
Abstract
The scavenger receptor class B type I (SR-BI) is a versatile HDL receptor protein. It is highly expressed in liver and steroidogenic tissues. SR-BI regulates selective uptake of cholesterol ester (CE) from HDL, revealing its role in mediating reverse cholesterol transport (RCT) and steroid hormone synthesis. In addition, SR-BI is involved in cholesterol transport, cellular inflammatory response, platelet reactivity, and HDL-initiated signaling in the vascular system in several mouse models. Mutations in the human SR-BI gene (SCARB1) have been found to be associated with abnormally high plasma HDL-C levels and an increased risk of atherosclerotic cardiovascular disease. At present, the key regions of SR-BI transmembrane structure and the regulatory mechanisms of SR-BI expression still need to be further studied. In this chapter, the structural, functional, and regulatory characteristics of SR-BI are reviewed, and the importance of SR-BI in related metabolic diseases was expounded.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Scavenger receptor class B type I (SR-BI)
- HDL metabolism
- Reverse cholesterol transport
- Cholesterol transport
- Cellular inflammatory response
- Platelet reactivity
- HDL-initiated signaling
- Atherosclerotic cardiovascular disease
The scavenger receptor class B type I (SR-BI), a member of the CD36 superfamily, is a membrane glycoprotein that functions as a HDL receptor [1]. SR-BI is well conserved between species and abundantly expressed in the liver and steroidogenic tissues where it mediates selective uptake of cholesteryl ester (CE) from HDL. Hepatic SR-BI plays an important role in reverse cholesterol transport (RCT) and is closely related to HDL metabolism [2, 3]. Meanwhile, several studies from mouse models support that SR-BI regulates cholesterol trafficking, cell inflammatory responses, platelet reactivity, and HDL-initiated signaling in the vasculature, which is related to its role in anti-atherogenesis [4, 5]. Recently, some large population based studies have demonstrated that the human SR-BI gene (SCARB1) has single-nucleotide polymorphism (SNP) and subjects heterozygous for the P376L mutant form of SR-BI have abnormally high levels of HDL-C and increased risk of cardiovascular disease [6, 7]. Here, we summarize the structural, functional, and regulatory characteristics of SR-BI for a better understanding that SR-BI-mediated pathway may be a potential gateway for therapy and diagnosis in related diseases.
6.1 Structural Features of SR-BI
6.1.1 SR-BI Gene Structure
The SR-BI gene sequence was originally obtained from a Chinese hamster ovary (CHO) cell variant [8]. Mouse and rat SR-BI genes were found to be localized on chromosomes 5 and 12q15–16, respectively. The human SCARB1 gene is located on chromosome 12, with a full-length of 86 kb and consisting of 13 exons and 12 introns. The exon structure of SR-BI is very similar to that of CD36. Exon 1 comprises the 5’-UTR of cDNA and a short coding sequence, and exon 13 contains all the 3’-UTR. The length of human SR-BI mRNA is about 2.8 kb. Due to exon alternative splicing, SR-BI gene can be transcribed to produce multiple mRNA species. For example, Exons 2 and 3 of human SR-BI can be selectively spliced, resulting in deletion of 300 bp mRNA. The jump of exon 12 (129 bp) also generates a C-terminal variant mRNAs encoding SR-BII, in which three residues at C-terminus are shorter than SR-BI and the last 39 amino acids (467–506) are different.
At present, studies found a number of essential regulation elements in the SCARB1 promoter. The human SCARB1 promoter has two E-box consensus sequences (CANNTG) at the proximal (−160 bp) and the distal (−1145 bp) regions of the transcription start site, respectively, which bind to transcription factors with the basic helix-loop-helix (bHLH) motif and sterol regulatory element (SRE)–binding protein (SREBP). There are also two conserved CAAT elements that bind enhancer-binding protein (C/EBP) at −476 bp and −1046 bp regions upstream of the transcription starting site, and a proximal (−217 bp) binding site for steroidogenic factor-1 (SF-1). Meanwhile, a liver receptor homolog-1 (LRH-1) response element and a liver X receptor (LXR) response element are located in the promoter at −77 bp and − 1139 bp, respectively. Additionally, two SREs, four estrogen response element (ERE) half sites, seven simian virus 40 protein-1 (Sp1)–binding sites, and eleven E-boxes in proximal (−238 bp) and distal (−1958 bp) regions are found in the rat SR-BI promoter [9].
6.1.2 SR-BI Protein Structure and Localization
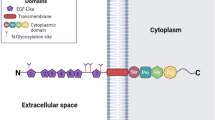
Human SR-BI protein was early named CLA-1 (CD36- and LIMPII-analogous-1) because of its structural similarity. UniProt predictions indicated that selective splicing of human SCARB1 mRNA might produce five SR-BI isomers. Isoform 1 was the first to be identified and named SR-BI, which contains 509 amino acid residues and has a predicted molecular weight (MW) of 56.9 kDa. In fact, due to glycosylated modification, it is usually detected with a relative MW of 82–83 kDa. SR-BI has a hairpin-like topological structure, the 409 amino acid residues in the middle segment form the extracellular domain, which is anchored to the plasma membrane by two transmembrane domains (aa 12–32 and 441–461), and is adjoined two short cytoplasmic domains at the N-termini (aa 1–11) and C-termini (aa 462–509), respectively [9].
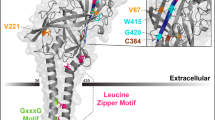
Structure-function analyses identify that the extracellular domain binding to HDL particle contains several Cys residues that form disulfide bridges and four proven N-glycosylation sites (Asn-102, Asn-108, Asn-173, Asn-330). Nearly all Cys (251, 280, 321–323, 274–329, 334, 384) are conserved in a variety of species, such as human, cow, pig, rabbit, hamster, and mouse, among which four Cys in the ectodomain (280, 321, 323, and 334) are important for structural integrity, the intracellular transport, HDL-binding, and lipid-transporter activity of SR-BI [10]. It has been reported that Cys-384 is involved in the binding site for block lipid transport 1 (BLT-1), an inhibitor of SR-BI synthesis [11]. Mutational studies in human SR-BI have shown that mutation of Asn-108 or Asn-173 causes an inability of SR-BI to locate to the plasma membrane and reduce lipid transfer from HDL to cells. In addition, the glycine dimerization motif (G15_G18_G25) exists in the N-terminal transmembrane domain of SR-BI, which is a necessity for normal homo-oligomerization and lipid-transporter activity [9]. The N-terminus and C-terminus of the extracellular domain of SR-BI, as well as Trp-415 combined with other Trp residues, are also critical for mediating lipoprotein binding and HDL-C selective uptake [12]. Moreover, membrane localization and SR-BI-mediating signaling are highly dependent on the VLQEAKL motif at the tail of the C-terminal cytoplasmic domain, which interacts with PDZK1, a multi-PDZ domain–containing adaptor protein [13]. It is worth noting that human and rodent SR-BI motifs share a number of common features, including three potential phosphorylation sites (Ser481, Ser4, and Ser477), a leucine zipper (aa 427–455), and a possible peroxisomal-targeting sequence (PTS-1) (aa507–509), which may be modulated by phosphorylation and by dimerization. Recently, it has been reported that the C-terminal cytosolic domain contains a Ser-496 that is phosphorylated by the kinase SIK-1, which modulates SR-BI efficiency for selective HDL-CE uptake [14]. Additionally, early studies found that mouse SR-BI could undergo fatty acylated at Cys462 and Cys470, which may be palmitoylation or myristoylation.
