
Abstract
Wilms’ tumor in horseshoe kidney (HSK) and solitary kidney is an uncommon occurrence, and their management poses a daunting surgical challenge. Imaging aids in diagnosis and delineating the anatomy and blood supply making resection safe and reducing complications. Neoadjuvant chemotherapy downstages the tumor and reduces the incidence of spill, thereby making inoperable tumors amenable to resection and reduces chances of recurrence. Pre-operative chemotherapy is recommended in Wilms’ tumor in solitary kidney before nephron sparing surgery (NSS).
Learning objectives of this chapter are to understand the presentation, origin, the role of imaging and neo-adjuvant chemotherapy, surgical principles, and outcome in Wilms’ tumor in HSK and solitary kidney. Special emphasis is on imaging detailing the varied anatomical variations in the blood supply of HSK which helps the surgeon in planning resection. The role of pre-operative chemotherapy followed by NSS in Wilms’ tumor in solitary kidney and subsequent development of end stage renal disease (ESRD) in some patients is also highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

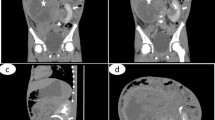
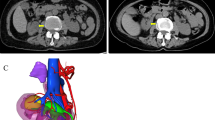
Keywords
- Wilms’ tumor
- Horseshoe kidney (HSK)
- Solitary kidney
- Nephron sparing surgery (NSS)
- End stage renal disease (ESRD)
25.1 Wilms’ Tumor in Horseshoe Kidney
Wilms’ tumor (WT) in horseshoe kidney (HSK) presents as a unique diagnostic and a surgical challenge. The incidence of HSK in general population is 1 in 400 [1]. WT occurs uncommonly in HSK and the incidence is 0.4–0.9% of all WT [1, 2]. Renal cell carcinoma and other renal pelvis tumor are more common in HSK than WT [1, 3]. Still, a child with HSK has twofold increased risk to develop WT than the general population [1, 3, 4]. WT can arise in either moiety or the isthmus in HSK and can be unifocal or multifocal. In a National Wilms Tumor Study Group (NWTSG) report, WT was present in both moieties of HSK in 7% of patients and was considered bilateral disease [2]. Metanephric blastemal elements sequestered in the isthmus have malignant potential and cause WT. Other theory is the embryologic alteration causing HSK itself, after second event may give rise to WT [1, 3, 4]. The NWTS did not identify any WT1 mutations in patients with WT and HSK. The incidence of nephrogenic rests in HSK with WT was also found similar to that in unilateral WT [2, 5]. Routine screening of people with HSK for WT is not warranted as the overall incidence of WT development is less than 0.001% [1].
Presentation of WT in HSK is the same, as for any other case of WT with respect to age, sex, clinical stage at presentation, and histological pattern [4]. Asymptomatic abdominal mass is the most common presenting symptom. Others can be abdominal pain, hematuria, anemia, and hypertension [6]. Other congenital anomalies like chromosomal (Edward syndrome, Turner syndrome), cardiac, genito-urinary, and musculoskeletal abnormalities were present in 44% patients of WT with HSK [2].
Pre-operative imaging including ultrasound and computerized tomography (CT) scan for WT can miss the diagnosis of HSK. More often than not, the diagnosis of HSK is made intra-operatively. In the NWTS, the diagnosis of HSK was missed pre-operatively in 13/41 WT patients (32%). A large size tumor crossing midline may obscure the isthmus and HSK may be missed. Also, it is difficult to distinguish a tumor in the isthmus from a lower pole tumor [2, 7, 8]. HSK has a lot of anatomical variations in shape, blood supply, and collecting system. There may be duplicated or triplicated renal arteries, unilaterally or bilaterally. The isthmus may get its blood supply from either of the renal arteries, aorta, inferior mesenteric artery, or iliac arteries [4]. The ureters may be displaced anteriorly and medially or laterally depending upon the origin of tumor. Hydronephrosis may be present due to compression of one or more ureter [3]. Pre-operative knowledge of vascular anatomy and collecting system can help in planning safe resection and minimizing complications. CT-angiography with 3-D reconstruction or a magnetic resonance (MR) imaging with MR angiogram with arterial and venous phase study can assist in localization of the tumor and clearly defining vascular anatomy in the presence of HSK [1, 2].
