
Abstract
Stress proteins (SPs) are groups of protein/RNA chaperones that respond fast to intracellular and extracellular stress stimuli to maintain cell homeostasis. Among them, heat-shock proteins (HSPs) play major roles in chaperoning misfolded or unfolded polypeptides, protecting cells from toxic stress, and regulating innate immunity, adaptive immunity, and even inflammatory response; while RNA chaperones are composed of a great number of heterogeneous nuclear ribonucleoproteins (hnRNPs), essential for manipulating both the functions and metabolisms of pre-mRNAs/heterogeneous nuclear RNAs (hnRNAs), including chromatin remodeling, transcription regulation, RNP assembly and stabilization, RNA export, virus replication, histone-like nucleoid structuring, and even intracellular immunity. Dysregulation or dysfunction of stress proteins causes many human diseases, such as human cancer, cardiovascular diseases and neurodegenerative diseases, and infectious diseases. In this chapter, we briefly discuss their biological function in human diseases, particularly the diseases caused by virus infections. Stress proteins have attracted huge interest as targets for combating SARS-CoV-2 (COVID-19) and many other virus infections.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Stress Proteins (SPs)
When an organism (human beings, animals, plants, and even bacteria) meets a stress, they usually find out a way to reduce or eradicate the stress. For example, people would think of a way and execute it, be it tackling the stress source or mediating the harm caused by the stress. Likewise, when cells are exposed to either intracellular or extracellular stressful stimuli, they also have to do similar things. Groups of protein, referred to as stress proteins (SPs), including heat-shock proteins (HSPs), RNA chaperones, and proteins that mainly function in the endoplasmic reticulum (ER), namely, peptidyl-prolyl isomerases, the lectin-binding chaperone system, and protein disulfide isomerases (PDIs), are quickly synthesized to remove or limit the cell damage caused by stresses [1]. This is a common stress response phenomenon for cells to protect their physiological functions and survival by either neutralizing or eradicating stresses from both extracellularly (e.g., UV irradiation, toxic chemicals, hypoxia, pathogen invasion) and intracellularly (e.g., starvation, stimulation by cytokines/chemokines or hormones, oxidative stress, DNA damage). The increase of stress proteins triggers signaling cascades that may promote cell survival, initiate cell death (i.e., apoptosis, necrosis, pyroptosis or autophagic cell death), or modulate innate and adaptive immune response for the elimination of damaged cells to protect a particular organ/tissue under given conditions. Many human diseases, including cardiovascular diseases, stroke, neurodegenerative diseases (e.g., Parkinson’s diseases, Alzheimer’s disease), cancers, and infectious diseases, are well known to be associated with the dysfunction or dysregulation of stress proteins. In this chapter, we will briefly introduce the functions of stress proteins in human diseases and particularly focus on their functions in diseases caused by viral infections.
4.2 Heat-Shock Proteins (HSPs)
Heat-shock proteins belong to a large chaperone family that is classified as Hsp100, Hsp90, Hsp70, Hsp60, Hsp40, and small HSP (15–40 kDa, sHSP) subfamilies according to their molecule weights. HSPs rapidly increase when a cell is under a physiological or environmental stress such as exposure to UV, irradiation, high temperature, starvation, malnutrition, chemicals, hypoxia or hyperoxia, and pathogen invasion. Heat-shock proteins and their co-chaperones form extensive networks to promote or stabilize the correct folding of newly synthesized peptides and substrate proteins [2]. They participate in many biological processes including cell survival, stemness, transformation and carcinogenesis, differentiation, migration, and death. Accumulating evidence shows that some HSPs participate not only in intracellular innate immunity but also in antigen presentation in adaptive immune response [3, 4]. In this chapter, we mainly focus on the function of Hsp90, Hsp70, and sHSP family members.
Hsp90s are very abundant chaperones located in different cellular compartments of all eukaryotic cells. Hsp90α and Hsp90β are in the cytoplasm, accounting for the greatest proportion in humans. Glucose-related protein GRp94 and TRAP1 are located in the endoplasmic reticulum (ER) and mitochondria, respectively [5]. The functions of Hsp90s are ATP-dependent through three distinct regions: an N-terminal ATPase-dependent hydrolytic domain, a middle linker region, and a C-terminal dimerization domain [6]. Hsp90 binds unfolded peptides or misfolded proteins to prevent their aggregation and degradation. Upon binding with ATP, the N-terminal domain forms a transient dimer to facilitate the substrate binding. Following the hydrolysis of ATP and energy release, a conformational change of the N-terminal domain occurs to correct the folding of the substrate peptides or proteins (Fig. 4.1). In fact, this process is collaborated with or regulated at large by Hsp90 co-chaperones, including CDC37 and CDC37-associated kinase, for inhibiting ATPase activity, E3 ubiquitin ligase carboxyl terminus of Hsp70-interacting protein (CHIP) for protein degradation, and Hsc70/Hsp90-organizing protein (HOP) for inhibiting the N-terminal domain dimerization [7, 8]. Hsp90 also contributes to telomere maintenance, cell survival, growth, cell cycle progression and apoptosis, etc.

