
Abstract
Grade 2 gliomas constitute a heterogeneous cohort of central nervous system (CNS) tumors of glial origin with variable clinical presentation and long-term outcomes. Despite inclusion into low grade gliomas, some of them may exhibit locally aggressive behavior and potential for malignant transformation into high grade gliomas. They usually impact children or young adults. Surgery is the primary treatment and extent of resection has prognostic implications. The World Health Organization (WHO) 2016 CNS classification has introduced several changes in the categorization of tumors with incorporation of molecular and genetic factors which has impact on prognosis as well as treatment decisions. Surgery is the main treatment modality—it relieves symptoms (most commonly seizures) and gives pathologic information to plan further adjuvant therapy. In astrocytic and oligodendroglial tumors, several randomized trials till date have defined the role of adjuvant therapy types, doses, timing, etc., to maximize survival. Incorporation of new molecular information for post hoc analysis of prior studies and stratification of future trials will help determine if data from randomized trials over last three decades still hold value. Since most of these tumors have potential for long-term survival, quality of life considerations are as important as local control in choosing between therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
This chapter deals with grade 2 diffuse astrocytic and oligodendroglial tumors, “other” astrocytic tumors and mixed neuronal and neuronal-glial tumors, as defined by World Health Organization (WHO) 2016 Central nervous system (CNS) classification. For the ease of discussion, low grade gliomas (LGGs) mentioned in this chapter would essentially refer to grade 2 tumors. Grade 1 tumors have been discussed in a separate chapter.
2 Epidemiology
LGGs comprise about 2000 cases per annum and represent nearly 10% of primary brain tumors. They have a slight male preponderance, with a mean age of diagnosis at 37 years (range 20–50 years), the incidence decreasing with advancing age. The presenting complaints for a patient with LGG may depend on the location of disease and may span several years before they are reported; these include seizures (65%, higher in LGG than glioblastoma, GBM), headache (35%), focal weakness (30%), visual disturbances (15%), personality changes (14%), and nausea or vomiting (10%).
3 Pathology and Molecular Characteristics
Traditionally, they were defined as astrocytic, oligodendroglial, and mixed tumors, but now, with advent and incorporation of molecular characteristics in WHO 2016 CNS classification, the “mixed” group is no longer valid. Apart from infiltrative histopathology, low mitotic rate and absence of necrosis/microvascular proliferation, molecular characteristics are now crucial for diagnosis, treatment decisions, and prognostication. Astrocytomas and oligodendrogliomas (ODG) are clubbed together based on growth pattern, behavior, and isocitrate dehydrogenase (IDH) mutation. Grade 2 astrocytic/oligodendroglial tumors are divided into three groups: (a) IDH-mutant diffuse astrocytoma, (b) IDH-mutant and 1p19q codeleted ODG, (c) IDH wild-type diffuse astrocytoma. “Other” astrocytic grade 2 tumors with a more circumscribed growth pattern, lack of IDH gene changes, and BRAF mutations include pleomorphic xanthoastrocytoma (PXA); grade 2 glioneuronal tumors include central neurocytoma, extraventricular neurocytoma, and cerebellar liponeurocytoma, which will also be discussed briefly.
Incorporation of both phenotypic and genotypic characteristics makes the new classification more objective and integrates histologic information, grade, and molecular information into one diagnosis [1].
Diffuse astrocytomas are lesions having tumor cells with wide infiltration into brain parenchyma; cellular density and anaplasia increase with the grade of tumor. Grade 2 diffuse astrocytomas have moderate increase in cellularity with highly fibrillar processes extending from neoplastic cells. There is normal CNS parenchymal architectural distortion with splaying of neuropil and microcysts. Nuclear enlargement, oblong shape, heterogeneous hyperchromatism, and irregular contours differentiate them from normal astrocytes. In contrast, classic ODGs have round regular nuclei with small nucleoli, clear perinuclear haloes, and bland chromatin. Glial fibrillary acid protein (GFAP) is positive. IDH-mutant diffuse astrocytoma (grade 2), IDH-mutant anaplastic astrocytoma (grade 3), and IDH-mutant GBM (grade 4) all lie on a continuum of increasing aggressiveness of similar origin cells. Similarly, grade 2 and grade 3 ODGs line on the same continuum with no grade 4 counterpart.
The main molecular markers used in the diagnostic workup are isocitrate dehydrogenase (IDH-1 and IDH-2), alpha-thalassemia/mental retardation syndrome X-linked (ATRX), Tumor protein p53 (p53), 1p19q codeletion, O6-methylguanine-DNA methyltransferase (MGMT), and telomerase reverse transcriptase (TERT), of which the first three are essential.
IDH can help distinguish gliomas from gliosis and may be present in astrocytomas, ODG, and 10% GBM. IDH-1 mutations (R132H) are more common and usually detectable by immunohistochemistry (IHC). If IHC is negative, IDH-1/IDH-2 genotyping may be needed if there is clinical suspicion. IDH wild-type tumors are those without IDH-1 or IDH-2 mutations. IDH-1 mutation confers a good prognosis in gliomas but is also a driver mutation promoting tumorigenesis. If IDH testing is inconclusive or cannot be performed, it is called IDH NOS (not otherwise specified).
ATRX: This gene has several important roles in cell division. Mutations will lead to loss of ATRX staining and is seen in several high grade gliomas (HGG).
p53: This is a tumor suppressor gene located on 17p chromosome. Loss of p53 promotes tumorigenesis in several CNS tumors, including GBM, medulloblastoma, and >50% IDH-mutant astrocytomas.
1p19q codeletion detectable by fluorescent in situ hybridization (FISH) is a diagnostic hallmark for ODGs. It has a strong prognostic value as well as predictive for response to chemotherapy and radiotherapy (RT).
MGMT gene encodes a DNA repair enzyme that may counter effect of alkylating agents such as temozolomide (TMZ). Methylation of MGMT promoter silences the gene. This promoter methylation is an established prognostic and predictive factor in GBM but has emerging value in LGGs as well.
TERT promoter mutations: They predict poor survival and treatment resistance in gliomas. This may be found in GBM and ODG.
IDH-mutant astrocytoma is characterized by a combination of IDH mutation, TP53 mutation, and ATRX loss.
If IDH mutation is absent by both IHC and genotyping, it qualifies as IDH wild-type diffuse astrocytoma, but only once IDH-1 and IDH-2 both have been deemed non-mutated. This group is an uncommon diagnosis and other alternative diagnoses such as ganglioglioma and IDH wild-type GBM should be ruled out.
IDH mutation with retained ATRX, limited p53 positivity and 1p19q codeletion define ODGs.
