
Abstract
Asthma is the most prevalent chronic respiratory disease worldwide and the leading serious chronic illness in children. Clinical characteristics are wheezing, reversible airway obstruction, airway inflammation, and airway hyperreactivity. Asthma susceptibility is influenced by genes and environment. 17q12–21 is the most significant genetic asthma susceptibility locus and single nucleotide polymorphisms (SNPs) within that high-risk locus are linked to increased expression of the Ormdl sphingolipid biosynthesis regulator (ORMDL) 3. ORMDL3 is an endoplasmic reticulum protein that stabilizes the serine palmitoyl transferase (SPT) complex that regulates sphingolipid de novo synthesis. Sphingolipids essential for formation and integrity of cellular membranes and bioactive molecules that regulate key cellular processes can be synthesized de novo and through recycling pathways. Their metabolism is tightly regulated through feedback regulation. ORMDL3 inhibits de novo synthesis when it engages subunit 1 of the SPT complex. This chapter focuses on the effect of decreased sphingolipid synthesis on asthma features and summarizes studies in mouse models and in children with and without asthma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Asthma Is a Common Disease
Asthma affects more than 300 million people worldwide and is the most common chronic respiratory medical condition in childhood, affecting six million children in the United States [1, 2]. Asthma is a complex disease and childhood and adult asthma differ with respect to severity and comorbidity [3]. In order to improve consistency in genetic and environmental correlations, the observable characteristics of the asthma phenotype have been further classified into endotypes that are defined by specific biological mechanisms [4]. Current thought is that childhood asthma is a developmental disorder in which interactions between common genetic variants and environmental exposure to viruses, allergens, pollution at critical times during the development of the immune system and the airways play a critical role [5]. And, that genetic risk factors of adult-onset asthma are largely a subset of the genetic risk for childhood asthma suggesting a greater role for non-genetic factors in adult-onset asthma [6]. Typical asthma phenotypic features are airway hyperreactivity, inflammation, mucus overproduction, and smooth muscle hypertrophy [7, 8]. The majority of patients have allergic asthma that presents with eosinophilic airway inflammation, increased total and antigen-specific IgE levels, blood eosinophilia, and sensitization to various allergens of which house dust mites and Alternaria alternata are very prevalent [9, 10]. Treatment options consist of β-2 agonists to decrease airway smooth muscle contraction, corticosteroids to reduce inflammation, and monoclonal antibodies to antagonize inflammatory mediators.
10.2 17q12–21 Is a High-Risk Asthma Locus
A GWAS study in 2007 identified 17q12–21 as a high-risk asthma locus and showed that single nucleotide polymorphisms (SNPs) that increase ORMDL3 expression contribute to the risk of childhood asthma [11]. Consecutive studies confirmed the significance of 17q21, extended the association to additional gene candidates, specifically to the adjacent gasdermin-B (GSDMB) and replicated the results in children and adults in different ethnicities and localities [12,13,14,15,16,17]. SNPs associated with childhood asthma were consistently associated with increased transcript levels of ORMDL3 [11, 18,19,20,21,22] and with wheezing illness, a characteristic of very early onset asthma [22, 23].
10.2.1 ORMDL Proteins Are Evolutionary Conserved Endoplasmic Reticulum Proteins
There are three human ORMDL proteins (ORMDL1, ORMDL2, and ORMDL3) that are encoded on chromosomes 2q32.2 (ORMDL1), 12q13.2 (ORMDL2), 17q21.1 (ORMDL3). These proteins have the same size (153 aa acids), similar molecular weight (ORMDL1 17,371 Da, ORMDL2 17,363 Da, ORMDL3 17,495 Da), and a high homology with orthologues in yeast, plants, invertebrates and vertebrates, and with each other. Human ORMDL1 shares 83% identity with ORMDL2 and 84% identity with ORMDL3 [24]. ORMDL 2 and ORMDL 3 expression is highest in myocytes, immune and epithelial cells, while ORMDL1 is more widely expressed (heart, brain, lung, liver, skeletal muscle and kidney) [25].
10.2.2 ORMDLs Mediate Sphingolipid Homeostasis by Regulating De Novo Synthesis
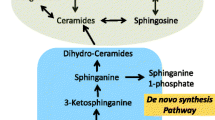
Serine palmitoyl transferase (SPT) is the first and rate limiting step of de novo synthesis. The SPT complex converts a fatty acid, mostly the C16:0 palmitoyl-CoA with an amino acid (mostly serine) to generate 3-keto sphinganine that in turn is rapidly degraded to sphinganine, the substrate for sphingosine kinases that generate sphinganine-1-phosphate, and ceramide synthases that generate dihydroceramides. Introduction of a 4,5-bond in the sphingosine backbone of dihydroceramides generates ceramides that are metabolized to more complex sphingolipids such as glucosylceramides and sphingomyelins [26].
Yeast studies showed that Orm1 and Orm2 that physically interact with long-chain base subunit 1 (Lcb1) regulate sphingolipid homeostasis and protein quality control [27, 28].
Deletion of Orm proteins increases sphingolipid de novo synthesis and induces a constitutive unfolded protein response that is reversed when sphingolipid synthesis is inhibited with myriocin [28]. Endoplasmic reticulum stress in turn induces Orm2 transcription through activation of calcium and calcineurin dependent pathways [29]. Regulation occurs post-transcriptional through Orm phosphorylation in response to sphingolipid depletion. Phosphorylated Orm1 and Orm2 can no longer inhibit sphingolipid de novo synthesis [27].