The general chaperone cycle of heat-shock proteins. The unfolded or misfolded client protein binds to Hsp70 and its co-chaperone, Hsp40 chaperones, and then interacts with the free Hsp90 to form a complex. ATP (implicated with a red P) binds Hsp90 and then induces the client protein to be transferred from Hsp70 to Hsp90, followed by Hsp70 and Hsp40 release from the complex. After the hydrolysis of ATP, Hsp90 undergoes additional conformation change, leading to the correctly folded client protein release from Hsp90-client complex
Hsp70s account for the majority of molecular chaperones in cells [7], including Hsp72, Hsp70-2, Hsp70B′, and Hsc70 mainly in the cytosol; GRp75 in the mitochondria, and GRp78 mainly in the ER [9]. Some Hsp70 members such as GRp78 can also be presented on the cell surface and even be secreted out. Hsp70 contains a 44-kDa nucleotide-binding domain (NBD) in the N-terminal region and a 28-kDa substrate-binding domain (SBD) in the C-terminal region [10, 11]. Similar to Hsp90, Hsp70s are ATP-dependent molecular chaperones and play key roles in cellular protein surveillance without or under stress. They participate in almost all folding processes, including folding of newly synthesized polypeptides; refolding of misfolded or aggregated proteins, solubilizing or degrading proteins, and transporting proteins; and assembling or disassembling of oligomeric protein complexes [6, 12,13,14].
The main co-chaperones of Hsp70 are the nucleotide exchange factors (NEFs) and the J-domain proteins (JDPs) of the Hsp40 family. In the process of protein folding, the unfolded peptide substrates first bind JDPs and then form a complex with Hsp70. The Hsp70’s ATPase activity is activated for folding and preventing unfolded protein aggregation. NEFs help to release substrates when the correct and active conformation is formed. Therefore, Hsp70s always cooperate with co-chaperones in the process of protein folding or refolding [15, 16].
The small heat-shock protein (sHSP) family contains ubiquitous (HspB5, Hsp20, Hsp22, and Hsp27) and tissue-specific (HspB2, HspB3, HspB4, HSPB7, HspB9, and HspB10) ones [17]. They exist as monomers, dimers, or even large multimeric complexes [18]. The structure of sHSPs is quite different from the other HSPs because of less conserved sequences. The basic structure of sHSPs is a conserved α-crystallin domain (ACD) flanked by a non-conserved N-terminal sequence (NTS) and a C-terminal sequence (CTS). sHSPs are important players and regulators of stress tolerance, apoptosis, aging, and longevity [19,20,21,22]. The sHSPs may exhibit different functions when they undergo phosphorylation and oligomerization. The phosphorylated sHSPs normally form monomers or small oligomers that bind F-actin to regulate actin polymerization [23], while the hypophosphorylation of sHSPs promotes large homo- and hetero-oligomer formation crucial for chaperone activity [24].
4.3 Transcriptional Regulation of the HSPs
The expressions of HSPs are mainly regulated by four members of heat-shock factors (HSFs) in vertebrates [25]. Among them, HSF1 plays a major role in regulating HSP expression. Under stress conditions, the monomeric HSF1 trimerizes and translocates from the cytoplasm into the nucleus and binds on the heat-shock elements (HSEs) in the promoter region, therefore activating the hsp expression [26]. Experiments show that the fibroblasts without knockout (hsf1−/−) undergo apoptosis at a high temperature stress due to no hsp transcription [27].
4.4 Protein Disulfide Isomerase (PDI)
Chaperone protein PDIs are multifunctional oxidoreductases mainly locating in the ER. They catalyze the formation, isomerization, and reduction of disulfide bonds of newly synthesized peptides or malformation of disulfide bonds within or among protein subunits. During disulfide bond formations, cysteine residues at the catalytic CGHC active site accept two electrons from the cysteine residues in the polypeptide that causes the PDI reduction and substrate oxidation. Then, the electrons are transferred from the PDI molecule to an acceptor and start a new cycle of disulfide bond formation [28]. Of course, PDIs also keep the molecular chaperone activity to ensure the quality control of glycosylated proteins [29]. ERp57 is one of the most studied PDIs. It shares a similar structure to other PDIs, i.e., four domains (namely, a-b-b′-a′) and two localization sequences (namely, ER retention signal (QDEL) and nucleus localization signal (KKKKKPK) at the C-terminus). The b domains have a high binding affinity for calreticulin and calnexin [30]. When a protein is not correctly folded, UDP-glucose:glycoprotein glucosyltransferase (UGGT) is recruited to ERp57/CRT/CNX complex for reglycosylation [29, 31, 32]. ERp57 is also well known to play crucial roles in antigen presentation during adoptive immune response to infections.
4.5 RNA Chaperones
The 3D structure is the basis of RNA functions. The process of pre-RNA folding into 3D structures, i.e., the multitude of possible RNA base pairings together with the high stability of RNA duplexes that generates alternative secondary and tertiary structures, is highly dependent on RNA chaperones. They promote RNA folding through accelerating the escape from kinetic folding traps and prevention of RNAs from being trapped in nonfunctional conformations. RNA chaperones participate in many biological processes, such as chromatin remodeling, 3'-end formation, mRNA packaging, RNA silencing, RNA transport, transcription regulation, RNP assembly and stabilization, RNA export, translation, DNA repair, telomere biogenesis, histone-like nucleoid structuring, virus replication, and even intracellular immunity. Heterogeneous nuclear ribonucleoproteins (hnRNPs) are essential factors for manipulating both the functions and metabolisms of pre-mRNAs and other heterogeneous nuclear RNAs (hnRNAs) transcribed by RNA polymerase II. An hnRNP molecule normally contains common RNA binding motifs like RGG boxes (arginine glycine boxes), RNA recognition motifs (RRMs), hnRNP K homology (KH)-domains, and zinc finger (ZF)-domains [33]. They are mainly in the nucleus and are redistributed in the cytosol under certain conditions, such as virus infections [34].
