
Abstract
Non-alcoholic fatty liver disease (NAFLD) is a metabolic disorder manifested in hepatic fat accumulation (hepatic steatosis) in the absence of heavy alcohol use. NAFLD consists of four major stages ranging from simple steatosis or non-alcoholic fatty liver (NAFL) to more advanced stages, non-alcoholic steatohepatitis (NASH), fibrosis, and cirrhosis. NFLAD may further advance to hepatocellular carcinoma (HCC). Primary causes of NAFLD are obesity and obesity-associated insulin resistance (IR). As a result of the obesity pandemic, NAFLD has become one of the most common liver disorders worldwide and both the incidence and mortality rate of HCC that develops from NAFLD are increasing steadily. As treatment options are not available for advanced NAFLD, a better understanding of the molecular mechanisms for NAFLD development and progression is urgently needed. Emerging evidence suggests that dysregulation of the metabolism of sphingolipids contributes to development and progression of NAFLD and NAFLD-associated HCC. The present chapter summarizes roles of bioactive sphingolipids, ceramides, sphingosine, and sphingosine-1-phosphate (S1P) and their metabolizing enzymes in NAFLD and HCC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Ceramide
- Non-alcoholic fatty liver disease (NAFLD)
- Non-alcoholic steatohepatitis (NASH)
- Hepatocellular carcinoma (HCC)
- Insulin resistance
11.1 Overview of NAFLD and HCC
Non-alcoholic fatty liver disease (NAFLD) is a metabolic disorder manifested in hepatic fat accumulation (hepatic steatosis) in the absence of heavy alcohol consumption in past medical history. NAFLD exhibits a spectrum of conditions ranging from simple steatosis or non-alcoholic fatty liver (NAFLD) to non-alcoholic steatohepatitis (NASH) or cirrhosis, which may further advance to hepatocellular carcinoma (HCC) [1]. Primary causes of NAFLD are obesity and obesity-associated insulin resistance (IR). As a result of the obesity pandemic, NAFLD has become one of the most common liver disorders worldwide and currently affects around 25% of the general population of Western countries [2, 3]. NASH is expected to become the leading cause of liver transplant [3]. However, NASH currently has no effective treatment apart from lifestyle interventions [4]. Therefore, there are unmet medical needs for understanding the molecular and cellular mechanisms for the progression of NAFLD to NASH and cirrhosis and developing novel approaches to treating advanced NAFLD based on an understanding of the pathogenesis of this disease.
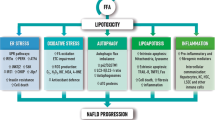
Obesity [2] and IR [5] have been suggested to be major risk factors for NAFLD. Obesity and IR result in accumulation of free fatty acids (FFAs) in the liver [2]. Hepatic FFAs are metabolized into many lipotoxic species that cause various forms of hepatocellular stress, including oxidative stress, endoplasmic reticulum (ER) stress, and cell death [1], thus leading to liver damage, inflammation, and fibrosis, which are the hallmark comorbidities of NASH [6]. The ER plays central roles in calcium ion storage, lipid biosynthesis, and protein sorting and processing [7]. Accumulation of unfolded proteins in the ER lumen or impairment of the ER membrane integrity results in ER stress which in turn activates a series of signaling processes called the unfolded protein response (UPR) [8]. Transient activation of the UPR restores the homeostasis of the ER while chronic UPR activation due to persistence of ER stress results in cell death and cellular injury. Chronic UPR activation has been observed in liver and/or adipose tissue of dietary and genetic murine models of obesity, and in human obesity and NAFLD [8]. The chronic responses to cell death and cellular injury lead to chronic hepatocyte turnover, the recruitment of immune cells, and activation of hepatic stellate cells (HSCs), thus contributing to the development of liver fibrosis and cirrhosis [9,10,11]. It has become evident that, besides apoptosis, necroptosis is a highly relevant form of programmed cell death (PCD) in the liver [9, 12, 13]. In addition to PCD, increasing studies have implicated autophagy in the pathogenesis of NAFLD [14]. Autophagy is a lysosomal degradative pathway that promotes cell survival by supplying energy under the stress of energy crisis or by removing damaged organelles and proteins after cellular injury [15]. An initial study suggests that autophagy mediates the breakdown of lipids in hepatocytes [16]. Subsequent studies have implicated autophagy in regulating several pathological effects of NAFLD, such as insulin sensitivity, hepatocellular injury, innate immunity, fibrosis, and liver carcinogenesis [14].
However, lipotoxic species that mediate ER stress, cell death, and autophagy in the context of NAFLD have not been completely identified. Increasing studies have implicated bioactive sphingolipids, such as ceramides, sphingosine, and sphingosine-1-phosphate (S1P), in both initiation of NAFLD and its progression to HCC.