The cellular and tissue distribution of SR-BI protein is closely related to the function of lipid transport and metabolism. Early studies have been reported that SR-BI protein in human, mouse, and rat showed a similar tissue distribution. SR-BI is predominantly expressed in the liver and steroidogenic cells of the adrenal gland, ovary, and testis, little expression in testes, mammary gland, heart, and intestine, but no expression in the uterus, muscles, brain, kidney, spleen, and lungs. Later studies have shown that unlike in rodents, SR-BI distribution in humans is characterized by low expression in ovaries and high expression in placenta. Besides parenchymal hepatic cells and steroidogenic cells, SR-BI is also expressed in various cell types, including monocytes, macrophages, smooth muscle cells, endothelial cells, platelets, adipocytes, keratinocytes, and various epithelial cells [9].
At present, it has been recognized that SR-BI is located on the plasma membrane with a unique structure for mediating the flux of CE, free cholesterol, and related lipids into and out of cells [15]. Studies using electron microscopy indicated that SR-BI could preferentially locate in the microvillar domains to form hydrophobic channels for capturing various lipoproteins, including HDL. Of interest, about 60% of SR-BI is present in caveolae-rich membrane invaginations, which is usually associated with cellular signaling, potocytosis, and transcytosis. Some SR-BI also existed within the caveolin-poor membrane component [16].
6.2 Functional Features of SR-BI
Early experiments evidenced that scavenger receptors, as cell surface membrane proteins, could bind native lipoproteins and chemically modified lipoproteins, including oxidized LDL (ox-LDL), acetylated LDL (ac-LDL), phospholipids, phosphatidylserine, and many other types of ligands [17]. However, after the discovery of the SCARBI gene encoding the SR-BI in 1994, SR-BI was first identified by Krieger’s laboratory as a physiological HDL receptor, which could selectively remove cholesterol esters from HDL through high-affinity binding to HDL [1, 5]. Subsequently, a series of studies demonstrated that mice with SR-BI deficiency had abnormally high plasma levels of HDL-C, while the adrenal cholesterol content in mice decreased by 72%, and SR-BI knockout (SR-BI−/−) mice showed the increased diet-induced atherosclerosis susceptibility, which was paralleled by pathological phenomena such as reticulocytosis, anemia, thrombocytopenia, splenomegaly, and female infertility [3, 18]. Recently, the growing evidence reveals that SR-BI not only has a crucial role in HDL metabolism, steroid metabolism, and cholesterol homeostasis in the body, but also has an important impact on cell homeostasis, immune inflammation, and reproductive development [5].
6.2.1 SR-BI and HDL Metabolism
Numerous studies have identified that SR-BI plays an important role in cholesterol and steroid metabolism, especially in RCT and HDL metabolism. SR-BI regulates the first step and the last step of RCT by promoting cholesterol efflux from peripheral tissues and the selective delivery of HDL-CE to the liver. Through the selective uptake of HDL-CE, hepatic SR-BI mediates plasma cholesterol clearance for routing to the bile, thereby modulating HDL composition and function [5]. Because the lipoproteins as donors of CE for SR-BI-regulated selective CE uptake differ in characteristics, the processes for SR-BI selective uptake of CEs from lipid-rich lipoproteins have not been completely understood. Based on several lines of evidence obtained using CD36/SR-BI chimeras, SR-BI mutant (N173Q) and apoAI-deleted HDL particles, it seems that the initial binding of HDL to SR-BI and subsequent selective CE transport into cells can be divided into two independent steps: the first step involves the binding of cholesterol-rich lipoprotein to the extracellular domain of SR-BI, and the second step refers to SR-BI-mediated transfer of CEs from HDL to the plasma membrane and the release of the cholesterol-poor HDL particles back into the circulation. Among this process, SR-BI mediates selective uptake of HDL-CE without degradation of HDL protein via a holoparticle internalization and HDL recycling way, which differs from that of endocytosis through the caveolae and vesicle structure [5, 19]. Although the exact mechanism is not fully clear, a proposed model suggests that SR-BI forms a hydrophobic channel between HDL and the cell membrane, and then CEs move through the channels in a concentration-gradient manner.
Studies from mouse model have revealed that hepatocytes in SR-BI−/− mice completely lose the ability to selectively uptake CE from plasma HDL, and the secretion of biliary cholesterol is impaired due to RCT pathway blocking, resulting in a 2.5-fold increase in plasma levels of HDL-C and total cholesterol (TC), and accumulation of abnormally large HDL particles in mice. Especially, SR-BI−/− mice have a high ratio of plasma free cholesterol (FC) to TC increased by 2–3 times versus wild-type mice, which may be closely related to the decreased activity of lecithin-cholesterol acyltransferase (LCAT) [5, 20]. A series of studies demonstrated that compared with the relative amount of protein in HDL from wild-type mice, that in SR-BI−/−, HDL was reduced by about one quarter, and the capacity of functional component apoAI and paraoxonase l (PON1) in SR-BI−/− HDL was significantly decreased although the relative level of plasma apoAI in mice was not changed, suggesting that abnormally high levels of HDL-C and unesterified properties of HDL are closely related to the accumulation of dysfunctional HDL in SR-BI−/− mice [21, 22]. Other studies also demonstrated that overexpression of SR-BI in mouse livers with adenoviral vectors or transgenes decreased HDL-C levels and promoted secretion of cholesterol into the bile, resulting in reduced atherosclerosis [23]. Besides CEs, SR-BI also mediates the bidirectional flux of other lipids, including FC, phospholipids, and triglycerides (TGs) between HDL and cell plasma membranes.
Human SR-BI is also abundantly expressed in the liver for regulating RCT and HDL-C levels, but the plasma lipoprotein profile in humans is different from those in mice. In contrast with rodents, humans express cholesteryl ester transfer protein (CETP) that can transfer CE from HDL to VLDL for conversion to LDL, and LDL is then internalized via hepatic LDL receptors mediating route, providing an alternative pathway for HDL-CE delivery from the circulation to the liver. It had been considered that functional defects of human SR-BI do not cause serious consequences of abnormal HDL metabolism, evidenced by the study that about one-third of HDL-CE may be directly taken up by hepatic SR-BI in human subjects. However, a number of clinical studies have reported the association between SR-BI polymorphisms and alterations in plasma HDL-C levels, HDL functional changes, and cardiovascular diseases. Vergeer et al. identified that an SCARB1 missense mutation (889C > T) led to a functional mutation SR-BI (P297S), resulting in elevated HDL-C levels in carriers and a 56% decrease in HDL-CE uptake in hepatocytes [6]. The earliest identified genetic variation of SR-BI gene (SCARB1) is a single-nucleotide polymorphism (SNP) of exon 1 (rs4238001), which results in an SR-BI variant (G2S), the subjects who carried this variant show a significant negative correlation between SR-BI protein levels and HDL-C levels, and have an association with incident coronary heart disease (CHD) in multiethnic populations [24]. Subsequently, carriers of SR-BI missense mutations (S112F and T175A) also showed increased HDL-C levels and impaired function of SR-BI binding to HDL [25]. In 2016, large population-based studies reported by Zanoni et al. revealed that individuals who are heterozygous carriers harboring a P376L missense mutation in SR-BI are associated with an increased risk of CHD albeit increased plasma levels of HDL-C. The P376L variant disrupts posttranslational processing of SR-BI and abrogates selective uptake of HDL-C in hepatocyte-like cells [7]. Therefore, SR-BI is atheroprotective through promoting RCT and affecting HDL metabolism.
