
Abstract
Macroautophagy (autophagy) is an evolutionally conserved cytoplasmic degradation system in which varieties of materials are sequestered by a double membrane structure, called autophagosome, and delivered to the lysosomes for the degradation. Due to the wide varieties of targets, autophagic activity is essential for cellular homeostasis and survival. Accumulating evidences suggest that the activity of autophagy decreases with age, whereas several interventions which induce activation of autophagy promote longevity and prevents age-related diseases. Here we summarize recent progress regarding the role of autophagy in animal aging and life span regulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Overview of Autophagy
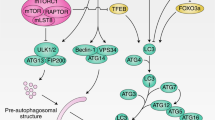
Autophagy is a conserved lysosomal degradation essential for cellular homeostasis and stress resistance. Autophagy can be classified into three distinct types depending how cytoplasmic materials are delivered to lysosomes: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Among these, this review particularly focuses on macroautophagy, since its roles and regulation in aging and age-related diseases are well documented. Macroautophagy, hereafter referred to as autophagy, is a catabolic process targeting wide varieties of cellular contents. Autophagy occurs at basal level in normal condition but is accelerated by several stresses such as starvation, accumulation of abnormal proteins, organelle damage, and pathogen infection. During autophagy, a small cisterna, called isolation membrane (also called isolation membrane or phagophore), elongates and surrounds a portion of cytoplasm to form a double-membraned structure, called the autophagosome. Autophagosomes are then transported and fuse with lysosomes to form autolysosomes for the digestion of sequestered contents (Fig. 11.1). Autophagy is originally considered to be a bulk and nonselective degradation system. But subsequent studies show autophagy selectively degrades cargos and by doing so contribute to the intracellular homeostasis. Many cargos such as damaged mitochondria, damaged lysosomes, invading bacteria, lipid droplet, and aggregated proteins are selectively sequestered and degraded by specific selective autophagy, called mitophagy, lysophagy, xenophagy, lipophagy, and aggrephagy, respectively. During autophagy, several autophagy-related (ATG) genes are engaged sequentially in a highly regulated manner. Genetic studies in yeast have identified more than 30 ATG genes that are required for autophagy, most of which are conserved from yeast to mammals. Essential ATG genes are organized into at least six functional groups that allow for the nucleation, elongation, maturation, and fusion of the autophagosome. These functional groups are the Atg1/ULK initiation complex, the class III PI3 kinase nucleation complex, Atg9 vesicles, the phosphatidylinositol 3-phosphate (PI3P)-binding Atg18/Atg2 complex, the Atg5-Atg12 conjugation system, and the Atg8/LC3-PE (Atg8/LC3-phosphatidylethanolamine) conjugation system. The first step of autophagy initiates from the activation of Atg1/ULK complex, which leads to the formation of isolation membrane. The next step involves membrane nucleation by the class III Vps34/PI3 kinase nucleation complex (consisting of Vps34, Atg6/Beclin1, Atg14L, and Vps15/p150) via the production of PI3P, to start the formation of a double-membrane structure, isolation membrane (or phagophore). In mammals, the isolation membrane originates from the endoplasmic reticulum (ER), the mitochondrial contact site, and from others including the Golgi, endosomes, and plasma membrane (Chan and Tang 2013; Hamasaki et al. 2013). To start elongation, the isolation membrane recruits the PI3P-binding complex consisting of Atg18/WIPI and Atg2, which regulates the distribution of Atg9, a transmembrane protein that has been proposed to deliver lipids to the isolation membrane and the growing autophagosome. During the next step, the isolation membrane expands into a double-membrane structure called the autophagosome. Autophagosome elongation is dependent on two ubiquitin-like conjugation systems, the Atg5-Atg12 conjugation system and the Atg8/LC3-PE. In Atg5-Atg12 conjugation system, Atg7 and Atg10 (E1- and E2-like enzymes, respectively) conjugate Atg12 to Atg5, and this complex associates with Atg16. Then, the Atg12–Atg5 conjugate promotes the conjugation of phosphatidylethanolamine (PE) to cytosolic Atg8/LC3, which is formed by the cleavage of the ubiquitin-like protein Atg8/LC3 by the protease Atg4. During this process, PE-conjugated LC3 associates with the autophagosomal membrane, and therefore LC3 is most commonly used as an experimental marker of autophagosomes (Fujita et al. 2008; Kabeya 2000; Mizushima and Levine 2010). The autophagosome eventually matures into a closed cargo-containing vesicle, which then fuses with the lysosome to become the autolysosome, and its contents are finally degraded for recycling. Autophagosome-lysosome fusion step is mediated by HOPS complex, phosphoinositides, Rab proteins, and SNEREs. In addition, autophagosome lysosome fusion step is negatively regulated by Rubicon which comprises different class III PI3K complex including Beclin1, UVRAG, Vps34, and Vps15 (Matsunaga et al. 2009). The detailed molecular mechanism of autophagosome formation and autophagosome-lysosome fusion is summarized in recent specific review paper (Nakamura and Yoshimori 2017; Nakatogawa 2020). As described in the following section, recent genetic evidence indicates that autophagy has a crucial role in the regulation of animal life span. The basal level of autophagic activity is elevated in many longevity paradigms, and importantly its activity is required for life span extension. On the other hand, the activity of autophagy decreases with age in many organisms. Pharmacological treatments have been shown to extend life span through the activation of autophagy, indicating autophagy could be a potential and promising target to modulate animal life span.