4.6 Stress Proteins and Human Diseases
Protein aggregation hallmarks quite a few diseases caused by either loss-of-function or toxic gain-of-function of stress proteins. Cellular homeostasis is mainly maintained by stress proteins. Upon stress stimuli, stress response is rapidly activated to release the pressure through increasing the level of stress proteins. The elevated accumulation of Hsp27, Hsp70, and Hsp90 has been extensively observed in many types of cancer cells [35,36,37]. When a cell undergoes carcinogenesis, fast growth for escaping the immune response is needed. Under long-term stresses, the constant high level of HSPs not only helps the mutated cell’s survival, but it also participates in or promotes anti-apoptosis, angiogenesis, cell stemness, metastasis, and invasion as well [38]. On the other hand, the expression level of HSPs and RNA chaperones is downregulated in nearly all age-related neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, several polyglutamine diseases (such as Huntington’s disease), and different forms of spinocerebellar ataxias [39, 40] (Fig. 4.2). The decrease of RNA chaperones disturbs and even hinders RNA metabolism [39], while the attenuated HSPs make cells lose protein quality control (PQC) or timely degrade proteins in neurologic disease [40]. The protein aggregations caused by the dysregulation or dysfunction of stress proteins are the hallmarks of age-related neurodegenerative diseases. Stress proteins involved in other diseases are also observed. For examples, cystic fibrosis and Gaucher’s disease are associated with loss of functions by either mutation or downregulation of HSPs, whereas diseases (cancer, hepatitis, sever acute respiratory syndrome) caused by pathogen infections may result from the elevated stress proteins’ stimulated excessive immune response or enhanced pathogen replication. In the following sections, we mainly discuss the functions of stress proteins in virus infections.

Diseases caused by the dysfunction or dysregulation of stress proteins. The left panel shows the expression level of stress proteins in diverse human diseases, including cancers and neurodegenerative diseases. The expression level of stress proteins is significantly elevated in cancer cells but dramatically decreased in neurodegenerative disease. The right panel shows the virus infection-caused disease from mild to severe
4.7 Stress Proteins and Virus Infections
Disease caused by viruses is a major threat to public health. Throughout human history, pandemic virus infections have caused dozens of million deaths and tremendous economic disasters. In the past, each periodic smallpox virus outbreak killed multiple million people in both Europe and Asia. In 1918, the outbreak of Spanish flu (influenza H1N1) resulted in approximately 50 to 100 million deaths worldwide. In the twenty-first century, we have already experienced several large-scale outbreaks of virus infection which severely damaged the global health and economy, including severe acute respiratory syndrome (SARS) caused by SARS-coronavirus (SARS-CoV) outbreak from Guangzhou-Hong Kong, China, in 2003; swine flu pandemic (2019 H1N1) outbreak from California, United States, in 2009, Middle East respiratory syndrome caused by MERS-CoV outbreak from Saudi Arabia in 2012; avian influenza A virus (H7N9 and H7N10) outbreak in China in 2013; and SARS-CoV-2 outbreak from Wuhan, China, in 2019. Particularly, the SARS-CoV-2 pandemic spread very quickly to almost all countries, infected over 115 million people, and caused over 2.5 million deaths worldwide by February 26, 2021. The quarantine of cities (Wuhan, Beijing, Shanghai, etc.), province (Hubei province), and even countries (e.g., Italy) is used as an emergency measure (China, United States). Almost all public social activities, such as government service, schools, colleges and universities, entertainment, business in companies, international conferences, and travel through airlines or trains, are suspended or arranged online only in many regions and countries (e.g., Hong Kong, China, Italy, Spain, United States). For example, to shut off the virus transmission pathway and better protect citizens’ health, all the schools and universities were closed from February 2020 until further notice from government in China, Hong Kong, and United States. Since March 10 of 2020, a great number of prestigious and active research universities have been closed in the United States, including Harvard University, MIT, Stanford University, and the University of California at Berkeley. The reopen date of universities could even not be assured March, 2021.
The virus genome is composed of either DNA or RNA. Based on the composition of virus genome, the virus is classified into seven types (Baltimore classification), i.e., double-stranded DNA virus (dsDNA virus, Type I), single-stranded DNA virus (ssDNA virus, Type II), double-stranded RNA virus (dsRNA virus, Type III), positive single-stranded RNA virus ((+) ssRNA virus, Type IV), negative single-stranded RNA virus ((-) ssRNA virus, Type V), retrovirus (Type VI), and DNA retrovirus (Type VII). The replication of DNA virus occurs in the nucleus, while the RNA virus may replicate its genome in either the cytosol (Type III, Type IV) or in the nucleus (Type V). In the following sections, we will separately discuss the function of stress proteins in virus infection according to the feature of genetic materials.