Consequent to WHO 2016 classification, the boundary between grade 2 and grade 3 gliomas has become less well defined in terms of survival outcomes. Instead of grade, IDH-1 mutation and 1p19q codeletion have become major drivers of disease categorization and eventual prognosis. Many tumors earlier defined as grade 3, such as anaplastic ODG, may have a good prognosis if IDH mutant and 1p19q codeleted, while a tumor previously defined as a diffuse astrocytoma (grade 2) may turn out to be IDH wild type and have an aggressive course akin to GBM. Epidermal growth factor receptor (EGFR) amplification, chromosome 7 gain, chromosome 10 loss, TERT promoter mutations, if present, would qualify grade 4 behavior and though not officially part of WHO classification, this entity has a proposed designation of “diffuse astrocytic glioma, IDH wild type, with molecular features of GBM, WHO grade 4.”
Hence the term “high risk lower grade gliomas” is sometimes used to encompass the IDH-mutant anaplastic gliomas along with the traditional definition of age, tumor, and surgical extent. Since most phase 2 and 3 trials did not use these molecular characteristics for inclusion in trials until recently, we do not have clear-cut data for decision-making based on these molecular groupings. However, general principles of prognostic categorization and post hoc analysis for some of the earlier trials may assist in treatment choices.
Malignant transformation can occur in up to 70% of grade 2 glioma patients, the main risk factors being initial contrast enhancement, large tumor size, and subtotal resection (STR). In EORTC 22845 trial, early RT after surgery did not confer a higher risk for malignant transformation in LGGs compared to observation. Patients with malignant transformation have a shortened lifespan.
Astrocytic and oligodendroglial tumors may occur anywhere in the neuraxis. IDH-mutant gliomas are more common in cerebral subcortical white matter of frontal lobe. IDH wild-type astrocytomas arise in supratentorial region, and may involve multiple lobes or deep grey matter. Astrocytomas which appear grade 2 on histology but are in midline/thalamic location should be tested for H3 K27M mutation to rule out grade 4 diffuse midline glioma, which is particularly aggressive.
4 Imaging in LGGs
4.1 Computed Tomography (CT) Scan
Grade 2 gliomas are slow growing tumors and may produce calvarial erosion, expansion, or remodeling on plain CT scan. Tumors are seen as ill-defined homogenous hypodense masses. Most of them show no to minimal contrast enhancement and do not have significant perilesional edema. ODGs may have characteristic heterogeneous density with calcification in nearly 70%. Enhancement in previously known LGGs may raise suspicion of secondary transformation to HGG.
4.2 Multiparametric Magnetic Resonance Imaging (MRI)
This is now the standard and preferred imaging modality for brain tumors. It helps distinguish LGGs from HGGs better and gives fine anatomic detail of tumor location and extent that helps to plan local therapies such as surgery and RT, and also monitor treatment response. Diffuse astrocytomas are hypointense on T1 weighted sequences and hyperintense on T2/FLAIR, with no to minimal enhancement with gadolinium. ODGs may sometimes be isointense on T1 and may show heterogeneous enhancement. Calcification in ODGs appears as areas of low intensity blooming on GRE, SWI, or T2 sequences. MR spectroscopy may help distinguish brain tumors from non-neoplastic lesions and also to determine grade. N-acetylaspartate (NAA) is a surrogate marker of neuronal viability; it is present in higher levels in LGGs than HGGs due to preservation of neuronal integrity and astrocyte function. Choline (Cho) is a marker of cell membrane density and is raised in higher grade tumors and tumors with high Ki-67. Creatine (Cr) denotes energy metabolism and is largely stable. LGGs have higher NAA/Cr ratios and lower Cho/Cr ratios than HGGs. Cho peak may be higher in LGGs compared to background normal brain. Lactate and lipid peaks usually denote necrosis and myelin sheath disruption and are almost never seen in LGGs. Diffusion tensor imaging (DTI) helps visualize the path of white matter tracts owing to physiologic water molecule anisotropy within them. LGGs have more discrete boundaries and rarely invade along or into white matter tracts, unlike HGG. Surgeries are often performed with MRI guidance where preoperative MRI is co-registered with the operative field to help define planes for resection between normal brain and tumor tissue; T2 and FLAIR sequences are particularly useful in LGGs. CT guidance may be useful in some ODGs with calcification or CT angiography for navigation of critical vascular structures in tumor vicinity. Ultrasound, on the other hand, may be a useful real-time navigation tool, especially in the setting of brain shift following tumor removal. Intraoperative MRI in the operating suite itself also helps ensure the completeness of resection, though more useful in HGGs.
4.3 Positron Emission Tomography (PET)-CT Scans
They have a limited role in brain tumors but C-11 methionine and 18F-fluoro-ethyl-L-tyrosine (FET)-PET may have some value in distinguishing gliomas from non-neoplastic lesions and in stereotactic biopsy planning.
5 Treatment
Most evidence on LGG management derives from seminal trials that did not segregate patients based on molecular characteristics. Additionally, incompletely resected pilocytic astrocytomas (PA) which are grade 1, and some aggressive forms of diffuse astrocytomas (IDH wild, H3K27M mutant) were included, diluting the results somewhat. However, post hoc analyses of several trials suggest that majority of patients do belong to the recently defined grade 2 category and the trial interpretations would still be valid.
5.1 Surgery
This is the first-line treatment for LGGs, even when detected incidentally and neurologic signs are absent. This relieves the mass effect over surrounding normal brain, relieves seizures if they were the presenting symptom, gives tissue for histopathology and molecular characterization and prognostication, reduces risk of malignant transformation, and even gives information for planning adjuvant therapy. Early resection is favored over watchful waiting as it shows a survival advantage [2]. A greater extent of resection (mostly based on volumetric studies comparing the pre- and postoperative tumor extent) correlates with better overall survival (OS). Gross total resection (GTR, defined as >90% removal based on postoperative MRI taken within 48 hours) improves survival. In a retrospective series, 8-year OS was 91% for patients with >90% resection versus (vs.) 60% for STR [3, 4]. A maximal safe resection is advocated to avoid short-term and long-term neurologic deficits; GTR is the goal but may not always be possible due to large size, proximity to eloquent brain and vascular involvement or encasement. Intraoperative electrostimulation mapping/functional mapping on awake patients helps in improving resection extent (and possibly long-term control and survival) without increased risk of neurologic deficits.
5.2 Adjuvant Radiotherapy and Chemotherapy
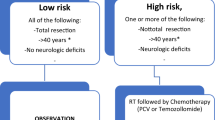
Indications for RT traditionally include all high risk patients—diffuse astrocytoma or ODGs occurring in patients over 40 years of age or those with STR. Additional factors that may have a role in decision-making include tumor size, midline extension and baseline neurologic deficits; importance of these shall be discussed in detail citing individual studies incorporating these factors. As evidence discussed subsequently will show, RT is no longer supported as monotherapy and should be followed up with adjuvant chemotherapy. Most of the available evidence does not take into account the molecular characteristics of the tumor which may change treatment recommendations in coming years. At present, the evidence is agnostic to molecular subtype, and observation after STR should be offered only to the subset of young patients with extremely favorable tumor characteristics willing for regular imaging.