Overview of autophagy. Upon induction of autophagy by stress, cytoplasmic materials are sequestered by a double-membraned structure, called an autophagosome. These autophagosomes fuse with lysosomes to become autolysosomes, in which the sequestered cargos are degraded and recycled for the maintenance of cellular homeostasis. Autophagy can be divided into several steps: formation of the isolation membrane (nucleation), elongation of the isolation membrane (elongation), completion and transport of the autophagosome (maturation), docking and fusion between autophagosome and lysosome (fusion), and degradation of the cargos inside the autolysosome (degradation)
2 Activation of Autophagy Is One of the Convergent Mechanisms of Animal Longevity
Aging represents the functional deterioration of an organism. For a long time, aging is not considered as a tightly regulated process. During last twenty decades, the evolutionally conserved molecular mechanisms which delay animal aging and extend life span have been identified using several model organisms, including yeast, worms, fly, and mice. These pathways, for instance, include reduced insulin/IGF-1 signaling, dietary restriction, reduced mTOR signaling, germline removal, and reduced mitochondrial respiration. Extensive efforts to identify the downstream mechanism in each longevity pathway reveals that numerous but different sets of factors or biological processes mediate in each longevity pathways, although some factors work in common. Notably, recent studies suggest that autophagy is one of the convergent downstream mechanisms of all these longevity paradigms. The activity of autophagy is elevated by several transcription factors in long-lived animals and is required for their longevity (Fig. 11.2, Table 11.1).

Activation of autophagy is one of the convergent mechanisms of several longevity pathways. Several transcription factors such as HLH-30/TFEB and MML-1/Mondo are commonly activated in multiple longevity pathways. These regulate autophagic activity and extend its life span
Reduced insulin/IGF-1 signaling has been shown to extend the life span in several species (Kenyon 2010). The first connection between autophagy and longevity has been reported in this insulin/IGF-1 signaling pathway in C. elegans (Meléndez et al. 2003). In long-lived daf-2 (encoding C. elegans insulin/IGF-1 receptor) mutants, autophagy activity is elevated, as reflected by increased autophagic vesicles by electron microscopy and GFP::LGG-1 (a homolog of LC3 in C. elegans) puncta, a C. elegans autophagosome marker. Importantly, RNAi knockdown of bec-1/Beclin1 shortens daf-2 life span, indicating that the activity of autophagy is essential for daf-2 longevity. Reduction of insulin/IGF-1 signaling pathway extends the life span in Drosophila and mice as well. In Drosophila, life span extension with deletion of the insulin receptor substrate chico was completely abrogated by the knockdown of Atg5 (Bjedov et al. 2020). Moreover, human centenarian has mutations in this pathway, suggesting that this longevity pathway seems to be conserved up to human. The exact mechanisms of autophagic activation in daf-2 mutants are unclear, but they could include posttranslational and transcriptional regulation. For instance, the catalytic subunit of the energy regulator AMPK (AAK-2 in C. elegans) is essential for life span extension in daf-2 mutants (Apfeld et al. 2004), and it regulates autophagy in both C. elegans and mammals (Egan et al. 2011). It is possible that Ampk/aak-2-regulated autophagy contributes to life span, since AMPK overexpression is sufficient to increase the longevity of Drosophila in an Atg1/Ulk1/unc-51-dependent manner (Ulgherait et al. 2014). daf-2 mutants also displays lower expression of key autophagy-related genes. They require a master regulator of autophagy and lysosomal biogenesis, hlh-30/TFEB, for their long life span, to display nuclear-localized HLH-30 and have elevated levels of several autophagy-related and lysosomal genes (Lapierre et al. 2013). HLH-30 translocates to the nucleus of intestinal cells following knockdown of mTOR and daf-2 (Lapierre et al. 2013). Since mTor RNAi inhibition in daf-2 mutants do not extend C. elegans life span in an additive manner (Vellai et al. 2003), they mediate life span extension through at least partially overlapping mechanisms. What is the autophagy cargo relevant for longevity conferred by reduced insulin/IGF-1 signaling? A recent study suggested that mitophagy is induced in daf-2 mutants because mitochondria accumulate upon bec-1 and mitophagy gene inhibition and daf-2 mutants require mitophagy genes, including adaptor protein Bnip3/dct-1, the E3 ligase Park/pdr-1, and the kinase pink-1 for full life span extension (Palikaras et al. 2015).