11.2 Metabolism of Ceramides in NAFLD
11.2.1 Dysregulation of Metabolism of Sphingolipids in the Liver of Patients with NAFLD
Increasing studies have demonstrated that ceramides, a subclass of sphingolipids, are potential lipotoxic mediators of saturated FFAs in driving NAFLD onset and progression [17, 18] . Palmitic acid, the most abundant hepatic FFA in NAFLD patients, is an essential precursor of ceramides and other sphingolipids [19, 20] (Scheme 11.1). Palmitic acid is activated to form palmitoyl-CoA, which is condensed with serine into keto-dihydrosphingosine by the action of serine palmitoyltransferase (SPT) [19]. Keto-dihydrosphingosine is reduced to form dihydrosphingosine [20], which is acylated by various fatty acids to form dihydroceramides with various acyl-chains by the action of 6 (dihydro)ceramides synthases (CERS1–6) [21, 22]. Dihydroceramides are then converted to ceramides by the action of dihydroceramide desaturase. These enzymatic steps occur in the ER and belong to so-called the de novo pathway for ceramide formation. Once generated in the ER, ceramides are transported from the ER to the Golgi complex where they are then incorporated into more complex sphingolipids such as sphingomyelins and glycosphingolipids. Sphingomyelins on the plasma membrane can be hydrolyzed to form ceramides by the action of neutral sphingomyelinase. This metabolic pathway is termed the sphingomyelin-hydrolysis pathway for ceramide formation. Complex sphingolipids can also be transported back to the lysosomes where they are converted back to ceramides, which are hydrolyzed into sphingosine by the action of ceramidases [23, 24]. Sphingosine can be converted back to ceramides by the action of CERS, which is termed the salvage pathway for ceramide formation. Therefore, there are three major metabolic pathways that lead to the formation of ceramides in cells, including the de novo, sphingomyelin-hydrolysis, and salvage pathways. In vitro cellular studies demonstrated that oversupply of palmitate markedly increases the levels of ceramides in hepatocytes [25]. Several clinical studies demonstrated that the hepatic content of ceramides is elevated in patients with NAFLD. Apostolopoulou et al. [26] found that the hepatic levels of ceramides were increased in NASH patients compared to NAFL patients or healthy individuals. Luukkonen et al. [27] demonstrated that the hepatic levels of ceramides and dihydroceramides were elevated in patients with steatosis plus IR compared to patients with steatosis. However, Vvedenskaya et al. [28] showed that the hepatic levels of ceramides were similar between healthy individuals and patients with NAFL or NASH. Several other studies demonstrated that the hepatic levels of ceramides were also increased in murine models of NASH [29,30,31,32]. Accumulated ceramides are derived from both de novo pathway and sphingomyelin-hydrolysis pathway. Enzymes in both ceramide-generating pathways have been shown to be upregulated in the liver of mice with NAFLD, including SPT, CERS1, CERS2, CERS4, CERS6, and SMPD1 [33,34,35,36,37]. These results indicate that NAFLD is indeed associated with increased hepatic levels of ceramides.

Metabolism of sphingolipids. Cer ceramide, CGT galactosylceramide synthase, Dhcer dihydroceramide, DHS dihydrosphingosine, E1P phosphoethanolamine, Gal-Cer galactosylceramide, GLS glycosphingolipid, Glu-cer glucosylceramide, KDSR ketodihydrosphingosine reductase, Pal-CoA palmitoyl-CoA, SPH sphingosine, S1P sphingosine-1-phosphate, SM sphingomyelin
As mentioned earlier, ceramides can be hydrolyzed into sphingosine by the action of 5 different ceramidases including acid ceramidase (ASAH1) [38], neutral ceramidase (ASAH2) [39], alkaline ceramidase 1 (ACER1) [40], alkaline ceramidase 2 (ACER2) [41], and alkaline ceramidase 3 (ACER3) [42]. As such, the hepatic content of sphingosine may be increased with increasing FFAs and ceramides in NAFLD patients and animal models of NAFLD. Indeed, the content of sphingosine was also increased in human hepatocytes treated with palmitate, a cellular model of steatosis [43, 44]. Hepatic sphingosine has been shown to be elevated in several mouse models of NAFLD [26, 29, 30, 45] although it remains unclear whether this is also the case in patients with NAFLD.
As sphingosine can be phosphorylated to form S1P by the action of SPHK1 and/or SPHK2 in liver cells, several studies have shown that hepatic levels of S1P are increased in patients with NAFLD [30] and murine models of NAFLD [46,47,48].
11.2.2 Role of Ceramide and Sphingosine in Hepatocyte Injury
In addition to structural components of membranes of mammalian cells, ceramides are bioactive lipids implicated in regulating oxidative stress [49], ER stress [50], and PCD [51, 52], which are key drivers of hepatic injury in NASH. This indicates that increased hepatic ceramides may contribute to the pathogenesis of NAFLD. In line with this notion, several animal studies demonstrated that inhibiting the generation of ceramides protected mice from diet-induced NASH. Treatment with myriocin, a specific and potent SPT inhibitor that inhibits the biosynthesis of ceramides, reduced liver damage and fibrosis, and expression of the inflammation markers, IL-1β, MCP-1, and TNFα [53]. Myriocin has also been found to be effective in suppressing high-fat diet (HFD)-induced steatosis, inflammation, fibrosis, and apoptosis in LDLr −/− mice, a genetic mouse model of NASH [54]. Moreover, myriocin was also shown to reduce the severity of NAFLD in rats fed diet enriched in fat and cholesterol [55]. In contrast to myriocin, treatment of ceramide per se was found to induce hepatic steatosis by enhancing hepatic lipogenesis [56]. These results suggest that ceramides or their metabolites accumulated in the liver may drive NASH onset and progression.
Indeed, several recent studies revealed the distinct roles of specific ceramide species in the pathogenesis of NAFLD. We showed that knocking down ACER3 increased the levels of C18:1-ceramide and alleviated oxidative stress and cell death in human hepatocytes oversupplied with palmitic acid [57]. We also demonstrated that knocking out the mouse Acer3 protected mice from hepatic oxidative stress and death of hepatocytes in a mouse model of NAFLD likely by increasing hepatic levels of C18:1-ceramide [57]. These results suggest a protective role of the unsaturated long-chain ceramide in NAFLD. CerS2 haploinsufficiency decreased very-long-chain ceramides while inducing a compensatory increase of C16-ceramide in mice, resulting in inhibition of beta-oxidation and advanced steatohepatitis [58], suggesting either a protective role of very-long-chain ceramides or a pathological role of C16-ceramide in NAFLD. A series of studies demonstrated that increased C16-ceramide due to Cers6 deficiency promoted lipogenesis and impaired mitochondrial respiration in NAFLD with obesity background [58,59,60]. Additionally, C16-ceramide treatment per se induced steatosis in mice by upregulating lipogenesis [56]. Interestingly, Cers5 knockout decreased the levels of C16-ceramide in various tissues including the liver in a feeding-independent manner and protected mice from weight gain, white adipose tissue inflammation, and hyperglycemia after HFD challenge [61]. These results suggest that C16-ceramide produced by the action of different CERS isozymes has distinct roles in regulating NAFLD.