In addition to CE uptake from HDL, liver SR-BI may also have roles in the clearance of VLDL and atherogenic lipoprotein (a) [Lp(a)]. Studies identified that VLDL clearance was decreased by two-thirds in SR-BI−/− mice compared to wild-type mice, and that hepatic SR-BI binds VLDL. SR-BI internalizes VLDL through mechanisms involving heparin sulfate proteoglycans and lipoprotein lipase. Recently, the genetic role of SCARB1 gene in impacting plasma levels of apoB was found between rs4765615 and apoB, whereas rs11057844 demonstrated the strongest association with HDL-C level in a population-based investigation [26].
6.2.2 SR-BI Action in Vascular Cells
SR-BI, as a HDL receptor in the cell surface, plays different roles in a variety of cells. Besides SR-BI regulates selective HDL-CE uptake by high-affinity binding HDL/apoAI in hepatocytes and steroidogenic cells, SR-BI in other cells from the vasculature also mediates pleiotropic effects. For example, SR-BI is expressed in endothelial cells and macrophages where it functions to reduce atherosclerosis.
6.2.2.1 SR-BI in Endothelial Cells
SR-BI expressed by endothelial cells plays a crucial role in HDL-induced activation of endothelial nitric oxide synthase (eNOS) through HDL binding to SR-BI in endothelial cell caveolae, which is evidenced by the experiments wherein antibodies against SR-BI or apoAI completely abrogate HDL effects [27]. Frank et al. found that SR-BI expression markedly increased the stabilization of caveolin-1, an integral protein of caveolae, which contributes to the regulation of cellular cholesterol homeostasis and participates in transcytosis and cell signaling [16]. In endothelial cells, the role of SR-BI in the regulation of cholesterol efflux is involved in the activation of eNOS by HDL. It has been found that the interaction of SR-BI C-terminal PDZ-interacting domain with Src leads to Src phosphorylation, which further activates PI3K, Akt, and eNOS [28]. In vivo data from SR-BI−/− mice are consistent with increased endothelial cell apoptosis, suggesting that SR-BI has a protective effect against apoptosis of endothelial cells. Kimura et al. have demonstrated that SR-BI plays an important role in the inhibition of adhesion molecule expression (VCAM-1, ICAM-1) induced by HDL. A recent study demonstrates that SR-BI binds with apoAI/HDL and negatively regulates sphingosine 1-phosphate (S1P) and its receptor-mediated inflammation in endothelial cells through activating PI3K/Akt signaling pathway [29]. Taken together, these effects contribute to decreasing monocyte adhesion and recruitment into the intima, thus preventing the progression of atherosclerotic lesions.
In addition, SR-BI has an important role in HDL-induced endothelial cell migration and re-endothelialization. Seetharam et al. have determined that HDL promotes migration of endothelial cells in a nitric oxide (NO)–independent manner through SR-BI-mediated Rac GTPase activation. Rapid initial stimulation of lamellipodia formation by HDL via SR-BI, Rac, and Src kinases has been demonstrated. Moreover, impaired re-endothelialization was observed in SR-BI−/− mice. Therefore, HDL promotes the migration of endothelial cells through SR-BI-initiated signaling, and these mechanisms enhance endothelial monolayer integrity in vivo [30].
Studies have revealed that endothelial SR-BI is actively participated in transcytosis of HDL from the apical to basolateral side of a cell to stimulate cholesterol efflux from intimal macrophages and lymphatic vessels. However, SR-BI regulates the transmembrane effect of HDL in brain microvascular endothelial cells, which differs from the receptor-mediated lipid uptake and signaling pathways in other cells [31].
Additionally, SR-BI is also expressed in endothelial progenitor cells (EPCs) and plays a critical role in HDL-mediated EPC number and function. It has been shown that EPC migration and mobilization is mediated via NO and that enhanced NO production requires signaling via SR-BI [32].
6.2.2.2 SR-BI in Macrophages
SR-BI is also expressed in macrophages in tissues and atherosclerotic lesions. Macrophage SR-BI protects against atherosclerosis by affecting cellular cholesterol homeostasis, inflammation, apoptosis, and efferocytosis. Early evidence has shown that macrophage SR-BI acts as a multiple ligand receptor and regulates the uptake of native (HDL, LDL, and VLDL) and modified (oxLDL and acetylated LDL) lipoproteins, resulting in foam cell formation [17]. On the other hand, SR-BI interacts with HDL with high affinity to regulate intracellular FC efflux to HDL particles, which is a passive, nonenergy, concentration-dependent transport. The study from bone-marrow-derived macrophages (BMMs) revealed that SR-BI expression could enhance both cell cholesterol influx and efflux from HDL in the early stage of cell differentiation, but did not result in altered cellular cholesterol mass [33]. Therefore, SR-BI plays a unique role in promoting bidirectional cholesterol flux in nonlipid-loaded cells and facilitates net efflux from the cell when a favorable cholesterol gradient exists.
Linton et al. conducted BMT studies in the apoE−/− mouse model to examine the role of macrophage SR-BI in the pathogenesis of atherosclerosis in vivo. The results demonstrated that the loss of macrophage SR-BI expression increased atherosclerotic lesion area without alterations in plasma lipid and lipoprotein profiles. The removal of cholesterol from macrophages is one mechanism through which macrophage SR-BI could alleviate atherosclerosis [5, 34]. Some evidence suggests that macrophages could not restrain the excess uptake of cholesterol, and therefore, macrophages rely on an active cholesterol efflux process to sustain cholesterol homeostasis within the cell. Moreover, macrophage SR-BI acts similarly to ABCA1 and might account for the cholesterol efflux to HDL. SR-BI overexpression in mouse macrophages significantly enhanced the efflux of cholesterol to HDL, whereas inhibition of cholesterol transport by SR-BI decreases cholesterol efflux of macrophages [4, 33]. A recent report found that macrophages enriched in cholesterol from human carriers of the SCARB1 gene P297S variant displayed significantly decreased efflux of cholesterol to HDL, in comparison with macrophages from control subjects [6]. However, previous researches have demonstrated that ABCG1 and ABCA1 are the main mechanisms of cholesterol efflux from macrophages in mice, whereas SR-BI only makes a very limited contribution [35]. These inconsistent findings suggest that cholesterol flux between HDL and cellular SR-BI is modulated by the cholesterol status as well as phospholipid species of both the macrophages and HDL.
