https://doi.org/10.1093/NAR/GKQ388
Mithöfer A, Boland W (2012) Plant defense against herbivores: chemical aspects. Annu Rev Plant Biol 63:431–450. https://doi.org/10.1146/ANNUREV-ARPLANT-042110-103854
Nagaraju M, Kumar SA, Reddy PS et al (2019) Genome-scale identification, classification, and tissue specific expression analysis of late embryogenesis abundant (LEA) genes under abiotic stress conditions in Sorghum bicolor L. PLoS One 14:e0209980. https://doi.org/10.1371/JOURNAL.PONE.0209980
Nagaraju M, Reddy PS, Kumar SA et al (2020) Genome-wide identification and transcriptional profiling of small heat shock protein gene family under diverse abiotic stress conditions in Sorghum bicolor (L.). Int J Biol Macromol 142:822–834. https://doi.org/10.1016/J.IJBIOMAC.2019.10.023
Oshlack A, Robinson MD, Young MD (2010) From RNA-seq reads to differential expression results. Genome Biol 1112 11:1–10. doi:https://doi.org/10.1186/GB-2010-11-12-220
Pasam RK, Sharma R, Malosetti M et al (2012) Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol 121(12):1–22. https://doi.org/10.1186/1471-2229-12-16
Planell N, Lagani V, Sebastian-Leon P et al (2021) STATegra: multi-omics data integration–a conceptual scheme with a bioinformatics pipeline. Front Genet 0:143. https://doi.org/10.3389/FGENE.2021.620453
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5. https://doi.org/10.3835/PLANTGENOME2012.05.0005
Rohart F, Gautier B, Singh A, Cao K-AL (2017) mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol 13:e1005752. https://doi.org/10.1371/JOURNAL.PCBI.1005752
Salem MA, de Souza LP, Serag A et al (2020) Metabolomics in the context of plant natural products research: from sample preparation to metabolite analysis. Meta 10. https://doi.org/10.3390/METABO10010037
Shen R, Olshen AB, Ladanyi M (2009) Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 25:2906–2912. https://doi.org/10.1093/BIOINFORMATICS/BTP543
Sim S-C, Durstewitz G, Plieske J et al (2012) Development of a large SNP genotyping Array and generation of high-density genetic maps in tomato. PLoS One 7:e40563. https://doi.org/10.1371/JOURNAL.PONE.0040563
Tardieu F, Cabrera-Bosquet L, Pridmore T, Bennett M (2017) Plant Phenomics, from sensors to knowledge. Curr Biol 27:R770–R783. https://doi.org/10.1016/J.CUB.2017.05.055
Ubbens JR, Stavness I (2017) Deep plant Phenomics: a deep learning platform for complex plant phenotyping tasks. Front Plant Sci 0:1190. https://doi.org/10.3389/FPLS.2017.01190
Ulfenborg B (2019) Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinforma 201(20):1–10. https://doi.org/10.1186/S12859-019-3224-4
Van Bel M, Proost S, Wischnitzki E et al (2012) Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol 158:590–600. https://doi.org/10.1104/PP.111.189514
Vanderschuren H, Lentz E, Zainuddin I, Gruissem W (2013) Proteomics of model and crop plant species: status, current limitations and strategic advances for crop improvement. J Proteome 93:5–19. https://doi.org/10.1016/J.JPROT.2013.05.036
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57. https://doi.org/10.1038/NRG2484
Wee Y, Bhyan SB, Liu Y et al (2019) The bioinformatics tools for the genome assembly and analysis based on third-generation sequencing. Brief Funct Genomics 18:1–12. https://doi.org/10.1093/BFGP/ELY037
White JW, Andrade-Sanchez P, Gore MA et al (2012) Field-based phenomics for plant genetics research. F Crop Res 133:101–112. https://doi.org/10.1016/J.FCR.2012.04.003
Xiong H, Guo H, Xie Y et al (2017) RNAseq analysis reveals pathways and candidate genes associated with salinity tolerance in a spaceflight-induced wheat mutant. Sci Reports 71(7):1–13. https://doi.org/10.1038/s41598-017-03024-0
Yan J, Shah T, Warburton ML et al (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One 4:e8451. https://doi.org/10.1371/JOURNAL.PONE.0008451
Yasunori I, Sinha NR (2014) From genome to phenome and back in tomato. Curr Opin Plant Biol 18:9–15. https://doi.org/10.1016/J.PBI.2013.12.004
Ye J, Duan Y, Hu G et al (2017) Identification of candidate genes and biosynthesis pathways related to fertility conversion by wheat KTM3315A transcriptome profiling. Front Plant Sci 0:449. https://doi.org/10.3389/FPLS.2017.00449
Zebelo SA, Maffei ME (2012) Signal transduction in plant–insect interactions: from membrane potential variations to metabolomics. Plant Electrophysiol Signal Responses 143–172. https://doi.org/10.1007/978-3-642-29110-4_6
Zhang Y, Gao P, Xing Z et al (2013) Application of an improved proteomics method for abundant protein cleanup: molecular and genomic mechanisms study in plant defense. Mol Cell Proteomics 12:3431. https://doi.org/10.1074/MCP.M112.025213
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mishra, D.C., Guha Majumdar, S., Budhlakoti, N., Kumar, A., Chaturvedi, K.K. (2022). OMICS Tools and Techniques for Study of Defense Mechanism in Plants. In: Kumar, R.R., Praveen, S., Rai, G.K. (eds) Thermotolerance in Crop Plants. Springer, Singapore. https://doi.org/10.1007/978-981-19-3800-9_11
Download citation
DOI: https://doi.org/10.1007/978-981-19-3800-9_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-3799-6
Online ISBN: 978-981-19-3800-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)