
Abstract
Amyloidosis is a rare disease that induces systemic amyloid deposition in multiple organs. Gastrointestinal (GI) amyloidosis can induce various symptoms depending on the type of proteins and invasion site and is very difficult to diagnose in the absence of a physician’s clinical suspicion. Amyloidosis can be diagnosed when the amyloid deposition is proved by histologic examination of the involved organs. Abdominal fat or gingival biopsy may be helpful in patients with inaccessible target organs. On endoscopy, multiple elevated mucosae and fine granular patterns may be present in various forms depending on the type and distribution of the deposited proteins. Treatment depends on the underlying cause and type of protein. Chemotherapy and stem cell transplantation are useful for AL amyloidosis. As for SAA amyloidosis, it is important to correct the underlying disease. Symptomatic therapy can be useful.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Gastrointestinal (GI) amyloidosis can induce various symptoms depending on the type of proteins and invasion site and is very difficult to diagnose in the absence of a physician’s clinical suspicion.
-
The most common invasion site of GI amyloidosis is the small bowel and various symptoms may appear depending on invasion depth.
-
Systemic chemotherapy, symptomatic therapy, and surgical treatment can be applied depending on the type of deposited protein.
Introduction
Amyloidosis represents a rare disease that induces systemic amyloid deposition in multiple organs, particularly the heart and kidneys, as well as the nerves, soft tissues, and digestive tract, which are among the most frequently affected sites. Amyloid proteins are abnormally deposited in the extracellular matrix, leading to organ dysfunction. Gastrointestinal (GI) amyloidosis can induce various symptoms depending on the type of proteins and invasion site and is very difficult to diagnose in the absence of a physician’s clinical suspicion. More than 31 types of proteins are recognized as the precursors of amyloid protein; nonetheless, only some types of proteins are considered to be of clinical significance. This article aimed to review the pathologic, clinical, and endoscopic features of GI amyloidosis.
Types of Amyloidosis
Amyloidosis is classified into two types according to the cause of amyloid deposition—namely, primary and secondary amyloidosis. Primary amyloidosis is the most common type of monoclonal immunoglobulin light chain (AL) deposition and is associated with plasma cell abnormalities, including multiple sclerosis [1]. Secondary amyloidosis mainly results from the deposition of serum amyloid A proteins (SAAs), which are circulating acute-phase reactants, and is often due to chronic inflammatory diseases such as rheumatoid arthritis, Crohn’s disease, leprosy, and tuberculosis [2]. Furthermore, transthyretin amyloidosis, which is associated with β2 microglobulin amyloidosis or familial amyloidotic polyneuropathy (FAP), is commonly encountered in clinical practice. Amyloidosis is often difficult to diagnose because of its diverse clinical manifestations, which can depend on the deposition patterns of each protein.
Clinical Features and Histopathology
Determination of clinical features may vary according to the amount and distribution of amyloid deposits. If protein distribution is chiefly confined to the mucosal layer, diarrhea or absorptive disorder may be a major symptom. If proteins are deposited in the muscle layer, this may lead to intestinal obstruction. GI bleeding can mainly occur when protein deposition invades the mucosal and submucosal layers. The primary mechanism of GI bleeding is vascular occlusion and weakening of the blood vessel. Vascular occlusion results from protein deposition in the vessel, and amyloid deposition may lead to ischemia, ulceration, wall infarction, and finally weakening of the blood vessel.
In primary amyloidosis, the AL protein mainly invades the mucosal layer and proper muscle layer. Therefore, it may appear as a protrusion of the mucous membrane or thickened wrinkles on endoscopic examination. The AL protein is predominantly involved in the proximal small bowel in older male patients and is thus more likely to appear as a duodenal lesion on duodenoscopy (Fig. 1).