A supra-umbilical wide transverse incision is considered the best. HSKs are usually located lower than normal kidneys within the abdomen. In the majority, the isthmus lies just in front of the aorta and IVC, at the level of the 3rd to 5th lumbar vertebral bodies. Pre-operative insertion of ureteric stents helps identifying the ureters and averting ureteric injury [9].
WT arising in either moiety of HSK is staged and treated the same as WT in a unilateral normal kidney, other than that there is a global consensus about administering neoadjuvant chemotherapy (ChT) in every patient [10]. Removal of the involved kidney with isthmus, along with meticulous hemostasis, is recommended followed by the stage appropriate ChT [1,2,3]. Removal of isthmus is important because if the remaining kidney does not drain its urine, a urinary fistula may form. If the tumor is arising from the isthmus, then isthmusectomy along with removal of both lower poles is done. Lymph node sampling protocol also remains the same as in unilateral WT.
In cases with multifocal disease, an accurate estimation of normal uninvolved renal parenchyma needs to be done pre-operatively, and the patient should be treated with NSS as per protocols for bilateral WT. The aim of surgery would be maximal preservation of unaffected renal parenchyma without sacrificing oncological control [11].
Neo-adjuvant ChT can downstage and reduces the bulk of the disease enabling safe and a complete resection, lesser chances of tumor rupture/spill, and maximal preservation of renal parenchyma and function. Of the 41 cases of HSK in NWTS, 37% were deemed inoperable at the time of initial exploration. But after ChT, all were amenable to resection [2]. Pre-treatment FNAC/biopsy is recommended before starting neo-adjuvant ChT.
Abnormal anatomy and blood supply makes the resection difficult. There is increased risk of urine leak and ureteral injury. Any injury to urinary collecting system must be repaired.
The prognosis of WT in HSK depends on the stage and histology. The incidence of anaplasia in WT in HSK is similar to general WT population [2]. The estimated 4-year overall survival (OS) of WT in HSK was 86%, similar to the patients with unilateral WT. [2]
25.2 Wilms’ Tumor in Solitary Kidney
Management of WT in solitary kidney is a difficult proposition. Other kidney may be non-functional due to unilateral renal agenesis, dysplasia due to severe reflux, or nephrectomy of previously involved kidney and development of metachronous WT in the remaining kidney [12, 13]. Children’s oncology group (COG) recommends nephron sparing surgery (NSS) after neo-adjuvant ChT with Vincristine, Actinomycin-D, and Doxorubicin under certain special circumstances such WT in a solitary kidney, bilateral WT, suprahepatic inferior vena caval (IVC) thrombus, and severe respiratory distress due to extensive pulmonary metastatic disease [14]. It reduces the amount of tumor burden, thus enabling NSS, and decreases the incidence of tumor spillage [15]. Pre-treatment biopsy is advisable, but is not mandatory. After 4–6 weeks of ch ChT, partial nephrectomy, i.e., complete excision of tumor with a rim of healthy renal parenchyma is performed. Partial nephrectomy is preferred over enucleation, which involves bluntly dissecting outside the pseudocapsule of the tumor. It may be rapid but has a higher risk of tumor spillage and positive margins [16, 17]. Davidoff et al. reported positive margins in 7/51 resected specimens (14%) after enucleation in a series of NSS in bilateral WT [15].
NSS may still not be feasible following neo-adjuvant ChT in certain patients. Polar tumors localized within the kidney but outside the hilum and sparing the sinus, not involving the renal vein or IVC in whom more than two-thirds to half of renal parenchyma can be preserved and have favorable histology, are amenable to NSS [18]. A technique of performing longitudinal partial nephrectomy for central tumors involving the renal hilum has also been described [19]. The patients can develop end stage renal disease (ESRD) after NSS. A functioning remnant of at least 25–33% of one kidney is sufficient to avoid ESRD [16]. In most series with bilateral WT, progression to ESRD occurred in 8–18% of patients [15, 18]. ESRD may develop immediately following surgery because of the removal of bulk of renal parenchyma or may develop later due to chronic kidney disease (CKD) as a result of hyperfiltration injury in remaining nephrons [20]. Risk might increase during puberty due to decrease in glomerular filtration rate and increased deterioration of renal function in CKD [21, 22]. The patients in the second group who develop ESRD late following CKD had good opportunity to receive transplant (79% within 5 years) and have higher overall survival (81% at 5 years) [20].