5.2.1 Timing of Radiotherapy
One randomized controlled trial (RCT) determined that early RT may delay tumor growth and control seizures better but does not improve survival, hence there is an option for deferring RT in patients with good general condition and compliance for follow-up.
The EORTC 22845 (“Non-Believers Trial,” 1986–1997) recruited supratentorial LGGs (excluded completely resected PA, brainstem and optic pathway gliomas) between the ages of 16 and 65 years, post-surgery for randomization to early RT (54 gray, Gy) vs. delayed RT at progression, with follow-up contrast CT every 4 months. At a median follow-up of 7.8 years, the study concluded that early RT delayed local progression (median progression free survival, PFS, 5.3 vs. 3.4 years; p < 0.0001) and better seizure control (1 year, 25% vs. 41%) but did not improve OS (median OS, 7.4 vs. 7.2 years; p = 0.872). Nearly 65% patients in delayed RT group eventually received salvage RT at progression, and this probably contributed to lack of OS advantage. Both groups had similar cognitive function, performance status (PS), and focal deficits on follow-up [5].
5.2.2 Dose of Radiotherapy
Two randomized studies established that adjuvant RT doses of 45–50.4 Gy at 1.8 Gy per fraction (Fr) may be sufficient for LGGs, and contrary to popular belief, focal RT at given doses does not cause any major cognitive decline in adults.
EORTC 22844 trial (“Believers Trial,” 1985–1991) investigated the adjuvant RT dose for LGG (excluding completely resected PA) patients aged 16–65 years, comparing two schedules: 45 Gy/25 Fr vs. 59.4 Gy/33 Fr. Both groups had similar OS (58% vs. 59%) and PFS (47% vs. 50%) at a median follow-up of 6.2 years, suggesting no dose response above 45 Gy [6]. The high dose group also had worse quality of life (QOL) and symptom burden (fatigue, malaise, and insomnia at RT completion and leisure time and emotional functioning after 7–15 months of randomization) [7]. This trial proved a shorter RT schedule was more convenient and comfortable while not compromising on disease control. Tumor size was deemed prognostic for survival.
The Intergoup (NCCTG/RTOG/ECOG) trial (1986–1994) also randomized patients adults with supratentorial LGGs (other than PA) to adjuvant RT doses of 50.4 Gy vs. 64.8 Gy. At a median follow-up of 6.43 years, this trial also reported no survival advantage with high RT doses (5-year OS, 64% vs. 72%; p = 0.48). In addition to tumor size (<5 cm), this trial also noted histologic subtype (oligo component) and age (under 40 years) as significant prognostic factors for better survival (5-year OS, 82% for <40 years and ODG vs. 32% for >40 years and astrocytoma). High dose arm had higher incidence of grade 3–5 RT necrosis (5% vs. 2.5%) [8]. Cognitive testing of these patients and follow-up for >7 years discovered that most patients had a stable neurocognitive status after RT irrespective of dose or baseline patient or tumor characteristics. Some patients with abnormal cognition at baseline showed improvement after RT [9].
5.2.3 Radiotherapy Target Volume and Technique
Limited RT fields are used for treatment. Gross tumor volume (GTV) is the postoperative tumor bed and any residual tumor usually defined by the T2 or FLAIR abnormality on postoperative MRI. Clinical tumor volume (CTV) is generated by 1.5 cm anatomically constrained expansion around GTV. Planning target volume (PTV) is dependent on the expected setup errors with a treatment technique as well as frequency of verification imaging, and is usually 0.3–1.0 cm. Organs at risk would include optic structures, brainstem, cochlea, temporal lobes, and possibly hippocampi. The dose is variable across regions. European institutions prefer a dose of 45 Gy @ 1.8 Gy per fraction due to lack of benefit at higher doses in EORTC trials. American institutions typically use 54 Gy at same dose per fraction. Three-dimensional conformal radiation therapy suffices for most patients; however, when the target is close to critical structures or hippocampal avoidance is desired, intensity modulation with image guidance may help reduce high dose spillage with smaller PTV margins.
Figure 13.1 shows (a) the postoperative magnetic resonance image with outlined tumor bed (gross tumor volume), and (b) the three-dimensional conformal radiotherapy plan for a patient with grade 2 oligodendroglioma, post gross total resection.

A 50-year-old male who underwent gross total resection for grade 2 oligodendroglioma of right parietal lobe was advised adjuvant radiotherapy (RT) followed by adjuvant chemotherapy. (a) RT planning computed tomography (CT) scan co-registered with magnetic resonance imaging (MRI) of brain shows the T2 hyperintense tumor bed with minimal residual tumor. (b) Three-dimensional conformal radiotherapy plan for the same patient. A total dose of 54 gray (Gy) in 27 fractions was planned in two phases (46 Gy in phase 1 followed by 8 Gy in phase 2 with a shrinking field technique)
Protons have been advocated to reduce long-term morbidity, especially neurocognitive decline in pediatric patients, by reducing dose spillage beyond the target, but large clinical data or direct comparisons with photons are not available. Additionally, limited availability and cost of proton therapy preclude wider use, especially in developing countries. A randomized trial (NRG BN005) comparing proton beam with intensity modulated radiation therapy (IMRT) to preserve brain function in IDH-mutant grade 2–3 gliomas opened in 2017. Primary outcome is change in cognition and secondary outcomes include QOL, treatment toxicity, OS, and PFS.
5.2.4 Addition of Chemotherapy Following Adjuvant RT
Experience with chemotherapy agents procarbazine/lomustine (CCNU)/ vincristine combination (PCV) and TMZ in recurrent LGG gave encouraging response rates though without a demonstrable survival advantage [10].
One of the initial studies evaluating addition of chemotherapy to RT was by the Southwest Oncology Group (SWOG, 1980–1985). They randomized adult LGGs following STR or biopsy to receive RT (55 Gy) with or without chemotherapy (CCNU 100 mg/m2 every 6 weeks, starting 2 days before RT and continuing for 2 years). Median OS was 4.45 years for RT alone vs. 7.4 years for RT + CCNU (p = 0.7). However, the 10-year OS of the chemotherapy arm was much lower (40% for RT and 20% for CCNU). Age and PS had the maximum impact on OS [11].