A 59-year-old male patient with small bowel amyloidosis. Elevated mucosal change was detected on duodenoscopy
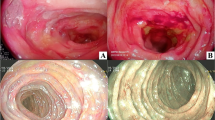
In secondary amyloidosis, the SAA protein is mainly confined to the mucosal and submucosal layers and may cause diarrhea, absorption disorder, GI bleeding, and intestinal obstruction. Secondary amyloidosis can occur mainly in the form of fine granular mucosal pattern and mucosal friability on endoscopy (Fig. 2). Secondary amyloidosis accompanying FAP may lead to diarrhea, constipation, or early satiety owing to protein deposition in the submucosal layer and intestinal nerve plexus. Normal or nonspecific mucosal change can be observed, but fine granular mucosal change or mucosal friability, as seen with the SAA protein, may also occur.

Fine granular mucosal change, as detected on capsule endoscopy
There exist few reports on amyloidosis in Korea. In one Korean single-center study, 24 (15.5%) out of 155 patients showed GI involvement, whereas only 1 patient had small bowel involvement. Diagnosing amyloidosis based solely on patients’ symptoms is difficult because various nonspecific GI symptoms (e.g., GI bleeding, weight loss, dyspepsia, abdominal pain, constipation, diarrhea) may appear [3].
Diagnosis and Treatment
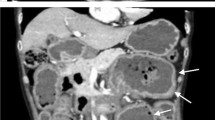
As mentioned above, diagnosing amyloidosis is very difficult if there exists no positive suspicion of amyloidosis because most patients present with nonspecific symptoms. Active diagnostic evaluation is needed for patients with unexplained GI symptoms and underlying amyloidosis-associated disease. Amyloidosis can be diagnosed when the amyloid deposition is proved by histologic examination of the involved organs. Amyloid deposition can be clearly observed using Congo red stain (Fig. 3). Abdominal fat or gingival biopsy may be helpful in patients with inaccessible target organs. On endoscopy, multiple elevated mucosae and fine granular patterns may be present in various forms depending on the type and distribution of the deposited proteins (Figs. 1 and 2). Computed tomography (CT) may be helpful in some cases. On CT, the blood supply is reduced; hence, the mucosa is thickened and the center of the lesion may appear as a low-density area (Fig. 4) [4].

Amyloid deposition, which was confirmed by Congo red staining

A 46-year-old female patient. (a) Luminal narrowing due to elevated mucosal change was observed. (b, c) Low-attenuation polypoid lesion with calcification was detected on CT scan
Treatment depends on the underlying cause and type of protein. Chemotherapy and stem cell transplantation are useful for AL amyloidosis. As for SAA amyloidosis, it is important to correct the underlying disease. Prokinetic agents can be useful in patients with nausea and dysmotility symptoms. In case of severe diarrhea and protein-losing enteropathy, octreotide, corticosteroid, loperamide, and opiate may be helpful. Furthermore, antibiotics for small intestinal bacterial overgrowth may be a therapeutic option for severe diarrhea, and surgical resection for a localized disease may be considered.
Conclusion
Because small bowel amyloidosis is a rare disease and may have nonspecific symptoms, there is no way to make a quick diagnosis, except when the treating doctor suspects the disease. As nonspecific mucosal changes alone can be observed on endoscopy, an aggressive biopsy may aid in diagnosing amyloidosis in case of suspicious endoscopic findings.
References
Bansal R, Syed U, Walfish J, Aron J, Walfish A. Small bowel amyloidosis. Curr Gastroenterol Rep. 2018;20:11.
Hokama A, Kishimoto K, Nakamoto M, et al. Endoscopic and histopathological features of gastrointestinal amyloidosis. World J Gastrointest Endosc. 2011;3:157–61.
Lim AY, Lee JH, Jung KS, et al. Clinical features and outcomes of systemic amyloidosis with gastrointestinal involvement: a single-center experience. Korean J Intern Med. 2015;30:496–505.
Kala Z, Valek V, Kysela P. Amyloidosis of the small intestine. Eur J Radiol. 2007;63:105–9.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kang, S.H. (2022). Amyloidosis of the Small Bowel. In: Chun, H.J., Seol, SY., Choi, MG., Cho, J.Y. (eds) Small Intestine Disease. Springer, Singapore. https://doi.org/10.1007/978-981-16-7239-2_49
Download citation
DOI: https://doi.org/10.1007/978-981-16-7239-2_49
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-7238-5
Online ISBN: 978-981-16-7239-2
eBook Packages: MedicineMedicine (R0)