
Abstract
A malignant tumor has ability to invade the nearby tissues and colonize distant sites (organs) through the lymphatic/vascular system. From intravasation of cancer cells into the vascular system to extravasation from the vascular system to a new organ involve basic processes and concepts of biology, chemistry, and physics. Cancer cells migrate either singly or collectively using actin deformation proteins (RhoGTPase family), which help to create cell protrusions to move to different parts of a cancer-affected body. For moving to secondary locations, cancer cells breach the surrounding extracellular matrix and the basement membrane of vascular systems with the help of tissue-degrading proteins (proteolytic enzymes and matrix metalloproteinases).
Once tumour cells enter into the vascular system, they are controlled by the blood flow pattern, blood vessels’ diameter, shear flow, and intercellular adhesion. They are also affected by hemodynamic forces, immunological stress, collisions with the blood cells, and the endothelial cells of the vessel wall. The shear flow in the blood vessels influences the rotational and translational motion of circulating tumor cells. These two motions decide the orientation of the cell with respect to the receptor–ligand interactions with the vessel’s wall. During circulation, cancer cells bind to platelets, leukocytes, and fibrin with the help of adhesion proteins. Circulating cancer cells, which survive from fluid shear force and immune surveillance, arrest the microvascular endothelium of a secondary location using physical occlusion and/or cellular adhesion. The probability of cell arrest depends on the collision rate between the membrane-bound receptors and endothelial ligands, and residence time of the cell. The chances of cell arrest are much higher if the shear force is at intermediate level. The location of secondary site for a tumor cell line is preferential and not random. This chapter provides an outline of the processes involved in cancer cell motility and metastasis based on the basic concepts of biology, chemistry, and physics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cancer cell motility
- Single cell motility
- Collective cell motility
- Metastasis
- Intravasation
- Extravasation
1 Introduction
An adult human body may contain nearly 100 trillion (≈1014) cells. The growth of multicellular human body occurs due to cell divisions, which are aptly controlled and regulated by some factors. Noncancerous cells obey the cellular signals that instruct whether a cell should divide, differentiate into another cell, or die. Due to some inheritable (successive rounds of mutation) or epigenetic changes at the time of cell division, some cells may be transformed and divide at abnormally faster rate. The abnormal growth of cells in an uncontrollable and uncoordinated manner forms a lump/tumor, which may lead to cancer. If this proliferation is allowed to continue and spread, it may become life-threatening to the host.
Tumor development and its progress are a combination of several correlated biochemical processes. Genetic irregularities that affect mainly oncogenes and tumor suppressor genes lead to tumor development. While responsibility of oncogenes is expressing growth factors, which stimulate mitosis and lead to cell survival and proliferation, tumor suppressor genes restrain cell cycle and lead to apoptosis (programmed cell death) if any cellular abnormality arises. When oncogene is affected by gain in functions during mutations and becomes either constitutively expressed or overexpressed, the cell begins to proliferate regardless of the amount or even the absence of growth factors, resulting in uncontrolled cell growth and proliferation (Croce 2008). On the other hand, if a cell becomes damaged or mutated, the tumor suppressor gene arrests the progression of the cell cycle in order to carry out DNA repair or to induce apoptosis. When tumor suppressor genes lose its functionality, the cells are able to avoid apoptosis, enabling the propagation of mutated and damaged DNA to the daughter cells (Barnes et al. 1993). A successive number of mutations in oncogenes and tumor suppressor genes initiate the development of carcinogenesis.
The growth of a tumor may be classified into three distinct phases: avascular phase, angiogenesis, and vascular phase. In avascular phase, tumor does not have any blood supply, and it has to depend on nutrients and oxygen transported to and from extracellular matrix (ECM) through passive diffusion (Sutherland 1988) and consists of approximately 106 to 107 cells. The avascular nodules grow to 1–2 mm3 in volume. Being deprived of vital nutrients, cells near the center of a tumor may die and create a necrotic core of dead cells (Sherratt and Chaplain 2001). Proliferative cells are present on the surface area of a tumor. Between the necrotic and the proliferative layers, another layer of cells exists (quiescent cells), which are recruited into the proliferative layer as required (Ang and Tan 2008).
To continue its growth, the tumor needs nutrient in proportion to its volume, but its ability to absorb nutrient is proportional to its surface area (Orme and Chaplain 1996). This phenomenon limits maximum size of tumor to which it can grow before it experiences nutrient deficiency. So, it diffuses a number of chemical substances (tumor angiogenesis factors (TAFs)) into the nearby blood vessels. These diffusible TAFs stimulate the endothelial cells of local vessels to form capillary sprouts that grow toward the tumor. The newly developed sprouts are often fused together to form loops (anastomoses) that are also fused with the other loops and build a complex network of vessels. The newly developed networks eventually supply necessary nutrients to the tumor for continuing growth. This phenomenon is known as angiogenesis or neovascularization.
After angiogenesis, tumor enters into the vascular phase, and it rapidly increases in mass. This massive growth in tumor is self-limiting due to breakdown of the vascular system and again a necrotic core is developed at the center of a tumor surrounded by a layer of proliferative and quiescent cells (Paweletz and Knierim 1989). Thus, in order to support constant progression, the vascular system untiringly modifies itself, and this process lasts until a tumor is removed or the host dies.
A malignant tumor is distinguished from a benign tumor by its ability to invade nearby tissues. The rapidly growing tumor cells (in vascular phase) create tremendous pressure to the adjacent cells and the ECM and penetrate into the nearby tissues (invasion). Some of the tumor cells may detach from its origin and spread through the blood/lymphatic vessels to the distant parts of the body (metastasis). The circulating tumor cells form distant colonies depending upon its ability to induce new blood vessels from the nearby tissues to sprout toward the tumor. This results in adequate blood supply and microcirculation in the tumor cells.
The rest of the chapter elaborates the processes of cancer cell motility and metastasis in detail and is organized as follows: Sect. 5.2 gives the biological backgrounds; Sect. 5.3 illustrates the biological and chemical processes of cancer cell motility and metastasis; and Sect. 5.4 describes the physical interactions and mechanical forces involved in cancer development and its progression. Section 5.5 concludes the chapter. Some of the biological terms are briefly described in the appendix.
2 Biological Background
Cells are the unit of life. Most of the activities in living organisms are performed inside the cells. A cell contains a nucleus, bounded by cytosol and wrapped in a bilipid membrane. Cell membrane protects the cell from the surrounding environment, and it is porous to passive diffusion of small size molecules like oxygen and glucose. It helps to maintain pH and other chemical balance within the cell by actively pumping other molecular species and also exchange complex signals with the microenvironment. These complex signals play a key role to maintain equilibrium with the surrounding environment; that is, the cell cycle speed is regulated by growth-inhibitory signals, which are further controlled through the interaction of cyclin and cyclin-dependent kinases (CDKs). Surface receptors regulate gene expression through complex signals. Also, the gene expression pattern dictates the production and balance of proteins. Hence, a complex interaction exists between a cell and its surroundings that control the cell cycle and cell growth (Clyde et al. 2006).
2.1 Equilibrium in Cell Population
The population of each cell type in a tissue is maintained by balancing cell proliferation and apoptosis. If differentiated cell dies, somatic (adult nongermline) stem cells divide asymmetrically into a stem cell and a progenitor cell. The progenitor cell then further splits or terminally differentiates into the preferred cell type, which migrates to the correct place and replaces the dead cell. The whole process is regulated and controlled by a complex system of biochemical signals that are monitored by all the cells in the structure. Whenever a signal is received, each cell activates the corresponding receptors on its surface. The activated receptors are responsible for activation and/or deactivation of genes within the nucleus. The transformed genes then start to express proteins within the cells that control the cell cycle and all other cell functions. Stromal cells (connective tissue cells of any organ) play a major part in creating and responding to growth factors and biochemical signals (Zipori 2006).