In the life cycle of viruses, stress proteins play fundamental roles in almost every stage of infections. The dysregulation of stress proteins is often observed in the course of pathogenesis upon virus infections, for example, gastroenteritis [41,42,43,44], respiratory symptoms [45, 46], and hemorrhagic fevers [47, 48]. It is well known that at least 20% of human cancers are induced by chronic virus infections such as human papillomaviruses (HPVs) for cervical cancer and several other epithelial malignancies, hepatitis B and C viruses for liver cancer, and Epstein-Barr virus (EBV) for B-cell lymphoma and nasopharyngeal carcinoma. The recently accumulated data indicate that neurodegenerative diseases and neurobehavioral disorders may be caused by chronic and neuropathic viral infections that lead to neural cell deaths or loss of function in the brain followed by ageing [49], such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and autism spectrum disorders [50, 51].
4.7.1 Hsp90s in Virus Infection
During an RNA virus infection, Hsp90 may be involved in every step of the virus life cycle. At the entry step, Hsp90 functions as a receptor or a co-receptor of enterovirus [52], Japanese encephalitis virus (JEV), and dengue virus (DENV) [53]. The membrane lipid rafts are associated with Hsp90 and Hsp70 in response to DENV infection. Treatment with anti-Hsp90 antibodies or inhibitors blocks the majority of DENV entries into host cells.
At the viral RNA replication step, Hsp90 may exhibit functions in different means. The viral RNA replication is processed by an RNA-dependent RNA polymerase (RdRp). Firstly, Hsp90 stabilizes and extends the RdRp’s half-life of influenza virus, vesicular stomatitis virus (VSV), human parainfluenza viruses-2 (HPIV-2), human parainfluenza viruses-3 (HPIV-3), and respiratory syncytial virus (RSV). In addition, Hsp90 also maintains the stability of proteins in the viral replication complex (VRC), e.g., the nonstructural proteins (nsPs) NSP3 of chikungunya virus (CHIKV) and the nonstructural protein NS3 (protease) of HCV. Secondly, Hsp90 promotes viral replication by enhancing the RdRp activity through its chaperone activity. Hsp90 directly binds NS5 (RdRp) to form VRC for HCV replication. In addition, Hsp90 also stabilizes phosphoinositide-dependent kinase l (PDK1) that indirectly enhances NS5 activity for HCV replication. Hsp90 inhibitors significantly decrease viral replication by disrupting the Hsp90-NS5 VRC formation [54]. Thirdly, Hsp90 helps redistribute RdRp’s location in the host cells. During an influenza virus infection, the viral RdRp subunits (PB1 and PB2) interact with Hsp90 to form a complex and then co-translocate into the nucleus [55, 56]. In the process of nuclear import, Hsp90 maintains the stability of PB1 and PB2. After entry into the nucleus, Hsp90 dissociates from the Hsp90/PB1/PB2 complex and forms a new VRC complex with polymerase acidic protein (PA) [57]. During the viral replication, the function of Hsp90 is often regulated by phosphorylation. Results from our study show that the replication of enterovirus A71 (EV-A71) is promoted by PIM1 kinase [58]. Ectopic expression of PIM1 or Hsp90β promotes viral replication, whereas inhibition of PIM1 or Hsp90β suppresses viral replication. When Hsp90β is knocked out, the sensitivity of viral replication is lost, no matter if PIM1 is overexpressed or knocked down [59]. Hsp90 inhibitors also effectively inhibited SARS-CoV-2 replication in culture cells.
At viral protein maturation, assembly, and release steps, Hsp90 monitors the proper folding of viral proteins. For example, NS2/NS3 is cleaved into two separate proteins right after translation, a key step of HCV NS2/NS3 protein maturation. Hsp90 strictly regulates the proper folding of newly synthesized NS2/3 protein [60]. Hsp90 and its co-chaperone p23 form a complex to assist the proper folding of capsid precursor polyprotein P1 of poliovirus, rhinovirus, and coxsackievirus [61]. Hsp90 binds with neuraminidase (NA) and stabilizes Hsp90-NA complex for facilitating virion release of influenza [62]. The Hsp90-NA complex formation is also necessary for promoting cell survival to generate and release more virions.
During DNA virus infection, the viral DNA replication occurs in the nucleus. Hsp90 may assist the viral nuclear import. This step is dependent on the microtubule (MT)-dependent molecular motor dynein/dynactin complex. Viruses strictly modulate the status of tubulin acetylation that is critical for transportation. In cells infected by herpes simples virus (HSV), Hsp90 forms a complex with the acetylated tubulin and viral capsid protein VP5 [63]. Treatment with Hsp90 inhibitors disrupts the complex, thereby inhibiting the nuclear transport of HSV capsid protein [63]. Similar to that in the RNA virus replication, Hsp90s mainly maintain and promote the function of viral DNA polymerase or reverse-transcriptase (RT). Reverse transcription is the key step for hepatitis B virus (HBV) replication. RT recognizes and interacts with an RNA signal (the packaging signal, ε) on the pregenomic RNA to initiate the reverse transcription. Meanwhile, GRp94 also stabilizes and activates RT by binding to the pregenomic RNA during HBV replication. In addition, Hsp90 also regulates the location of virus DNA polymerase in host cells. When treated with Hsp90 inhibitors, the polymerases of HSV is mis-distributed from the nucleus to the cytoplasm and subsequently degraded in a proteasome-dependent manner [64, 65]. It is observed that Hsp90 is also essential for the nuclear translocation of DNA polymerase of EBV [66, 67]. VP16 is a potent HSV-encoded transcription activator. At viral RNA transcription step, Hsp90 stabilizes and enhances viral protein VP16 activity. Hsp90 also promotes the protein translation of a variety of viruses, including HSV, varicella-zoster virus (VZV), EBV, and Kaposi’s sarcoma-associated herpesvirus (KSHV).