RTOG 9802 (1992–2002) randomized adults with high risk supratentorial LGGs to receive RT alone (54 Gy/30 Fr) or RT followed by chemotherapy (6 cycles PCV). Eligible patients were those older than 40 years with any surgical extent, or those younger than 40 years with STR or biopsy. Initial results of this trial reported PFS advantage with PCV (5-year PFS, 63% vs. 46%; p = 0.005) but no OS advantage (5-year OS, 72% vs. 63%; p = 0.13). 2-year survivors had a survival advantage with PCV on post hoc analysis suggesting a delayed benefit with chemotherapy [12]. Updated results with 12 year follow-up indeed proved the delayed survival advantage with PCV (10-year OS, 60% vs. 40%; median OS 13.3 vs. 7.8 years; HR 0.59; P = 0.003, no difference in initial 4 years) and delayed progression (10-year PFS, 51% vs. 21%, no difference in initial 2 years). This difference of 5.5 years in OS happened despite the 77% cross-over rate to receive PCV in patients who progressed in RT alone arm. Favorable prognostic factors for OS were age under 40 years, ODG histology, and chemotherapy addition [13]. A post hoc study done on 106 patients to see impact of molecular subgroups on prognosis showed that 35% patients had both IDH-1 mutation and 1p19q codeletions and 41% had IDH-1 mutation without 1p19q codeletion. PCV was associated with longer PFS (HR 0.32 for IDH-mutant/non-codeleted, HR 0.13 for IDH-mutant with 1p19q codeletion) and OS (HR 0.38 for IDH-mutant/non-codeleted; HR 0.21 for IDH-mutant with 1p19q codeletion) in both these groups, thus confirming that PCV had a benefit for all IDH-mutant patients [14]. TERT promoter and Capicua transcriptional repressor (CIC) mutations also have a significant association with OS, while IDH-1/2 mutations have a significant association with PFS [15]. MGMT promoter methylation has emerged as an important prognostic marker for PFS in this group of high risk LGG treated with RT ± PCV [16].
RTOG 0424 (2005–2009) was a single phase 2 study that attempted to replace PCV with TMZ as adjuvant therapy for high risk LGGs using Pignatti criteria (described later, in prognostic factors), and compared the data with a historical cohort. Patients who had at least 3 of the 5 high risk factors were treated with RT (54 Gy/30 Fr) along with concurrent (75 mg/m2) and adjuvant TMZ (150–200 mg/m2 D1–5 q4 weeks for 12 cycles) with pneumocystis carinii pneumonia (PCP) prophylaxis. Long-term results from this study with a median follow-up of 9 years show a median OS of 8.2 years, higher than historical controls treated with RT alone (median OS for intermediate risk, 7.2–7.6 years; high risk, 4.8–5.5 years) with acceptable toxicity [17]. MGMT methylation status was available for nearly 60% of patients in this study; 76% of these had methylated MGMT. Unmethylated MGMT promoter had a statistically worse OS and PFS association, independent of IDH1/2 mutation status, signifying MGMT promoter methylation as an important prognostic biomarker for grade 2 gliomas treated with RT and TMZ, possibly highlighting the effect of TMZ as radiosensitizer in this group as well [18].
Eastern Cooperative Oncology Group (ECOG) had started a randomized study (E3F05) in 2009 comparing RT with or without concurrent and adjuvant TMZ for 12 cycles for symptomatic or progressive LGGs. However, the study recruitment was suspended after RTOG 9802 results were published, assuming that RT alone would be an inferior arm.
EORTC 26951 and RTOG 9402 trials compared adjuvant RT with or without 6 cycles of PCV in patients with anaplastic ODGs and demonstrated higher OS in chemotherapy arm. Although traditionally grade 3 tumors, the new WHO 2016 classification places them in the better prognostic category of “high risk lower grade gliomas.” Post hoc analysis on these trials suggested that the OS benefit of PCV was limited to patients with 1p19q codeletion group and IDH mutation group, potentially identifying the true molecularly defined ODG subgroup [19,20,21,22].
5.2.5 Radiotherapy Versus Chemotherapy
Though RT has been established as an adjuvant therapy choice in LGGs with unfavorable factors, some patients who are not candidates for RT or unwilling for the same may be offered adjuvant TMZ alone at the risk of higher hematologic toxicity. This option may not be valid in high risk LGGs since combinations of both RT and chemo have shown better survival in that cohort.
EORTC 22033–26033 (2005–2010) was a phase 3 trial that compared RT (50.4 Gy/28 Fr) with TMZ (75 mg/m2 OD X 21 days q4 weeks for 12 cycles) in adult LGG patients with at least one of the five unfavorable factors mentioned by Pignatti et al. Primary endpoint was PFS, and secondary endpoints included OS, toxicity, neurocognition, and correlation of PFS with several biomarkers. At a median follow-up of 4 years, both arms had similar PFS (3.8 years with RT vs. 3.25 years with TMZ; p = 0.22). Median OS was not reached. Patients with IDH mutation without 1p19q codeletion had longer PFS with RT (HR 1.86) while the two arms had similar PFS in patients with (a) IDH mutation with 1p19q codeletion, and (b) IDH wild-type tumors. TMZ arm had higher grade 3–4 hematologic toxicity, infections, and fatigue [23]. There was no difference between the two groups in neurocognition or QOL in first 3 years of follow-up [24]. Genomic studies have tried to define an MGMT methylation score in IDH-mutant tumors with 1p19q codeletion that may help identify patients who merit TMZ as first-line adjuvant therapy for PFS benefit [25].
A retrospective National cancer database (NCDB) analysis of 1054 patients with high risk LGGs (age ≥40 years or STR as per RTOG 9802 criteria) who had received chemotherapy or chemoradiation also revealed no significant OS difference between the two groups (8-year OS, 52.8% vs. 56.7%) but tumor size >6 cm, older age, and astrocytoma histology predicted worse OS. Younger patients with ODG histology or 1p19q codeletion were offered chemotherapy more often [26].
5.2.6 Choice of Adjuvant Chemotherapy: PCV Versus TMZ
There are no direct comparisons between the two treatment regimes. RTOG 9802 used PCV in a randomized study while RTOG 0424 used TMZ in a single arm study. Median PFS in the former study with PCV was 10.4 years (3 years PFS 75–80%) while it was 4.5 years in the latter (3 years PFS 59%) giving some indication that PCV may be superior to TMZ, notwithstanding the inherent bias and challenges of such cross-trial comparisons.
The CODEL trial (NCT00887146) is a randomized phase 3 study that initially started to compare addition of TMZ vs. PCV to RT in adjuvant management of anaplastic ODGs with 1p19q codeletion and IDH mutation, but has now extended its inclusion to grade 2 gliomas as well; the trial is still recruiting and results will take a long time to mature.
5.3 Which Patients with LGG may Do Well with Observation Only
RTOG 9802 also had a low risk arm (111 patients) which consisted of patients younger than 40 years having neurosurgeon-determined GTR; these patients were followed up without any adjuvant therapy after surgery. This group had an excellent 5-year OS of 93% but 48% PFS rate over this period. However, it was discovered on review of postoperative MRI that almost all these patients had residual disease; nearly 31% had residual more than 1 cm. Preoperative tumor diameter ≥4 cm, astrocytoma component and residual ≥1 cm predicted poorer PFS [27].