Dietary restriction is one of the most prominent ways to slow aging and extend the life span in many species. Dietary restriction was first observed to slow down aging in rat about 100 years ago. Since then the beneficial effects to extend life span was confirmed in numerous species including yeast, worms, fly, fish, dogs, mice, and apes (Mair and Dillin 2008). Multiple molecular mechanisms have been proposed to mediate the effect of dietary restriction on longevity, including TOR and insulin/IGF-1 signaling. The life span of the budding yeast S. cerevisiae can be measured by two methods: replicative life span (RLS) and chronological life span (CLS). Both RLS and CLS can be modulated in S. cerevisiae by reducing nutrients in the growth media (Smith et al. 2007). One method to induce dietary restriction is by amino acid limitation, which has been shown to extend CLS and also induce autophagy (Alvers et al. 2009a). Similarly, the inhibition of the nutrient sensor mTOR by rapamycin (a compound discovered in a soil bacterium on the Easter Island Rapa Nui) increases CLS and autophagy, and autophagy genes are required for rapamycin to extend life span (Alvers et al. 2009b). However, the role of autophagy in yeast aging seems complex. Intriguingly deletion of only ATG15, but not other autophagy genes tested, blocks RLS extension induced by glucose limitation (Tang et al. 2008) which is another method of dietary restriction in yeast. Several models of dietary restriction exist in C. elegans (Greer and Brunet 2009), including eat-2 mutants, which carry an acetylcholine receptor mutation that impairs pharyngeal pumping and reduces food intake. eat-2 mutants show increased numbers of GFP::LGG-1 in hypodermal seam cells. The longevity of eat-2 mutants are also abolished when several autophagy genes including unc-51/ULK1, bec-1/Beclin1, vps-34, atg-18, and atg-7 are inactivated (Hansen et al. 2008; Jia and Levine 2007). In eat-2 animals, some autophagy genes are transcriptionally induced by several transcription factors, including hlh-30, pha-4, and nhr-62 (Hansen et al. 2008; Heestand et al. 2013; Lapierre et al. 2013). Recently it has been shown that intestinal autophagy is essential for life span extension during dietary restriction (Gelino et al. 2016). How these transcription factors contribute to activation of autophagy and longevity in spatial and temporal manners need to be clarified in future study. Similar to yeast, in C. elegans, life span extension induced by dietary restriction may be at least partly mediated through TOR, because TOR inhibition in eat-2 mutants does not further extend life span (Hansen et al. 2007). In line with this, similar to dietary-restricted worms, the inhibition of TOR extends life span in a transcription factor pha-4- or hlh-30-dependent manner (Lapierre et al. 2013; Sheaffer et al. 2008). In Drosophila, rapamycin treatment results in a modest life span extension, and this effect requires the autophagy gene Atg5 (Bjedov et al. 2010), suggesting that the reduction of TOR extends the life span in Drosophila at least partially through autophagy similar to yeast and worms. Rapamycin extends mammalian life span and ameliorates neurodegeneration and osteoarthritis in mice (Harrison et al. 2009; Li et al. 2014). Other groups also confirmed the positive effect of rapamycin on the life span in mice using different genetic backgrounds (Lamming et al. 2013). However, the contribution of autophagy to these mice is unclear.
Reproduction is negatively correlated with longevity in many species. Removal of germline stem cells by laser microsurgery or genetic mutation extends life span in C. elegans and Drosophila. In worms, temperature-sensitive mutant, glp-1(e2141), which encodes C. elegans Notch receptor shows the reduction of germline stem cells and life span extension. It has been shown that the numbers of GFP::LGG-1 puncta are increased in germline-deficient glp-1 animal and autophagy genes are essential for their longevity (Lapierre et al. 2011). In germline-deficient animal, several transcription factors including hlh-30, mml-1/mxl-2, and pha-4 have been shown to induce autophagy genes (Lapierre et al. 2011, 2013; Nakamura et al. 2016). Interestingly, intestine-specific knockdown of autophagy genes abolishes glp-1 longevity, while it is not the case in daf-2 mutants, indicating critical differences of autophagy regulation in individual tissues between conserved longevity paradigms (Chang et al. 2017). glp-1 animals have increased lipase activity, and lipl-4 is required for glp-1 animals to live long (Wang et al. 2008). Lipl-4 overexpression increases autophagy and life span, and this animal requires autophagy gene for longevity (Lapierre et al. 2011). These studies indicate lipid turnover by autophagy is essential for longevity.
The free radical theory proposes that aging is the cumulative result of oxidative damages to cells and tissues over time. These molecular damages are caused by reactive oxygen species (ROS) which is generated primarily from mitochondrial respiration. Although oxidative damages increase with age, it is still unclear if this is indeed a causative effect to organism aging. Importantly, reduced mitochondrial respiration is known to extend the life span of many organism from yeast to mice (Hur et al. 2010; Kirchman et al. 1999). In worms, the reduction of electron transport chain components extends life span, when they are inhibited during larval stages. Several mitochondrial mutants including ubiquinone synthetase mutant clk-1 and iron-sulfur mutant isp-1 also show longevity. Larval inhibition of autophagy genes (vps-34, atg-18, and lgg-1) specifically shortens the life span of clk-1 and isp-1 mutants (Lapierre et al. 2013; Tóth et al. 2008). Consistent with a role for autophagy, these mutants display increased numbers of GFP::LGG-1 punctae in the hypodermal cells during larval stage L3 (Lapierre et al. 2013). Frataxin is a nuclear-encoded mitochondrial protein involved in the biogenesis of iron-sulfur (Fe-S) cluster-containing proteins and also involved in the function of the mitochondrial respiratory chain. Partial depletion of frh-1 has been shown to increase autophagic activity and extends the life span of wild-type animals, but not bec-1 mutants (Schiavi et al. 2013). Moreover, a recent report showed that the longevity of frh-1 mutants requires mitophagy genes for its longevity (Schiavi et al. 2015).