During the chronic infection by some DNA viruses, viral proteins together with the long-term elevated Hsp90 family proteins promote cell growth and tumorigenesis. The increased Hsp90 also attenuates apoptosis, induces drug resistance, and causes cancer recurrence. Interestingly, the virus-hijacked Hsp90 may also display antiviral activities by modulating host immunity, instead, under certain conditions. In EBV-infected B cells, the expression of Hsp90 is induced to present in the cell surface at the acute infection stage. The cell surface Hsp90 stimulates the expanding gamma delta T cells (γδ T cells) [68], which exhibit potent abilities in combating infections of HIV, influenza, Sendai, coxsackie, vaccinia, VSV, and HSV-1. It works as both early sentinels of the immune system by providing immediate protection and as bridging elements between innate immunity and adaptive immunity [69].
4.7.2 Hsp70s in Virus Infection
In the course of RNA virus infections, Hsp70s may participate in every step of virus reproduction, i.e., the virus entry, RNA replication, viral protein translation, virion assembly, secretion, and even host immune response to limit virus infection or escape host defense.
At the entry step, many viruses (e.g., families of Flaviviridae, Picornaviridae, and Reoviridae) use Hsp70s to help their entry. Hsp70 functions as a virus receptor to facilitate virus attachment and entry through interacting with the envelope protein of DENV and JEV on the cell surface [70]. Hsp70 participates in multiple steps of Zika virus (ZIKV) infection. Hsp70 inhibitors impair virus entry, RNA replication, and capsid assembly of different ZIKV strains in diverse cell lines [70, 71]. GRp78 is one of the receptors of coxsackievirus A9 (CAV-9) because 50% of the virus trying to bind to the cell membrane is blocked when treated with antibodies against GRp78 [72]. Recently study shows that GRp78 is a reciptor of SARS-CoV-2, sugesting a potential idea drug target for combating COVID-19. GRp78 also interacts with major histocompatibility complex (MHC) I molecules to help with viral internalization into host cells. During the rotavirus infection, Hsc70 helps viral internalization but does not involve in virus attachments. At the replication step, Hsp70s employ different means to facilitate viral RNA replication. Similar to Hsp90s, as a molecule chaperone, Hsp70s facilitate VRC formation and maintain the stability of VRC proteins. Hsc70s can also regulate VRC activity by interacting with other components of the complex. For example, Hsp72 physically interacts with NS5A, NS3, and NS5B (RdRp) of HCV in VRC, while Hsc70 promotes HCV replication by interacting with VRC through attachment on the 3’ polyU/UC motif of HCV RNA genome. In addition, Hsp70s may facilitate virus replication through modulating nuclear import of polymerase or nuclear capsid. Influenza virus replicates its genome in the nucleus. The nuclear entry of RdRp subunits PB2 and PB1 depends on the complex formation of PB1/PB2 monomers or PB2/PB1 heterodimers with Hsp70 [73]. At the translation step, Hsp70s employ different mechanisms to promote viral protein translation. The genome of (+) ssRNA virus (such as enterovirus, flavivirus, and coronavirus) encodes a polypeptide. After entry, the first step for initiating the viral life cycle is to translate the polypeptide, followed by autocleavage into functional viral proteins including RNA polymerase. The viral protein translation is often driven by internal ribosome entry site (IRES) for many RNA viruses; in the meantime, the host cellular cap-dependent translation is inhibited by virus-encoded protein(s). The interaction of Hsc70 and 2A protease of EV-A71 enhances the cleavage on eukaryotic translation initiation factor EIF4G for favoring viral protein translation and impairing host cell cap-dependent translation [74]. For some viruses without IRES, plenty of dsRNAs are generated during replication. The dsRNAs activate protein kinase-RNA-activated (PKR) eIF2a signaling to shut down global cellular protein translation for releasing stresses [75, 76]. To avoid the blockage of viral proliferation, influenza A virus downregulates PKR in an Hsp70-dependent way [77,78,79,80,81]. At virion assembly and release steps, Hsp70 participates in the assembly of reovirus trimeric sigma 1 protein, a main viral component for the interaction with the host cell receptor [82]. Hsp70 also interacts with the capsid precursor P1 to form the Hsp70-P1 complex, which is believed to be an assembly intermediate of picornaviruses [83]. Both Hsp70 and Hsc70 interact with NS5A protein to regulate virion release of HCV. Hsp70 only enhances viral protein expression. Hsc70 not only promotes virion release but is also embedded in the viral capsid for ensuring infectivity, although it has no effects on viral protein expression [84, 85].