2.2 Oncogene and Tumor Suppressor Genes
Gene carries genetic information of growth, development, functioning, etc. Gene expression pattern expresses the behavior of a cell, which receives growth signals and growth inhibitory signals. These two signals are vital to maintain the health of a tissue. There are mainly two types of genes that are responsible for promoting the cell cycle advancement: oncogenes and tumor suppressor genes. While the former is responsible for generating and endorsing the cell cycle progression, the later guarantees proper DNA repair, react to the growth inhibitory signals, and may control apoptosis due to some special circumstances. The malfunctioning DNA is the key factor for developing carcinogenesis (Hanahan and Weinberg 2000). A single erroneous mutation is enough to affect the functionality of an oncogene (Malumbres and Barbacid 2003), or neutralize the functionality of tumor suppressor gene (Horowitz et al. 1989). Sometimes, at the time of cell division (during mitosis phase), any kind of errors can produce a faulty oncogene. Thus, signals change their routes to the oncogene, which regulate the activity. There are other possibilities too. Errors may occur at the time of cell division if the offspring get extra copies of the oncogenes or fewer copies of the tumor suppressor genes from the parent cell (Castro et al. 2006). The increment in the functions of the oncogenes and decrement in functions of tumor suppressor genes in the daughter cell can significantly damage the normal behavior and accelerate the chances of completing a multistep carcinogenesis path (Quon and Berns 2001). Latest study (Kashkin et al. 2013) says that key sponsor of unrestricted cell growth is the over- and underexpressed genes. As the gene expression product transfers from one generation of a cell to its next generation, the fundamental changes in gene expression potentially affect the malignant transformation of a cell, similar to a genetic mutation. When a cell with defective genes malfunctions and interacts with the body in an abnormal, proliferative manner, we term it cancer.
2.3 Cellular Adhesion
Human body is constructed with a community of cells. Each cell occupies a suitable place to perform its tasks (white blood cell is an exception). Every cell has its own address or area code system-written domain that can either interact with the cellular address molecules (CAMs) of the same kind (hemophilic binding) or interact with the other kind of CAMs or the extracellular matrix (heterophilic binding). In hemophilic binding, receptor molecules on the cell surface knot with similar target molecules of adjacent cells (in cell–cell adhesion), the ECM, or the basement membrane in the microenvironment (in cell–ECM or cell–basement membrane adhesion), while in heterophilic binding, surface adhesive molecules of one type knot with dissimilar molecules in the ECM or the basement membrane or the adjacent cells.
Adhesion is crucial to multicellular structure and cell motility. There are mainly three types of cellular adhesive forces: cell–cell adhesion, cell–basement membrane adhesion, and cell–matrix adhesion. Cell–cell adhesion controls the tissue structure, while cell–basement membrane adhesion and cell–matrix adhesion are necessary for adhesive friction during cell motility. Cell–cell adhesion helps a cell to cooperate and attach to its adjacent cells and act as intermediator for communication among molecules of the cell surface. It controls the epithelial cell growth and cell division. The proliferation rate is increased to fill up the gap in the epithelium due to apoptosis and the rate is reduced whenever the epithelium is fully populated (Hansen and Bissell 2000). These interactions and the ability to transfer signals among cells are essential for the survival of the tissues.
Cell–matrix and cell–basement membrane adhesive forces are transient or temporary such as adhesion between cells of the immune system or the interactions involved in tissue inflammation. Cell–basement membrane and cell–matrix adhesion govern the traction during cell motility. After losing the adhesive interaction with the basement membrane, epithelial cells are often converted to a specialized type of apoptosis, named anoikis (Frisch and Ruoslahti 1997). Anoikis helps to suppress overgrowth of separated cells into the lumen. ECM controls the growth, differentiation, and apoptosis of stromal cells (Selam et al. 2002). Cellular adhesive force governs the multicellular arrangement and cell motility. The loss of adhesive forces between cells can result in uncontrollable cell growth and carcinogenesis. Cell–cell adhesion is an important factor in normal tissues, but in the cancerous environment, cell–cell adhesion molecules seem to disappear or compromised. Losing cell–cell adhesion ability between cells seems to be the first step in cancer invasion.
2.4 Cell Signaling
Cell signaling regulates the basic functionalities of cells and synchronizes all cell activities. The basic developments like tissue repair, wound healing, and tissue homeostasis all depend on it. A cell can be able to receive a signal from the surrounding and produce an appropriate reply to the microenvironment; it is the basis of the cellular developments. Malfunctions in cell signaling system are the key factors for the diseases such as cancer, diabetes, and autoimmunity (Wang et al. 2013).
Cell signals can be categorized into two groups: mechanical type of signals and biochemical type of signals. In the case of mechanical type signal, forces are generated by the cell and it is applied on the surrounding cells. Also, these forces can be sensed and responded by the other cells. In case of biochemical signals, signals are the molecules of proteins, lipid, gases, and ions. Based on the distance between the sender and the receiver cells, biochemical signals can also be classified as intracrine, autocrine, juxtacrine, paracrine, and endocrine. In a multicellular organism, signaling between cells transmits either through extracellular space (paracrine signals over short distances, endocrine signals over a longer distance) or through direct contact (juxtacrine signals). Cells receive information from its surroundings through some proteins known as receptors. Notch is one of the cell surface proteins that act as a receptor. A set of signaling proteins interact with the Notch receptors and generate response to the cells. The receptor activation molecules (ligand receptors) can be classified as hormones, neurotransmitters, cytokines, and growth factors. Hormones are the main signaling molecules of the endocrine system; however, they often control each other’s secretion through local signaling. Neurotransmitters (including neuropeptides and neuromodulators) are signaling molecules of the nervous system, and cytokines (paracrine or juxtacrine in nature) are signaling molecules of the immune system. Signaling molecules communicate with a target cell (through ligands to cell surface receptors, and/or by entering through the cell membrane (endocytosis)) for intercrine signals. Ligand receptors are responsible for cell signaling mechanisms and communications. Notch acts as a receptor for ligands. Some receptors are cell surface proteins, but others are found inside the cells.
Several signaling factors activate surface receptors, which regulate gene expressions. Internal chemical molecules (e.g., oxygen) also affect gene expressions. Failures in these signaling processes are involved in several cancers. A few examples of signaling are hypoxia-induced factor (HIF)-1α signaling, epidermal growth factor (EGF) signaling, and E-cadherin/β-catenin signaling. These signals are directly or indirectly responsible for tumor growth and progression. HIF-1α is generated inside cells and is affected by the presence of oxygen (Semenza 2001). A cell enters into hypoxic state due to insufficient oxygen levels. Whenever a cell enters into hypoxic state, HIF-1α activates the target genes, which are responsible for the increment of motility rate, secretion of angiogenic factors, anaerobic glycolysis, and reduction in cell–cell and cell–matrix adhesion, and also reduces the effectiveness of apoptotic signals (Pouysségur et al. 2006). Epidermal growth factor (EGF) activates the EGF receptors. When two activated EGF receptors bind together, it can transmit signals that increase the secretion of HIF-1α, cell proliferation, and cell motility and reduce apoptosis (Allen et al. 2006). Among the signal receptors, some of them can perform in multiple roles, like E-cadherin acts as arbitrating in the intracellular domain as it binds to α-catenin (using β-catenin) to mechanically bind an adhered cell to its actin cytoskeleton, and also suppress cell cycle progression (Seidensticker and Behrens 2000). These signaling systems play a crucial role to sustain tissue microarchitecture (Wei et al. 2007).
3 Cancer Cell Motility and Metastasis: Biology and Chemistry
While the biological notion is required to understand the processes behind tumor formation, growth, progression, and transformation into cancer, chemistry helps to recognize the microscopic interactions of thousands of genes, molecules, and conditions that lead to aggressive cellular growth and the chemical processes involved at the time of cancer cell motility and metastasis. On the other hand, physics expresses the key physical and mechanical forces applied on the cancer cells and their role in cancer cell motility and metastasis. It also explains the physical properties of cell interaction, the role of inflammation in cancer, and connective tissue microenvironment.
3.1 Cancer Cell Motility
In a biological environment, different types of cell movements can be seen. Cell movement is the fundamental property of living cells that have been adapted throughout the development of multicellular organisms including immune response, embryogenesis, and adult tissue homeostasis. Cells move due to some external signals in the direction of higher concentration of the substrate, this is, a primitive feature of biological systems. It is called chemotaxis. Motile cells require mechanical interaction between the microenvironment and the cell membrane bulges. Individual cells may move through the stroma in an amoeboid motion by squeezing between the ECM fibers or by extending a finger-like pseudopod that forms focal adhesions with the ECM to create space for motion, and is accomplished by forming tiny invadopodia on the filopodium surface that secretes proteases to degrade the ECM (Weaver 2006).