Additionally, SR-BI has been reported to mediate inflammatory responses through modulating the conversion of macrophages to inflammatory M1 versus anti-inflammatory M2 phenotype. SR-BI mediates cholesterol efflux from macrophages to HDL, contributing to the anti-inflammatory effect of HDL. Recent lines of evidence have showed that the anti-inflammatory effects of HDL in macrophages depend on SR-BI [36]. SR-BI knockdown or inhibition of SR-BI ligand binding abrogated the anti-inflammatory effects of HDL. When binding to its ligand HDL, SR-BI can activate Akt signaling and inhibit the inflammatory responses to LPS by dramatically decreasing NF-κB activation, resulting in elevated anti-inflammatory cytokines such as transforming growth factor-beta (TGF-β) and interleukin (IL)-10. Exposure of SR-BI-deficient macrophages to LPS showed increased expression of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6) and reduced expression of TGF-β [37]. Although SR-BI is a multiligand receptor that can bind to modified lipoproteins, oxidized phospholipids, and native HDL, it has been reported that inhibiting SR-BI expression or function potentiated glycated HDL induced generation of TNF-α, indicating an important anti-inflammatory effect for SR-BI. Meanwhile, SR-BI also regulates macrophage inflammation by reducing P38, and JNK signaling.
Recent lines of evidence have demonstrated that SR-BI also affects apoptosis susceptibility and efferocytosis in macrophages [5, 38]. Akt-mediated SR-BI activation plays a role in inhibiting macrophage apoptosis, since Akt promotes survival by activating Bad phosphorylation, which prevents Bad from inhibiting the anti-apoptotic factor BCL-XL. SR-BI interaction with HDL also inhibits cell apoptosis by suppressing ox-LDL-induced endoplasmic reticulum stress (ERS) [39]. In agreement with a role for SR-BI in inhibiting cell death in atherosclerotic lesions, Tao et al. demonstrated that transplantation with SR-BI-deficient bone marrow to either apoE−/− or LDLR−/− mice led to remarkably elevated numbers of lesion TUNEL-positive cells, compared to lesions in mice with control bone marrow transplantation. Macrophage SR-BI can also regulate the removal of apoptotic cells or efferocytosis via the Src/PI3K/Akt/Rac1 signaling pathway, leading to enhanced survival of phagocytes [38].
Furthermore, SR-BI appears to have a role in autophagy in macrophages [5]. Recent lines of evidence showed that in the setting of infection, impaired autophagy was observed in SR-BI−/− mouse macrophages, and the formation of subcellular membrane cholesterol domains participates in the SR-BI-induced autophagy. This raises the possibility that SR-BI mediates cytoplasmic cholesterol mobilization. Interestingly, it was found that SR-BI trafficked to lysosomes with Rab7, a key protein, which promotes autophagosome-lysosome fusion and localizes to lysosomes [40]. Lysosomes enriched in FC and engorged in neutral lipid are contained in SR-BI-deficient macrophages.
Additionally, SR-BI also exists in platelets and lymphocytes. As a functional HDL-binding receptor on platelets, the direct interaction between SR-BI and HDL is a necessity for maintaining normal platelet function. SR-BI deficiency leads to platelets’ hyperreactivity and high sensitivity of thrombosis [41]. Meanwhile, SR-BI−/− mice also exhibit an imbalance of lymphocyte homeostasis, such as the proliferation of spleen T and B lymphocytes, and led to an imbalance of interferon γ (INF-γ) and IL-4 in cells [42]. Taken together, further research applying SR-BI−/− mice will discover more about SR-BI’s role in the control of vascular cells and other cell lines.
6.2.3 SR-BI-Mediating Signaling Pathway of HDL
Accumulated studies have confirmed that SR-BI is involved in regulating HDL-dependent activation of signaling pathways, which may lead to its atheroprotective function. As previously mentioned, SR-BI on cell membrane interacts with PDZK1 (multi-PDZ-domain-containing adaptor protein) by its C terminus and directly participates in HDL-mediated signaling pathways. SR-BI is located in caveolae, and the alterations of cholesterol content and distribution in plasma membrane can affect the activities of the serine/threonine phosphatase PP2A and the tyrosine phosphatase HePTP, which stimulate Erk1/2 phosphorylation, influencing the resulting intracellular signaling governing processes in cells. Mutation of a highly conserved C-terminal transmembrane domain (SR-BI-Q445A) is incapable of HDL-induced signaling, suggesting that SR-BI serves as a plasma membrane sensor of cholesterol. In addition, SR-BI is required for HDL-induced angiogenesis in vivo and HDL activation of endothelial NO synthase and migration in cultured endothelial cells [15]. SR-BI also serves as a mediator in the uptake of bioactive lipids (phospholipids, sphingosine, sterol hormones, etc.) from HDL [27]. For example, SR-BI participates in eNOS activation by HDL-bound sphingosine 1-phosphate (S1P). Therefore, there are at least three predominant characteristics of SR-BI, which are necessary for its signal initiation: its ability to invoke cholesterol flux; the tail of its C-terminal intracellular domain that interacts with PDZK1; and its C-terminal transmembrane domain, which uniquely interacts with plasma membrane cholesterol [15].
It has been reported that HDL-induced activation of AMP-activated protein kinase (AMPK) is dependent on both S1P receptors and SR-BI through calcium−/calmodulin-dependent protein kinase kinase (CaMKK). In endothelial cells, serine-threonine kinase LKB1 may participate in SR-BI signaling. HDL-stimulated endothelial cell migration, activation of Akt and endothelial NO synthase, and suppression of monocyte adhesion and expression of adhesion molecules are dependent on AMPK activation [43].
In addition, SR-BI has also been reported to interact with downstream signaling molecules, for example, the nonreceptor tyrosine kinase, Src, which causes the activation of PI3K and further leads to the independent activation of both mitogen-activated protein kinase (MAPK) /ERK1/2 and Akt kinase (also known as protein kinase B, PKB) signaling pathways, resulting in phosphorylation of eNOS and NO production [44]. Several studies suggest that apoAI of HDL binding to SR-BI could also activate Src kinase–PI3K/ERK-Rac pathway, causing migration and subsequently repairing the endothelial injuries. In endothelial cells, SR-BI-mediated PI3K/Akt/eNOS signaling pathway participated in HDL-induced cyclooxygenase-2 (COX-2) expression and PGI2 release. Besides, HDL-C stimulates the proliferation of bone-derived mesenchymal stem cells by binding SR-BI and activation of PI3K/Akt, MAPK/ERK1/2 pathway [45]. Moreover, HDL augments angiogenesis in hypoxia through hypoxia-inducible factor 1α (HIF-1α)/vascular endothelial growth factor (VEGF) pathway via SR-BI-mediated PI3K/Akt signaling and modulation of the posttranslational regulators of HIF-1α [46].
6.3 Regulations of SR-BI
6.3.1 Regulating Factors of SR-BI
Numerous studies have established that SR-BI expression in hepatocytes can be regulated by various diets, drugs, hormones, glucose metabolism, and lipid metabolism [9, 47]. Studies in hamsters have shown that polyunsaturated fatty acids (PUFAs) in diet promote hepatic SR-BI expression and HDL-CE uptake, and combined treatment of obeticholic acid (OCA) and LXR agonist GW3965 increases the mRNA level and protein expression of SR-BI in the liver. Nevertheless, dietary myristic acid reduces SR-BI expression in hamster liver. It has been reported that the atherogenic diet of cholesterol loading may lead to posttranslational downregulation of hepatic SR-BI expression in mice and rats. Accumulating evidence in vivo and in vitro has indicated that hepatic expression of SR-BI is transcriptionally regulated by endogenous compounds such as insulin-like growth factors, glucose, lipopolysaccharide, leptin, vitamin A, and several synthetic compounds such as disodium ascorbyl phytostanol phosphate (FM-VP4), and isoflavones. All of these compounds are neither primary nor direct activators of transcriptional factors. Interestingly, some studies have identified new upregulators of SR-BI from 6000 microbial secondary metabolite crude extracts, including trichostatin A, pratensein, genistein, daidzein, etc.