During DNA virus infection, Hsp70s also play crucial roles in each step. At the step of SV40 entry into the cytosol from the ER, Hsc70, as well as its family member, Hsp105, interacts with and is regulated by Hsc70 co-chaperone SGTA. The Hsc70/Hsp105/SGTA complex facilitates SV40 cytosol entry, a key step in SV40 life cycle. In this process, GRp78 interacts with its co-chaperone Hsp40 family member DNAJB-11 to help SV40 disassembly and enter into the cytoplasm. In the case of adenovirus infection, Hsp70/Hsc70 and co-chaperone Bag3 form a complex with the penton base protein, the viral capsid constituent responsible for the import of viral capsid and DNA into the nucleus [86, 87]. At the viral gene transcription and translation steps, the chaperone activity of Hsp70s guarantees the viral gene expression by stabilizing transcription factors and correct folding of newly synthesized viral proteins. More importantly, Hsp70/Bag1 complex stimulates the general viral gene transcription in an Hsp70-dependent manner upon cells infected by different viruses, such as human polyomavirus and Human cytomegalovirus (HCMV) [88, 89]. Many DNA viruses start the virion assembly in the nucleus. Hsp70 and Hsc70 translocate from the cytosol into the nucleus and form a complex with all three viral capsid proteins (VP1, VP2, and VP3) of polyomavirus for initiating virion assembly.
Cellular transformation, survival, and apoptosis: Some DNA viruses without encoding their own polymerase have to depend on host DNA replication machinery. To ensure replication in quiescent cells, the virus transforms the host cells to keep host cell division by overcoming the cell cycle restriction points. A well-known example is the SV40-induced cell transformation. The large and small T antigens (TAg) are responsible in this process. The J-domain of both types of TAg binds with Hsc70 through its Hsp70-co-chaperone signature motif. Then, the TAg-Hsc70 complex sequesters the retinoblastoma protein (pRb) from the pRb-E2F complex to release free E2F. The free E2F activates transcription of the S-phase genes for DNA replication in an Hsc70-ATP hydrolysis-dependent manner [90, 91]. Alternatively, TAg induces Hsc70 to disassemble an inhibitory silencer complex or to remodel the chromatin. The E7 protein of HPV and E1A protein of adenovirus also exhibit a similar behavior to target the pRb-E2F complex through recruiting Hsc70-interacting protein hTid-1 or directing binding with Hsc70, respectively [92]. In summary, most double-stranded DNA viruses use Hsp70s to reprogram the cell cycle for ensuring virus replication.
In the early stage of infection, the virus has to undergo fast replication to escape host defense, keep host cell alive, and resist cell death. Hsp70s function as chaperones to correct the misfolding of proteins and remove protein aggregations under the stress of fast protein synthesis. In the early stage of EBV infection, the virus-encoded nuclear oncoprotein EBNA3A helps Hsp70 nuclear translocation and Hsp70 chaperone complex formation to immortalize B cells by inhibiting apoptosis [93]. In the late stage of infection, apoptosis is beneficial for virus’s spreading. Virions are often observed in apoptotic bodies and subsequently engulfed by phagocytic cells. This makes it possible for the virus to infect neighbor cells without exposure to the host immune system [94, 95]. It has been demonstrated that the decrease of Hsp70 would help induce the timed apoptosis for virion release at the late stage of adenovirus infections.
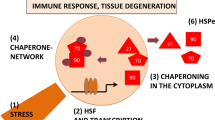
Innate immunity: It is well known that Hsp70s play an important role in innate immunity and inflammatory responses. Hepatocyte was believed to have a lack of innate immunity for a long time. Instead of promoting virus replication, the elevated GRp78 expression stimulated by HBV replication restricts virus replication through the activation of intracellular innate immunity by inducing the expression of IFNβ and its downstream antiviral proteins [96]. Other studies show that Hsp70s greatly contribute to the innate immunity of host cells either infected by bacteria or virus. Hsp70 interacts with IκB kinase (IKK) and disturbs the function and stability of the NF-κB/IkBα/IKK complex. Hsp70 also impairs IkBα phosphorylation, affecting IkBα proteasomal degradation and nucleus translocation of NF-κB complex [97]. Moreover, Hsp70 also regulates inflammation in an NF-κB independent pathway through impacting the NLRP3/ASC inflammasome formation [98]. More interestingly, Hsp70 can be secreted out, and the extracellular Hsp70 binds to dendritic cells and macrophages before being recognized by innate immune receptors [99, 100]. Upon infection, Toll-like receptors (TLRs) can immediately detect virus and trigger cellular innate immune response. TLR2/TLR4 interact with Hsp70 and participate in the extracellular Hsp70-stimulated pro-inflammatory signaling cascade in a CD14-dependent manner, thereby promoting pro-inflammatory cytokine production through MyD88/IRAK/NF-κB axis signaling cascade [101]. In addition, many other innate immunity-related signal transduction pathways are also activated in the process, such as p38 MAPKs, JNK, and ERK pathways. Then, the downstream transcription factors CREB and AP-1 are activated, resulting in pro-inflammatory cytokine expression, including TNFα, IL-6, IL-1β, and a number of chemokines [102,103,104,105].