In case of cancer cells, though the mechanism is not fully understood, it can be said that oncogenes are responsible for this phenomenon. The proteins, that is made by the oncogenes carry a wrong signal to misguide the nucleus that the cell is appropriately attached to its neighbors, though actually, it is not. Thus, the cell tries to stop its own growth and die due to apoptosis. Epithelial cells are the most common cancer sources in the human body. It is separated from the rest of the body by a basement membrane and a skinny layer of the specialized ECM. Basement membrane develops an obstacle that most of the normal cells cannot breach, but the cancer cells can break the wall, penetrating it by secreting metalloproteinases enzyme (Liotta 1992). Basement membrane and other extracellular lamina are dissolved by the matrix metalloproteinases (MMPs). After puncturing the basement membrane, tumor cells shortly meet another basement membrane, surrounding blood vessels/lymphatic vessels. MMPs also break the second basement membrane by penetrating the wall and forming a finger-like shape to get access to the bloodstream directly. After breaching the basement membrane of blood vessels/lymphatic vessels, cancer cells are free to spread throughout the whole body through those vessels, though the tumor cells must undergo some changes/transformation (till now not clearly understood) to adapt, survive, and proliferate in the new locality. If the invasion occurs within the primary organ, then tumor cells do not require any transformation. Eventually, a tumor cell may lodge in a capillary. If it penetrates the capillary wall again, it can create a secondary tumor. Perhaps less than 1 of 10,000 tumor cells is able to escape a primary tumor and survive to colonize at another location. So, the cell motility mechanism is primitively knotted with cell survival, which is the key to metastasis.
Tumor cell motility and invasion to the other organ or blood/lymphatic vessels are the pillars of metastasis. Motile cancer cells remodel their cell–cell or cell–matrix adhesion and their actin cytoskeleton, which are involved in cell signaling. There can be various ways of cancer cell migration depending on cell types and degree of differentiation. These different types of cell migration are controlled by various mechanisms. The main factor of these mechanisms is a dynamic reformation of the actin cytoskeleton, and this reformation yields the necessary force for cell migration. The actin reformation is governed by RhoGTPase family, which includes Rho, Rac, and Cdc42. The RhoGTPase carries the extracellular chemotactic signals to activate the downstream enzyme family: Wiskott–Aldrich syndrome proteins (WASPs) that are the major factors in cell migration. These WASPs initiate the development of protrusion in the cellular structures that are involved in the motility process and degradation of the ECM.
3.1.1 RhoGTPase Family
Rho, Rac, and Cdc42 are members of RhoGTPase family. They are the key factors for cell migration and invasion, as it has the ability to alter the polymerization of actin. They are activated by guanosine triphosphate (GTP)-binding proteins. Their stimulation is controlled by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). GEFs stimulate RhoGTPase through the interchange of Rho-bound guanosine diphosphate (GDP) by GTP, and it is further neutralized by GAPs that accelerate the performance of RhoGTPase and hydrolysis of bounded GTP to GDP. Lastly, GDIs bind to Rho-GDP and avoid their contact with RhoGEFs (Fig. 5.1). RhoGTPases regulate the effector protein serine/threonine kinases, P21-activated kinases (PAK1–3), and myosin light-chain kinase (for Rac1); PAK1–6, WASP, N-WASP, and mDia2 (for Cdc42); and ROCK kinases I and II, Citron, and mDia 1 and mDia2 (for RhoA). Activated Rac1 and Cdc42 initiate the reformation of the actin cytoskeleton at the leading edge (Small et al. 2002).

Activation of RhoGTPase family
Actin polymerization impulses at the foremost edge of the cell membrane and forms finger-like protuberances (filopodia) or sheet-like bulges (lamellipodia) (Fig. 5.2a, b). These protuberances provide locomotive forces, which help cells to move to a different location. On the other hand, RhoA controls the contractile actomyosin filaments using downstream enzyme ROCK/Rho-kinase. Actomyosin endorses locomotive forces in the cell body and the trailing edge (Fig. 5.2c). While Rac1 and Cdc42 motivate the development of cellular bulge at the leading edge, RhoA makes retraction at the trailing edge. These synchronized efforts make a cell to move toward a direction (Fig. 5.2d).

Cell migration consists of these four processes: (a) protrusions, (b) adhesion, (c) translocation, and (d) retraction (Yamazaki et al. 2005)
3.1.2 Single Cell Motility
There are many types of cell motility observed in cancer cells (Friedl and Wolf 2003), and these movements are simultaneous. In epithelial tissues, the cell moves as a sheet-like formation. This type of motility in epithelial cells is not only seen in angiogenesis but also seen in other biological phenomena including wound healing (Friedl 2004). At the time of epithelial cell differentiation, the utility of cadherin is inhibited that leads to abrogation cell–cell adhesion. The suppression of cell–cell adhesive force separates the cells from each other and makes them ready for individual or collective migration (Lozano et al. 2003). This phenomenon is known as epithelial-to-mesenchymal translation (EMT). Cancer cells are able to migrate and invade in the absence of EMT too (Friedl 2004). The reason behind EMT, cell motility, and invasions is the damage of the cell–cell adhesion molecules. E-cadherin is the key element of epithelial adherens junctions. With the loss of E-cadherin functions in mesenchymal cell–cell adhesion, N-cadherin increases (cadherin switch) leading to cell motility and invasion (Lehembre et al. 2008). This functional loss can happen due to germline and somatic gene mutations, chromosomal abnormality, transcriptional repression, DNA hypermethylation of E-cadherin gene (cdh1), signal transduction of a large number of growth factors, and Notch signaling. Several growth factors like transforming growth factor β, hepatocyte growth factor, epidermal growth factors, fibroblast growth factors, and Notch signaling stimulate the E-cadherin loss. Also, hypoxic condition in tumor cells influence them for migration and spreading. These changes in the microenvironment stimulate one or several receptors of E-cadherin gene expression (e.g., Snail1, Snail2, ZEB1, ZEB2, E47, and Twist). Binding of the transcriptional repressor to the E-cadherin gene promoter causes silencing of the gene (for details, one may refer to Herranz et al. 2008; Hou et al. 2008).
Cell motility can be either single (amoeboid or mesenchymal) or collective (in cell sheets, strands, tubes, or clusters). As an example, cells in colorectal cancer transform into motile form and can roam alone after losing E-cadherin, whereas squamous cell carcinomas migrate and invade collectively. Different factors like extracellular protease activities, integrin-mediated cell–matrix adhesion, cadherin-mediated cell–cell adhesion, cell polarity, and cytoskeletal organization decide the different types of cell motility and invasion. Single or collective cell migration can be seen in stromal cells. In EMT, during embryonic development in morphogenic procedures, loss of epithelial polarity or gain of mesenchymal morphology is responsible for single cell migration.
Tyrosine kinases at the endosome receptor arbitrate motogenic activation of Rac. To limit Rac to the plasma membrane and to induce the development of migratory actin flange, Rab-5-dependent endocytosis is needed. The control of endocytosis on migrating cells is needed for the fast recovering of β1-integrin at pseudopodal membrane flanges and the retaining of a pool of β1-integrin at the cell front by the GTPase Rab25. Mutant p53 endorses the transferring of β1-integrin and of EGFR, which increases the β1-integrin/EGFR signaling, cell invasion, and metastasis (Muller et al. 2009). p120-catenin is present at the cell membrane of epithelial cells, and it regulates the activity of RhoA and Rac1 with E-cadherin. RhoA degrades cell–cell adhesion. p120-catenin-reliant RhoGEF enzymes, for example, Vav2 or RhoGAP enzymes, are required for activation and deactivation of RhoA. p190-RhoGAP activates Rac1 for stabilizing E-cadherin junction through suppression of the actions of IQGAP1 (a Rac1 effector protein and a mediator of E-cadherin endocytosis). Cytoplasmic p120-catenin endorses the activity of Rac1 at the cell membrane, and it leads to the formation of lamellipodia (Fig. 5.3a, b). So, cytoplasmic p120-catenin and membrane-sequenced p120-catenin both collaborate with each other during EMT that leads to cell motility. Rac1 suppresses the actions of RhoA through the generation of responsive oxygen species (increases the expression of Snail1) that also inhibit lower molecular weight protein: tyrosine phosphatase (it stimulates p190RhoGAP). All the above-mentioned activities may be found in the single cell migration. (For the detailed study, one may refer to Kleer et al. 2006).