Additionally, it has been reported that numerous drugs, such as thyromimetics, dexamethasone, fibrates, probucol, ezetimibe, tamoxifen, pioglitazone, and natural medicine rutaecarpine (RUT), quercetin, etc., affect SR-BI expression in different cells through different mechanisms [47, 48]. For example, RUT triggers the promoters of CLA-1 genes, upregulating SR-BI to inhibit atherosclerosis in apoE−/− mice.
6.3.2 Regulating Mechanism of SR-BI
A series of studies have revealed that many nuclear transcription factors, such as liver receptor homolog-1 (LRH-1), liver X receptors (LXRs), steroidogenic factor-1 (SF-1), sterol-regulatory-element-binding proteins (SREBPs), and peroxisome-proliferator-activated receptors (PPARs), affect SR-BI gene transcription. Several consensus sequences for nuclear receptor binding have been determined in the flanking promoter region of the human SCARB1 gene [5, 9, 47]. For example, adrenocorticotropic hormone (ACTH) has been found to upregulate SR-BI expression in steroidogenic tissues of both human subjects and rats. The suppression of ACTH by dexamethasone, a synthetic corticosteroid, decreased SR-BI levels. Human SR-BI expression can be stimulated by steroidogenic hormones through the cellular cAMP-dependent regulation, and SF-1 is one of the primary transcription factors involved in this pathway via binding to its consensus sequence in the SCARB1 promoter. In hepatocytes, LRH-1 binds to a proximal response element on the human SR-BI promoter in an overlapping manner with SF-1 and activates the SR-BI promoter. Overexpression of LRH-1 induces SR-BI gene expression, which may be related to histone H3 acetylation on the promoter. Moreover, LRH-1 also regulates CETP, which participates in the remodeling of HDL particles and RCT [47, 49].
LXRs, members of the nuclear hormone receptor superfamily, participates in controlling hepatic SR-BI expression [9]. The oxysterols induce activation of both LXRα and LXRβ and increase the expression of SR-BI both in human and murine cell lines. Since LXRα is upregulated by TGF-β1, TGF-β1 significantly increases SR-BI expression in a dose- and time-dependent manner at the transcriptional and translational levels. PPARs, a family of ligand-activated transcription factors, such as PPARα and PPARγ, heterodimerize with retinoid X receptor (RXR) to bind to distal PPARE on the SR-BI promoter and regulate human SR-BI gene expression. It has been found that macrophage SR-BI expression can be promoted by activators of PPARs [50]. Some PPAR ligands, including fatty acids, glitazones, and fibrates, regulate SR-BI expression at the liver level. Mitogen-activated protein kinase (MAPK) Erk1/2 is involved in the regulation of nuclear receptor activity, including PPARs and LXR, to alter the capacity of the cell to export cholesterol, while inhibition of Mek1/2 enhances PPARα-dependent degradation of SR-BI in hepatocytes [51]. It has been reported that SREBP-1 modulates human SCARB1 gene expression in response to altered levels of intracellular sterols. Cholesterol depletion can trigger an SREBP1a-regulated induction of SR-BI gene transcription in HepG2 cells. Nonetheless, SR-BI mRNA and protein expressions are normal in livers obtained from a mouse model that lacks mature nuclear SREBPs due to gene knockout of the SREBP cleavage-activating protein (SCAP) and from SREBP transgenic mice, indicating that the SR-BI gene in the liver is not under transcriptional regulation by SREBPs [47].
Studies have shown that estrogen can bind to three different estrogen response elements (EREs) on the SR-BI promoter through estrogen receptor α/β to regulate SR-BI gene transcription. In endothelial cells, 17β-estradiol (E2) increased the mRNA expression of the human SR-BI gene as well as the activity of the SR-BI promoter in a protein kinase C (PKC)–dependent manner [52]. Interestingly, rats supplemented with estrogens and HepG2 treated with 17β-estradiol showed a noted reduction in SR-BI expression combined with an increase of SR-BII. Meanwhile, treatment with the synthetic estrogen 17α-ethinylestradiol in rats stimulates a reduction in hepatocyte SR-BI expression combined with an increase in Kupffer cell SR-BI expression.
Another nuclear receptor in association with the regulation of hepatic SR-BI expression is Farnesoid X Receptor (FXR) [53]. The mRNA level of SR-BI gene was reduced in the liver of FXR-deficient (FXR−/−) mice. When treating mice with a diet containing 0.4% of the FXR agonist cholic acid, elevated mRNA and protein levels of hepatic SR-BI were observed in the wild-type but not in the FXR−/− mice, suggesting that bile acids could upregulate SR-BI gene expression via FXRs. A recent study found a novel pathway in which FXR activation upregulated SR-BI expression in association with alterations in hepatic regulatory factors such as hepatocyte nuclear factor 4α (HNF4α) and p-JNK. It is likely that the contradiction of these results may be due to the activation of other regulatory pathways of gene expression besides selective agonists of FXR. In addition, pregnane X receptor (PXR), known as a xenobiotic receptor in human subjects, was shown to reduce SR-BI mRNA level in response to the PXR agonists, lithocholic acid, rifampicin, or pregnenolone 16α-carbonitrile, although no consensus sequences for the binding of PXR have been identified in the SR-BI promoter [9, 47].
Additionally, Krüppel-like factor 4 (KLF4), a conserved zinc finger transcription factor, can be induced by HDL and upregulate SR-BI expression, leading to the binding to a putative KLF4-binding element (−342/−329 bp) on SR-BI promoter in macrophages [54]. Another study was reported that Sp1 and Sp3 are also essential transcriptional factors for SR-BI gene transcription through binding GC box at the proximal promoter.
Of note, the inhibitors, which act on the promoter of SR-BI, have been found. Yin Yang protein-1 (YY1), a zinc finger transcription factor that can play both positive and negative regulatory roles, decreases SREBP-regulated induction of SR-BI transcription by directly binding to two YY1-binding sites in the promoter as well as to SREBPs [55]. The sex adrenal hypoplasia congenital critical region on the X-chromosome, gene-1 (DAX-1), an adrenal-specific inhibitor and orphan nuclear receptor (NR0B1), can bind to SREBP-1α and inhibit its binding to SREs. A p21-activated kinase-1(PAK-1) interacted with small GTPases and regulates lipopolysaccharide (LPS)-induced decrease of the activity of human SR-BI promoter in macrophages.
Recent studies revealed that small noncoding RNAs, including microRNAs (miRNAs, miRs), are implicated in the posttranscriptional regulation of SR-BI gene expression [56]. Analysis of the SR-BI 3′ UTR sequence identified potential binding sites for a number of miRs. When either miR-125a or miR-455 was overexpressed, the level of SR-BI was reduced, leading to a decrease of both HDL-C uptake and steroidogenesis. Both miR-125a and miR-455 as expressed in steroidogenic cells were downregulated by ACTH and cAMP treatment. Studies demonstrated that miR-24, miR-96, miR-185, and miR-223 could bind the 3’-UTR of SR-BI and negatively regulate the expression level of SR-BI in the liver and macrophages, thereby reducing HDL uptake and selective lipid uptake [57, 58]. Therefore, these observations suggest that certain miRs may have potential therapeutic value for atherosclerosis via targeting SR-BI and HDL.