4.7.3 The Function of Small Heat-Shock Proteins (sHSPs) in Virus Infection
Small heat-shock proteins are the most upregulated proteins responding to stress stimuli such as oxidative stress (elevated reactive oxygen species (ROS) level, hypoxia, etc.), high temperature, UV, starvation, malnutrition, toxic material, or pathogen invasion [106]. sHSPs are capable of recognizing the misfolded proteins and transferring them to other ATP-dependent chaperones for correction or proteasomes/autophagosomes for degradation [107, 108]. Hsp27 is one of the most extensively studied sHSPs. It is ubiquitously expressed at high level in skeletal, smooth, and cardiac muscles. Because of its particular importance in viral infection, in this chapter, we use Hsp27 as a model to address the functions of sHSPs.
During RNA virus infection, Hsp27 may participate in every step of the viral life cycle. Hsp27 is fast elevated to promote viral replication and protein expression during EV-A71 infections. The viral replication is significantly inhibited when Hsp27 is either silenced by small interfering RNA (siRNA) or knocked out by CRISPR/cas9-mediated gene editing. The suppressed viral activities are recovered after Hsp27 is restored in the host cells. Further studies revealed that Hsp27 promotes 2Apro-mediated EIF4G cleavage that favors the internal ribosome entry site (IRES)-mediated viral protein translation. In addition, Hsp27 is translocated from the cytosol into the nucleus and reversely correlated with hnRNP A1 redistribution from the nucleus to the cytosol, an important step to initiate viral protein translation [109]. Surprisingly, the knockout of Hsp27 blocks the hnRNP A1 cytosol redistribution, indicating that Hsp27 is an important player for the import/export of some proteins in virus infection. Swine fever virus (CSFV) is a member of the family Flaviviridae. Hsp27 binds with NS5A, an essential protein for viral replication and assembly. Porcine epidemic diarrhoea virus (PEDV) infection causes high fatality in swine. Hsp27 is quickly elevated in response to PEDV infection by triggering antiviral immunity through the activation of NF-κB signaling that stimulates the expression of IFNβ and its downstream interferon stimulated genes (ISGs) [110].
Hsp27 also contributes to DNA virus replication. It may regulate virus replication either in a positive way or a negative way. The increase of phosphorylated Hsp27 is associated with the upregulated virus replication when cells are infected by porcine circovirus type 2 (PCV2). PCV2 is a single-stranded DNA virus responsible for the postweaning multisystemic wasting syndrome (PMWS) in pigs. The phosphorylated Hsp27 is also upregulated in EBV-positive cells via the activation of Akt signaling. However, similar to GRp78, Hsp27 displays antiviral effect against HBV replication through enhancing IFN production in hepatocytes [111]. Hsp27 can alter viral infections through regulating pro-inflammatory cytokine expression via a variety of signaling pathways. The knockdown of Hsp27 causes an increased release of IL-8 from keratinocytes infected by adenovirus and stimulated with UV or TNFα. The increased IL-8 level is correlated with enhanced IκB-α degradation and phosphorylated IκB-α accumulation in Hsp27-depleted cells. It is also found that Hsp27 is associated with the inhibitory protein IκB kinase (IKK) complex. Hsp27 also activates p38 MAPK/MK2 kinase signaling to increase cyclooxygenase-2 and IL-6 expressions. Besides, Hsp27 is of importance in regulating stress caused by reactive oxygen species (ROS) [107]. Hsp27 regulates enzyme activities by upholding glutathione in reduced form, such as glutathione reductase and glucose-6-phosphate dehydrogenase [112]. Hsp27 also greatly contributes to the degradation of oxidized proteins during virus infection, although the underlying mechanisms are not well understood.
4.7.4 The Function of PDIs in Virus Infection
The function of PDIs on virus entry and uncoating: The entry of some viruses into eukaryotic cells is controlled by redox-regulated processes. In the case of the bird newcastle disease virus (NDV) infection, it enters cells through large conformational changes in its F fusion protein via thiol/disulfide exchange because the overexpression of PDIs increases the viral membrane fusion. In endothelial cell surface, PDI decreases β1 and β3 integrins to facilitate DENV entry [113, 114]. The entry of rotavirus into MA104 cells is blocked by thiol blockers and PDI inhibitors [115]. PDIs also participate in HIV entry. The cleavage of two disulfide bonds of gp120 is needed for virion entry into CD4+ cells. PDI inhibitors effectively block the cleavage, leading to loss of entry activity and defect of HIV infection [116].
PDIs regulate viral protein translation: Only little data show that PDIs may participate in viral protein translation. The viral protein synthesis of some (+) ssRNA virus is dependent on IRES-mediated translation. EV-A71 infection is potently inhibited by an ERp57 inhibitor Oblongifolin M (OM) through suppressing the viral IRES activity. This finding is further confirmed because the overexpression of ERp57 enhances the IRES activity and partially rescues the OM-inhibited IRES activity and viral replication [117]. It is generally accepted that PDIs ensure the correct formation and activity of viral glycoproteins.