Cell motility: (a) mesenchymal cell migration (b) amoeboid motility (Sahai 2005)
Single cell motility is classified into mesenchymal cell migration (Fig. 5.3a) and amoeboid migration (Fig. 5.3b) (Friedl and Wolf 2010), and anyone or both of it can be seen in cancer cells. Mesenchymal cell motility is usually exercised by spindle-shaped, fibroblast-like cells (such as fibroblasts, endothelial cells, smooth muscle cells, and cancer cells). It can be categorized by Rac-induced cell protrusion-driven cellular movements and actin polymerization-based cellular movements. Amoeboid cell migration can be seen among round-shaped cells such as hematopoietic stem cells and leukocytes. The driving force of amoeboid cell movement is RhoA/ROCK-mediated bleb-like protrusions with active myosin/actin contractions.
3.1.3 Motility of Collective Cells
In collective cell migration (Fig. 5.4), cells preserve their cell–cell junctions and move with the connection to their originating tissue or separately in the form of a sheet, strands, tubes, and clusters. Migrating cells develop a cone or finger-like cell membrane bulge; it then intravasate and circulate like cell clusters that are very proficient in embolizing (it refers to the path and accommodation of an embolus within the bloodstream) lymphatic or blood vessels. Collectively migrating cells differ from the solitary migrating cells in many ways, like collectively migrating cells only use their pulling forces on its neighboring cells that are connected through adhesion junctions instead of reacting to their cellular trails. Though collective migrating cells have few similarities with solitary migrating cells on the cellular level, like both of the migratory processes develop protrusions that use β1-integrin and/or β3-integrin to develop focal adhesive force connected with actin cytoskeleton, and use actin contractile for cell motility and local shrinkage. Collective migration can be seen in many physical morphogenic processes (it causes an organism to develop its shape), where cells move in groups. Motile cells use cell membrane bulges that help to distinguish motile cells from their adjacent followers that move in a particular direction.

Collective motility is characterized by collection of cells invading with many still retaining cell–cell contacts (Sahai 2005)
Collective migration needs synchronization between cell–cell adhesion and cell contractibility (Montell 2008). Collective migration is seen in various types of cancers (like breast cancer, prostate cancer, lung cancer, and melanoma). Motile cells collectively penetrate the ECM; the leading cells develop an invasion path using β1-integrin-mediated focal adhesions and MMPs to rupture the collagen fibers. It forms a tube-like structure in which the following cells can migrate. Invasive migration and proteolytic matrix remodeling control collective cell motility. It can be explained in detail through the inspection of cell surface glycoprotein podoplanin (38 kilodalton) that define various cell types like kidney podocytes, lymphatic endothelial cells, and platelets and various cancer types including squamous cell carcinoma of oral cavity, lung, and skin. In the above-mentioned cancer types, collectively motile cells form a finger or cone-like shape to penetrate the neighboring tissue.
3.2 Metastasis
If a normal cell has undergone repetitive genetic and epigenetic changes, then it will transform into cancerous with uncontrolled proliferation potential. These features of the cancerous cells are passed on to its daughter cells. This uncontrolled proliferation helps to produce primary heterogenetic tumor (different cells showing distinct morphological and phenotypic profile). The cells of a developing tumor ultimately undergo metaplasia, dysplasia, and then anaplasia. This results in malignant phenotype. Cancer cells break away from a primary tumor and degrade the surrounding ECM for entering the vascular and lymphatic systems. These circulating tumor cells develop a second site of tumorigenesis.
Using the tissue-degrading proteins, the cancerous cells can be able to penetrate the ECM and escape from its primary location. These circulating tumor cells may be able to breach the lymphatic or blood vessels walls and then reach to other sites of the body through the lymphatic channels or blood vessels (lymphatic spread/hematogenous spread). After reaching the secondary site, they might repenetrate the vessel wall and continue to grow leading to a cancer cell colony. This new tumor is known as a metastatic/secondary tumor. From carcinogenesis to metastasis, a series of interconnected biochemical processes are involved (Fidler 1990). The processes are as follows: primary tumor formation and its growth, vascularization, invasion, and entry of tumor cells into blood/lymphatic vessels. Interaction of tumor cells with fibrin and platelets, sticking on capillary bed of secondary organs, and extravasation into the secondary organs are also involved. It is important to state that the cells of this secondary tumor are the same as the cells of a primary tumor.
Not all malignant tumors give rise to metastasis. Basal cell carcinoma of the skin is capable of local invasion, but it rarely produces secondary growths. Most other cancers give rise to metastasis at some point in their evolution. Though metastasis has a great clinical importance, till now this process is very poorly understood (Kaplan et al. 2006). The time period between the initial diagnosis of primary cancer and the first detection of metastasis varies widely from patient to patient. This phenomenon is very common in case of breast cancer. Different tumors have different metastatic capabilities. The presence of malignant cells in circulation does not necessarily lead to metastasis. Researches showed that after 24 h in circulation, less than 1 percent of cancerous cells are viable and less than 0.1 percent survived to form metastasis (Fidler 1970).
Spread of cancer cells takes place through three different routes: transcoelomic, lymphatic, and hematogenous. In transcoelomic spread, malignant cells reach into body cavities by crossing the surface of the peritoneal, pleural, pericardial, or subarachnoid areas. In lymphatic spread, tumor cells move into the lymph nodes and then spread into other parts of the body, and in the hematogenous spread, tumor cells spread through blood vessels. While the lymphatic spread is the usual route for carcinoma metastasis, it is an unusual route for sarcoma metastasis (suitable route is blood vessels). On the contrary, hematogenous spread is also suitable for a few types of carcinoma like renal cell carcinoma. Lymphatic vessel is the most common pathway for the initial spread of carcinomas. The vessel walls of the lymphatic system provide less resistance to the tumor cells during spread. Cancerous cells very easily break these vessel walls and reach the regional lymph nodes. Within the lymph nodes, the malignant cells can halt and proliferate further and develop a secondary tumor/metastatic tumor. The malignant cells may move away from the lymph nodes and enter into the bloodstream, ultimately reaching other tissues (Fig. 5.5).

Metastatic process leading to secondary tumor development (Wirtz et al. 2011)
The most common organs affected by metastasis are lungs, liver, brain, and the bones (Berenson et al. 2006). Different types of cancer tend to spread to specific organs and tissue in preferential way at a rate that is higher than expected. There is a famous Paget postulate (Paget 1989) regarding this phenomenon, known as seed and soil hypothesis. It says that cancer cells spread to different organs throughout the body but grow only in “fertilized soil.” The “fertilized soil” defines the presence of favorable growth factors and absence of anti-growth factors for metastasis.
3.2.1 Proteolytic Enzymes
During cancer cell invasion and metastasis, a number of tissue walls require to be breached. These walls are made with basement membrane or interstitial connective tissues. In invasion and metastasis, basement membrane is invaded at least thrice: first, when the cancerous cells invade these walls to escape from its primary location; and second during entering into and third at time of exiting from the blood vessels. The most important elements of basement membrane are collagen (type IV), laminin, proteoglycans (heparin sulfate and chondroitin sulfate), entactin, and osteonectin; the key elements of interstitial connective tissue are collagen fibers (type I, II, III, or interstitial collagen), glycoproteins (e.g., fibronectin), proteoglycans, and hyaluronic acid. During degradation of walls, cancer cells secrete a number of enzymes that degrade proteases like urokinase form of plasminogen activator (uPA), cathepsin B (CB), cathepsin D (CD), and various metalloproteases that perform as a cascade (see appendix).
Several proteases are involved in tissue degradation processes. Though these processes partly look like normal cell invasion, proteolysis is self-limiting and organized, whereas in cancer the regulatory mechanisms are absent. There is a correlation between the different proteases and metastatic attributes of a tumor. According to Duffy (1992), malignant cells’ capability to metastasize not only depends on the levels of the different proteases but also depends on the density of appropriate endogenous substrate. An inversely proportional relation is present between the metastatic potential and the density of a specific substrate as the low level of substrate may boost the metastatic process, whereas the higher level may suppress metastasis, though in the case of some proteases, higher density of substrate helps to upregulate metastasis (Nekarda et al. 1994).