Similar to ceramides, previous studies indicate that sphingosine also functions as a bioactive lipid to mediate PCD in human cells in response to different stressful insults, such as the pro-death cytokine TNF-α [62], glucocorticoid [63], serum deprivation [64], or oxidative stress [65]. However, which ceramidase is responsible for generating the pro-death sphingosine remained unclear until our recent studies have firmly established that ACER2 is a major ceramidase responsible for production of sphingosine that mediates cell death in response to a variety of stress stimuli. We demonstrated that genotoxic cancer chemotherapeutic agents [66] or ionizing radiation increased the levels of sphingosine in human tumor cells in a p53-dependent manner by upregulating ACER2 [67]. Knocking out ACER2 inhibited not only the generation of sphingosine but also PCD in response to DNA-damaging agents, whereas overexpression of ACER2 was sufficient to induce both sphingosine generation and PCD in cells [66, 67]. Furthermore, we identified ACER2 as a novel transcriptional target of p53, explaining why DNA-damaging agents and ionizing radiation upregulate both ACER2 and sphingosine in cells in a p53-dependent manner [67]. Many previous studies suggest that ceramides act as bioactive lipids to mediate PCD in tumor cells in response to different forms of stress, including DNA damage [68,69,70]. However, we demonstrated that an increase in the levels of ceramides due to ACER2 knockout or knockdown in cells failed to induce cell death [66], suggesting that ceramides that serve as endogenous substrates of ACER2 do not directly mediate PCD. These results suggest that sphingosine converted from ceramides by the action of ACER2 may also mediate the pathogenesis of NAFLD by inducing PCD of hepatocytes. Gulibositan et al. recently reported that hepatocyte-specific knockout of Sphk2, which phosphorylates sphingosine into S1P, significantly impaired insulin sensitivity and glucose tolerance in HFD-fed mice [71]. Intriguingly, the mechanistic study by the same group found that hepatocyte-specific ablation of Sphk2 increased the hepatic levels of sphingosine without affecting those of S1P, suggesting that sphingosine may be a bona fide mediator of HFD-induced insulin resistance and glucose intolerance [71].
11.2.3 Role of S1P in Steatosis and Inflammation in NAFLD
Emerging evidence suggests that S1P plays roles in inflammation and fibrosis in the context of NAFLD. In vitro study reported that SPHK1 expression protected hepatocytes from lipotoxicity by inhibiting IRE1α activation and JNK phosphorylation and thereby ER-stress-associated apoptosis of hepatocytes [72]. However, in vivo studies found that knocking out Sphk1 alleviated steatosis and hepatic inflammation in a mouse model of NAFLD, suggesting a role of S1P in NAFLD progression [48, 73]. In contrast, Sphk2 knockout mice were found to be more susceptible to NAFLD induction likely due to the compensatory upregulation of Sphk1 [47], further confirming the pathological role of S1P in NAFLD.
11.3 Sphingolipid Metabolism in HCC
11.3.1 Dysregulation of Ceramide Metabolism in HCC
Several studies demonstrated that ceramide metabolism is dysregulated in HCC in humans and animals. Krautbauer et al. found that most ceramide species (C16-C24:1) were decreased in human HCC tissues compared to their matched nontumor hepatic tissues [74]. In line with this study, Ismail et al. [75] and Li et al. [76] showed that the hepatic levels of ceramides were significantly decreased in human HCC tumors compared to paired nontumor hepatic tissues. In contrast to the above studies, Miura et al. found that the hepatic levels of several subclass sphingolipids, including ceramides, were significantly increased in human HCC tumors compared with their matched nontumor hepatic tissues [77]. The discrepancies among these studies might be attributed to variations in the etiology of HCC patients. Preclinical studies also indicate that hepatic levels of ceramides are altered in chemically induced liver tumors compared to nontumor liver tissues in mouse models of HCC. Haberl et al. [78] demonstrated that the levels of ceramides were higher in liver tumors than in nontumor hepatic tissues in mice treated with the carcinogen N-nitrosodiethylamine (DEN).
11.3.2 Role of Ceramides in HCC
Ceramides derived from either the de novo or sphingomyelin-hydrolysis pathway have been implicated in regulating HCC development and progression. Targeting specific CERS with a genetical approach has revealed that ceramides with different acyl-chains may have distinct roles in regulating HCC in mice. It has been shown that Cers2 deficiency decreased the hepatic levels of very-long-chain (C22–24) ceramides while increasing those of C16-ceramide and promoted sporadic liver tumor formation and chemical-induced hepatocarcinogenesis in mice [79, 80]. These results suggest that very-long-chain ceramides have a tumor suppressor role in HCC or that long-chain ceramide has an oncogenic role in HCC. Silencing CERS6, which synthesizes C16-ceramide, was found to mediate the cytotoxicity of antifolate methotrexate in HCC cell, suggesting that C16-ceramide might be a mediator of chemotherapeutic agents in HCC [81]. CERS4 was found to be upregulated in HCC tissues and its upregulation promoted the proliferation and survival of HCC cells [82]. These data suggest that ceramides with different chain lengths and saturation degrees, which are produced by specific CERSs, may function distinctly in HCC.