Human ORMDLs function similarly, although they are not regulated by phosphorylation. With adequate SPT subunit concentrations ORMDL proteins become regulatory [30, 31]. Overexpression of each ORMDL inhibits sphingolipid synthesis and deletion of all ORMDLs increases sphingolipid synthesis [32,33,34]. Deletion of ORMDL3, increases SPT activity and synthesis of C16 ceramides and sphingomyelin in A549 cells more than deletion of ORMDL1 and ORMDL2 [33].
10.2.3 ORMDLs Stabilize the Multi-Dimeric SPT Homocomplex
Structure and assembly of the SPT complex, elucidated by two independent groups in 2021, reveal that SPT is a double dimer complex composed of two SPTLC1 subunits, two SPTLC2 (or SPTLC3) subunits, and two ssSPTa units (SPT small subunit A). ORMDLs bind the N-terminus of subunit SPTLC1 and thus stabilize the complex while the ssSPTa units anchor SPTLC2 to the endoplasmic reticulum membrane and form a substrate binding tunnel [35, 36]. De novo synthesis proceeds when the ssSPT subunit engages a long-chain substrate in the catalytic tunnel. De novo synthesis is inhibited when the catalytic site within the tunnel is occupied by myriocin or by the N-terminal of ORMDL that blocks accessibility to the tunnel. The experiments suggest however that an additional, currently unknown factor is likely required for ORMDL to mediate inhibition. Because levels of ORMDL and SPT are the same in high and low sphingolipid conditions, SPT regulation could be mediated by conformational changes [30]. The structural data are consistent with the prior observation that ORMDL regulates SPT activity through post-transcriptional mechanisms that are independent of ORMDL mRNA expression [32, 37]. These studies also showed that inhibition occurs in response to increased ceramide levels and that it depends on functional SPT. Interestingly, free cholesterol promotes degradation of ORMDL1 in macrophages, but it is not known whether this mechanism extends to different cell types and the other ORMDLs [38]. It is thus still unclear what regulates ORMDL to function as an inhibitor, i.e., occupy the substrate binding tunnel, and what regulates de-repression.
10.3 ORMDL3 and Asthma
Genetic variations in 17q21 that increase ORMDL3 have prompted multiple studies in mice and humans with the obvious goal to understand whether or not ORMDL3 is causally related to asthma pathology. Early ORMDL3 overexpression and knockout studies suggested that inflammation, mainly through activation of the unfolded protein response, could be the functional link to asthma [39]. The identification of ORMDL3 as a regulator of mammalian sphingolipid de novo synthesis 3 years after the initial GWAS study [27] suggested that this large lipid class of membrane constituents and bioactive signaling molecules may also be involved in asthma pathogenesis. Sphingolipid synthesis is decreased in mouse models that overexpress human or murine ORMDL3, but phenotypes differ starkly [40, 41]. Overexpression of human ORMDL3 induces airway remodeling and airway responsiveness characteristic of asthma, while overexpression of murine ORMDL3 does not alter or induce key asthma features in mice [40]. By the same token however, ORMDL3 knockout protects mice from developing allergic airways disease triggered by Alternaria alternans [42]. The model shows a marked decrease in pathophysiology, including airway hyperreactivity and airway eosinophilia induced by Alternaria without activation of the unfolded protein response. The protection was lost when ORMDL3 was reconstituted in bronchial epithelial cells of these ORMDL3 knockout mice.
A complicating factor of transgenic models is that unphysiological and unregulated (over)expression could affect the stoichiometry of ORMDL3 and SPTLC1, and thus feedback regulation [30]. In-vitro experiments in lung epithelial cells and macrophages demonstrate that small increases in ORMDL3 expression decrease ceramide levels, while higher expression increases ceramide production through recycling pathways resulting in sphingolipid overload that might also characterize ORMDL knockout models [32, 43, 44].
10.4 Decreased Sphingolipid Synthesis and Asthma
The following highlights experimental and clinical asthma cohort studies that link decreased sphingolipid synthesis to the 17q21 genotypes and asthma and how sphingolipid synthesis might be a target for asthma therapies.
SPT Inhibition Increases Airway Reactivity
To understand the relevance of decreased sphingolipids, asthma features were assessed in wild-type mice treated intranasally with SPT inhibitor myriocin and in haploinsufficient SPTLC2+/− mice in which sphingolipid synthesis and mass are decreased by 40–50%. In contrast to complete SPTLC knockout that is embryonically lethal, haploinsufficient mice are healthy [45]. We evaluated the effects of acute and chronic decrease of sphingolipid synthesis on inflammation, mucus production, and airway remodeling and assessed methacholine-mediated airway constriction in mice and explanted murine and human bronchial airways. All experiments were carried out without prior sensitization to allergens (i.e., house dust mites, ovalbumin, Alternaria alternata).
Myriocin inhibits sphingolipid synthesis by physically blocking the substrate channel of SPT that is formed by SPTLC2 and ssSPTa [46, 47]. Of note is that myriocin is also a potent immunosuppressive that alters S1P receptor-mediated lymphocyte egress and inhibits lymphocyte proliferation and generation of allo-reactive T lymphocytes [36, 46, 48, 49].
In first experiments, myriocin was directly administered to the respiratory tract of wild-type mice or added to incubation media of murine and human bronchial rings. Within 3 h after installation, myriocin decreased lung sphinganine and total ceramides and increased airway reactivity that was determined by changes in central airway resistance in response to nebulized methacholine. Baseline lung mechanical properties such as static compliance were not altered. Application of myriocin for 90 min to human or murine bronchial rings equally showed a dose-dependent increase in bronchoconstriction [50].