Recent studies have found that in the setting of infection, autophagy of macrophages is impaired in SR-BI−/− mice, and the SR-BI-induced autophagy involved the formation of subcellular membrane cholesterol domains. These findings raise the possibility that SR-BI mediates cytoplasmic cholesterol mobilization for availability to ABCA1 for efflux. Interestingly, it was found that SR-BI could traffic to lysosomes with Rab7, a protein, which is crucial in promoting the fusion of autophagosomes with lysosomes, and localize to macrophage lysosomeshepatic SR-BI protein levels. The stability, localization, and function of SR-BI in the hepatocyte plasma membrane are primarily controlled by PDZK1 containing four PDZ (PDZ1 to PDZ4) domains [59]. PDZK1 is expressed in the liver, gut, and kidney, but not in steroidogenic tissues. Small PDZK1-associated protein (SPAP) overexpression downregulates PDZK1 in a liver-specific fashion, resulting in the subsequent downregulation of hepatic SR-BI. In PDZK1-null mice, protein levels of hepatic SR-BI are markedly decreased by 95%, whereas mRNA levels remain unaltered, leading to an increase in plasma total cholesterol carried in abnormally large HDL particles [13]. The PDZK1 homologs can serve as physiological translation/posttranslational regulators of the functional expression of SR-BI. In addition, it was shown that posttranslationally activated PI3K/Akt promotes hepatic SR-BI function through mediating the subcellular localization of SR-BI. The Ras/MEK/ERK signaling cascade regulates the protein level of hepatocyte SR-BI, which is associated with PPARα-induced degradation pathways [51].
Other studies have shown that the synonymous variant of rs5888 (C > T), which is frequently found in humans, can alter the RNA secondary structure of SR-BI, and the translation of SR-BI protein in macrophages is significantly reduced, which may be related to SR-BI protein expression and function [60].
6.4 SR-BI and Atherosclerosis
The increasing evidence involving the manipulation of SR-BI expression in mouse models, either using SR-BI−/− mice or using adenovirus-mediated or transgenic hepatic overexpression, indicates that SR-BI expression protects against atherosclerosis [5]. SR-BI−/− mice fed with a Western diet for 20 weeks showed a significant increase of dysfunctional HDL, which contributes to an increased susceptibility to atherosclerosis. Deletion of SR-BI in apoE−/− mice leads to accelerated atherosclerosis causing coronary artery occlusion, myocardial infarction, and premature death at the age of 5–8 weeks. When fed with a modified Western diet, SR-BI/LDLR double knockout mice also shown abnormal profiles of lipoproteins resulting in severe coronary atherosclerosis, myocardial infarction, and reduced survival rate. On the contrary, transgenic mice overexpressing hepatic SR-BI have accelerated clearance of HDL-C, enhanced selective uptake of hepatic cholesterol, and increased content of biliary cholesterol, which reduces atherosclerosis. Meanwhile, studies employing the bone marrow transplantation showed that macrophage SR-BI inactivation promotes the development of atherosclerotic lesion in apoE−/− mouse model. Interestingly, SR-BI deficiency in bone marrow-derived macrophages inhibited the initial formation of atherosclerotic lesions in LDLR−/− mice, but stimulated the atherogenic process in later stages in LDLR−/− mice, suggesting that the double effects of macrophage SR-BI depend on multiple roles in the different stages of atherosclerosis.
As previously mentioned, SR-BI protects against atherosclerosis via one or more of the following mechanisms [5, 9, 19, 61]: hepatic SR-BI is the main effective way to inhibit atherosclerosis by selective uptake of HDL-CE and promoting RCT pathway, stimulating cholesterol secretion from bile. Additionally, hepatic SR-BI is also involved in the clearance of apoB-containing lipoprotein; the anti-inflammatory effect of HDL depends on the expression of SR-BI in cells, which can be indirectly regulated by lipid transport activity or directly by SR-BI as a receptor for cellular inflammatory signaling; SR-BI directly affects the function and status of cells in vessel wall by minimizing the formation of foam cells and by modulating signaling pathways implicated in efferocytosis, apoptosis, cell migration, and inflammation. It mediates the bidirectional flux of unesterified cholesterol between cells and HDL, and modulates the inflammatory response in macrophages. In endothelial cells, SR-BI plays a critical role in cell migration and vascular protection by mediating HDL-dependent eNOS activation and PI3K/Akt signaling. Moreover, SR-BI promotes the release and migration of endothelial progenitor cells. SR-BI modulates lymphocyte homeostasis via regulating cell proliferation, production of INF-γ and IL-4 cytokine, and HDL function [42]. The direct interaction between SR-BI and HDL on platelets is necessary for maintaining its normal function; otherwise, it will lead to platelet hyperreactivity and high sensitivity of thrombosis [41]. Taken together, SR-BI serves as a multifunctional receptor against atherosclerosis, making it a potential therapeutic target.
6.5 SR-BI and Cancer
Given that cholesterol regulates essential signaling pathways involved in cell proliferation, migration, and survival, accumulated evidence has shown that in addition to mediating the transfer of cholesterol between HDL and healthy cells, SR-BI also promotes the selective cholesterol uptake by malignant cells. Interestingly, compared to normal tissue, SR-BI expression is increased in multiple cancers, such as Leydig cell tumors, breast cancer, prostate cancer, metastatic melanoma, nasopharyngeal carcinoma (NPC), and clear cell renal cell carcinoma (ccRCC) [62]. Therefore, recent lines of evidence have focused on the role of SR-BI in the progression of cancers.
Studies have shown that overexpression of SR-BI can stimulate HDL-induced proliferation of the breast cancer cell line MCF-7 through the PI3K/AP-1 pathway, and a mutant of SR-BI could inhibit proliferation of human breast cancer cells. Meanwhile, higher expression of SR-BI in breast cancer tissue is associated with an increased disease aggressiveness and mortality of patients. Knockdown of SR-BI reduces HDL-mediated activation of MAPK and PI3K/Akt pathways in the two human breast cancer cell lines, MCF7 and MDA-MB-231. Twiddy et al. showed that SR-BI knockdown decreases viability and prostate-specific antigen (PSA) secretion of prostate cancer cells [63]. Research by Kinslechner et al. revealed that SR-BI knockdown perturbs the metastasis-associated epithelial-to-mesenchymal transition (EMT) phenotype, in association with reduced invasion and migration of melanoma cells and decreased xenograft tumor growth [64]. In recent studies, it has been discovered that SR-BI is overexpressed in all investigated NPC cell lines and 75% of NPC biopsies, and has a great effect on cell motility but does not affect cell proliferation [65]. Of note, high expression of SR-BI was closely associated with aggressive features of ccRCC and could predict a poor clinical outcome. Using specific siRNA to inhibit SR-BI in ccRCC cell lines in vitro successfully impaired the growth, colony formation, migration, and invasion, and HDL-C uptake. Furthermore, SR-BI-targeting siRNA was also reported to yield potent in vivo effects [66]. Taken together, the above promising preliminary studies suggest that SR-B1 may serve as a potential biomarker for cancer diagnosis and therapeutic target in the pathogenesis of cancer [67].