The sphingomyelin-hydrolysis pathway is also involved in the pathogenesis of HCC. Neutral sphingomyelinase 1 encoded by the gene SMPD2 was found to be downregulated in HCC tissues and its downregulation negatively correlates with poor long-term survival of patients with HCC [83]. Neutral sphingomyelinase 2 encoded by the gene SMPD3 was identified as a tumor suppressor-like gene that negatively correlates with early recurrence of human HCC after curative surgery [84]. Smpd3 deficiency in mice promoted survival and proliferation of cancer stem-like cells, resulting in spontaneous HCC. Unexpectedly, Smpd3 deficiency increased both sphingomyelin and C16-ceramide levels in mouse HCC tissues, especially in cancer stem-like cells. The increased sphingomyelin and C16-ceramide were attributed to the compensatory upregulation of Cers5 [85]. These results suggest that sphingomyelinases play an anti-cancer role in HCC.
On the other hand, inhibition of glucosylceramide synthesis was also found to suppress HCC. Jennemann et al. found that glucosylceramide synthase encoded by the UGCG gene was significantly overexpressed in human HCC tissues as compared to nontumor liver tissues and that knockout of the mouse Ucgc specifically in the hepatocyte inhibited HCC initiation and progression in a chemically induced HCC model [86]. Su et al. found that ganglioside synthesis was increased in the livers of an animal model of activation and expansion of liver cancer cells, and pharmacological inhibition of ganglioside synthesis suppressed proliferation and sphere growth of liver cancer cells [87]. Guri et al. [88] demonstrated that activating the mTOR signaling pathway specifically in the liver through the liver-specific knockout of both Tsc1 and Pten increased synthesis of fatty acids and sphingolipids including glucosylceramides in the liver, resulting in hepatosteatosis and HCC, which occurred sequentially. Inhibiting sphingolipid biosynthesis with myriocin, which specifically inhibits SPT, or knocking down hepatic Ugcg by RNA interference markedly reduced liver tumor numbers in Tsc1 and Pten double knockout mice. This study provides compelling evidence that increasing glucosylceramides or more complex glycosphingolipids may promote NAFLD development and progression to HCC.
The breakdown of ceramide catalyzed by ceramidases plays important role in regulating cancer-related pathobiology [24]. Pharmacological and siRNA inhibition of acid ceramidase was found to inhibit growth of liver tumor xenografts of HepG2 cells and enhanced the cytotoxicity of daunorubicin on HCC cells by upregulating oxidative stress and apoptosis [89]. The expression of ACER3 was found to be upregulated in different liver cancer cell lines compared to normal liver cells and its increased expression inversely correlates with the overall and disease-free survival of HCC patients [90]. Knockdown of ACER3 inhibited cell growth and promoted apoptosis in HCC cells but had no influence on growth or apoptosis in normal hepatocyte cells. Similar to ACER3, Liu et al. found that ACER2 was also upregulated in HCC and its upregulation promoted HCC cell survival and migration [91]. These results suggest that increased alkaline ceramidases promotes liver tumorigenesis likely by increasing hydrolysis of ceramides.
11.3.3 Role of S1P in HCC
SPHK-derived S1P has been shown to function as an oncogenic lipid of HCC by promoting survival, migration, and proliferation in HCC cells. SPHK1 was found to be upregulated in human HCC tissues compared to adjacent non-tumorous liver tissues, and the overexpression of SPHK1 was associated with advanced malignancy and poor prognosis of HCC [92,93,94,95]. Bao et al. found that the SphK1-induced migration and invasion of HCC cells was mediated by the S1P receptor S1PR1 [92]. Mu et al. demonstrated that SPHK1 could mediate the migration of hepatoma cells induced by hepatocyte growth factor (HGF) [96]. SPHK1-mediated invasion and metastasis were also attributed to induction of the mesenchymal transition (EMT) in HCC cells as increased SPHK1 accelerates lysosomal degradation of the cell-cell adhesion molecule E-cadherin (CDH1) [97]. Notably, this study found that SPHK1-produced S1P bound to TRAF2 and stimulated lysine 63-linked ubiquitination and beclin1 activation, resulting in autophagic degradation of E-cadherin and thereby EMT [97]. SPHK1 has been shown to mediate HCC proliferation by activating the Ras/ERK, MEK1/2, FAK/MLC-2, Wnt5A/b-catenin, Akt/GSK3β, and Akt/NF-kb signaling pathways [98,99,100]. In addition to regulating the malignancy of HCC, Sphk1 deletion in mice was found to suppress carcinogenesis in a chemically induced HCC model, supporting a role of SPHK1 in promoting hepatocarcinogenesis [101]. SPHK2 has a similar role in HCC. SPHK2 mRNA levels were found to be increased in HCC tissues and positively correlated with intra- and extra-hepatic recurrence [95]. Shi et al. reported that SPHK2 overexpression was associated with regorafenib resistance in HCC by activating NF-κB and STAT3 [102]. Beljanski et al. also reported that inhibition of SPHK2 in combination with sorafenib suppressed cell growth through the MAPK pathway in HCC [103]. Interestingly, S1P lyase was also increased in HCC tissues, and higher S1P lyase mRNA levels in HCC were associated with increased proliferation and poorer differentiation of HCC [95]. These results suggest that S1P may have an important role in liver tumorigenesis and that targeting the S1P pathway may improve the therapy of HCC.