Decreased SPTLC2 Activity Increases Airway Reactivity
To ensure that the effects are not mediated by mechanisms related to the immune suppressive function of myriocin and to further distinguish acute inhibition of sphingolipid de novo synthesis from a habitually decreased sphingolipid de novo synthesis and decreased sphingolipid mass, we used SPTLC2+/− mice. Airway resistance was equally increased in response to methacholine in Sptlc2+/− compared to Sptlc2+/+ controls and no differences were detected in the baseline mechanical lung parameters, including static compliance. Contractile response to methacholine was also increased in bronchial rings isolated from Sptlc2+/− mice compared to bronchial rings isolated from Sptlc2+/+ controls. Notably, very similar results were obtained in an allergic asthma model in which co-administration of myriocin with house dust mites during a 2-week sensitization period dramatically increased methacholine stimulated airway constriction compared to house dust mites alone [51].
In the SPTLC+/− and SPTLC+/+ controls or myriocin-treated wild-type and wild-type controls, there were no differences with regard to lung histology, cell composition of bronchioalveolar lavage, or expression of tumor necrosis factor-α (TNF-α), inflammatory cytokines interleukin-6 (IL-6) and IL-1β that are found in allergic and chronic asthma. These characteristics are consistent with the association of ORMDL3 polymorphisms with non-allergic asthma.
10.5 Blood Sphingolipids Are Decreased in Children with Non-Allergic Asthma
Plasma and serum sphingolipids have been investigated in different asthma pheno- and endotypes in patients of different ages using different methodological approaches. In a study of adult patients with allergic asthma, serum ceramides (C16, C18, C18:2, C24, C24:1) were higher in allergic asthma patients than in controls, and higher ceramide C16 and C24:0 distinguished uncontrolled from controlled patients [52]. In a study of house dust mite allergized adult asthma patients, ceramide levels were not different compared to controls, but asthma patients failed to mount an increase in sphinganine in response to allergic stimulation [53]. In another adult cohort, no effect of annotated ORMDL3 asthma SNPs was found on total plasma long-chain bases albeit by a method that cannot distinguish whether analytes originate from the de novo or the recyling pathway [54].
In a study of children with clinically mild disease and releative normal lung function but lower FEV1/FVC and atopy increased serum dihydroceramide C18 and ceramide C20 at ages 7 and 8 years predicted asthma persistence at ages 10 to 11 years [55]. In another study that measured ceramides and sphingomyelin in children with different asthma endotypes, decreased sphingomyelin distinguished non-allergic childhood asthma from allergic childhood asthma [56]. Moreover, plasma sphingolipid analysis of 500 children enrolled in the ‘Copenhagen prospective studies on asthma in childhood’ (COPSAC) birth cohort found that lower concentrations of ceramides and sphingomyelins at the age of 6 months were associated with an increased risk of developing asthma before age 3. At the age of 6 years, lower concentrations of sphinganine-1-phosphate were associated with increased airway resistance [57].
Our group investigated the effect of asthma-associated SNPs on sphingolipid synthesis and mass in children with non-allergic and allergic asthma and controls. To this end we enrolled 61 children with physician diagnosed asthma and 59 children without asthma and measured sphingolipids in whole blood and plasma and determined sphingolipid de novo synthesis in peripheral blood monocytes (PBMC). Eosinophilia was used to stratify patients with allergic asthma (>300 eosinophils/μL). Patients with non-allergic asthma (<300 eosinophils/μL) had lower whole blood dihydroceramides (C18, C18:1, C24:1), ceramides (C18, C20, C22, C24, C24:1), and sphingomyelins (C18, C18:1, C24:1) compared to controls. When comparing patients with allergic asthma to patients with non-allergic asthma, we found significantly higher dihydroceramides (C18, C18:1), ceramides (C18, C24), and sphingomyelins (C18, C18:1, C24:1) in the allergic asthma patients but no difference when compared to controls with eosinophilia. Similar results were obtained when the asthma group was stratified by IgE as a marker for atopy. Strikingly, these results were seen only in whole blood but not in plasma that is devoid of erythrocytes that have a high membrane content and PBMC in which ORMDL3 expression is high. In plasma there was a trend for increased dihydroceramide C24:1 for asthma patients (non-allergic and allergic) that did not reach significance, and significantly increased C20 ceramide in patients with allergic asthma compared to non-asthma controls with high eosinophilia, confirming earlier studies that showed high plasma ceramide C20 in exercise-induced wheezing [55].
Together the strong association of non-allergic asthma with decreased sphingolipids is consistent with the original report that associated non-allergic asthma with increased ORMDL3, and with results obtained in the COPSAC study that indicate a sphingolipid-associated childhood asthma endotype with an early onset of symptoms and increased airway resistance by the age of 6 years that is already present in infancy and is associated with 17q21 genetic variants and expression of SPT enzymes [57].
10.5.1 Plasma Sphingolipids Are Higher Than Controls in Non-Allergic and Allergic Asthma
Notably, in our study, plasma sphingolipids in the same population showed significantly increased ceramide C20 in asthma patients with high eosinophils, and a trend for increased dihydroceramides C24:1 in asthma patients with both low and high eosinophils (p < 0.035 by t-test but not significant when correcting for a false discovery rate of 0.05). The strikingly different results obtained in whole blood and plasma are not understood well. They alert on the one hand to the difficulty in comparing studies in different matrixes and cohorts but also to the possibility that decreased whole blood and increased plasma sphingolipids in the same individual reflect different sphingolipid pools originating possibly through compensatory mechanisms.