NCDB data for RTOG 9802-defined low risk LGG patients between 2010 and 2013 were analyzed. Within this cohort, patients with tumor size >5 cm and non-ODG histology had an inferior OS compared to other low risk patients (80.5% vs. 94.0%). There are recommendations to include these patients into an “intermediate risk” group and investigate appropriate adjuvant therapy strategies for them [28].
5.4 Prognostic Factors
The following factors have prognostic value as seen in several randomized studies:
-
1.
Patient factors
-
(a)
Patient age ≥40 years—unfavorable
-
(b)
Baseline neurologic deficits—unfavorable
-
(c)
Seizures at diagnosis—favorable
-
(a)
-
2.
Tumor factors
-
(a)
Size ≥6 cm—unfavorable
-
(b)
Astrocytoma histology—unfavorable
-
(c)
Tumors crossing midline—unfavorable
-
(d)
Molecular studies
-
IDH mutation—favorable
-
1p19q codeletion—favorable
-
MGMT promoter methylation—favorable
-
p53 mutation—unfavorable
-
TERT mutation—unfavorable
-
CIC mutation—favorable
-
-
(a)
-
3.
Extent of surgery: subtotal resection or biopsy—unfavorable
Based on the patient data in EORTC trials 22844 and 22845, Pignatti et al. proposed the following five factors as unfavorable for survival: age ≥40 years, neurologic deficit present before surgery, preoperative tumor diameter ≥6 cm, tumor crossing midline, and astrocytoma histology. They grouped the patients into categories of low risk if up to two factors were present (median OS, 7.8 years), and high risk if three or more factors (median OS, 3.4 years) were present, known as Pignatti criteria for risk grouping [29]. RTOG 9802, however, defined only two factors: age above 40 years and STR, presence of either of which was considered a high risk disease.
The data from EORTC trials 22844 and 22845 were reanalyzed by only including patients who had undergone a central pathology review and confirmed to be grade 2 gliomas; this excluded 21% patients. This was believed to be a more homogenous group after excluding HGGs, and had median PFS and OS of 4.6 years and 7.2 years, respectively. These were further modeled based on several factors including some of Pignatti criteria along with time since LGG symptoms, to generate three risk groups: low, intermediate, and high, which were later validated on patient data from intergroup and RTOG 9802 trials. The median OS stratified by risk groups was 11.6–12.7 years for low risk, 7.2–7.6 years for intermediate risk, and 4.8–5.4 years for high risk groups [30].
5.5 Reirradiation/Salvage
Recurrence in LGGs may manifest as symptomatic worsening or progression on MRI. Nearly 70% may have malignant transformation on long-term follow-up. Salvage treatment option should include surgery, if feasible—to verify histology and transformation, reduce mass effect and thereby relieve symptoms. Reirradiation, either with conventional fractionation or SRS to small residual disease may be decided on the merit of an individual case; risks of critical structure damage and radiation necrosis should be evaluated carefully. Systemic therapy may include PCV, TMZ, lomustine alone or bevacizumab, with variable results, depending on type of prior therapy given and resultant response/toxicity. The longer the time since prior therapy, the better is the expected outcome from retreatment.
6 “Other” Astrocytic Tumors
6.1 Pleomorphic Xanthoastrocytoma Grade 2
These rare tumors account for 1% of astrocytic neoplasms, first incorporated in 1993 in WHO classification. They may affect children or young adults, with median age at diagnosis of 29 years, without any gender predilection. Most common location is supratentorial in cerebral hemispheres (usually temporal lobe) but other sites such as sella, ventricles, spine, retina, etc., have also been reported. On CT, they appear as well-circumscribed superficial solid-cystic masses, usually hypo- or isodense, with areas of calcification in solid component. The solid component and cyst wall enhance with contrast. On MRI, the lesions are T1 hypointense, T2 hyperintense and heterogeneously enhancing on T1 contrast with rim enhancement of the cyst wall as well. Minimal perilesional edema can be seen. The lesion is often in contact with leptomeninges and there may occasionally be leptomeningeal enhancement [31]. In histology, pleomorphic lipidized cells may be seen with multinucleated giant cells, eosinophilic granular bodies in an inflammatory background. Necrosis is absent and mitoses are rare. The histologic appearance is often confused with high grade astrocytomas such as GBM. On IHC, S-100, GFAP, and oligodendrocyte transcription factor-2 (Olig2) are positive [32]. Neuronal markers such as synaptophysin and neurofilament (NF) are occasionally seen. Ki-67 is low. Reticulin fiber staining is commonly seen, suggesting an origin potentially from subpial fibrous astrocytes. Nearly 60–70% PXA have BRAF V600E mutation and this confers a better outcome [33]. Nearly 25% have p53 mutations. Grade 2 PXAs have a favorable prognosis with 5-year survival >75% and 5-year PFS >60%. Treatment of choice is surgery and extent of surgery (GTR vs. STR) has prognostic value. Adjuvant RT has been tried more often for salvage in recurrent settings than in adjuvant setting after STR at a dose of 45–54 Gy with or without concurrent TMZ with a possible PFS benefit but no OS advantage [34]. Systemic therapy (TMZ, carmustine, lapatinib, bevacizumab) has been used in salvage settings but with inconsistent outcomes.
7 Other Gliomas
7.1 Chordoid Glioma of Third Ventricle
These are rare neuroepithelial tumors with a slight female preponderance and median age of presentation at 48 years, originating in midline in suprasellar region or anterior part of third ventricle, and containing both glial and chordoid elements [35]. Due to their location, presenting features are due to raised intracranial tension (ICT) with obstructive hydrocephalus (headache, nausea, vomiting, ataxia), hypothalamic dysfunction, memory deficits, mental status changes, and visual impairment. On CT, they appear most commonly as well-circumscribed, solid, hyperdense lesions with homogenous contrast enhancement. MRI shows a mass that is T1 iso- or hypointense, T2 hyperintense and homogenously enhancing on T1 contrast sequences [36].
On histology, they appear as clusters and cords of ovoid or polygonal epithelial cells in a mucinous background. GFAP is strongly positive; other positive IHC include epithelial membrane antigen (EMA), CD34, cytokeratin (CK), S100, and vimentin. Mitoses are low or absent with MIB-LI ≤5%, and necrosis is absent [37].