11.3.4 Therapeutic Role of Exogenous Ceramide in HCC
Exogenous treatment of ceramide and sphingosine have been demonstrated to exert cytotoxic effects in HCC. Short-chain ceramide, including C2- and C6-ceramide, are well-studied antitumor lipids that induce growth arrest and cell death in various types of cancer [104]. Since Obeid et al. demonstrated for the first time that C2-ceramide induces PCD [105], short-chain ceramides have come into the spotlight of cancer research. C2-ceramide was later shown to induce cell death in HCC cell, including apoptosis and necrosis, possibly by downregulating Bcl-2 and inhibiting respiratory chain to produce reactive oxygen species (ROS), exhausting ATP, and impairing mitochondria function [106, 107]. C6-ceramide was shown to induce apoptosis in HCC cells concurrent with release of cytochrome c and activation of caspase-3 without affecting mitochondrial respiratory chain [107]. Co-administration of C6-ceramide was also found to enhance cytotoxic and pro-apoptotic effects of mTOR complex 1/2 (mTORC1/2) dual inhibitor AZD-8055 in a panel of HCC cell lines and primary cultured human HCC cells, with no adverse effect on growth and survival of normal human hepatocytes [108]. Nanoliposomal C6-ceramide, which can be administered intravenously and has improved bioavailability and solubility [109], was then applied in several preclinical studies to evaluate its therapeutic efficiency in HCC. Tagaram et al. reported that nanoliposomal C6-ceramide administration suppressed HCC growth in mice engrafted with HCC cells by reducing tumor vascularization and proliferation, inducing tumor cell apoptosis, and inhibiting phosphorylation of AKT [110]. More recently, in addition to tumor growth suppression, nanoliposomal C6-ceramide was found to improve antitumor immune response [111]. Nanoliposomal C6-ceramide injection could reduce numbers of tumor-associated macrophages and their production of ROS while inducing differentiation of tumor-associated macrophages into M1 phenotype of macrophages, which reduce immune suppression and increase activity of anti-cancer CD8+ T cell [111]. Similar to liposomal C6-ceramide, liposomal C8-ceramide was also found to induce apoptosis in HCC by activating caspase pathways and apoptosis signal-regulating kinase 1 (ASK1)-Jun N-terminal protein kinase (JNK) signaling, and injection of liposomal C8-ceramide inhibited HepG2 xenograft growth in severe combined immuno-deficient mice and improved their survival [112]. The liposomal short-chain ceramides were also found to enhance the effect of several chemotherapeutic drugs. Co-treatment with nanoliposomal C6-ceramide and vinblastine synergistically inhibited growth in HCC cells, probably by inhibiting autophagy flux and increasing apoptosis [113]. Similarly, liposomal C6-ceramide augmented the growth inhibitory effects of mTOR complex 1/2 inhibitor AZD-8055 in HCC mice xenografts with HCC cells [108]. Notably, co-loading C6−ceramide with sorafenib into liposomes synergistically increased antitumor activity in HCC cells with reduced systemic toxicity of sorafenib [114]. Similar to ceramides, sphingosine treatment was found to induce apoptosis by suppressing the activation of AKT kinase and upregulating caspase pathways in HCC cells [115,116,117]. All the data highlight the anti-cancer activities of ceramides and sphingosine mainly by inducing cell death in HCC, which warrants further clinical studies to evaluate the potential of ceramide and sphingosine treatment for HCC.
11.4 Conclusions
Recent findings strongly suggest that specific ceramide species, sphingosine, sphingosine-1-phosphate, and other sphingolipids play prominent roles in NAFLD and liver tumorigenesis. Targeting the metabolism of these bioactive sphingolipids may represent a novel approach to halting NAFLD development and/or its progression to HCC. Therefore, an important area of future study in the sphingolipid field is to develop novel drugs targeting ceramide metabolism to enhance therapeutic response and improve survival outcome in HCC patients.
References
Friedman, S. L., et al. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine, 24(7), 908–922.
Polyzos, S. A., Kountouras, J., & Mantzoros, C. S. (2019). Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism, 92, 82–97.
Younossi, Z., et al. (2018). Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nature Reviews. Gastroenterology & Hepatology, 15(1), 11–20.
Cole, B. K., et al. (2018). Non-alcoholic fatty liver disease (NAFLD) models in drug discovery. Expert Opinion on Drug Discovery, 13(2), 193–205.
Leite, N. C., et al. (2014). Non-alcoholic fatty liver disease and diabetes: From physiopathological interplay to diagnosis and treatment. World Journal of Gastroenterology, 20(26), 8377–8392.
Kim, J. Y., et al. (2018). ER stress drives lipogenesis and steatohepatitis via Caspase-2 activation of S1P. Cell, 175(1), 133–145.e15.
Wang, L., et al. (2018). Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic fatty liver disease (NAFLD). Current Drug Targets, 19(9), 1087–1094.
Henkel, A., & Green, R. M. (2013). The unfolded protein response in fatty liver disease. Seminars in Liver Disease, 33(4), 321–329.
Schwabe, R. F., & Luedde, T. (2018). Apoptosis and necroptosis in the liver: A matter of life and death. Nature Reviews. Gastroenterology & Hepatology, 15(12), 738–752.
Kanda, T., et al. (2018). Apoptosis and non-alcoholic fatty liver diseases. World Journal of Gastroenterology, 24(25), 2661–2672.
Luedde, T., Kaplowitz, N., & Schwabe, R. F. (2014). Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology, 147(4), 765–783.e4.
Afonso, M. B., et al. (2015). Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clinical Science (London, England), 129(8), 721–739.
Gautheron, J., et al. (2014). A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Molecular Medicine, 6(8), 1062–1074.
Czaja, M. J. (2016). Function of autophagy in nonalcoholic fatty liver disease. Digestive Diseases and Sciences, 61(5), 1304–1313.
Parzych, K. R., & Klionsky, D. J. (2014). An overview of autophagy: Morphology, mechanism, and regulation. Antioxidants & Redox Signaling, 20(3), 460–473.
Singh, R., et al. (2009). Autophagy regulates lipid metabolism. Nature, 458(7242), 1131–1135.
Nikolova-Karakashian, M. (2018). Alcoholic and non-alcoholic fatty liver disease: Focus on ceramide. Advances in Biological Regulation, 70, 40–50.
Montefusco, D. J., et al. (2018). Non-alcoholic fatty liver disease: Insights from sphingolipidomics. Biochemical and Biophysical Research Communications, 504(3), 608–616.
Hannun, Y. A., & Obeid, L. M. (2011). Many ceramides. The Journal of Biological Chemistry, 286(32), 27855–27862.