In addition to the role of autophagy in longevity, the loss of autophagic activity has been shown to cause premature aging phenotypes in many species. An unbiased screening for genes involved in chronological life span in yeast identified several short-lived mutants which have mutation in macroautophagy genes (Matecic et al. 2010). Decreased life span is also observed in C. elegans Atg1/unc-51, Atg7, Atg18, and Beclin1/bec-1 loss of function mutants (Tóth et al. 2008). Similar findings are reported in Drosophila as well (Simonsen et al. 2008). Although whole-body knockout of Atg genes in mice leads to postnatal death, conditional tissue-specific knockouts of Atg7 or Atg5 show several age-associated phenomena including aggregation of inclusion bodies in neurons, accumulation of lysosomes containing lipofuscin pigments, disorganized mitochondria, increased protein oxidation, and decreased muscle mass (Rubinsztein et al. 2011).
3 Autophagic Activity Declines with Age
Autophagic activity is known to decrease with age in several species (Chang et al. 2017; Chapin et al. 2015; Del Roso et al. 2003; Donati et al. 2001; Uddin et al. 2012). Interestingly, the study using centenarians shows the general increase of autophagy genes (Xiao et al. 2018) and also increased circulating Beclin1 (Emanuele et al. 2014). Based on these correlations between autophagy and aging, it is reasonable to test if the forced activation of autophagy suffices to extend animal life span (Table 11.1). Indeed, the overexpression of HLH-30/TFEB, a master regulator of autophagy and lysosomal biogenesis, extends worm life span (Lapierre et al. 2013). Consistent with this, the inhibition of HLH-30/TFEB nuclear export or the treatment of HLH-30/TFEB agonists have been recently shown to extend the life span in worms and mitigate metabolic syndromes in mice (Silvestrini et al. 2018; Wang et al. 2017). In addition, ATG5 overexpression or Beclin1 gain of function in mice extends life span (Fernández et al. 2018; Pyo et al. 2013). Moreover, the neuronal overexpression of Atg8 or mild upregulation of Atg1 is sufficient to extend life span in Drosophila (Bjedov et al. 2020; Simonsen et al. 2008). Although the molecular mechanism underlying age-dependent autophagic decline has remained elusive, the recent study suggests that age-dependent accumulation of autophagy negative regulators; Rubicon is one of such mechanisms (Matsunaga et al. 2009; Nakamura et al. 2019). Rubicon is increased in C. elegans, Drosophila, and mouse tissues such as the liver and kidney, and importantly knockdown of Rubicon increases life span in an autophagy-dependent manner and/or ameliorate several age-associated phenotypes, such as kidney fibrosis and α-synuclein pathology in these animals (Fig. 11.3). Specific microRNA is also involved in the autophagic decline across tissues during aging in C. elegans (Zhou et al. 2019b). mir-83 is upregulated by the transcription factor hsf-1/HSF1 in the intestine during aging and transported across tissues. mir-83 disrupts autophagy in intestines and muscles by downregulating lysosomal calcium channel cup-5/TRPML1 essential for the induction of autophagy. Lysosomal morphology and activity decrease age in C. elegans and are regulated in many longevity pathways (Sun et al. 2020), which partly explain autophagic decline with age. In addition, a key autophagy-negative regulator, the mTOR activity, has been shown to increase over time in some mouse tissues, which might also affect age-dependent autophagic impairment (Baar et al. 2016). On the other hand, another upstream regulator of autophagy, the AMPK activity, is constant during aging, but AMPK activation by an activator, such as AICAR or exercise, is blunted by aging (Reznick et al. 2007).

The increase of Rubicon is a signature of aging and autophagic decline. Rubicon expression increases with age. This causes a decrease in autophagic activity, which curtails life span and leads to aging. Knockdown of Rubicon increases life span and improves age-associated phenotypes in many species
4 Autophagy and Age-Related Neurodegenerative Diseases
Autophagy is essential to prevent many age-associated diseases. Among them autophagy activity in neuronal cells is particularly essential, since neuronal cells are post-mitotic and they cannot segregate and dilute the proteotoxic damage. Therefore, proteostasis function by autophagic activity is essential to prevent the accumulation of several aggregation-prone proteins which lead to neurodegeneration. Indeed, the impairment of the autophagy pathway is involved in many age-associated neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson disease (PD) (Hara et al. 2006; Komatsu et al. 2006; Mizushima et al. 2008).
AD involves the deposition of extracellular β-amyloid (Aβ) plaque and intracellular neurofibrillary tangles containing hyperphosphorylated tau. Both Aβ and Tau are known to be substrates of autophagy. The loss of autophagy by conditional knockout of ATG7 in mouse brain leads to phosphor-tau accumulation (Inoue et al. 2012). On the other hands, autophagic activation decreases tau levels (Krüger et al. 2012). The heterozygous deletion of Beclin1 increases both intracellular and extracellular Aβ (Pickford et al. 2008). The accumulation of autophagic vacuoles is observed in AD neurons (Nixon et al. 2005), suggesting that autophagy is impaired in this pathological condition.
PD is characterized by the accumulation of α-synuclein. α-synuclein overexpression impairs autophagy due to mislocalization of ATG9 (Winslow et al. 2010). Accumulation and propagation of α-synuclein aggregation in the brain can be suppressed by autophagy activation by an autophagy-negative regulator, Rubicon (Nakamura et al. 2019). The loss of function of Parkin, an E3 ubiquitin ligase, and PINK1, a mitochondrial protein kinase, causes autosomal-recessive and sporadic juvenile onset PD (Kitada et al. 1998; Valente et al. 2001, 2004). Parkin and PINK1 are known to regulate mitophagy to clear the damaged mitochondria in the cell culture. PINK1 recruits Parkin on damaged mitochondria to induce mitophagy. However, PINK1 and Parkin knockout mice fail to develop PD-associated phenotypes (Gautier et al. 2008; Palacino et al. 2004), suggesting that PD could be caused by other mechanism.