6.6 Conclusions
It has been recognized that SR-BI is a multifunctional HDL receptor, whose prominent function is to play a key role in RCT by regulating selective uptake of HDL-CE in the liver. SR-BI also performs many other important functions involved in the atheroprotective effect: hepatic SR-BI plays a key role in the elimination of remnant lipoproteins and atherogenic Lp(a). In endothelial cells and macrophages, SR-BI minimizes foam cell formation through regulating cellular cholesterol flux and participates in HDL-initiated signaling involved in inflammation, cell survival, and efferocytosis. Recent studies using genomic analyses suggest that the SNPs of human SCARBI gene could lead to loss-of-function variants of SR-BI and may be involved in the occurrence of abnormal circulating HDL-C and the increased risk of atherosclerotic cardiovascular disease. It is becoming increasingly clear that a bright future for SR-BI as a therapeutic target can be foreseen not only in cardiovascular disease, but certainly also in inflammatory diseases as well as in cancer. However, it remains obscure whether the HDL derived from human carriers of loss-of-function SCARBI variants is dysfunctional, and whether SR-BI mutation is an independent risk factor for atherosclerosis in general population. Secondly, how the key domain of SR-BI transmembrane structure regulates the selective transport of cholesterol and signal transduction is still unknown. Next, very limited information is currently available about the mechanisms of SR-BI expression and its regulation in vivo. The direct regulatory elements and key transcription factors of SCARB1 gene still need to be clarified. Finally, considering a supposed negative effect in cancer versus a positive effect in inflammation and cardiovascular disease, future efforts should focus on increasing the functional efficiency of SR-BI using genetic or pharmacological approaches, and consequently assist the prevention and treatment of cancer/atherosclerotic cardiovascular disease.
References
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271(5248):518–520
Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M (1997) Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 387(6631):414–417
Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M (1997) A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A 94(23):12610–12615
Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC et al (1997) Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem 272(34):20982–20985
Linton MF, Tao H, Linton EF, Yancey PG (2017) SR-BI: a multifunctional receptor in cholesterol homeostasis and atherosclerosis. Trends Endocrinol Metab 28(6):461–472
Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK et al (2011) Genetic variant of the scavenger receptor BI in humans. N Engl J Med 364(2):136–145
Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M et al (2016) Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351(6278):1166–1171
Acton SL, Scherer PE, Lodish HF, Krieger M (1994) Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem 269(33):21003–21009
Shen WJ, Azhar S, Kraemer FB (2018) SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol 80:95–116
Hu J, Zhang Z, Shen WJ, Nomoto A, Azhar S (2011) Differential roles of cysteine residues in the cellular trafficking, dimerization, and function of the high-density lipoprotein receptor, SR-BI. Biochemistry 50(50):10860–10875
Yu M, Romer KA, Nieland TJ, Xu S, Saenz-Vash V, Penman M et al (2011) Exoplasmic cysteine Cys384 of the HDL receptor SR-BI is critical for its sensitivity to a small-molecule inhibitor and normal lipid transport activity. Proc Natl Acad Sci U S A 108(30):12243–12248
Holme RL, Miller JJ, Nicholson K, Sahoo D (2016) Tryptophan 415 is critical for the cholesterol transport functions of scavenger receptor BI. Biochemistry 55(1):103–113
Trigatti BL (2017) SR-B1 and PDZK1: partners in HDL regulation. Curr Opin Lipidol 28(2):201–208
Hu Z, Hu J, Shen WJ, Kraemer FB, Azhar S (2015) A novel role of salt-inducible kinase 1 (SIK1) in the posttranslational regulation of scavenger receptor class B type 1 activity. Biochemistry 54(46):6917–6930
Saddar S, Carriere V, Lee WR, Tanigaki K, Yuhanna IS, Parathath S et al (2013) Scavenger receptor class B type I is a plasma membrane cholesterol sensor. Circ Res 112(1):140–151
Frank PG, Marcel YL, Connelly MA, Lublin DM, Franklin V, Williams DL et al (2002) Stabilization of caveolin-1 by cellular cholesterol and scavenger receptor class B type I. Biochemistry 41(39):11931–11940
Krieger M (2001) Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest 108(6):793–797
Dole VS, Matuskova J, Vasile E, Yesilaltay A, Bergmeier W, Bernimoulin M et al (2008) Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 28(6):1111–1116
Mineo C, Shaul PW (2012) Functions of scavenger receptor class B, type I in atherosclerosis. Curr Opin Lipidol 23(5):487–493
Lee JY, Badeau RM, Mulya A, Boudyguina E, Gebre AK, Smith TL et al (2007) Functional LCAT deficiency in human apolipoprotein A-I transgenic, SR-BI knockout mice. J Lipid Res 48(5):1052–1061
Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK et al (2007) Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol 27(11):2413–2419
Cao J, Xu Y, Li F, Shang L, Fan D, Yu H (2018) Protein markers of dysfunctional HDL in scavenger receptor class B type I deficient mice. J Transl Med 16(1):155
Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ (2005) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 115(10):2870–2874
Manichaikul A, Wang XQ, Musani SK, Herrington DM, Post WS, Wilson JG et al (2015) Association of the lipoprotein receptor SCARB1 common missense variant rs4238001 with incident coronary heart disease. PLoS One 10(5):e0125497
Chadwick AC, Sahoo D (2012) Functional characterization of newly-discovered mutations in human SR-BI. PLoS One 7(9):e45660
Niemsiri V, Wang X, Pirim D, Radwan ZH, Hokanson JE, Hamman RF et al (2014) Impact of genetic variants in human scavenger receptor class B type I (SCARB1) on plasma lipid traits. Circ Cardiovasc Genet 7(6):838–847
Al-Jarallah A, Trigatti BL (2010) A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochim Biophys Acta 1801(12):1239–1248
Mineo C, Shaul PW (2013) Regulation of signal transduction by HDL. J Lipid Res 54(9):2315–2324
Ren K, Lu YJ, Mo ZC, Liu X, Tang ZL, Jiang Y et al (2017) ApoA-I/SR-BI modulates S1P/S1PR2-mediated inflammation through the PI3K/Akt signaling pathway in HUVECs. J Physiol Biochem 73(2):287–296
Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL et al (2006) High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res 98(1):63–72
Fung KY, Wang C, Nyegaard S, Heit B, Fairn GD, Lee WL (2017) SR-BI mediated transcytosis of HDL in brain microvascular endothelial cells is independent of Caveolin, Clathrin, and PDZK1. Front Physiol 8:841
Zhang Y, Liao B, Li M, Cheng M, Fu Y, Liu Q et al (2016) Shear stress regulates endothelial cell function through SRB1-eNOS signaling pathway. Cardiovasc Ther 34(5):308–313