Surgery in hypothalamic location is tricky, with higher risk of neuroendocrine dysfunction (including diabetes insipidus) when attempting GTR. GTR is feasible in less than half of patients although it is the most crucial factor for local control. Hence, for most tumors, STR only is feasible but there are reports where the surgical approach (transcortical and transcallosal vs. trans-lamina terminalis) is linked to increased complications rather than extent of surgery [38]. RT has been used occasionally for residual tumors in the form of stereotactic radiosurgery (SRS) with marginal doses of 12–20 Gy or fractionated RT with 54 Gy in 30 fractions, with variable results [39, 40]. Adjuvant chemotherapy has no demonstrable role. The disease carries high postoperative morbidity and mortality as well as high recurrence rates following STR, hence a lot of attention has to be directed to surgical planning and postoperative care.
8 Mixed Neuronal and Neuronal-Glial Tumors
8.1 Central Neurocytoma
Central neurocytomas (CN), described first in 1982, are uncommon neuroepithelial tumors with neuronal differentiation, possibly originating from neuroglial precursor cells. They constitute less than 0.5% of all adult intracranial tumors, most commonly presenting in the third decade. Presenting features are commonly due to raised ICT (headache, vomiting) but vision changes, gait imbalance, and memory disturbances may also occur. The lesions originate in subependymal plate of ventricles. CT demonstrated a well-circumscribed enhancing ventricular mass, most commonly in lateral ventricles. Calcification is seen in 50%. The lesion is iso- to hyperintense on T2 MRI with heterogeneous contrast enhancement on T1 contrast sequences. Histology demonstrates small round cells, rounded nuclei, and scant cytoplasm. IHC is positive for synaptophysin. Some cases may show features such as high mitotic activity, microvascular proliferation, necrosis, and Ki-67 >3%, and are referred as atypical CN with higher recurrence potential [41].
Most cases have a favorable prognosis with benign biological behavior. Maximal safe resection is the dictum of management. GTR gives 5-year survival of 93% and needs no further adjuvant management. For patients with STR or high MIB-LI, adjuvant RT (dose ~56 Gy) may help improve local control as well as OS [42, 43]. For small recurrent lesions, SRS has also been used with peripheral doses of 10.5–17.0 Gy, with local control rates exceeding 90% [44]. TMZ and PCV have been tried as salvage therapies in recurrent inoperable tumors with variable outcomes [45].
8.2 Extraventricular Neurocytoma
Extraventricular neurocytoma (EVN) was first described as an entity in 1997, having the same histopathological characteristics as CN but located outside the ventricular system. It is a rare glioneuronal tumor, with two incidence peaks in the second and fifth decades [46]. Most common presenting complaints are seizure and headache. There are no specific imaging characteristics. Typically it manifests as a large circumscribed mass, most commonly frontal, with a solid portion and variable cystic regions, calcification, hemorrhage, and perilesional edema. More than 80% show contrast enhancement of solid portion, mostly heterogeneous [47].
The standard therapy is surgery; GTR is curative. In case of STR when tumors lie in proximity to critical structures or eloquent cortex, or when histopathology shows atypical features, recurrence risk is high, often 50% or more; in these situations, adjuvant RT (50.4–54.0 Gy) may help improve control. Salvage RT may also be considered in inoperable recurrences or after incomplete removal of recurrent tumor. Overall outcome is poorer compared to CN. This may be due to low GTR frequency and difficulty of salvage surgeries in the same critical locations. There is no role of adjuvant chemotherapy but salvage TMZ has been used anecdotally for recurrences [48].
8.3 Cerebellar Liponeurocytoma
This rare tumor was first described by Bechtel et al. in 1978. It was earlier regarded as grade 1 but upgraded to grade 2 in WHO 2007 classification due to reported atypia and recurrence patterns. The mean age of presentation is 49 years with a female preponderance [49]. Most common location is hemispheric, though direct intraspinal extension to cervical spine and lumbar spine metastases have also been reported [50]. Presenting features are due to cerebellar involvement (gait ataxia, frequent falls, dizziness) or obstructive hydrocephalus (headache, vomiting, nausea). On CT imaging, the fatty part is detected as a hypodense lesion. MRI shows well-demarcated T1 hyperintense lesion with inversion on fat suppressed images and minimal contrast enhancement; T2/FLAIR imaging shows heterogeneous intensity and diffusion weighted imaging (DWI) shows high intensity with minimal perilesional edema. There is a possible familial association with autosomal dominant inheritance [51]. Germline mutations have also been postulated. Pathology shows variable neuronal/neurocytic differentiation and focal lipomatous and astrocytic differentiation. Histologic features include isomorphic oval or round cells with focal accumulation of lipid-filled neuroepithelial cells. IHC shows synaptophysin, neuron-specific enolase (NSE), neurospecific nucleoprotein (NeuN) and microtubule associated protein 2 (MAP 2) positivity and IDH-1, EMA, CK and nuclear Olig2 negativity. The lipid vacuole membranes of lipomatous cells are positive for synaptophysin, MAP 2, vimentin, and S100. GFAP is limited to reactive astrocytes. 1p19q codeletion is absent, differentiating them from ODG. MIB-LI is low (<5%) [52].
Surgery is the primary treatment of choice. Adjuvant RT (dose 54 Gy) reduces recurrence risk by nearly 50%, even in the setting of GTR. Nearly 30% patients recur even after long follow-up periods of 8–10 years. For recurrent tumors, resurgery followed by RT should be considered [52].
9 Response to Treatment
Since LGGs have a long follow-up with potential for progression as well as conversion to HGG, changes in tumor characteristics over time are monitored using several criteria. Uniformity of these criteria is necessary to ensure comparison of results across institutions and treatment strategies. Macdonald criteria were developed for supratentorial gliomas in 1990. They were initially based on CT and later, adapted to MRI [53]. They defined four groups: Complete response (CR): Complete disappearance of tumor on consecutive imaging, at least a month apart, without steroids, Partial response (PR): ≥50% decrease in the enhancing area of tumor without neurologic deterioration or increasing steroids; Progressive disease (PD): ≥25% increase in size of enhancing lesion; Stable disease (SD): Not fitting any of the previous three criteria. This system is unable to account for changes such as radiation necrosis, pseudoprogression, response to bevacizumab, TMZ or immune changes, and is largely not applicable in non-enhancing LGGs. The Response Assessment in Neuro-Oncology (RANO) Working Group defined MRI-based criteria for LGGs in 2011 with more emphasis on T2/FLAIR sequences which have more relevance in LGGs. The new criteria also incorporate new lesions, enhancement pattern changes, and clinical changes [54]. Five groups as per RANO criteria include CR: Full resolution of T2/FLAIR changes, no new radiographic findings, clinically improved or stable, and off steroids, sustained for 4 weeks; PR: >50% decrease in product of perpendicular diameters of pre-treatment T2 or FLAIR change, no new lesions, sustained for 4 weeks, clinically improved or stable, and stable steroid dose; Minor response: >25% but <50% reduction in product of perpendicular diameters of pre-treatment T2 or FLAIR change, no new lesions, sustained for 4 weeks, clinically improved or stable, and stable steroid dose; Stable response: Imaging unchanged, clinically stable, stable steroid dose; PD: New lesions, new enhancement (evidence of progression), or >25% increase in T2/FLAIR change, clinical decline not due to causes other than tumor.