5 Molecular Mechanism Regulating Autophagy and Longevity
In many cases, autophagic activation at the transcript level seems essential for longevity. Several autophagy and lysosomal genes are regulated by different transcription factors. The bHLH transcription factor, TFEB originally identified as a master regulator of lysosomal biogenesis, is subsequently shown to regulate autophagy and fat metabolism (Sardiello et al. 2009; Settembre et al. 2011, 2013). TFEB is known to be negatively regulated by nutrient sensor mTOR. At nutrient-rich condition, TFEB is phosphorylated on lysosome. Phosphorylated TFEB is bound to 14-3-3 and is mainly localized on cytosol. Upon starvation, TOR becomes inactivated, and TFEB is then dephosphorylated and translocated in the nucleus to initiate the transcription of target genes (Martina et al. 2012; Settembre et al. 2012). The dephosphorylation of TFEB is mediated by calcineurin activated upon calcium efflux through TRPML1 (Medina et al. 2015). The C. elegans homolog of TFEB, HLH-30, has been shown to regulate genes involved in autophagy and lysosomal function. Essentially, HLH-30 is translocated to the nucleus by inhibition of insulin/IGF-1 signaling, mitochondrial respiration, TOR signaling, translation, and germline removal and is required for their longevity (Lapierre et al. 2013). Moreover, the overexpression and activation of hlh-30 is sufficient to extend the life span of wild-type animals (Lapierre et al. 2013). These results indicate that HLH-30/TFEB is a master transcription factor downstream of many longevity pathways possibly through the transcriptional activation of target genes involved in autophagy and lysosomal function.
Other bHLH transcription factor complex, MML-1/MXL-2, has been identified as a novel regulator of longevity (Johnson et al. 2014; Nakamura et al. 2016). MML-1/MXL-2 belongs to Myc and Mondo family member and their homologs, MondoA/MLX or ChREBP/MXL functions as a glucose sensor. MML-1/MXL-2 is required for the longevity conferred by germline removal, reduced insulin/IGF-1 signaling, reduced mitochondrial respiration, and reduced TOR signaling in C. elegans. Interestingly, the inhibition of MML-1/MXL-2 impairs HLH-30 nuclear localization and activation of autophagy in germline less long-lived animals, glp-1. This is partly through the regulation of lars-1, a positive regulator of TOR signaling. Interestingly, in glp-1, MML-1/MXL-2 and HLH-30 mutually regulated each other. Comprehensive transcriptome analysis reveals that they have many shared target genes including lysosomal genes but also have preferential targets. Some autophagy genes including atg-2/ATG2, atg-9/ATG9, and epg-9/ATG101 are preferentially regulated by MML-1/MXL-2, while unc-51/ULK1 and lgg-1/LC3 are regulated by HLH-30. Thus, they might distribute the responsibilities to reinforce autophagy and longevity in germline less animals.
In C. elegans, Drosophila, and mouse, the reduction of insulin/IGF-1 signaling ultimately activates DAF-16/FOXO function and extends life span. DAF-16 could regulate autophagy partly through regulating autophagy and lysosomal genes (Li and Zhang 2016). Consistent with this, the overexpression of DAF-16 increases the number of autophagosomes during bacterial infection (Jia et al. 2009). However, although daf-2 and daf-16 double mutants do not show longevity, these mutants still have increased numbers of autophagosomes (Hansen et al. 2008). Possibly other factors compensate the activity of autophagy, or DAF-16 regulates autophagy at other timing. Interestingly, DAF-16 interacts with HLH-30 and cooperates target gene expression, promoting stress resistance and longevity (Lin et al. 2018). Other forkhead transcription factor, PHA-4/FOXA, binds to the promoter region of unc-51/Ulk1, bec-1/Becn1, and lgg-1/LC3 which work in the early stage of autophagosome formation and upregulates these genes in worms, leading to autophagic activation. pha-4 is required for the longevity by mTOR inhibition, germline removal, and calorie restriction through the activation of autophagy (Hansen et al. 2008; Lapierre et al. 2011).
6 Intervention of Aging via Modulating Autophagy
In addition to the abovementioned rapamycin, several pharmacological treatments have been shown to extend animal life span and health span through the activation of autophagy (Table 11.2). Administration of a natural polyamine, spermidine, is beneficial for the health in a number of species and extends the life span of yeast, worms, flies, and mice (Eisenberg et al. 2009, 2016). The survival of cultured mammalian cells is also promoted by treatment with spermidine, and this is accompanied by epigenetic hypoacetylation of histone H3 via the inhibition of histone acetyltransferase activity. This, in turn, correlates with the transcriptional upregulation of multiple autophagy-related genes, including Atg5 and Lc3/ATG8/lgg-1/2 (Eisenberg et al. 2009). In keeping with this observation, spermidine fails to extend the life span of C. elegans subjected to bec-1 RNAi, whereas it increases the expression of DsRed::LGG-1 (Eisenberg et al. 2009) in a sir-2-independent fashion (Morselli et al. 2011). In flies, spermidine alters the expression of autophagy markers, protects against age-induced memory loss in an autophagy-dependent manner and extends the life span in an Atg7-dependent manner (Gupta et al. 2013). Collectively, these data suggest that the positive effects of spermidine on health and longevity are mediated, at least in part, via autophagy induction.