Ji A, Meyer JM, Cai L, Akinmusire A, de Beer MC, Webb NR et al (2011) Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis 217(1):106–112
Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S et al (2003) Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 108(18):2258–2263
Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH et al (2007) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117(8):2216–2224
Song GJ, Kim SM, Park KH, Kim J, Choi I, Cho KH (2015) SR-BI mediates high density lipoprotein (HDL)-induced anti-inflammatory effect in macrophages. Biochem Biophys Res Commun 457(1):112–118
Cai L, Wang Z, Meyer JM, Ji A, van der Westhuyzen DR (2012) Macrophage SR-BI regulates LPS-induced pro-inflammatory signaling in mice and isolated macrophages. J Lipid Res 53(8):1472–1481
Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L et al (2015) Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res 56(8):1449–1460
Song G, Wu X, Zhang P, Yu Y, Yang M, Jiao P et al (2016) High-density lipoprotein inhibits ox-LDL-induced adipokine secretion by upregulating SR-BI expression and suppressing ER stress pathway. Sci Rep 6:30889
Pfeiler S, Khandagale AB, Magenau A, Nichols M, Heijnen HF, Rinninger F et al (2016) Distinct surveillance pathway for immunopathology during acute infection via autophagy and SR-BI. Sci Rep 6:34440
Ma Y, Ashraf MZ, Podrez EA (2010) Scavenger receptor BI modulates platelet reactivity and thrombosis in dyslipidemia. Blood 116(11):1932–1941
Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z et al (2011) Deficiency of scavenger receptor BI leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol 31(11):2543–2551
Kimura T, Tomura H, Sato K, Ito M, Matsuoka I, Im DS et al (2010) Mechanism and role of high density lipoprotein-induced activation of AMP-activated protein kinase in endothelial cells. J Biol Chem 285(7):4387–4397
Zhang QH, Zu XY, Cao RX, Liu JH, Mo ZC, Zeng Y et al (2012) An involvement of SR-B1 mediated PI3K-Akt-eNOS signaling in HDL-induced cyclooxygenase 2 expression and prostacyclin production in endothelial cells. Biochem Biophys Res Commun 420(1):17–23
Xu J, Qian J, Xie X, Lin L, Ma J, Huang Z et al (2012) High density lipoprotein cholesterol promotes the proliferation of bone-derived mesenchymal stem cells via binding scavenger receptor-B type I and activation of PI3K/Akt, MAPK/ERK1/2 pathways. Mol Cell Biochem 371(1–2):55–64
Tan JT, Prosser HC, Vanags LZ, Monger SA, Ng MK, Bursill CA (2014) High-density lipoproteins augment hypoxia-induced angiogenesis via regulation of posttranslational modulation of hypoxia-inducible factor 1alpha. FASEB J 28(1):206–217
Leiva A, Verdejo H, Benitez ML, Martinez A, Busso D, Rigotti A (2011) Mechanisms regulating hepatic SR-BI expression and their impact on HDL metabolism. Atherosclerosis 217(2):299–307
Shen D, Li H, Zhou R, Liu MJ, Yu H, Wu DF (2018) Pioglitazone attenuates aging-related disorders in aged apolipoprotein E deficient mice. Exp Gerontol 102:101–108
Malerod L, Sporstol M, Juvet LK, Mousavi SA, Gjoen T, Berg T et al (2005) Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1. Biochem Biophys Res Commun 336(4):1096–1105
Lopez D, McLean MP (2006) Activation of the rat scavenger receptor class B type I gene by PPARalpha. Mol Cell Endocrinol 251(1–2):67–77
Wood P, Mulay V, Darabi M, Chan KC, Heeren J, Pol A et al (2011) Ras/mitogen-activated protein kinase (MAPK) signaling modulates protein stability and cell surface expression of scavenger receptor SR-BI. J Biol Chem 286(26):23077–23092
Fukata Y, Yu X, Imachi H, Nishiuchi T, Lyu J, Seo K et al (2014) 17beta-estradiol regulates scavenger receptor class BI gene expression via protein kinase C in vascular endothelial cells. Endocrine 46(3):644–650
Li G, Thomas AM, Williams JA, Kong B, Liu J, Inaba Y et al (2012) Farnesoid X receptor induces murine scavenger receptor class B type I via intron binding. PLoS One 7(4):e35895
Yang T, Chen C, Zhang B, Huang H, Wu G, Wen J et al (2010) Induction of Kruppel-like factor 4 by high-density lipoproteins promotes the expression of scavenger receptor class B type I. FEBS J 277(18):3780–3788
Lopez D, McLean MP (1999) Sterol regulatory element-binding protein-1a binds to cis elements in the promoter of the rat high density lipoprotein receptor SR-BI gene. Endocrinology 140(12):5669–5681
Dai Y, Condorelli G, Mehta JL (2016) Scavenger receptors and non-coding RNAs: relevance in atherogenesis. Cardiovasc Res 109(1):24–33
Wang L, Jia XJ, Jiang HJ, Du Y, Yang F, Si SY et al (2013) MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol Cell Biol 33(10):1956–1964
Ren K, Zhu X, Zheng Z, Mo ZC, Peng XS, Zeng YZ et al (2018) MicroRNA-24 aggravates atherosclerosis by inhibiting selective lipid uptake from HDL cholesterol via the post-transcriptional repression of scavenger receptor class B type I. Atherosclerosis 270:57–67
Tsukamoto K, Wales TE, Daniels K, Pal R, Sheng R, Cho W et al (2013) Noncanonical role of the PDZ4 domain of the adaptor protein PDZK1 in the regulation of the hepatic high density lipoprotein receptor scavenger receptor class B, type I (SR-BI). J Biol Chem 288(27):19845–19860
Constantineau J, Greason E, West M, Filbin M, Kieft JS, Carletti MZ et al (2010) A synonymous variant in scavenger receptor, class B, type I gene is associated with lower SR-BI protein expression and function. Atherosclerosis 210(1):177–182
Hoekstra M (2017) SR-BI as target in atherosclerosis and cardiovascular disease - a comprehensive appraisal of the cellular functions of SR-BI in physiology and disease. Atherosclerosis 258:153–161
Rajora MA, Zheng G (2016) Targeting SR-BI for cancer diagnostics, imaging and therapy. Front Pharmacol 7:326
Gutierrez-Pajares JL, Ben Hassen C, Chevalier S, Frank PG (2016) SR-BI: linking cholesterol and lipoprotein metabolism with breast and prostate cancer. Front Pharmacol 7:338
Kinslechner K, Schorghofer D, Schutz B, Vallianou M, Wingelhofer B, Mikulits W et al (2018) Malignant phenotypes in metastatic melanoma are governed by SR-BI and its association with glycosylation and STAT5 activation. Mol Cancer Res 16(1):135–146
Zheng Y, Liu Y, Jin H, Pan S, Qian Y, Huang C et al (2013) Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics 3(7):477–486
Xu GH, Lou N, Shi HC, Xu YC, Ruan HL, Xiao W et al (2018) Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer 18(1):88
Mooberry LK, Sabnis NA, Panchoo M, Nagarajan B, Lacko AG (2016) Targeting the SR-B1 receptor as a gateway for cancer therapy and imaging. Front Pharmacol 7:466
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yu, H. (2022). HDL and Scavenger Receptor Class B Type I (SRBI). In: Zheng, L. (eds) HDL Metabolism and Diseases. Advances in Experimental Medicine and Biology, vol 1377. Springer, Singapore. https://doi.org/10.1007/978-981-19-1592-5_6
Download citation
DOI: https://doi.org/10.1007/978-981-19-1592-5_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-1591-8
Online ISBN: 978-981-19-1592-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)