10 Treatment-Related Morbidity
10.1 Early/Acute Effects
The acute effects of cranial RT include nausea, appetite loss, and somnolence. Patchy alopecia and dry desquamation, dryness of eyes, and fatigue may be present, but these problems are usually self-limiting. PCV or TMZ may cause hematologic toxicity and increased risk of infections such as pneumocystis, for which appropriate monitoring and prophylaxis are recommended.
10.2 Delayed Effects: Hearing, Neurocognition, and Neuroendocrine Dysfunction
Patients with temporal lobe lesions may experience conductive or sensorineural hearing loss in the long term if the middle ear or cochlea receive mean doses exceeding 45 Gy. Due to the prolonged survival in LGGs, neurocognition is an important issue that may be affected and impacts function and QOL. With the RT doses used in EORTC 22033–26033 trial, neurocognitive deficits and QOL issues did not differ between the treatment arms of RT vs. chemotherapy till 3 years follow-up; they rather improved in some people with baseline deficits, pointing to the fact that largely the disease itself contributes to this issue. Reducing the volume of RT has helped preservation of intelligence quotient (IQ) and neuroendocrine function in children [55]. Hippocampal sparing is now being attempted for most low grade tumors and even for whole brain RT for brain metastases to preserve memory [56]. Theoretically, proton therapy may contribute further to this goal.
10.3 Pseudoprogression and Radiation Necrosis
Pseudoprogression refers to increase in size or enhancement of a lesion in response to treatment; it is more common after combined RT and chemotherapy and in ODGs, and less often seen after RT alone. Median time to appearance varies from 8–12 months but delayed presentations are also seen. In LGGs, pseudoprogression is more often seen in volumes receiving doses over 45 Gy, and in 1p19q intact and IDH wild-type tumors [57].
Radionecrosis refers to coagulative necrosis of brain tissue in response to focal radiation. It is less common in LGGs than in HGGs, but may occasionally be seen after reirradiation for recurrent or progressive disease.
Both pseudoprogression and radionecrosis may be confused with disease progression and may need to be differentiated for management decisions. Pseudoprogression may have mismatch between worsening radiologic appearance and clinical features which may be stable or improved. Radiation necrosis may be suspected based on the history of radiation dose, treatment technique, and volume. Multiparametric MRI with perfusion and diffusion weighted sequences, and occasionally PET scan may help distinguish them from recurrent disease.
11 Current Status and Future Directions
The group LGG encompasses several heterogeneous primary CNS tumors with different natural history and treatments. Surgery (maximal safe resection) is the standard treatment with need for adjuvant therapy defined by extent of resection, tumor characteristics, and patient age. For most astrocytic and oligodendroglial tumors, both adjuvant radiation and chemotherapy are recommended. RT alone is not recommended after RTOG 9802 findings. RT delay or observation can only be justified in very small IDH-mutant completely resected tumors in young patients provided they continue rigorous follow-up. Molecular characterization and inclusion of these factors into clinical trial stratification and inclusion criteria will help fine tune treatment intensity to meet the dual goals of maximum control with minimum long-term morbidity.
References
Gupta A, Dwivedi T. A simplified overview of world health organization classification update of central nervous system tumors 2016. J Neurosci Rural Pract. 2017;8(4):629–41.
Jakola AS, Skjulsvik AJ, Myrmel KS, et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann Oncol. 2017;28(8):1942–8.
Aghi MK, Nahed BV, Sloan AE, Ryken TC, Kalkanis SN, Olson JJ. The role of surgery in the management of patients with diffuse low grade glioma: a systematic review and evidence-based clinical practice guideline. J Neuro-Oncol. 2015;125(3):503–30.
Smith JS, Chang EF, Lamborn KR, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26(8):1338–45.
van den Bent MJ, Afra D, de Witte O, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial [published correction appears in Lancet. 2006 Jun 3;367(9525):1818]. Lancet. 2005;366(9490):985–90.
Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36(3):549–56.
Kiebert GM, Curran D, Aaronson NK, et al. Quality of life after radiation therapy of cerebral low-grade gliomas of the adult: results of a randomised phase III trial on dose response (EORTC trial 22844). EORTC Radiotherapy Co-operative Group. Eur J Cancer. 1998;34(12):1902–9.
Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20(9):2267–76.
Brown PD, Buckner JC, O’Fallon JR, et al. Effects of radiotherapy on cognitive function in patients with low-grade glioma measured by the folstein mini-mental state examination. J Clin Oncol. 2003;21(13):2519–24.
Soffietti R, Rudà R, Bradac GB, Schiffer D. PCV chemotherapy for recurrent oligodendrogliomas and oligoastrocytomas. Neurosurgery. 1998;43(5):1066–73.
Eyre HJ, Crowley JJ, Townsend JJ, et al. A randomized trial of radiotherapy versus radiotherapy plus CCNU for incompletely resected low-grade gliomas: a Southwest Oncology Group study. J Neurosurg. 1993;78:909–14.
Shaw EG, Wang M, Coons SW, et al. Randomized trial of radiation therapy plus procarbazine, lomustine, and vincristine chemotherapy for supratentorial adult low-grade glioma: initial results of RTOG 9802. J Clin Oncol. 2012;30(25):3065–70.
Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med. 2016;374(14):1344–55.
Bell EH, Zhang P, Shaw EG, et al. Comprehensive genomic analysis in NRG oncology/RTOG 9802: a phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J Clin Oncol. 2020;2020:JCO1902983.
Bell EH, McElroy JP, Fleming J, et al. Comprehensive mutation analysis in NRG oncology/RTOG 9802: a phase III study of RT vs RT + PCV in high-risk low-grade gliomas (LGGs). J Clin Oncol. 2016;34(15_suppl)
Bell EH, Zhang P, Aldape K, et al. OS01.7 MGMT promoter methylation status independently predicts progression-free survival in NRG oncology/RTOG 9802: a phase III trial of RT vs RT + PCV in high-risk low-grade gliomas. Neuro-Oncology. 2017;19(Suppl 3):iii2–3.
Fisher BJ, Pugh SL, Macdonald DR, et al. Phase 2 study of a temozolomide-based chemoradiation therapy regimen for high-risk, low-grade gliomas: long-term results of radiation therapy oncology group 0424. Int J Radiat Oncol Biol Phys. 2020;107(4):720–5.
Bell EH, Zhang P, Fisher BJ, et al. Association of MGMT promoter methylation status with survival outcomes in patients with high-risk glioma treated with radiotherapy and temozolomide: an analysis from the NRG oncology/RTOG 0424 trial. JAMA Oncol. 2018;4(10):1405–9.
van den Bent MJ, Brandes AA, Taphoorn MJB, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–50.