Resveratrol is a naturally occurring polyphenolic compound found in grapes and an activator of the NAD-dependent histone deacetylase sirtuin (SIRT1). Administration of resveratrol is known to extend the life span of several model organisms (Park et al. 2013). The life spans of yeast, worms, and flies can be extended by the overexpression and/or pharmacological activation of SIRT1, and the life span of mice is extended by brain-specific overexpression of SIRT1 (Giblin et al. 2014). Especially, the life span extension in C. elegans seems to be dependent on autophagy since resveratrol fails to extend the life span of bec-1(RNAi)-treated animals. Additionally, resveratrol increases DsRed::LGG-1 levels in wild-type animals but not in SIR-2.1 loss-of-function mutants (Morselli et al. 2010). These observations are in agreement with findings in mammalian cells, where the pharmacological activation of SIRT1 by resveratrol treatment stimulates autophagic flux. In contrast, SIRT1−/− fibroblasts show suppressed autophagy during starvation and show elevated acetylation of key autophagy protein (Lee et al. 2008). Thus, SIRT1 promotes autophagy via the deacetylation of proteins involved in the autophagy pathways. Recent evidence suggests that Beclin1 is acetylated by p300 and deacetylated by SIRT1 (Sun et al. 2015). Acetylated Beclin1 recruits Rubicon, leading to the inhibition of autophagosome maturation, and SIRT1 might promote autophagy through deacetylation of Beclin1. SIRT1 is also known to co-immunoprecipitate with ATG5, ATG7, and LC3 and deacetylate these in vitro, and these interactions could be also essential for autophagy regulation (Lee et al. 2008).
Urolithin A as a first-in-class natural compound that induces mitophagy both in vitro and in vivo following oral consumption. In C. elegans, urolithin A prevents the accumulation of dysfunctional mitochondria with age and extends life span (Ryu et al. 2016). Likewise, Urolithin A prolongs normal activity during aging in C. elegans, including mobility and pharyngeal pumping, while maintaining mitochondrial respiratory capacity. These effects are observed in rodents, where Urolithin A improves exercise capacity in two different mouse models of age-related decline of muscle function, as well as in young rats.
Tomatidine, a natural compound abundant in unripe tomatoes, inhibits age-related skeletal muscle atrophy in mice. Recent study shows that tomatidine extends life span and healthspan in C. elegans (Fang et al. 2017). Tomatidine improves many C. elegans behaviors related to healthspan and muscle health, including increased pharyngeal pumping, swimming movement, and reduced percentage of severely damaged muscle cells. Microarray, imaging, and behavioral analyses reveal that tomatidine maintains mitochondrial homeostasis by modulating mitochondrial biogenesis and PINK-1/DCT-1-dependent mitophagy. A detailed analysis shows tomatidine induces mitochondrial hormesis by mildly inducing ROS production, which in turn activates the SKN-1/Nrf2 pathway and possibly other cellular antioxidant response pathways, followed by increased mitophagy. This mechanism occurs in C. elegans, primary rat neurons, and human cells.
The biguanide metformin extends healthspan and life span in several models including C. elegans and mice and the risk of dementia in humans (Barzilai et al. 2016). Metformin has pleiotropic roles and inhibits mitochondrial respiration, mTOR, and activates AMPK, leading to the activation of autophagy (Song et al. 2015; Xie et al. 2011). Whether metformin-mediated life span extension requires autophagy activity needs to be clarified.
7 Conclusion
Accumulating evidence suggest that the activation of autophagy is one of the common mechanisms of many longevity paradigms. Moreover, the forced activation of autophagy suffices to extend life span and ameliorates age-associated phenotype in model organism, implying autophagy activation is one of the promising methods to delay human aging. However, actual roles of autophagy contributing to longevity need to be clarified further. It is also critical to establish the method to measure autophagic activity in human. In addition, recent evidence also suggests that autophagy activation in some context becomes detrimental rather than beneficial. For instance, too much upregulation of autophagy genes becomes rather detrimental (Bjedov et al. 2020) and elevated autophagy shortens life span when the mitochondria permeability increases (Zhou et al. 2019a). The neuronal inhibition of some autophagy genes during post reproductive period extends life span (Wilhelm et al. 2017). In addition, autophagy inhibition is necessary in adipocyte for proper function (Yamamuro et al. 2020). Further studies to reveal tissue and timing-specific roles of autophagy are required to make autophagy modulation a promising antiaging therapy in the next decades.