10.5.2 Risk Alleles Correlate with Whole Blood Sphingolipids
To further investigate the relationship between genetic variations at 17q21 and sphingolipids, we determined five asthma-associated 17q21 SNPs (rs7216389, rs8067378, rs4065275, rs8076131) [11, 14, 20, 25, 58,59,60,61]. SNPs for rs8067378, rs4065275, and rs12603332 did not correlate with whole blood sphingolipids, but risk alleles in the originally identified rs7216389 and rs8076131 that are expression quantitative trait loci (eQTLs) for ORMDL3 correlated with decreased dihydroceramides (C16, C18, C24) and three ceramides (C16, C18,20) in a genotype phenotype specific manner.
10.5.3 Sphingolipid De Novo Synthesis Is Decreased in Asthma and Associated with an Asthma-Risk Genotype
Given the high expression of ORMDL3 in T and B cells, PMBCs were used to assess de novo sphingolipid synthesis by measuring incorporation of stable isotope labeled serine (C13N15) into sphinganine [59]. PBMC from all children with asthma compared to all controls generated significantly less stable labeled sphinganine and sphinganine-1-phosphate (p < 0.05). When these data were stratified by the rs7218369 genotype, de novo synthesized sphinganine and sphinganine-1-phosphate were lowest with the asthma-risk TT genotype (CC + CT > TT, p < 0.05), and risk allele A (GG + AG > AA) for rs8076131 and consistent with decreased sphinganine-1-phosphate found in the COPSAC study by Rago et al. [57].
10.5.4 Increasing Sphingolipid De Novo Synthesis Decreases Airway Reactivity
We also evaluated if increasing sphingolipid synthesis can normalize excessive airway reactivity. These experiments were carried out in Sptlc2+/− mice using the FlexiVent system and in precision cut lung slices (PCLS) from Sptlc2+/− mice. Sphingolipid de novo synthesis was increased with chloride channel inhibitor GlyH101 and with fenretinide, an inhibitor of ceramide desaturase. These substances were chosen based on our previous observation that expression of defective CFTR as well as inhibition of CFTR increase sphingolipid de novo synthesis [62]. Whether the mechanisms behind increased de novo sphingolipid synthesis are related to an increase in intracellular volume induced by inhibition of CFTR is not known [63, 64]. Fenretinide, on the other hand is a drug that inhibits ceramide desaturase, leading primarily to decreased synthesis of ceramides in the de-novo pathway. This inhibition triggers an increase of de novo sphingolipid synthesis and affects recycling pathways [65]. GLYH101 and fenretinide increased sphinganine and dihydroceramides in human bronchoepithelial and smooth muscle cells, and fenretinide also increased ceramides and sphingomyelins.
Experiments were carried out in PCLS from SPTLC+/− mice to specifically assess constriction of small airways in addition to global airway constriction that was evaluated with the FlexiVent system. Incubation with GlyH101 for 15 h decreased methacholine-induced airway contraction in PCLS. Incubation for 15 min, sufficient to inhibit the chloride channel, but insufficient to significantly increase sphingolipid synthesis, had no effect. By the same token, incubation with increasing concentrations of fenretinide for 15 h decreased methacholine-induced contraction in a dose-dependent manner in PCLS and bronchial rings isolated from SPTLC+/− mice.
The results strongly suggest that targeting the imbalance of sphingolipids in asthma to oppose airway hyperresponsiveness can serve as a therapeutic target for asthma [66].
10.6 Conclusion
GWAS studies alerted to a potential role of sphingolipid metabolism in asthma when genetic variation in the asthma locus 17q21 was associated with increased expression of ORMDL3 with childhood asthma. Early ORMDL3 overexpression and knockout studies suggested that inflammation, mainly through activation of the unfolded protein response, could be the functional link to asthma [39]. The identification of ORMDL3 as a regulator of mammalian sphingolipid de novo synthesis 3 years after the initial GWAS study suggested that this large lipid class of membrane constituents and bioactive signaling molecules may also be involved in asthma pathogenesis [27]. Mice that are haploinsufficient for SPTLC2 show that decreased sphingolipid synthesis is sufficient to increase airway reactivity without affecting airway inflammation, mucus production, eosinophilia that characterize allergic asthma. Sphingolipid de novo synthesis is decreased in children with asthma, and whole blood sphingolipds are decreased in children with non-allergic asthma compared to controls. Children with allergic asthma have higher whole blood sphingolipids than children with non-allergic asthma, suggesting an additional effect of allergy on sphingolipid mass. These differences were seen in whole blood but not in plasma. Results obtained in mice indicate that increasing sphingolipid synthesis can normalize airway reactivity. Collectively, the data link increased airway reactivity to decreased sphingolipid synthesis, and genetic variations in 17q21 asthma locus to decreased sphingolipid synthesis and decreased whole blood sphingolipids in non-allergic childhood asthma. The studies establish a role of sphingolipids in asthma and serve as a basis to explore whether increasing sphingolipid de novo synthesis and exogenously increasing metabolites of the de novo pathway affect airway reactivity [67].
Abbreviations
- FEV1/FVC:
-
Forced expiration in the first second/forced vital capacity
- GWAS:
-
Genome Wide Association Study
- ORMDL:
-
Ormdl sphingolipid biosynthesis regulator
- PBMC:
-
Peripheral blood monocytes
- PCLS:
-
Precision cut lung slices
- SNP:
-
Single nucleotide polymorphism
- SPT:
-
Serine palmitoyl transferase
- SPTLC1:
-
Serine palmitoyl transferase long-chain base subunit 1
- SPTLC2:
-
Serine palmitoyl transferase long-chain base subunit 2
- ssSPTa:
-
Small subunit SPT A
References
Zahran, H. S., Bailey, C. M., Damon, S. A., Garbe, P. L., & Breysse, P. N. (2018). Vital signs: Asthma in children—United States, 2001–2016. MMWR. Morbidity and Mortality Weekly Report, 67, 149–155.