Gault, C. R., Obeid, L. M., & Hannun, Y. A. (2010). An overview of sphingolipid metabolism: From synthesis to breakdown. Advances in Experimental Medicine and Biology, 688, 1–23.
Stiban, J., Tidhar, R., & Futerman, A. H. (2010). Ceramide synthases: Roles in cell physiology and signaling. Advances in Experimental Medicine and Biology, 688, 60–71.
Park, J. W., Park, W. J., & Futerman, A. H. (2014). Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochimica et Biophysica Acta, 1841(5), 671–681.
Mao, C., & Obeid, L. M. (2008). Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochimica et Biophysica Acta, 1781(9), 424–434.
Coant, N., et al. (2017). Ceramidases, roles in sphingolipid metabolism and in health and disease. Advances in Biological Regulation, 63, 122–131.
Hamada, Y., et al. (2014). Involvement of de novo ceramide synthesis in pro-inflammatory adipokine secretion and adipocyte-macrophage interaction. The Journal of Nutritional Biochemistry, 25(12), 1309–1316.
Apostolopoulou, M., et al. (2018). Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care, 41(6), 1235–1243.
Luukkonen, P. K., et al. (2016). Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. Journal of Hepatology, 64(5), 1167–1175.
Vvedenskaya, O., et al. (2021). Nonalcoholic fatty liver disease stratification by liver lipidomics. Journal of Lipid Research, 62, 100104.
Mauer, A. S., et al. (2017). Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. American Journal of Physiology. Gastrointestinal and Liver Physiology, 312(3), G300–G313.
Sanyal, A. J., & Pacana, T. (2015). A lipidomic readout of disease progression in a diet-induced mouse model of nonalcoholic fatty liver disease. Transactions of the American Clinical and Climatological Association, 126, 271–288.
Saito, K., et al. (2015). Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Scientific Reports, 5, 12466.
Alonso, C., et al. (2017). Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology, 152(6), 1449–1461.e7.
Koh, E. H., et al. (2021). Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut, 70(10), 1954–1964.
Deevska, G. M., et al. (2009). Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia in mice. The Journal of Biological Chemistry, 284(13), 8359–8368.
Raichur, S., et al. (2019). The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Molecular Metabolism, 21, 36–50.
Lyn-Cook, L. J., et al. (2009). Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. Journal of Alzheimer's Disease, 16(4), 715–729.
Garcia-Ruiz, C., et al. (2015). Acid sphingomyelinase-ceramide system in steatohepatitis: A novel target regulating multiple pathways. Journal of Hepatology, 62(1), 219–233.
Koch, J., et al. (1996). Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. The Journal of Biological Chemistry, 271(51), 33110–33115.
El, B. S., et al. (2000). Molecular cloning and characterization of a human mitochondrial ceramidase. The Journal of Biological Chemistry, 275(28), 21508–21513.
Sun, W., et al. (2008). Upregulation of the human alkaline ceramidase 1 and acid ceramidase mediates calcium-induced differentiation of epidermal keratinocytes. The Journal of Investigative Dermatology, 128(2), 389–397.
Xu, R., et al. (2006). Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. The FASEB Journal, 20(11), 1813–1825.
Mao, C., et al. (2001). Cloning and characterization of a novel human alkaline ceramidase. A mammalian enzyme that hydrolyzes phytoceramide. The Journal of Biological Chemistry, 276(28), 26577–26588.
Brown, M. V., et al. (2013). Metabolomic signatures in lipid-loaded HepaRGs reveal pathways involved in steatotic progression. Obesity (Silver Spring), 21(12), E561–E570.
Charytoniuk, T., et al. (2019). The effect of enterolactone on sphingolipid pathway and hepatic insulin resistance development in HepG2 cells. Life Sciences, 217, 1–7.
Presa, N., et al. (2019). Vitamin E alleviates non-alcoholic fatty liver disease in phosphatidylethanolamine N-methyltransferase deficient mice. Biochimica et Biophysica Acta—Molecular Basis of Disease, 1865(1), 14–25.
Lee, S. Y., et al. (2015). Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology, 62(1), 135–146.
Nagahashi, M., et al. (2015). Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology, 61(4), 1216–1226.
Geng, T., et al. (2015). SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. Journal of Lipid Research, 56(12), 2359–2371.
Andrieu-Abadie, N., et al. (2001). Ceramide in apoptosis signaling: Relationship with oxidative stress. Free Radical Biology & Medicine, 31(6), 717–728.
Liu, Z., et al. (2014). Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca(2+) homeostasis in human adenoid cystic carcinoma cells. Cell & Bioscience, 4, 71.
Obeid, L. M., & Hannun, Y. A. (1995). Ceramide: A stress signal and mediator of growth suppression and apoptosis. Journal of Cellular Biochemistry, 58(2), 191–198.
Nganga, R., Oleinik, N., & Ogretmen, B. (2018). Mechanisms of ceramide-dependent cancer cell death. Advances in Cancer Research, 140, 1–25.
Martinez, L., et al. (2015). Myristic acid potentiates palmitic acid-induced lipotoxicity and steatohepatitis associated with lipodystrophy by sustaning de novo ceramide synthesis. Oncotarget, 6(39), 41479–41496.
Kasumov, T., et al. (2015). Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS One, 10(5), e0126910.
Kurek, K., et al. (2014). Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver International, 34(7), 1074–1083.
Jiang, C., et al. (2015). Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. The Journal of Clinical Investigation, 125(1), 386–402.
Wang, K., et al. (2020). Targeting alkaline ceramidase 3 alleviates the severity of nonalcoholic steatohepatitis by reducing oxidative stress. Cell Death & Disease, 11(1), 28.
Raichur, S., et al. (2014). CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metabolism, 20(4), 687–695.