Proteases are responsible for accelerating cancer cell spread; most of the events would have been estimated at the invading point of tumor where damage of normal tissue has occurred. That is why certain proteases are found in those sites with highest density. As an example, in Lewis lung tumors, uPA is found in high density in the area of tumor where invasive growth can be seen and no tissue damage has occurred. In some areas where invasive growth with tissue degradation has taken place, that contain uPA of high density with PAI-1 of low density. In general, proteases help to spread cancer cells through accelerating the destruction of ECM. As ECM is developed with different elements, various types of proteases are needed to accomplish the metastasis and also to activate several precursors, for example, plasmin, cathepsin, and a trypsin kind protease activate pro-uPA, and plasmin activates some metalloproteases. Additionally, proteases have some extra utility to boost invasion and metastasis, like uPA and CD, both are the mitogens and they help to accelerate cell division, which indirectly endorses cell motility and metastasis. Also, proteases activate the positive growth factor and inactivate the metastasis suppressor to help metastasis.
3.2.2 Adhesion Proteins
At the time of cancer cell spread, tumor cells divide repeatedly and develop new attachments with their neighboring structures. Thus, in the early phase, invasive and metastatic cells degrade the adhesion with the surrounding cells and release themselves from the neighborhood to stick with the basement membranes. During circulation within blood/lymphatic vessels, cancer cells bind to platelets, leukocytes, and fibrin, and later, they adhere to microvascular endothelium in the secondary organ. The chemical substrates that permit these different cell–cell or cell–matrix attachments are called adhesion proteins. Adhesion proteins can be categorized into different groups: integrins, cadherins, selectins, CD44, and immunoglobulin superfamily (see appendix).
Some of the adhesion proteins help cancer spread by decreasing expressions, while others accelerate metastasis by increasing expressions. Specifically, integrins, E-cadherin, and CD44 directly interfere in metastasis. Scientist has shown that peptides with RGD sequence try to suppress metastasis in animal system, which proves that some integrin proteins interfere in cancer spread (Ruoslahti 1992). It has also been found that for different integrins, transfection (the process of inserting genetic material) of various cell types with cDNA (complementary DNA) raises the metastatic phenotype of inheritor cells. Evidence has been found for negative correlation between the expression of E-cadherin and cancer cell spread; invasion can be prevented through E-cadherin transfection with cDNA and antibodies with the power to inactivate E-cadherin causing the invasive phenotypes. These facts help to conclude that loss of E-cadherin raises the chances of metastasis, and CD44 helps in cells spread. It has been discovered that isoforms of CD44 are involved in metastasis process (Günthert et al. 1991).
Cell motility is very important as without cell motility neither invasion nor metastasis occur. Motility in cancer cells was discovered using time-lapse cinematography that was applied on V2 malignant cells. In time-lapse cinematography, it was observed that those malignant cells continuously moved at an average speed of 6-7 μm/min. This speed is the same as the normal leukocyte, but it is more than the speed of normal fibroblasts, histiocytes, or epithelial cells. Cell motility is governed by several features that are known as “motogens.” Some of the motogens also act as growth factors or vice versa (Table 5.1). Motogens are also involved in angiogenesis; among them, few motivate cell motility and inhibit growth, whereas the others inhibit motility as well as growth. There are some factors that act as motility factor primarily, including scatter factors, autocrine motility factors (AMFs), and migration stimulating factors (MSFs). Scatter factor, AMF, and MSF all are peptides. Scatter factor is produced by fibroblast; AMF is actually refined from human melanoma cells; and MSFs are produced by embryo and certain tumor-linked fibroblasts. AMFs and MSFs both can govern the movement in an autocrine manner, but scatter factors act in a paracrine way. Plasminogen activator (μPA) receptors also play an important role in cell motility (Gyetko et al. 1994).
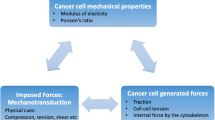
4 Physical and Mechanical Processes in Cell Motility and Metastasis
In metastasis, a series of steps are involved, which include cancer cell motility through blood vessels, lymphatic vessels, and the tissues at the secondary sites (organ). The ability to handle each phase and advancement toward the development of secondary tumor and its growth are completely dependent upon the mechanical forces and physical interactions between cancerous cells and its microenvironment. During the invasion through the basal membrane into the blood vessels, cancer cells must undergo elastic distortions to penetrate endothelial cell–cell joint. The interplay between the tumor cell velocity and adhesion helps to bind the cancerous cells to the vessel walls that lead to the formation of a secondary tumor and its growth (Wirtz et al. 2011).
4.1 Physical Interaction in Cell Motility
Loss of E-cadherin and cytokeratins causes reduction in adhesive forces between cells and morphological changes from cuboidal to mesenchymal. During this phase, cells undergo dramatic changes like, detachment of cells from the primary tumor, and acquisition of motile phenotypes as their physical and mesenchymal properties are altered. After leaving the primary site, the migrating cells find the ECM, which is structurally complex and rich in collagen I fiber and fibronectin (Hotary et al. 2003). As the ECM consists of enhanced collagen deposition and lysyl oxidase (LOX) cross-linking of collagen, it is more rigid in nature than the normal tissue. These collagen cross-linking improves integrin signaling and bundling of fibers, which promote cell proliferation and invasion. Despite recent advances, scientists have very little information regarding the molecular and physical mechanisms of cell motility and metastasis.
Stress fibers are either local to the cell cortex or emanates from the nucleus in the direction of the plasma membrane to develop pseudopodial protrusions (Bloom et al. 2008). In the case of tumor cells, cell–matrix adhesion molecules are absent in cells and matrix junctions and stress fibers are also reduced and re-localized. At the time of cell motility, cells generate contractile forces through cellular protrusions that influence the bundling of collagen fibers. These collagen bundles increase the available surface area, which helps to form larger adhesion (Smith et al. 2007). Collagen fibers help to form small-sized dynamic integrin clusters with less than a few seconds of life. Due to the absence of stress fibers and cell–matrix adhesion molecules in the junction of tumor cells and matrix, tumor cells and epithelial or endothelial cells cannot form wide lamellipodium and associated filopodial protrusions at the cell boundary. It is visible in 3D traction microscopy; cells, present in the matrix, only pull the local fibers, but it never pushes the local matrix (Bloom et al. 2008; Legant et al. 2010). These pseudopodial bulges pull the leading and trailing cell edges. Considerable matrix traction only happens in the neighborhood of pseudopodial protrusions (each cell contains one to five protrusions at a time) (Bloom et al. 2008). Though pseudopodia released from collagen fibers are asymmetric and develop a defect in the matrix during cancer cell migration, the pseudopodial protrusions at the trailing edge of cancer cell are released initially and then the back of the cell in the forward direction is dragged (biased motion). The cellular traction force on collagen may activate the MMPs (Ellsmere et al. 1999). Pseudopodial protrusion movement in the matrices is regulated by cell–matrix adhesion components like, scaffolding protein p130CAS and mechanosensing protein zyxin. p130CAS mediates a high number and high growth rate of protrusions. Scientists (Fraley et al. 2010) have shown that a strong correlation exists between the numbers of protrusion per unit time and the growth rate of protrusions and tumor cell motility in the matrices.
Experiments have been performed on mouse mammary tumors indicating that a few tumor cells diffuse from the primary tumor sites and go through a highly directed migration away from the tumor guided by collagen fibers (Sahai 2005). Intravital microscopy of GFP-labeled breast cancer cells in mice supports the fact that cancerous cells circulate as individual cells toward blood capillaries, whereas they migrate collectively in the direction of lymphatic vessels (Giampieri et al. 2009). Such type of collective migration requires the dominance of actomyosin contractility at intercellular adhesions (Hidalgo-Carcedo et al. 2011).
4.2 Cellular Mechanics in Intravasation
Tumor cells undergo shape changes during entry into and exit from the vascular system. This is driven by cytoskeletal remodeling for penetrating endothelial cell–cell junctions. Cytoplasm is a complex component of any cell; it is a rubber-like liquid material (viscous), and its distortion rate is very high (Wirtz 2009). The elasticity property of cytoplasm measures the ability to rebound from an applied force, whereas viscosity reflects its ability to go through under external shear of a flow. The nuclear lamina underlying the nuclear envelope, chromatin structure, and linkers of the nucleus and cytoskeleton (LINC) together determine the elastic property of the nucleus. But, the role of LINC and nuclear lamina in cell migration is still not properly understood.