Soriano, J. B., Abajobir, A. A., Abate, K. H., Abera, S. F., Agrawal, A., Ahmed, M. B., Aichour, A. N., Aichour, I., Aichour, M. T. E., Alam, K., Alam, N., Alkaabi, J. M., Al-Maskari, F., Alvis-Guzman, N., Amberbir, A., Amoako, Y. A., Ansha, M. G., Antó, J. M., Asayesh, H., … Vos, T. (2017). Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the global burden of disease study 2015. The Lancet Respiratory Medicine, 5, 691–706.
Larsen, G. L. (2001). Differences between adult and childhood asthma. Disease-A-Month, 47, 34–44.
Lötvall, J., Akdis, C. A., Bacharier, L. B., Bjermer, L., Casale, T. B., Custovic, A., Lemanske, R. F., Jr., Wardlaw, A. J., Wenzel, S. E., & Greenberger, P. A. (2011). Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. The Journal of Allergy and Clinical Immunology, 127, 355–360.
Martinez, F. D. (2021). Asthma as a developmental disorder. Annual Review of Developmental Psychology, 3, 229–248.
Pividori, M., Schoettler, N., Nicolae, D. L., Ober, C., & Im, H. K. (2019). Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: Genome-wide and transcriptome-wide studies. The Lancet. Respiratory medicine, 7, 509–522.
Bousquet, J., Jeffery, P. K., Busse, W. W., Johnson, M., & Vignola, A. M. (2000). Asthma. From bronchoconstriction to airways inflammation and remodeling. American Journal of Respiratory and Critical Care Medicine, 161, 1720–1745.
Papadopoulos, N. G., Arakawa, H., Carlsen, K. H., Custovic, A., Gern, J., Lemanske, R., Le Souef, P., Mäkelä, M., Roberts, G., Wong, G., Zar, H., Akdis, C. A., Bacharier, L. B., Baraldi, E., van Bever, H. P., de Blic, J., Boner, A., Burks, W., Casale, T. B., … Zeiger, R. S. (2012). International consensus on (ICON) pediatric asthma. Allergy, 67, 976–997.
Raedler, D., Ballenberger, N., Klucker, E., Böck, A., Otto, R., Prazeres da Costa, O., Holst, O., Illig, T., Buch, T., von Mutius, E., & Schaub, B. (2015). Identification of novel immune phenotypes for allergic and nonallergic childhood asthma. The Journal of Allergy and Clinical Immunology, 135, 81–91.
Zuiani, C., & Custovic, A. (2020). Update on house dust mite allergen avoidance measures for asthma. Current Allergy and Asthma Reports, 20, 50.
Moffatt, M. F., Kabesch, M., Liang, L., Dixon, A. L., Strachan, D., Heath, S., Depner, M., von Berg, A., Bufe, A., Rietschel, E., Heinzmann, A., Simma, B., Frischer, T., Willis-Owen, S. A., Wong, K. C., Illig, T., Vogelberg, C., Weiland, S. K., von Mutius, E., … Cookson, W. O. (2007). Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature, 448, 470–473.
Moffatt, M. F., Gut, I. G., Demenais, F., Strachan, D. P., Bouzigon, E., Heath, S., von Mutius, E., Farrall, M., Lathrop, M., & Cookson, W. (2010). A large-scale, consortium-based genomewide association study of asthma. The New England Journal of Medicine, 363, 1211–1221.
Valette, K., Li, Z., Bon-Baret, V., Chignon, A., Bérubé, J. C., Eslami, A., Lamothe, J., Gaudreault, N., Joubert, P., Obeidat, M., van den Berge, M., Timens, W., Sin, D. D., Nickle, D. C., Hao, K., Labbé, C., Godbout, K., Côté, A., Laviolette, M., … Bossé, Y. (2021). Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK biobank. Communications Biology, 4, 700.
Stein, M. M., Thompson, E. E., Schoettler, N., Helling, B. A., Magnaye, K. M., Stanhope, C., Igartua, C., Morin, A., Washington, C., 3rd, Nicolae, D., Bønnelykke, K., & Ober, C. (2018). A decade of research on the 17q12-21 asthma locus: Piecing together the puzzle. The Journal of Allergy and Clinical Immunology, 142, 749–764.e743.
Zhao, Y. F., Luo, Y. M., Xiong, W., & Wu, X. L. (2014). Genetic variation in ORMDL3 gene may contribute to the risk of asthma: A meta-analysis. Human Immunology, 75, 960–967.
Daya, M., Rafaels, N., Brunetti, T. M., Chavan, S., Levin, A. M., Shetty, A., Gignoux, C. R., Boorgula, M. P., Wojcik, G., Campbell, M., Vergara, C., Torgerson, D. G., Ortega, V. E., Doumatey, A., Johnston, H. R., Acevedo, N., Araujo, M. I., Avila, P. C., Belbin, G., … Barnes, K. C. (2019). Association study in African-admixed populations across the Americas recapitulates asthma risk loci in non-African populations. Nature Communications, 10, 880.
Calışkan, M., Bochkov, Y. A., Kreiner-Møller, E., Bønnelykke, K., Stein, M. M., Du, G., Bisgaard, H., Jackson, D. J., Gern, J. E., Lemanske, R. F., Jr., Nicolae, D. L., & Ober, C. (2013). Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. The New England Journal of Medicine, 368, 1398–1407.