Hammerschmidt, P., et al. (2019). CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell, 177(6), 1536–1552.e23.
Turpin, S. M., et al. (2014). Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metabolism, 20(4), 678–686.
Gosejacob, D., et al. (2016). Ceramide synthase 5 is essential to maintain C16:0-ceramide pools and contributes to the development of diet-induced obesity. The Journal of Biological Chemistry, 291(13), 6989–7003.
Ohta, H., et al. (1994). A possible role of sphingosine in induction of apoptosis by tumor necrosis factor-alpha in human neutrophils. FEBS Letters, 355(3), 267–270.
Lepine, S., et al. (2004). Sphingosine contributes to glucocorticoid-induced apoptosis of thymocytes independently of the mitochondrial pathway. Journal of Immunology, 173(6), 3783–3790.
Suzuki, E., et al. (2004). Sphingosine-dependent apoptosis: A unified concept based on multiple mechanisms operating in concert. Proceedings of the National Academy of Sciences of the United States of America, 101(41), 14788–14793.
Abrahan, C. E., et al. (2010). Synthesis of sphingosine is essential for oxidative stress-induced apoptosis of photoreceptors. Investigative Ophthalmology & Visual Science, 51(2), 1171–1180.
Xu, R., et al. (2016). Alkaline ceramidase 2 and its bioactive product sphingosine are novel regulators of the DNA damage response. Oncotarget, 7(14), 18440–18457.
Wang, Y., et al. (2017). Alkaline ceramidase 2 is a novel direct target of p53 and induces autophagy and apoptosis through ROS generation. Scientific Reports, 7, 44573.
Haimovitz-Friedman, A., et al. (1994). Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. The Journal of Experimental Medicine, 180(2), 525–535.
Kolesnick, R., & Fuks, Z. (2003). Radiation and ceramide-induced apoptosis. Oncogene, 22(37), 5897–5906.
Jarvis, W. D., et al. (1994). Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proceedings of the National Academy of Sciences of the United States of America, 91(1), 73–77.
Aji, G., et al. (2020). Regulation of hepatic insulin signaling and glucose homeostasis by sphingosine kinase 2. Proceedings of the National Academy of Sciences of the United States of America, 117(39), 24434–24442.
Qi, Y., et al. (2015). Sphingosine kinase 1 protects hepatocytes from lipotoxicity via Down-regulation of IRE1alpha protein expression. The Journal of Biological Chemistry, 290(38), 23282–23290.
Chen, J., et al. (2016). Deletion of sphingosine kinase 1 ameliorates hepatic steatosis in diet-induced obese mice: Role of PPARgamma. Biochimica et Biophysica Acta, 1861(2), 138–147.
Krautbauer, S., et al. (2016). Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochimica et Biophysica Acta, 1861(11), 1767–1774.
Ismail, I. T., et al. (2020). Remodeling lipids in the transition from chronic liver disease to hepatocellular carcinoma. Cancers (Basel), 13(1), 88.
Fekry, B., et al. (2019). HNF4alpha-deficient fatty liver provides a permissive environment for sex-independent hepatocellular carcinoma. Cancer Research, 79(22), 5860–5873.
Miura, K., et al. (2021). Dysregulation of sphingolipid metabolic enzymes leads to high levels of sphingosine-1-phosphate and ceramide in human hepatocellular carcinoma. Hepatology Research, 51(5), 614–626.
Haberl, E. M., et al. (2021). Accumulation of cholesterol, triglycerides and ceramides in hepatocellular carcinomas of diethylnitrosamine injected mice. Lipids in Health and Disease, 20(1), 135.
Imgrund, S., et al. (2009). Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. The Journal of Biological Chemistry, 284(48), 33549–33560.
Chen, L., et al. (2014). Enhancement of DEN-induced liver tumourigenesis in hepatocyte-specific Lass2-knockout mice coincident with upregulation of the TGF-beta1-Smad4-PAI-1 axis. Oncology Reports, 31(2), 885–893.
Fekry, B., et al. (2016). Ceramide synthase 6 is a novel target of methotrexate mediating its antiproliferative effect in a p53-dependent manner. PLoS One, 11(1), e0146618.
Chen, J., et al. (2017). Ceramide synthase-4 orchestrates the cell proliferation and tumor growth of liver cancer in vitro and in vivo through the nuclear factor-kappaB signaling pathway. Oncology Letters, 14(2), 1477–1483.
Lin, M., et al. (2018). Exosomal neutral sphingomyelinase 1 suppresses hepatocellular carcinoma via decreasing the ratio of sphingomyelin/ceramide. The FEBS Journal, 285(20), 3835–3848.
Revill, K., et al. (2013). Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology, 145(6), 1424–35.e1–1424–35.25.
Zhong, L., et al. (2018). Increased liver tumor formation in neutral sphingomyelinase-2-deficient mice. Journal of Lipid Research, 59(5), 795–804.
Jennemann, R., et al. (2017). Inhibition of hepatocellular carcinoma growth by blockade of glycosphingolipid synthesis. Oncotarget, 8(65), 109201–109216.
Su, T., et al. (2021). Inhibition of ganglioside synthesis suppressed liver cancer cell proliferation through targeting kinetochore metaphase signaling. Metabolites, 11(3), 167.
Guri, Y., et al. (2017). mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell, 32(6), 807–823.e12.
Morales, A., et al. (2007). Pharmacological inhibition or small interfering RNA targeting acid ceramidase sensitizes hepatoma cells to chemotherapy and reduces tumor growth in vivo. Oncogene, 26(6), 905–916.
Yin, Y., et al. (2018). Alkaline ceramidase 3 promotes growth of hepatocellular carcinoma cells via regulating S1P/S1PR2/PI3K/AKT signaling. Pathology, Research and Practice, 214(9), 1381–1387.
Xu, R., et al. (2018). Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell Death and Differentiation, 25(5), 841–856.