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–43.
van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer P. J Clin Oncol. 2006;24(18):2715–22.
Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32(8):783–90.
Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–32.
Reijneveld JC, Taphoorn MJB, Coens C, et al. Health-related quality of life in patients with high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1533–42.
Bady P, Kurscheid S, Delorenzi M, et al. The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol. 2018;135(4):601–15.
Jhaveri J, Liu Y, Chowdhary M, et al. Is less more? Comparing chemotherapy alone with chemotherapy and radiation for high-risk grade 2 glioma: an analysis of the National Cancer Data Base. Cancer. 2018;124(6):1169–78.
Shaw EG, Berkey B, Coons SW, et al. Recurrence following neurosurgeon-determined gross-total resection of adult supratentorial low-grade glioma: results of a prospective clinical trial. J Neurosurg. 2008;109(5):835–41.
Jairam V, Kann BH, Park HS, et al. Defining an intermediate-risk group for low-grade glioma: a national cancer database analysis. Anticancer Res. 2019;39(6):2911–8.
Pignatti F, van den Bent M, Curran D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20(8):2076–84.
Gorlia T, Wu W, Wang M, et al. New validated prognostice models and prognostic calculators in patients with low-grade gliomas diagnosed by central pathology review: a pooled analysis of EORTC/RTOG/NCCTG phase III clinical trials. Neuro Oncol. 2013;15:1568–79.
Shaikh N, Brahmbhatt N, Kruser TJ, et al. Pleomorphic xanthoastrocytoma: a brief review. CNS Oncol. 2019;8(3):CNS39.
Liu XF, Du X, Zhang XT, Yang M, Han YM, Lin XY. Pleomorphic xanthoastrocytoma inside lateral ventricle: a rare case report and literature review. Int J Clin Exp Pathol. 2019;12(4):1118–23.
Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:e1794.
Mallick S, Benson R, Melgandi W, Giridhar P, Rath GK. Grade II pleomorphic Xanthoastrocytoma; a meta-analysis of data from previously reported 167 cases. J Clin Neurosci. 2018;54:57–62.
Brat DJ, Scheithauer BW, Staugaitis SM, Cortez SC, Brecher K, Burger PC. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. 1998;57:283–90.
Vanhauwaert DJ, Clement F, Van Dorpe J, Deruytter MJ. Chordoid glioma of the third ventricle. Acta Neurochir. 2008;150(11):1183–91.
Reifenberger G, Weber T, Weber RG, et al. Chordoid glioma of the third ventricle: immunohistochemical and molecular genetic characterization of a novel tumor entity. Brain Pathol. 1999;9:617–26.
Ampie L, Choy W, Lamano JB, et al. Prognostic factors for recurrence and complications in the surgical management of primary chordoid gliomas: a systematic review of literature. Clin Neurol Neurosurg. 2015;138:129–36.
Kobayashi T, Tsugawa T, Hashizume C, et al. Therapeutic approach to chordoid glioma of the third ventricle. Neurol Med Chir. 2013;53:249–55.
Hanbali F, Fuller GN, Leeds NE, Sawaya R. Choroid plexus cyst and chordoid glioma. Report of two cases. Neurosurg Focus. 2001;10:E5.
Goyal S, Kataria T, Gupta D, et al. Atypical central neurocytoma with leptomeningeal dissemination: a case report. J Egypt Natl Canc Inst. 2020;32:23.
Imber BS, Braunstein SE, Wu FY, et al. Clinical outcome and prognostic factors for central neurocytoma: twenty year institutional experience. J Neuro-Oncol. 2016;126(1):193–200.
Mallick S, Roy S, Das S, et al. Role of adjuvant radiation in the management of central neurocytoma: experience from a tertiary cancer care center of India. Indian J Cancer. 2015;52(4):590–7.
Bui TT, Lagman C, Chung LK, et al. Systematic analysis of clinical outcomes following stereotactic radiosurgery for central neurocytoma. Brain Tumor Res Treat. 2017;5(1):10–5.
Johnson MO, Kirkpatrick JP, Patel MP, et al. The role of chemotherapy in the treatment of central neurocytoma. CNS Oncol. 2019;8(3):CNS41.
Mallick S, Benson R, Rath GK. Patterns of care and survival outcomes in patients with an extraventricular neurocytoma: an individual patient data analysis of 201 cases. Neurol India. 2018;66(2):362–7.
Romano N, Federici M, Castaldi A. Imaging of extraventricular neurocytoma: a systematic literature review [published online ahead of print, 2020 Apr 25]. Radiol Med. 2020; https://doi.org/10.1007/s11547-020-01198-8.
El Demellawy D, Sur M, Ahmed AD, Provias J. Hemispheric extra-ventricular glioneurocytoma: a clinicopathological review with detailed immunohistochemical profile. Pathol Res Pract. 2012;208:444–51.
Oudrhiri MY, Raouzi N, El Kacemi I, et al. Understanding cerebellar liponeurocytomas: case report and literature review. Case Rep Neurol Med. 2014;2014:186826.
Deora H, Prabhuraj AR, Saini J, Yasha TC, Arimappamagan A. Cerebellar liponeurocytoma: a rare fatty tumor and its literature review. J Neurosci Rural Pract. 2019;10(2):360–3.
Wolf A, Alghefari H, Krivosheya D, et al. Cerebellar liponeurocytoma: a rare intracranial tumor with possible familial predisposition. Case report J Neurosurg. 2016;125:57–61.
Gembruch O, Junker A, Mönninghoff C, et al. Liponeurocytoma: systematic review of a rare entity. World Neurosurg. 2018;120:214–33.
Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–80.
van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12(6):583–93.
Jalali R, Mallick I, Dutta D, et al. Factors influencing neurocognitive outcomes in young patients with benign and low-grade brain tumors treated with stereotactic conformal radiotherapy. Int J Radiat Oncol Biol Phys. 2010;77(4):974–9.
Farjam R, Pramanik P, Aryal MP, et al. A radiation-induced hippocampal vascular injury surrogate marker predicts late neurocognitive dysfunction. Int J Radiat Oncol Biol Phys. 2015;93(4):908–15.
Lin AL, White M, Miller-Thomas MM, et al. Molecular and histologic characteristics of pseudoprogression in diffuse gliomas. J Neuro-Oncol. 2016;130(3):529–33.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Goyal, S., Madan, R. (2021). Grade 2 Gliomas. In: Mallick, S., Giridhar, P., Rath, G.K. (eds) Evidence based practice in Neuro-oncology. Springer, Singapore. https://doi.org/10.1007/978-981-16-2659-3_13
Download citation
DOI: https://doi.org/10.1007/978-981-16-2659-3_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-2658-6
Online ISBN: 978-981-16-2659-3
eBook Packages: MedicineMedicine (R0)