Lluis, A., Schedel, M., Liu, J., Illi, S., Depner, M., von Mutius, E., Kabesch, M., & Schaub, B. (2011). Asthma-associated polymorphisms in 17q21 influence cord blood ORMDL3 and GSDMA gene expression and IL-17 secretion. The Journal of Allergy and Clinical Immunology, 127, 1587–1594.e1586.
Berlivet, S., Moussette, S., Ouimet, M., Verlaan, D. J., Koka, V., Al Tuwaijri, A., Kwan, T., Sinnett, D., Pastinen, T., & Naumova, A. K. (2012). Interaction between genetic and epigenetic variation defines gene expression patterns at the asthma-associated locus 17q12-q21 in lymphoblastoid cell lines. Human Genetics, 131, 1161–1171.
Schedel, M., Michel, S., Gaertner, V. D., Toncheva, A. A., Depner, M., Binia, A., Schieck, M., Rieger, M. T., Klopp, N., von Berg, A., Bufe, A., Laub, O., Rietschel, E., Heinzmann, A., Simma, B., Vogelberg, C., Genuneit, J., Illig, T., & Kabesch, M. (2015). Polymorphisms related to ORMDL3 are associated with asthma susceptibility, alterations in transcriptional regulation of ORMDL3, and changes in TH2 cytokine levels. The Journal of Allergy and Clinical Immunology, 136, 893–903.e814.
James, B., Milstien, S., & Spiegel, S. (2019). ORMDL3 and allergic asthma: From physiology to pathology. The Journal of Allergy and Clinical Immunology, 144, 634–640.
Halapi, E., Gudbjartsson, D. F., Jonsdottir, G. M., Bjornsdottir, U. S., Thorleifsson, G., Helgadottir, H., Williams, C., Koppelman, G. H., Heinzmann, A., Boezen, H. M., Jonasdottir, A., Blondal, T., Gudjonsson, S. A., Jonasdottir, A., Thorlacius, T., Henry, A. P., Altmueller, J., Krueger, M., Shin, H. D., … Stefansson, K. (2010). A sequence variant on 17q21 is associated with age at onset and severity of asthma. European Journal of Human Genetics, 18, 902–908.
Hallmark, B., Wegienka, G., Havstad, S., Billheimer, D., Ownby, D., Mendonca, E. A., Gress, L., Stern, D. A., Myers, J. B., Khurana Hershey, G. K., Hoepner, L., Miller, R. L., Lemanske, R. F., Jackson, D. J., Gold, D. R., O'Connor, G. T., Nicolae, D. L., Gern, J. E., Ober, C., … Martinez, F. D. (2021). Chromosome 17q12-21 variants are associated with multiple wheezing phenotypes in childhood. American Journal of Respiratory and Critical Care Medicine, 203, 864–870.
Hjelmqvist, L., Tuson, M., Marfany, G., Herrero, E., Balcells, S., & Gonzàlez-Duarte, R. (2002). ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biology, 3, Research0027.
Human Genomics. (2015). The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science, 348, 648–660.
Williams, R. D., Wang, E., & Merrill, A. H., Jr. (1984). Enzymology of long-chain base synthesis by liver: Characterization of serine palmitoyltransferase in rat liver microsomes. Archives of Biochemistry and Biophysics, 228, 282–291.
Breslow, D. K., Collins, S. R., Bodenmiller, B., Aebersold, R., Simons, K., Shevchenko, A., Ejsing, C. S., & Weissman, J. S. (2010). Orm family proteins mediate sphingolipid homeostasis. Nature, 463, 1048–1053.
Han, S., Lone, M. A., Schneiter, R., & Chang, A. (2010). Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proceedings of the National Academy of Sciences of the United States of America, 107, 5851–5856.
Gururaj, C., Federman, R., & Chang, A. (2013). Orm proteins integrate multiple signals to maintain sphingolipid homeostasis*. Journal of Biological Chemistry, 288, 20453–20463.
Siow, D., Sunkara, M., Dunn, T. M., Morris, A. J., & Wattenberg, B. (2015). ORMDL/serine palmitoyltransferase stoichiometry determines effects of ORMDL3 expression on sphingolipid biosynthesis. Journal of Lipid Research, 56, 898–908.
Kiefer, K., Carreras-Sureda, A., García-López, R., Rubio-Moscardó, F., Casas, J., Fabriàs, G., & Vicente, R. (2015). Coordinated regulation of the orosomucoid-like gene family expression controls de novo ceramide synthesis in mammalian cells. The Journal of Biological Chemistry, 290, 2822–2830.
Gupta, S. D., Gable, K., Alexaki, A., Chandris, P., Proia, R. L., Dunn, T. M., & Harmon, J. M. (2015). Expression of the ORMDLS, modulators of serine palmitoyltransferase, is regulated by sphingolipids in mammalian cells. The Journal of Biological Chemistry, 290, 90–98.
Green, C. D., Weigel, C., Oyeniran, C., James, B. N., Davis, D., Mahawar, U., Newton, J., Wattenberg, B. W., Maceyka, M., & Spiegel, S. (2021). CRISPR/Cas9 deletion of ORMDLs reveals complexity in sphingolipid metabolism. Journal of Lipid Research, 62, 100082.
Clarke, B. A., Majumder, S., Zhu, H., Lee, Y. T., Kono, M., Li, C., Khanna, C., Blain, H., Schwartz, R., Huso, V. L., Byrnes, C., Tuymetova, G., Dunn, T. M., Allende, M. L., & Proia, R. L. (2019). The Ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. eLife, 8.