Bao, M., et al. (2012). Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver International, 32(2), 331–338.
Wang, F., & Wu, Z. (2018). Sphingosine kinase 1 overexpression is associated with poor prognosis and oxaliplatin resistance in hepatocellular carcinoma. Experimental and Therapeutic Medicine, 15(6), 5371–5376.
Shi, J., et al. (2015). The impact of sphingosine kinase 1 on the prognosis of hepatocellular carcinoma patients with portal vein tumor thrombus. Annals of Hepatology, 14(2), 198–206.
Uranbileg, B., et al. (2016). Increased mRNA levels of sphingosine kinases and S1P Lyase and reduced levels of S1P were observed in hepatocellular carcinoma in association with poorer differentiation and earlier recurrence. PLoS One, 11(2), e0149462.
Mu, Z., et al. (2008). KAI1/CD82 suppresses hepatocyte growth factor-induced migration of hepatoma cells via upregulation of Sprouty2. Science in China. Series C, Life Sciences, 51(7), 648–654.
Liu, H., et al. (2017). SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy, 13(5), 900–913.
Zhang, C., et al. (2013). The blockage of Ras/ERK pathway augments the sensitivity of SphK1 inhibitor SKI II in human hepatoma HepG2 cells. Biochemical and Biophysical Research Communications, 434(1), 35–41.
Liu, H., et al. (2016). SphK1 inhibitor SKI II inhibits the proliferation of human hepatoma HepG2 cells via the Wnt5A/beta-catenin signaling pathway. Life Sciences, 151, 23–29.
Grbcic, P., et al. (2017). Dual sphingosine kinase inhibitor SKI-II enhances sensitivity to 5-fluorouracil in hepatocellular carcinoma cells via suppression of osteopontin and FAK/IGF-1R signalling. Biochemical and Biophysical Research Communications, 487(4), 782–788.
Chen, J., et al. (2018). Deletion of sphingosine kinase 1 inhibits liver tumorigenesis in diethylnitrosamine-treated mice. Oncotarget, 9(21), 15635–15649.
Shi, W., et al. (2020). Targeting SphK2 reverses acquired resistance of Regorafenib in hepatocellular carcinoma. Frontiers in Oncology, 10, 694.
Beljanski, V., Lewis, C. S., & Smith, C. D. (2011). Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biology & Therapy, 11(5), 524–534.
Liu, J., Beckman, B. S., & Foroozesh, M. (2013). A review of ceramide analogs as potential anticancer agents. Future Medicinal Chemistry, 5(12), 1405–1421.
Obeid, L. M., et al. (1993). Programmed cell death induced by ceramide. Science, 259(5102), 1769–1771.
Zhu, X. F., et al. (2000). Apoptosis induced by ceramide in hepatocellular carcinoma Bel7402 cells. Acta Pharmacologica Sinica, 21(3), 225–228.
Gentil, B., Grimot, F., & Riva, C. (2003). Commitment to apoptosis by ceramides depends on mitochondrial respiratory function, cytochrome c release and caspase-3 activation in Hep-G2 cells. Molecular and Cellular Biochemistry, 254(1–2), 203–210.
Liu, M., et al. (2016). C6 ceramide sensitizes the anti-hepatocellular carcinoma (HCC) activity by AZD-8055, a novel mTORC1/2 dual inhibitor. Tumour Biology, 37(8), 11039–11048.
Watters, R. J., et al. (2012). Development and use of ceramide nanoliposomes in cancer. Methods in Enzymology, 508, 89–108.
Tagaram, H. R., et al. (2011). Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut, 60(5), 695–701.
Li, G., et al. (2018). Nanoliposome C6-ceramide increases the anti-tumor immune response and slows growth of liver tumors in mice. Gastroenterology, 154(4), 1024–1036.e9.
Lv, H., et al. (2016). Preclinical evaluation of liposomal C8 ceramide as a potent anti-hepatocellular carcinoma agent. PLoS One, 11(1), e0145195.
Adiseshaiah, P. P., et al. (2013). Synergistic combination therapy with nanoliposomal C6-ceramide and vinblastine is associated with autophagy dysfunction in hepatocarcinoma and colorectal cancer models. Cancer Letters, 337(2), 254–265.
Yin, X., et al. (2018). Ceramide-fabricated co-loaded liposomes for the synergistic treatment of hepatocellular carcinoma. AAPS PharmSciTech, 19(5), 2133–2143.
Hung, W. C., Chang, H. C., & Chuang, L. Y. (1999). Activation of caspase-3-like proteases in apoptosis induced by sphingosine and other long-chain bases in Hep3B hepatoma cells. The Biochemical Journal, 338(Pt 1), 161–166.
Chang, H. C., et al. (2001). Role of AKT kinase in sphingosine-induced apoptosis in human hepatoma cells. Journal of Cellular Physiology, 188(2), 188–193.
Hossain, Z., Sugawara, T., & Hirata, T. (2013). Sphingoid bases from sea cucumber induce apoptosis in human hepatoma HepG2 cells through p-AKT and DR5. Oncology Reports, 29(3), 1201–1207.
Acknowledgement
The work by Dr. Cungui Mao’s group is supported by the National Institutes of Health (NIH) grants P01CA097132 and R01GM130878.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive licence to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wang, K., Wei, Y., Xu, R., Li, Y., Mao, C. (2022). Manifold Roles of Ceramide Metabolism in Non-Alcoholic Fatty Liver Disease and Liver Cancer. In: Jiang, XC. (eds) Sphingolipid Metabolism and Metabolic Disease. Advances in Experimental Medicine and Biology, vol 1372. Springer, Singapore. https://doi.org/10.1007/978-981-19-0394-6_11
Download citation
DOI: https://doi.org/10.1007/978-981-19-0394-6_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-0393-9
Online ISBN: 978-981-19-0394-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)