Cancerous cells are much softer than normal cells and have strong correlation with metastatic potential (Cross et al. 2007). Also, the softer cytoplasm of cancerous cells correlates with a less-structured cytoskeleton. Physical properties of the microenvironment are very crucial for cancer cell migration as they control the cell movement (Lee et al. 2006). Some optimal mechanical properties like rigidness or softness are the key during cell migration through matrix or incursion into endothelium. If the surface is too rigid or too soft, cells are able to distort the cross-linked collagen fibers to migrate proficiently. However, individual cells of a specific cell type are heterogeneous and have shown a variation in mechanical properties, which illustrates that invasion and intravasation (Fig. 5.6) capable cells are likely to preserve this phenotype over several generations. The attributes of cancer cells alter dynamically during the metastasis to survive in the new environment of the vasculature system and the stromal space. These alterations in mechanical attributes might also be controlled by biochemical gradients (Tseng et al. 2004), interstitial flows (Swartz and Fleury 2007), and endogenous electric fields (Mycielska and Djamgoz 2004).

Invasion and intravasation mechanism (Wirtz et al. 2011)
4.3 Forces Acting on the Circulating Tumor Cells
During migration through the vascular system, cancerous cells are affected by hemodynamic forces, immunological stress, and also collisions with the blood cells and the endothelial cells of the vessel wall. The survival of these circulating tumor cells and establishment of metastatic foci are largely controlled by these stresses. The circulating cancer cells, which succeed in dealing with the fluid shear and immune observation of the body, will be stuck to the vascular endothelium (Fig. 5.7) of a distant site (organ) and exit from the vascular systems successfully to go into the tissue.

Circulating tumor cell getting arrested under the influence of local flow pattern and collisions with wall (Wirtz et al. 2011)
When the tumor cells enter into the vascular system, it is regulated by a number of physical and mechanical attributes: the blood flow pattern, blood vessels’ diameter, and complicated interaction between shear flow and intercellular adhesion. The rotational and translational motion of circulating tumor cells is influenced by the shear flow, and therefore, these two motions determine the orientation with the receptor–ligand interactions, which lead to adhesion. Also, shear flow influences distortion of circulating tumor cells on the way to the vessel walls. Though the amount of these effects and motivation on occlusion and adhesion remain to be determined; there is very less information regarding the properties of shear flow on the capability and proliferation of circulating tumor cells.
4.4 Extravasation of Circulating Tumor Cells
Whenever circulating cancerous cells enter into the blood vessels with smaller diameter than the cancerous cells, then an arrest can occur by physical occlusion. In general, circulating cancer cells of epithelial origin are more than 10 μm in size. So, physical occlusion occurs in the vessels with less than 10 μm in size.
At the time of extravasation, the tumor cell circulating through a larger blood vessel needs the adhesion to stick to the vessel wall. The probability of arrest of a cell at a large vessel is proportionate to the product of multiplication of the collision rate between membrane-bound receptors and endothelial ligands, and residence time of the cell (Zhu et al. 2008). The residence time of a cell is dependent on the ligand–receptor adhesive forces between the vessel wall and the circulating cell and the shear fluid force applied to the cell. If the shear force increases, then the collision frequency with the endothelium increases, but the residence time is decreased. The total adhesive force depends on the strength of the ligand–receptor bond and the number of ligand–receptor bonds involved.
When a cell is moving along the vessel wall, it has two velocities: translational velocity and angular velocity. The translational velocity is always greater than the angular velocity. As a result, a sliding movement can be seen in contrast to the motionless vessel wall. This sliding movement raises the chances of meeting a single receptor on a circulating tumor cell and ligands on the vessel wall. In the case of rotational motion, it brings consecutive receptors on the circulating tumor cell surface into interaction with ligands on the vessel wall (Fig. 5.8).

Capture and arrest of circulating tumor cells through transient and/or persistent adhesion (Wirtz et al. 2011)
The chances of cell arrest are much higher if the shear force is at the intermediate level. A bond at a particular shear stress level and cellular adhesion pattern will be formed or not is mainly regulated by the kinetic and other micromechanical properties of receptor–ligand. As an example, to form receptor-mediated cell adhesion in the presence of shear force, a comparatively fast on rate and relatively slow off rate are seen. Fast on rate helps receptor–ligand binding within a short span of interaction time, whereas slow off rates allow sustainable bond lifetime. Receptor–ligand bonds (such as selectins and their ligands) show high micromechanical strength; fast on rates and comparatively fast off rates influence binding in the presence of shear force. Molecules with slower on rates (such as integrin) can be involved at a very low shear force only after selectin-mediated cell binding or in the absence of selectin-dependent interactions. Integrins are engaged in the circulation of tumor cells and also regulate angiogenesis and metastatic growth.
4.4.1 Receptor–Ligand Interactions of Circulating Tumor Cells
Circulating tumor cells sometimes escape immune surveillance and exit from the circulatory system (Fig. 5.9). Activated platelets release a number of chemical substrates to promote angiogenesis and metastatic growth (Pinedo et al. 1998). To promote vascular hyperpermeability and extravasation, platelets secrete a number of chemical factors like VEGF at the points of attachments to the endothelium (Nash et al. 2002). Circulating tumor cells often capture the endothelium of the secondary organ with the help of polymorphonuclear leukocytes (PMNs). Close association with PMNs and metastatic tumor cells during tumor cell arrest and extravasation has been observed (Crissman et al. 1985). By directly binding to the vascular endothelium with the help of selectin-mediated tethering, circulating tumor cells behave like neutrophils. This binding can happen using cell rolling followed by strong cell adhesion. In fact, P-, L-, and E-selectins help cancer metastasis through arresting the circulating tumor cells in the microvasculature.

Circulating tumor cell arrest through platelet-mediated capture (Wirtz et al. 2011)
4.4.2 Physics behind the Location of Metastatic Site
The patterns of metastasis can be explained with two different hypotheses: seed and soil hypotheses (discussed in Sect. 5.3.2), and mechanical hypotheses and both of them have complementary roles in influencing the location of metastasis. According to seed and soil hypothesis, the chances of metastasis occurring at a particular site are proportional to the multiplication of three components: (i) probability of adhesion due to the collision at a particular site, (ii) probability of extravasation at that site, and (iii) probability of colonization. On the other hand, according to the mechanical hypothesis, cell catching, its extravasation, and colonization occur in a cascade; so, the chances of metastasis occurring at a particular site is proportional to the multiplication of three components: (i) probability of encountering a vessel with diameter less than the cell diameter, (ii) probability of extravasation at that site, and (iii) probability of colonization. Chances of metastasis occurrence in a specific organ have common elements related to extravasation and colonization, and both of them are dependent on local microenvironment.
According to mechanical hypothesis, the probability of occurrence of metastasis at a site is dependent on the blood flow pattern (Weiss 2000). Blood is pumped from the heart to different organs through the arterial system and is subsequently returned to the heart. If a tumor cell is found in a capillary with smaller diameter, then the chances of the cell catching through physical occlusion are higher for that site.
For metastasis to occur, a tumor cell must extravasate and colonize some neighboring tissue. At the time of circulation, tumor cells often collide with the vessel wall and have potential to stuck in that location due to adhesion. If the cell is stuck in that location for sufficiently long time, then it might extravasate. The chances that the residing time is sufficiently long are dependent on shear stress. Receptor–ligand adhesion varies depending on organs (Trepel et al. 2002).
Metastatic tumor cells need an optimal shear stress to stuck sufficiently long time in a particular position. If the stresses are high (12 dyn per cm2) like in arterial circulation, then the immune system destroys the tumor cells due to cell cycle arrest (Chang et al. 2008). On the other hand, if the stress is at a lower level like in venous system, it can create an opposite effect on intercellular signaling and tumor cell function.
5 Conclusions
Cancer cells can move to other locations through several ways either singly or collectively. They breach the basement membrane of the host tissue using metalloproteinase enzymes and also the nearby blood vessels using proteolytic enzymes. After breaching these membranes, they are free to spread to other organs in the body. In this journey, tumor cells undergo some transformation to adapt to the new environment. During motility, cell modifies their cell–cell or cell–ECM junctions. The primary reason of the cell movement is dynamic reformation of actin cytoskeleton (using RhoGTPase family), which provides the required force during cell motility. During actin reformation, cell membrane forms finger-like filopodia or lamellipodia protrusions. These protrusions help the cell to move to a different location. Also, RhoA regulates actomyosin filaments, which promote locomotive forces in the cell body. While Rac1 and Cdc42 motivate the growth of cellular protrusions at the leading edge, RhoA makes retraction at the trailing edge. These cooperative efforts make a cell to move.