Li, S., Xie, T., Liu, P., Wang, L., & Gong, X. (2021). Structural insights into the assembly and substrate selectivity of human SPT-ORMDL3 complex. Nature Structural & Molecular Biology, 28, 249–257.
Wang, Y., Niu, Y., Zhang, Z., Gable, K., Gupta, S. D., Somashekarappa, N., Han, G., Zhao, H., Myasnikov, A. G., Kalathur, R. C., Dunn, T. M., & Lee, C. H. (2021). Structural insights into the regulation of human serine palmitoyltransferase complexes. Nature Structural & Molecular Biology, 28, 240–248.
Davis, D. L., Gable, K., Suemitsu, J., Dunn, T. M., & Wattenberg, B. W. (2019). The ORMDL/Orm-serine palmitoyltransferase (SPT) complex is directly regulated by ceramide: Reconstitution of SPT regulation in isolated membranes. The Journal of Biological Chemistry, 294, 5146–5156.
Wang, S., Robinet, P., Smith, J. D., & Gulshan, K. (2015). ORMDL orosomucoid-like proteins are degraded by free-cholesterol-loading-induced autophagy. Proceedings of the National Academy of Sciences of the United States of America, 112, 3728–3733.
Miller, M., Tam, A. B., Cho, J. Y., Doherty, T. A., Pham, A., Khorram, N., Rosenthal, P., Mueller, J. L., Hoffman, H. M., Suzukawa, M., Niwa, M., & Broide, D. H. (2012). ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proceedings of the National Academy of Sciences of the United States of America, 109, 16648–16653.
Debeuf, N., Zhakupova, A., Steiner, R., Van Gassen, S., Deswarte, K., Fayazpour, F., Van Moorleghem, J., Vergote, K., Pavie, B., Lemeire, K., Hammad, H., Hornemann, T., Janssens, S., & Lambrecht, B. N. (2019). The ORMDL3 asthma susceptibility gene regulates systemic ceramide levels without altering key asthma features in mice. The Journal of Allergy and Clinical Immunology, 144, 1648–1659.e1649.
Miller, M., Rosenthal, P., Beppu, A., Gordillo, R., & Broide, D. H. (2017). Oroscomucoid like protein 3 (ORMDL3) transgenic mice have reduced levels of sphingolipids including sphingosine-1-phosphate and ceramide. The Journal of Allergy and Clinical Immunology, 139, 1373–1376.e1374.
Löser, S., Gregory, L. G., Zhang, Y., Schaefer, K., Walker, S. A., Buckley, J., Denney, L., Dean, C. H., Cookson, W. O. C., Moffatt, M. F., & Lloyd, C. M. (2017). Pulmonary ORMDL3 is critical for induction of Alternaria-induced allergic airways disease. The Journal of Allergy and Clinical Immunology, 139, 1496–1507.e1493.
Siow, D. L., & Wattenberg, B. W. (2012). Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. The Journal of Biological Chemistry, 287, 40198–40204.
Oyeniran, C., Sturgill, J. L., Hait, N. C., Huang, W. C., Avni, D., Maceyka, M., Newton, J., Allegood, J. C., Montpetit, A., Conrad, D. H., Milstien, S., & Spiegel, S. (2015). Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3) expression dysregulates ceramide homeostasis in cells and ceramide exacerbates allergic asthma in mice. The Journal of Allergy and Clinical Immunology, 136, 1035–1046.e1036.
Hojjati, M. R., Li, Z., & Jiang, X. C. (2005). Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochimica et Biophysica Acta, 1737, 44–51.
Chiba, K. (2020). Discovery of fingolimod based on the chemical modification of a natural product from the fungus, Isaria sinclairii. The Journal of Antibiotics, 73, 666–678.
Han, G., Gupta, S. D., Gable, K., Niranjanakumari, S., Moitra, P., Eichler, F., Brown, R. H., Jr., Harmon, J. M., & Dunn, T. M. (2009). Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proceedings of the National Academy of Sciences of the United States of America, 106, 8186–8191.
Fujita, T., Inoue, K., Yamamoto, S., Ikumoto, T., Sasaki, S., Toyama, R., Chiba, K., Hoshino, Y., & Okumoto, T. (1994). Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. The Journal of Antibiotics, 47, 208–215.
Fujita, T., Hirose, R., Yoneta, M., Sasaki, S., Inoue, K., Kiuchi, M., Hirase, S., Chiba, K., Sakamoto, H., & Arita, M. (1996). Potent Immunosuppressants, 2-Alkyl-2-aminopropane-1,3-diols. Journal of Medicinal Chemistry, 39, 4451–4459.
Worgall, T. S., Veerappan, A., Sung, B., Kim, B. I., Weiner, E., Bholah, R., Silver, R. B., Jiang, X. C., & Worgall, S. (2013). Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Science Translational Medicine, 5, 186ra167.
Edukulla, R., Rehn, K. L., Liu, B., McAlees, J. W., Hershey, G. K., Wang, Y. H., Lewkowich, I., & Lindsley, A. W. (2016). Intratracheal myriocin enhances allergen-induced Th2 inflammation and airway hyper-responsiveness. Immunity, Inflammation and Disease, 4, 248–262.
Kim, S. H., Jung, H. W., Kim, M., Moon, J. Y., Ban, G. Y., Kim, S. J., Yoo, H. J., & Park, H. S. (2020). Ceramide/sphingosine-1-phosphate imbalance is associated with distinct inflammatory phenotypes of uncontrolled asthma. Allergy, 75, 1991–2004.