The patients with ESRD after NSS/nephrectomy require permanent renal replacement therapy (dialysis + renal transplant). Renal transplantation can be done after completion of ChT. Matsukura et al. report a case of a 2-year-old girl with WT in a solitary kidney who after pre-operative ChT underwent resection of the tumor followed by hemodialysis and received a kidney transplant from her mother after completion of her ChT [23]. A delay of 1–2 years following WT treatment before doing a transplant was recommended because of deaths due to sepsis and tumor recurrence in patients who underwent transplant early [24, 25]. European best-practice guidelines recommend a 2-year waiting period before transplant [26]. Grigoriev et al. re-evaluated the recommended waiting time and proposed that patients of WT who subsequently develop ESRD should not be subject to a 2-year delay and can immediately be considered for kidney transplant [20].
WT in solitary kidney poses a unique surgical challenge, and the involvement and extent of disease may not allow application of generally applied principles. There are even reports of successfully treating WT in solitary kidney in whom surgery was not possible without rendering the child anephric with ChT alone [12, 13]. Such adversity mandates an individualized treatment plan. A long-term careful follow-up is necessary to support and safeguard future of such children.
References
Lee SH, Bae MH, Choi SH, Lee JS, Cho YS, Joo KJ, et al. Wilms’ tumor in a horseshoe kidney. Korean J Urol. 2012;53:577–80. https://doi.org/10.4111/kju.2012.53.8.577.
Neville H, Ritchey ML, Shamberger RC, Haase G, Perlman S, Yoshioka T. The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg. 2002;37:1134–7. https://doi.org/10.1053/jpsu.2002.34458.
Talpallikar MC, Sawant V, Hirugade S, Borwankar SS, Sanghani H. Wilms’ tumor arising in a horseshoe kidney. Pediatr Surg Int. 2001;17:465–6. https://doi.org/10.1007/s003830000472.
Lai A, Marwaha RK, Narshimhan KL, Yadav K. Wilms tumor arising in a horseshoe kidney. Indian Pediatr. 1995;32:689–93.
Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr Path. 1990;10:1–36. https://doi.org/10.3109/1551381900906709.
Huang EY, Mascarenhas L, Mahour GH. Wilms’ tumor and horseshoe kidneys: a case report and review of the literature. J Pediatr Surg. 2004;39:207–12. https://doi.org/10.1016/j.jpedsurg.2003.10.019.
Bauer SB, Perlmutter AD, Retik AB. Anomalies of upper urinary tract. In: Walsh PC, Retik AB, Stamey TA, Vaughan ED, editors. Campbell’s urology. 6th ed. Philadelphia: W.B. Saunders Company; 1992. p. 1376–81.
Gay BB Jr, Dawes RK, Atkinson GO Jr, Ball TI Jr. Wilms’ tumor in horseshoe kidneys: radiologic diagnosis. Radiology. 1983;146:693–7. https://doi.org/10.1148/radiology.146.3.6298856.
Ritchey ML, Shamberger RC, Haase G, Horwitz J, Bergemann T, Breslow NE. Surgical complications after primary nephrectomy for Wilms’ tumor: report from the National Wilms’ tumor study group. J Am Coll Surg. 2001;192:63–8.
Shamberger RC. Pediatric renal tumors. Semin Surg Oncol. 1999;16:105–20. https://doi.org/10.1002/(sici)1098-2388(199903)16:2<105::aid-ssu4>3.0.co;2-t.
Cox S, Büyükünal C, Millar AJW. Surgery for the complex Wilms tumour. Pediatr Surg Int. 2020;36:113–27. https://doi.org/10.1007/s00383-019-04596-w.
Iñón AE, Gallo G, Richard L, Martorelli J, Puigdevall JC. Wilms’ tumor treated with chemotherapy in a patient with a solitary kidney. J Pediatr Surg. 1996;31:1305–7. https://doi.org/10.1016/s0022-3468(96)90260-2.