The migrating tumor cells reach other sites of a body through the blood/lymphatic vessels. When a tumor cell is in the vascular system, its movement is regulated by different factors like blood flow pattern, diameter of blood vessels, shear flow of fluids, and intercellular adhesion. The properties of cancer cells (in metastasized condition) dynamically change to survive in the new environment of the vasculature system and are affected by hemodynamic forces, immunological stress, and collisions with the blood cells and the endothelial cells of vessel wall. The rotational and translational motions of circulating tumor cells are influenced by the shear flow; these two motions determine the orientation with respect to the receptor–ligand interactions and lead to adhesion. A sliding movement of cancer cells can be seen in contrast to the motionless vessel wall that raises the chances of meeting between a single receptor on a circulating tumor cell and the ligands on the vessel’s wall.
Circulating tumor cell in a larger blood vessel needs the requisite adhesion to stick to the vessel wall. The chance of cell arrest at a large vessel depends on the collision rate between the membrane-bound receptors and the endothelial ligands, and the residence time of the cell. With increasing shear force, the collision frequency with the endothelium increases and the residence time decreases. The total adhesive force depends on the strength of the ligand–receptor bond and the number of ligand–receptor bonds involved. Tumor cells can exit from the vascular system, but it first binds to the vessel’s wall through physical occlusion and/or cellular adhesion. The cells that succeed to survive with the fluid shear and immune observation of the body will be stuck to the microvascular endothelium of a secondary organ and exit from the vascular system successfully (extravasation) and enter into another tissue by invading the vessel’s wall and the tissue membrane. After reaching the secondary site, it penetrates the secondary organ and continues its growth leading to a new colony of cancerous cells. This cycle continues till the host dies or the cancer cells are completely removed from the body.
References
Allen JW, Khetani SR, Johnson RS, Bhatia SN (2006) In vitro liver tissue model established from transgenic mice: role of HIF-1alpha on hypoxic gene expression. Tissue Eng 12(11):3135–3147
Ang KC, Tan LS (2008) A numerical approach to modelling avascular tumour evolution with white noise. ANZIAM J 50:C569–C582
Barnes DM, Dublin EA, Fisher CJ, Levison DA, Millis RR (1993) Immunohistochemical detection of p53 protein in mammary carcinoma: an important new independent indicator of prognosis? Hum Pathol 24(5):469–476
Berenson J, Rajdev L, Broder M (2006) Pathophysiology of bone metastases. Cancer Biol Ther 5(9):1078–1081
Bloom RJ, George JP, Celedon A, Sun SX, Wirtz D (2008) Mapping local matrix remodeling induced by a migrating tumor cell using three-dimensional multiple-particle tracking. Biophys J 95(8):4077–4088
Castro P, Soares P, Gusmao L, Seruca R, Sobrinho-Simoes M (2006) H-RAS 81 polymorphism is significantly associated with aneuploidy in follicular tumors of the thyroid. Oncogene 25(33):4620–4627
Chang SF, Chang CA, Lee DY, Lee PL, Yeh YM, Yeh CR, Cheng CK, Chien S, Chiu JJ (2008) Tumor cell cycle arrest induced by shear stress: roles of integrins and Smad. Proc Natl Acad Sci U S A 105(10):3927–3932
Clyde RG, Bown JL, Hupp TR, Zhelev N, Crawford JW (2006) The role of modelling in identifying drug targets for diseases of the cell cycle. J R Soc Interface 3(10):617–627
Crissman JD, Hatfield J, Schaldenbrand M, Sloane BF, Honn KV (1985) Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab Invest 53(4):470–478
Croce CM (2008) Oncogenes and cancer. N Engl J Med 358(5):502–511
Cross SE, Jin YS, Rao J, Gimzewski JK (2007) Nanomechanical analysis of cells from cancer patients. Nat Nanotechnol 2(12):780–783
Duffy MJ (1992) The role of proteolytic enzymes in cancer invasion and metastasis. Clin Exp Metastasis 10(3):145–155
Ellsmere JC, Khanna RA, Lee JM (1999) Mechanical loading of bovine pericardium accelerates enzymatic degradation. Biomaterials 20(12):1143–1150
Fidler IJ (1970) Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst 45(4):773–782
Fidler IJ (1990) Critical factors in the biology of human cancer metastasis: twenty-eighth GHA Clowes memorial award lecture. Cancer Res 50(19):6130–6138
Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, Wirtz D (2010) A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol 12(6):598–604
Friedl P (2004) Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol 16(1):14–23
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3(5):362–374
Friedl P, Wolf K (2010) Plasticity of cell migration: a multiscale tuning model. J Cell Biol 188(1):11–19
Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin Cell Biol 9(5):701–706
Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E (2009) Localized and reversible TGFβ signaling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11(11):1287–1296
Günthert U, Hofmann M, Rudy W, Reber S, Zöller M, Hauβmann I, Matzku S, Wenzel A, Ponta H, Herrlich P (1991) A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 65(1):13–24
Gyetko MR, Todd RF, Wilkinson CC, Sitrin RG (1994) The urokinase receptor is required for human monocyte chemotaxis in vitro. J Clin Invest 93(4):1380–1387
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hansen RK, Bissell MJ (2000) Tissue architecture and breast cancer: the role of extracellular matrix and steroid hormones. Endocr Relat Cancer 7(2):95–113
Herranz N, Pasini D, Díaz VM, Francí C, Gutierrez A, Dave N, Escrivà M, Hernandez-Muñoz I, Di Croce L, Helin K, De Herreros AG (2008) Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol 28(15):4772–4781
Hidalgo-Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B, Sahai E (2011) Collective cell migration requires suppression of actomyosin at cell–cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol 13(1):49–59
Horowitz JM, Yandell DW, Park SH, Canning S, Whyte P, Buchkovich K, Harlow ED, Weinberg RA, Dryja TP (1989) Point mutational inactivation of the retinoblastoma antioncogene. Science 243(4893):937–940
Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ (2003) Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114(1):33–45
Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD, Rauscher FJ (2008) The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol 28(10):3198–3207
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66(23):11089–11093
Kashkin KN, Chernov IP, Stukacheva EA, Kopantzev EP, Monastyrskaya GS, Uspenskaya NY, Sverdlov ED (2013) Cancer specificity of promoters of the genes involved in cell proliferation control. Acta Naturae 5(3):79–83
Kleer CG, Teknos TN, Islam M, Marcus B, Lee JSJ, Pan Q, Merajver SD (2006) RhoC GTPase expression as a potential marker of lymph node metastasis in squamous cell carcinomas of the head and neck. Clin Cancer Res 12(15):4485–4490
Lee JS, Panorchan P, Hale CM, Khatau SB, Kole TP, Tseng Y, Wirtz D (2006) Ballistic intracellular nanorheology reveals ROCK-hard cytoplasmic stiffening response to fluid flow. J Cell Sci 119(9):1760–1768
Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS (2010) Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nat Methods 7(12):969
Lehembre F, Yilmaz M, Wicki A, Schomber T, Strittmatter K, Ziegler D, Kren A, Went P, Derksen PW, Berns A, Jonkers J (2008) NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J 27(19):2603–2615
Liotta LA (1992) Cancer cell invasion and metastasis. Sci Am 266(2):54–63
Lozano E, Betson M, Braga VM (2003) Tumor progression: small GTPases and loss of cell–cell adhesion. BioEssays 25(5):452–463
Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3(6):459–465
Montell DJ (2008) Morphogenetic cell movements: diversity from modular mechanical properties. Science 322(5907):1502–1505
Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139(7):1327–1341
Mycielska ME, Djamgoz MB (2004) Cellular mechanisms of direct-current electric field effects: galvanotaxis and metastatic disease. J Cell Sci 117(9):1631–1639
Nash GF, Turner LF, Scully MF, Kakkar AK (2002) Platelets and cancer. Lancet Oncol 3(7):425–430
Nekarda H, Siewert J, Schmitt M, Ulm K (1994) Tumour-associated proteolytic factors uPA and PAI-1 and survival in totally resected gastric cancer. Lancet (London, England) 343(8889):117–117
Orme ME, Chaplain MAJ (1996) A mathematical model of the first steps of tumour-related angiogenesis: capillary sprout formation and secondary branching. Math Med Biol 13(2):73–98
Paget S (1989) Distribution of secondary growths in cancer of the breast. Lancet I 571
Paweletz N, Knierim M (1989) Tumor-related angiogenesis. Crit Rev Oncol Hematol 9(3):197–242