Kowal, K., Żebrowska, E., & Chabowski, A. (2019). Altered sphingolipid metabolism is associated with asthma phenotype in house dust mite-allergic patients. Allergy, Asthma & Immunology Research, 11, 330–342.
Zhakupova, A., Debeuf, N., Krols, M., Toussaint, W., Vanhoutte, L., Alecu, I., Kutalik, Z., Vollenweider, P., Ernst, D., von Eckardstein, A., Lambrecht, B. N., Janssens, S., & Hornemann, T. (2016). ORMDL3 expression levels have no influence on the activity of serine palmitoyltransferase. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 30, 4289–4300.
Perzanowski, M. S., Ono, J. G., Acosta, L. M., Kim, B. I., Divjan, A., Miller, R., Rundle, A., Worgall, S., & Worgall, T. S. (2017). Distinct serum sphingolipid profiles among school-aged children with exercise-induced wheeze and asthma persistence. American Journal of Respiratory and Critical Care Medicine, 195, 1068–1070.
Guo, C., Sun, L., Zhang, L., Dong, F., Zhang, X., Yao, L., & Chang, C. (2021). Serum sphingolipid profile in asthma. Journal of Leukocyte Biology, 110, 53–59.
Rago, D., Pedersen, C. T., Huang, M., Kelly, R. S., Gürdeniz, G., Brustad, N., Knihtilä, H., Lee-Sarwar, K. A., Morin, A., Rasmussen, M. A., Stokholm, J., Bønnelykke, K., Litonjua, A. A., Wheelock, C. E., Weiss, S. T., Lasky-Su, J., Bisgaard, H., & Chawes, B. L. (2021). Characteristics and mechanisms of a sphingolipid-associated childhood asthma endotype. American Journal of Respiratory and Critical Care Medicine, 203, 853–863.
Hao, K., Bossé, Y., Nickle, D. C., Paré, P. D., Postma, D. S., Laviolette, M., Sandford, A., Hackett, T. L., Daley, D., Hogg, J. C., Elliott, W. M., Couture, C., Lamontagne, M., Brandsma, C. A., van den Berge, M., Koppelman, G., Reicin, A. S., Nicholson, D. W., Malkov, V., … Sin, D. D. (2012). Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genetics, 8, e1003029.
Schmiedel, B. J., Seumois, G., Samaniego-Castruita, D., Cayford, J., Schulten, V., Chavez, L., Ay, F., Sette, A., Peters, B., & Vijayanand, P. (2016). 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nature Communications, 7, 13426.
Galanter, J., Choudhry, S., Eng, C., Nazario, S., Rodríguez-Santana, J. R., Casal, J., Torres-Palacios, A., Salas, J., Chapela, R., Watson, H. G., Meade, K., LeNoir, M., Rodríguez-Cintrón, W., Avila, P. C., & Burchard, E. G. (2008). ORMDL3 gene is associated with asthma in three ethnically diverse populations. American Journal of Respiratory and Critical Care Medicine, 177, 1194–1200.
Acevedo, N., Reinius, L. E., Greco, D., Gref, A., Orsmark-Pietras, C., Persson, H., Pershagen, G., Hedlin, G., Melén, E., Scheynius, A., Kere, J., & Söderhäll, C. (2015). Risk of childhood asthma is associated with CpG-site polymorphisms, regional DNA methylation and mRNA levels at the GSDMB/ORMDL3 locus. Human Molecular Genetics, 24, 875–890.
Hamai, H., Keyserman, F., Quittell, L. M., & Worgall, T. S. (2009). Defective CFTR increases synthesis and mass of sphingolipids that modulate membrane composition and lipid signaling. Journal of Lipid Research, 50, 1101–1108.
Thiagarajah, J. R., Ko, E. A., Tradtrantip, L., Donowitz, M., & Verkman, A. S. (2014). Discovery and development of antisecretory drugs for treating diarrheal diseases. Clinical Gastroenterology and Hepatology, 12, 204–209.
Muanprasat, C., Sonawane, N. D., Salinas, D., Taddei, A., Galietta, L. J., & Verkman, A. S. (2004). Discovery of glycine hydrazide pore-occluding CFTR inhibitors: Mechanism, structure-activity analysis, and in vivo efficacy. The Journal of General Physiology, 124, 125–137.
Wang, H., Maurer, B. J., Liu, Y. Y., Wang, E., Allegood, J. C., Kelly, S., Symolon, H., Liu, Y., Merrill, A. H., Jr., Gouazé-Andersson, V., Yu, J. Y., Giuliano, A. E., & Cabot, M. C. (2008). N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Molecular Cancer Therapeutics, 7, 2967–2976.
Lam, M., & Bourke, J. E. (2020). Solving the riddle: Targeting the imbalance of sphingolipids in asthma to oppose airway hyperresponsiveness. American Journal of Respiratory Cell and Molecular Biology, 63, 555–557.
Johnson, E. L., Heaver, S. L., Waters, J. L., Kim, B. I., Bretin, A., Goodman, A. L., Gewirtz, A. T., Worgall, T. S., & Ley, R. E. (2020). Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nature Communications, 11, 2471.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive licence to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Worgall, T.S. (2022). Sphingolipids and Asthma. In: Jiang, XC. (eds) Sphingolipid Metabolism and Metabolic Disease. Advances in Experimental Medicine and Biology, vol 1372. Springer, Singapore. https://doi.org/10.1007/978-981-19-0394-6_10
Download citation
DOI: https://doi.org/10.1007/978-981-19-0394-6_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-0393-9
Online ISBN: 978-981-19-0394-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)