Prasoon PM, Akbar Sherif VS, Babu PR, Regi George AN, Anoop P. Wilms’ tumor in a solitary kidney complicated by chemotherapy induced obstructive uropathy. Indian J Pediatr. 2004;71:465–7. https://doi.org/10.1007/BF02725644.
Metzger ML, Dome JS. Current therapy for Wilms’ tumor. Oncologist. 2005;10:815–26. https://doi.org/10.1634/theoncologist.10-10-815.
Davidoff AM, Giel DW, Jones DP, Jenkins JJ, Kresin MJ, Hoffer FA, et al. The feasibility and outcome of nephron-sparing surgery for children with bilateral Wilms tumor. Cancer. 2008;112:2060–70. https://doi.org/10.1002/cncr.23406.
Harel M, Makari JH, Ferrer FA. Oncology: the role of partial nephrectomy in Wilms tumor. Curr Urol Rep. 2013;14:350–8. https://doi.org/10.1007/s11934-013-0330-0.
Cozzi DA, Zani A. Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg. 2006;15:3–9. https://doi.org/10.1053/j.sempedsurg.2005.11.002.
Moorman-Voestermans CG, Aronson DC, Staalman CR, Delemarre JF, de Kraker J. Is partial nephrectomy appropriate treatment for unilateral Wilms’ tumor? J Pediatr Surg. 1998;33:165–70. https://doi.org/10.1016/s0022-3468(98)90425-0.
Fuchs J, Szavay P, Seitz G, Handgretinger R, Schäfer JF, Warmann SW. Nephron sparing surgery for synchronous bilateral nephroblastoma involving the renal hilus. J Urol. 2011;186:1430–6. https://doi.org/10.1016/j.juro.2011.05.068.
Grigoriev Y, Lange J, Peterson SM, Takashima JR, Ritchey ML, Ko D, et al. Treatments and outcomes for end stage renal disease following Wilms tumor. Pediatr Nephrol. 2012;27:1325–33. https://doi.org/10.1007/s00467-012-2140-x.
Ardissino G, Testa S, Daccò V, Paglialonga F, Vigano S, Felice-Civitillo C, et al. Puberty is associated with increased deterioration of renal function in patients with CKD: data from the ItalKid project. Arch Dis Child. 2012;97:885–8. https://doi.org/10.1136/archdischild-2011-300685.
Westland R, Schreuder MF, Bökenkamp A, Spreeuwenberg MD, van Wijk JA. Renal injury in children with a solitary functioning kidney—the KIMONO study. Nephrol Dial Transplant. 2011;26:1533–41. https://doi.org/10.1093/ndt/gfq844.
Matsukura H, Ibuki K, Nomura K, Higashiyama H, Takasaki A, Miyawaki T, et al. Intracranial calcification in a uremic infant with Wilms’ tumor in a solitary kidney. CEN Case Rep. 2012;1:86–9. https://doi.org/10.1007/s13730-012-0019-0.
DeMaria JE, Hardy BE, Brezinski A, Churchill BM. Renal transplantation in patients with bilateral Wilm’s tumor. J Pediatr Surg. 1979;14:577–9. https://doi.org/10.1016/s0022-3468(79)80143-8.
Penn I. Renal transplantation for Wilms tumor: report of 20 cases. J Urol. 1979;122:793–4. https://doi.org/10.1016/s0022-5347(17)56607-0.
EBPG Expert Group on Renal Transplantation. European best practice guidelines for renal transplantation. Section IV: long-term management of the transplant recipient. IV.11 Paediatrics (specific problems). Nephrol Dial Transplant. 2002;17(Suppl 4):55–8. https://doi.org/10.1093/ndt/17.suppl_4.55.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Khanna, V. (2022). Wilms’ Tumor in Horseshoe Kidney and Solitary Kidney. In: Sarin, Y.K. (eds) Wilms’ Tumor. Springer, Singapore. https://doi.org/10.1007/978-981-19-3428-5_25
Download citation
DOI: https://doi.org/10.1007/978-981-19-3428-5_25
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-3427-8
Online ISBN: 978-981-19-3428-5
eBook Packages: MedicineMedicine (R0)