Pinedo HM, Verheul HMW, D’amato RJ, Folkman J (1998) Involvement of platelets in tumour angiogenesis? Lancet 352(9142):1775–1777
Pouysségur J, Dayan F, Mazure NM (2006) Hypoxia signaling in cancer and approaches to enforce tumour regression. Nature 441(7092):437–443
Quon KC, Berns A (2001) Haplo-insufficiency? Let me count the ways. Genes Dev 15(22):2917–2921
Ruoslahti E (1992) Control of cell motility and tumour invasion by extracellular matrix interactions. Br J Cancer 66(2):239–242
Sahai E (2005) Mechanisms of cancer cell invasion. Curr Opin Genet Dev 15(1):87–96
Seidensticker MJ, Behrens J (2000) Biochemical interactions in the wnt pathway. Bioch Biophys Acta Mol Cell Res 1495(2):168–182
Selam B, Kayisli UA, Garcia-Velasco JA, Arici A (2002) Extracellular matrix-dependent regulation of Fas ligand expression in human endometrial stromal cells. Biol Reprod 66(1):1–5
Semenza GL (2001) HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107(1):1–3
Sherratt JA, Chaplain MA (2001) A new mathematical model for avascular tumour growth. J Math Biol 43(4):291–312
Small JV, Stradal T, Vignal E, Rottner K (2002) The lamellipodium: where motility begins. Trends Cell Biol 12(3):112–120
Smith ML, Gourdon D, Little WC, Kubow KE, Eguiluz RA, Luna-Morris S, Vogel V (2007) Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol 5(10):e268
Sutherland RM (1988) Cell and environment interactions in tumor microregions: the multicell spheroid model. Science 240(4849):177–184
Swartz MA, Fleury ME (2007) Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng 9:229–256
Trepel M, Arap W, Pasqualini R (2002) In vivo phage display and vascular heterogeneity: implications for targeted medicine. Curr Opin Chem Biol 6(3):399–404
Tseng Y, Lee JS, Kole TP, Jiang I, Wirtz D (2004) Micro-organization and visco-elasticity of the interphase nucleus revealed by particle nanotracking. J Cell Sci 117(10):2159–2167
Wang K, Grivennikov SI, Karin M (2013) Implications of anti-cytokine therapy in colorectal cancer and autoimmune diseases. Ann Rheum Dis 72(Suppl 2):ii100–ii103
Weaver AM (2006) Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis 23(2):97–105
Wei C, Larsen M, Hoffman MP, Yamada KM (2007) Self-organization and branching morphogenesis of primary salivary epithelial cells. Tissue Eng 13(4):721–735
Weiss L (2000) Patterns of metastasis. Cancer Metastasis Rev 19(3–4):281–301
Wirtz D (2009) Particle-tracking microrheology of living cells: principles and applications. Annu Rev Biophys 38:301–326
Wirtz D, Konstantopoulos K, Searson PC (2011) The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer 11(7):512–522
Yamazaki D, Kurisu S, Takenawa T (2005) Regulation of cancer cell motility through actin reorganization. Cancer Sci 96(7):379–386
Zhu C, Yago T, Lou J, Zarnitsyna VI, McEver RP (2008) Mechanisms for flow-enhanced cell adhesion. Ann Biomed Eng 36(4):604–621
Zipori D (2006) The mesenchyme in cancer therapy as a target tumor component, effector cell modality and cytokine expression vehicle. Cancer Metastasis Rev 25(3):459–467
Acknowledgement
We are thankful to Debarpita Santra, PhD scholar, Department of Computer Science and Engineering, University of Kalyani, West Bengal, India; Soumyabrata Roy, postdoctoral research associate at School of Medicine, University of California, Irvine, USA; and Subhasis Barik, Senior Scientific Officer-II, Chittaranjan National Cancer Institute, Kolkata, West Bengal, India, for their cooperation and help.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Appendix
Appendix
1.1 Biological Notes
Anaerobic glycolysis: | It transforms glucose into lactate in the presence of limited amount of oxygen |
Anaplasia: | Cells with the least cellular differentiation lose the morphological properties of mature cells and destroy the orientation with the other cells and endothelial cells |
Autocrine signals: | Cells secrete hormones that bind to its corresponding receptors on the same cell |
Cadherins: | Cadherins are calcium-based transmembrane, cell–cell adhesive glycoprotein |
Cathepsin B: | It is normally found in lysosomes and activated by cathepsin D-like enzymes or metalloprotease and can also activate some of the collagenases and uPA |
Cathepsin D: | It is a lysosomal protease. Cathepsin D behaves like a mitogen for estrogen-deprived breast cancer cell lines and accelerates the damage of the ECM |
CD44: | It acts as receptors for hyaluronate and lymphocyte homing receptors and is coded by a single gene located on chromosome 11 in human cells |
Cell differentiation: | In this process, a less specialized cell transforms into a more specialized type of cell. It changes a size, shape, metabolic activity, and responsiveness to signals of a cell |
Complementary DNA: | It is DNA synthesized from a single-stranded RNA template in a reaction catalyzed by the enzyme reverse transcriptase and is often used to clone eukaryotic genes in prokaryotes |
Dysplasia: | It is abnormal growth of a tissue |
Endocytosis: | Transporting molecules into a cell by engulfing it is known as endocytosis; when molecules cannot pass through the membrane, cells use endocytosis |
Extracellular matrix: | It is a group of extracellular molecules providing structural and biochemical support to the adjacent cells (tissue) |
Extravasation: | It is the process during metastasis in which cells are come out from blood vessels and develop a colony into secondary tissue |
Filopodia: | It is narrow cytoplasmic projections beyond the lamellipodia of migrating cells |
Glycoprotein podoplanin: | It upregulates in the invasive front of several human cancers. It has been associated with EMT and increased cell migration and tissue invasion |
Hemodynamic forces: | It is the dynamics of blood flow within circulatory systems |
Homeostasis: | It is the activity of an organism to autoregulate and maintain its internal environment in a stable state |
Immunoglobulin superfamily: | Proteins belonging to this family consist of an immunoglobulin domain that is made of 90–100 amino acid molecules decorated within a sandwich of two antiparallel strands |
Immunological stress: | It is the status of human immune system when it is affected by bacteria, virus, and endocrine. |
Integrins: | Integrins are a set of heterodimeric glycoproteins |
Intracrine signals: | Signaling molecules binding to intracellular receptors |
Intravasation: | It refers to cancer cells entering through the basal membrane into blood or lymphatic vessels |
Lamellipodia: | It refers to large cytoplasmic projections found primarily at the leading edge of migrating cells |
Matrix metalloproteinases: | It is a group of enzymes responsible for damaging most of the extracellular matrix proteins during organogenesis, growth, and normal tissue turnover |
Metalloproteases: | The metalloproteases can be categorized as interstitial collagenases, type IV collagenases, and the stromelysins. Interstitial collagenases accelerate the damage of type I, II, and III collagens. It helps to accelerate the degradation of different elements of ECM |
Metaplasia: | It is a change in the nature of a tissue that is not normal for that tissue |
p120-catenin: | It has multiple roles like controlling cadherin stability, adhesion-induced signaling, and cancer progression |
p130CAS: | It regulates varieties of signal pathways related to cell adhesion, migration, and invasion |
p190-RhoGAP: | It controls cell spreading and migration |
p53: | It is a gene that codes for a protein that controls cell cycle and hence functions as tumor suppressor |
Plasminogen activators: | It acts as catalyst during the conversion of inactive plasminogen to the active plasmin. It can be available in two forms: Tissue-type plasminogen activators and urokinase-type plasminogen activators into different genes |
Polymorphonuclear leukocytes: | It is a kind of immune cells having small particles with enzymes released during infections, allergic reactions, and asthma |
RGD sequence: | The tripeptide Arg-Gly-asp (RGD) is the amino acid sequence within the ECM protein fibronectin-mediating cell attachment |
Selectins: | The proteins belong to selectin group contain lectin-type domain. These types of proteins regulate heterophilic communications between endothelium and blood cells |
Vascular hyperpermeability: | It defines the capacity of a blood vessel wall to permit the flow of small molecules or even cells in and out of the vessel |
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sadhukhan, S., Dey, S. (2022). Biology, Chemistry, and Physics of Cancer Cell Motility and Metastasis. In: Basu, S.K., Panda, C.K., Goswami, S. (eds) Cancer Diagnostics and Therapeutics . Springer, Singapore. https://doi.org/10.1007/978-981-16-4752-9_5
Download citation
DOI: https://doi.org/10.1007/978-981-16-4752-9_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4751-2
Online ISBN: 978-981-16-4752-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)