References
Alvers AL, Fishwick LK, Wood MS, Hu D, Chung HS, Dunn WA Jr, Aris JP (2009a) Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 8:353–369
Alvers AL, Wood MS, Hu D, Kaywell AC, Dunn WA Jr, Aris JP (2009b) Autophagy is required for extension of yeast chronological life span by rapamycin. Autophagy 5:847–849
Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18(24):3004–3009
Baar EL, Carbajal KA, Ong IM, Lamming DW (2016) Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell 15:155–166
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA (2016) Metformin as a tool to target aging. Cell Metab 23:1060–1065
Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11:35–46
Bjedov I, Cochemé HM, Foley A, Wieser D, Woodling NS, Castillo-Quan JI, Norvaisas P, Lujan C, Regan JC, Toivonen JM et al (2020) Fine-tuning autophagy maximises lifespan and is associated with changes in mitochondrial gene expression in Drosophila. PLoS Genet 16:e1009083
Chan SN, Tang BL (2013) Location and membrane sources for autophagosome formation - from ER-mitochondria contact sites to Golgi-endosome-derived carriers. Mol Membr Biol 30:394–402
Chang JT, Kumsta C, Hellman AB, Adams LM, Hansen M (2017) Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. elife 6:e18459
Chapin HC, Okada M, Merz AJ, Miller DL (2015) Tissue-specific autophagy responses to aging and stress in C. elegans. Aging 7:419–434
Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E (2003) Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol 38:519–527
Donati A, Cavallini G, Paradiso C, Vittorini S, Pollera M, Gori Z, Bergamini E (2001) Age-related changes in the autophagic proteolysis of rat isolated liver cells: effects of antiaging dietary restrictions. J Gerontol A Biol Sci Med Sci 56:B375–B383
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R et al (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461
Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L et al (2009) Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314
Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A et al (2016) Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 22:1428–1438
Emanuele E, Minoretti P, Sanchis-Gomar F, Pareja-Galeano H, Yilmaz Y, Garatachea N, Lucia A (2014) Can enhanced autophagy be associated with human longevity? Serum levels of the autophagy biomarker beclin-1 are increased in healthy centenarians. Rejuvenation Res 17:518–524
Fang EF, Waltz TB, Kassahun H, Lu Q, Kerr JS, Morevati M, Fivenson EM, Wollman BN, Marosi K, Wilson MA et al (2017) Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci Rep 7:46208
Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang W-C et al (2018) Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558:136–140
Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T (2008) An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 19:4651–4659
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364–11369
Gelino S, Chang JT, Kumsta C, She X, Davis A, Nguyen C, Panowski S, Hansen M (2016) Intestinal autophagy improves healthspan and longevity in C. elegans during dietary restriction. PLoS Genet 12:e1006135
Giblin W, Skinner ME, Lombard DB (2014) Sirtuins: guardians of mammalian healthspan. Trends Genet 30:271–286
Greer EL, Brunet A (2009) Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 8:113–127
Gupta VK, Scheunemann L, Eisenberg T, Mertel S, Bhukel A, Koemans TS, Kramer JM, Liu KSY, Schroeder S, Stunnenberg HG et al (2013) Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci 16:1453–1460
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393
Hansen M, Taubert S, Crawford D, Libina N, Lee S-J, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6:95–110
Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C (2008) A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4:e24
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS et al (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395
Heestand BN, Shen Y, Liu W, Magner DB, Storm N, Meharg C, Habermann B, Antebi A (2013) Dietary restriction induced longevity is mediated by nuclear receptor NHR-62 in Caenorhabditis elegans. PLoS Genet 9:e1003651
Hur JH, Cho J, Walker DW (2010) Aging: dial M for mitochondria. Aging 2:69–73
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A (2012) Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 7:48
Jia K, Levine B (2007) Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3:597–599
Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B (2009) Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A 106:14564–14569
Johnson DW, Llop JR, Farrell SF, Yuan J, Stolzenburg LR, Samuelson AV (2014) The Caenorhabditis elegans Myc-Mondo/Mad complexes integrate diverse longevity signals. PLoS Genet 10:e1004278
Kabeya Y (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512
Kirchman PA, Kim S, Lai CY, Jazwinski SM (1999) Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 152:179–190
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-I, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Krüger U, Wang Y, Kumar S, Mandelkow E-M (2012) Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 33:2291–2305
Lamming DW, Ye L, Sabatini DM, Baur JA (2013) Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 123:980–989
Lapierre LR, Gelino S, Meléndez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21:1507–1514
Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu C-C, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE et al (2013) The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105:3374–3379
Li Y-H, Zhang G-G (2016) Towards understanding the lifespan extension by reduced insulin signaling: bioinformatics analysis of DAF-16/FOXO direct targets in Caenorhabditis elegans. Oncotarget 7:19185–19192
Li J, Kim SG, Blenis J (2014) Rapamycin: one drug, many effects. Cell Metab 19:373–379
Lin X-X, Sen I, Janssens GE, Zhou X, Fonslow BR, Edgar D, Stroustrup N, Swoboda P, Yates JR III, Ruvkun G et al (2018) DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat Commun 9:4400
Mair W, Dillin A (2008) Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem 77:727–754
Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8:903–914
Matecic M, Smith DL, Pan X, Maqani N, Bekiranov S, Boeke JD, Smith JS (2010) A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet 6(4):e1000921
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T et al (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288–299
Meléndez A, Tallóczy Z, Seaman M, Eskelinen E-L, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301:1387–1391
Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol 12:823–830
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I et al (2010) Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 1:e10
Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Bénit P, Rustin P, Criollo A et al (2011) Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 192:615–629
Nakamura S, Yoshimori T (2017) New insights into autophagosome-lysosome fusion. J Cell Sci 130:1209–1216
Nakamura S, Karalay Ö, Jäger PS, Horikawa M, Klein C, Nakamura K, Latza C, Templer SE, Dieterich C, Antebi A (2016) Mondo complexes regulate TFEB via TOR inhibition to promote longevity in response to gonadal signals. Nat Commun 7:10944
Nakamura S, Oba M, Suzuki M, Takahashi A, Yamamuro T, Fujiwara M, Ikenaka K, Minami S, Tabata N, Yamamoto K et al (2019) Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun 10:847
Nakatogawa H (2020) Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol 21:439–458
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64:113–122
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in Parkin-deficient mice*. J Biol Chem 279:18614–18622
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528
Park S, Mori R, Shimokawa I (2013) Do sirtuins promote mammalian longevity? A critical review on its relevance to the longevity effect induced by calorie restriction. Mol Cell 35:474–480
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B et al (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118:2190–2199
Pyo J-O, Yoo S-M, Ahn H-H, Nah J, Hong S-H, Kam T-I, Jung S, Jung Y-K (2013) Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4:1–9
Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu Z-X, Dong J, Mustard KJ, Hawley SA et al (2007) Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5:151–156
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146(5):682–695
Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D et al (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22:879–888
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS et al (2009) A gene network regulating lysosomal biogenesis and function. Science 325:473–477
Schiavi A, Torgovnick A, Kell A, Megalou E, Castelein N, Guccini I, Marzocchella L, Gelino S, Hansen M, Malisan F et al (2013) Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp Gerontol 48:191–201
Schiavi A, Maglioni S, Palikaras K, Shaik A, Strappazzon F, Brinkmann V, Torgovnick A, Castelein N, De Henau S, Braeckman BP et al (2015) Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr Biol 25:1810–1822
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC et al (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Eur Mol Biol Organ J 31:1095–1108
Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ et al (2013) TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15:647–658
Sheaffer KL, Updike DL, Mango SE (2008) The target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol 18:1355–1364
Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, Marion SW, St Amand V, Reenan RA, Lapierre LR (2018) Nuclear export inhibition enhances HLH-30/TFEB activity, autophagy, and lifespan. Cell Rep 23:1915–1921
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4:176–184
Smith DL Jr, McClure JM, Matecic M, Smith JS (2007) Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell 6:649–662
Song YM, Lee Y-H, Kim J-W, Ham D-S, Kang E-S, Cha BS, Lee HC, Lee B-W (2015) Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 11:46–59
Sun T, Li X, Zhang P, Chen W-D, Zhang H-L, Li D-D, Deng R, Qian X-J, Jiao L, Ji J et al (2015) Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun 6:7215
Sun Y, Li M, Zhao D, Li X, Yang C, Wang X (2020) Lysosome activity is modulated by multiple longevity pathways and is important for lifespan extension in C. elegans. elife 9:e55745
Tang F, Watkins JW, Bermudez M, Gray R, Gaban A, Portie K, Grace S, Kleve M, Craciun G (2008) A life-span extending form of autophagy employs the vacuole-vacuole fusion machinery. Autophagy 4:874–886
Tóth ML, Sigmond T, Borsos E, Barna J, Erdélyi P, Takács-Vellai K, Orosz L, Kovács AL, Csikós G, Sass M et al (2008) Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4:330–338
Uddin MN, Nishio N, Ito S, Suzuki H, Isobe K-I (2012) Autophagic activity in thymus and liver during aging. Age 34:75–85
Ulgherait M, Rana A, Rera M, Graniel J, Walker DW (2014) AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep 8:1767–1780
Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 68:895–900
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426:620
Wang MC, O’Rourke EJ, Ruvkun G (2008) Fat metabolism links germline stem cells and longevity in C. elegans. Science 322:957–960
Wang C, Niederstrasser H, Douglas PM, Lin R, Jaramillo J, Li Y, Oswald NW, Zhou A, McMillan EA, Mendiratta S et al (2017) Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun 8:2270
Wilhelm T, Byrne J, Medina R, Kolundžić E, Geisinger J, Hajduskova M, Tursun B, Richly H (2017) Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev 31:1561–1572
Winslow AR, Chen C-W, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S et al (2010) α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190:1023–1037
Xiao F-H, Chen X-Q, Yu Q, Ye Y, Liu Y-W, Yan D, Yang L-Q, Chen G, Lin R, Yang L et al (2018) Transcriptome evidence reveals enhanced autophagy-lysosomal function in centenarians. Genome Res 28:1601
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R et al (2011) Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60:1770–1778
Yamamuro T, Kawabata T, Fukuhara A, Saita S, Nakamura S, Takeshita H, Fujiwara M, Enokidani Y, Yoshida G, Tabata K et al (2020) Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat Commun 11:4150
Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, Zhang Y, Li S, Kacergis MC, Webster CM et al (2019a) Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell 177:299–314.e16
Zhou Y, Wang X, Song M, He Z, Cui G, Peng G, Dieterich C, Antebi A, Jing N, Shen Y (2019b) A secreted microRNA disrupts autophagy in distinct tissues of Caenorhabditis elegans upon ageing. Nat Commun 10:4827
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Nakamura, S., Shioda, T., Yoshimori, T. (2022). Autophagy in Aging and Longevity. In: Mori, N. (eds) Aging Mechanisms II . Springer, Singapore. https://doi.org/10.1007/978-981-16-7977-3_11
Download citation
DOI: https://doi.org/10.1007/978-981-16-7977-3_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-7976-6
Online ISBN: 978-981-16-7977-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)