PDIs regulate viral activities by influencing oxidative stress and ER stress: The rapid and large amount of viral protein synthesis causes disruption of redox balance in the establishment of infection and in the process of pathogenesis. The accumulated ROS in turn modulates viral replication and cellular response to viral pathogenesis that occur during HIV [118], influenza virus [119], HBV [120], HCV [121], encephalomyocarditis virus (EMCV) [122], RSV [123], and JEV [124] infections. ERp57 participates in the mechanisms of cell protection against oxidative stress, regardless of its subcellular location. The expression level of ERp57 and GST is positively correlated with tissue redox status during HPV infection, indicating potential viral-induced oxidative DNA and protein damages [125]. In addition, viral infections may cause the exploitation of the ER membrane, accumulation of misfolded proteins, and imbalance of calcium concentration. Many viruses (influenza virus, hepatitis B virus, Zika virus, etc.) hijack host cell processes to enhance viral pathogenesis, for instances, facilitating viral protein folding, trafficking, and modulating host immune responses [126,127,128]. As a major effector in ER stress response, ERp57 is critical for viral protein glycosylation and maturation, which in turn affects virus release and infectivity.
4.7.5 The RNA Chaperones in Virus Infection
The function of RNA chaperones in RNA virus infection: The life cycle of most RNA viruses is completed in the cytoplasm. Therefore, the virus is capable of redistributing some nuclear proteins into the cytosol that involves in RNA metabolism and functional regulation in different means. hnRNPs, such as hnRNP A1, C1/C2, D, and I, mainly locate in the nucleus but often shuttle to the cytoplasm to stabilize the viral RNA and initiate the cap-independent translation. For example, VP24 of Ebola virus (EBOV) binds to karyopherin-alpha nuclear import proteins and prevents their interaction with phosphorylated STAT1 (pSTAT1) and hnRNP C1/C2, leading to the cytoplasmic retention of pSTAT1 and hnRNP C1/C2 [129]. The cytosolic retention of pSTAT1 facilitates the virus’ escape from the intracellular innate immunity. Rae1 is an mRNA export factor. VSV infection induces hnRNP A1 cytoplasm redistribution in a Rae1-dependent manner [130].
Several hnRNPs are involved in virus replication. hnRNP I/PTB binds UCUAA repeats at IRES and 3’-UTR to modulate coronavirus RNA synthesis [131]. During the HCV RNA replication, hnRNP I/PTB and hnRNP C selectively interact with the 3 ′ end of the HCV genomic RNA and possibly initiate the (-) strand RNA replication to stabilize the RNA genome and/or to regulate the encapsidation of genomic RNA [132, 133]. Besides, hnRNP C also binds at the 5′- end of negative-strand RNA of poliovirus to facilitate the (+) strand RNA synthesis [134], while miR-555 decreases the expression of hnRNP C, thereby inhibiting poliovirus replication [135, 136].
The main function of hnRNPs is to modulate RNA process and metabolism (splicing, stabilization, transportation, etc.). The genome of influenza A virus is comprised of eight negative-stranded RNA segments. Among them, NS1 and M segments undergo alternative splicing and yield several proteins including nonstructural proteins NS1 and NS2, as well as matrix proteins M1 and ion channel M2. The NS segment encodes NS1 and nuclear export protein (NEP)/NS2; the M segment encodes M1 and M2. hnRNP K bridges the interaction of NS1-BP (binding protein) complex and M mRNA. Lack of neither NS1-BP nor hnRNP K ensures the proper splicing of M mRNA [137, 138]. The mutation of either or both the hnRNP K and NS1-BP-binding sites of viral RNA results in M segment mis-splicing and attenuated virus replication [139].
Most hnRNPs positively regulate viral protein translation. hnRNPs bind on the IRES of different viruses to assist translation. hnRNP I/PTB3, hnRNP L, hnRNP D/AUF1 [140,141,142], hnRNP A1, and hnRNP K [143] all bind the 5’UTR sequence to regulate HCV protein translation. hnRNP I, one of the polypyrimidine tract-binding proteins (PTBs), binds the IRES of picornaviruses, including cardio-aphthovirus group (including EMCV and EV-A71) and entero-rhinovirus group (including poliovirus and HRV-2). PTBs also bind HCV UTR regions, calicivirus RNA, and coronavirus RNAs. PTBs help IRES to fold into a translation-competent structure [144]. hnRNP D shuttles between the cytosol and the nucleus. It interacts with the stem-loop II of HCV 5' UTR to promote the IRES-dependent translation [140]. PIM1 inhibitors induce hnRNP D relocalization from the nucleus to the cytosol, giving rise to the inhibition of EV-A71 protein translation [58]. hnRNP A1 binds with both 5′- and 3′-UTR of HCV RNA to form a complex with NS5b and septin 6 and assists with viral protein translation. The hnRNP A1 homologs (such as hnRNP A/B, hnRNP A2/B1, and hnRNP A2) may substitute it to modulate the IRES-dependent translation [145]. hnRNP A1 specifically binds on the 5'-UTR of EV-A71 and enhances IRES-dependent translation [146]. Recent studies demonstrate that the cytoplasmic redistribution of hnRNP A1 is strictly regulated by some signaling or stress proteins. P38 inhibitors or Hsp27 knockout dramatically block hnRNP A1 redistribution in the cytosol, leading to the inhibition of virus replication [109, 147].
Apart from binding with IRES and promoting viral protein translation, hnRNPs can directly bind with virus proteins. Upon a CVB3 infection, hnRNP M is cleaved by 3C protease that facilitates virus replication without affecting IRES activity and RNA stability [148]. The core protein of DENV interacts with hnRNP K, resulting in the release of hnRNP K’s inhibitory effects on transcriptional activator C/EBPb [149, 150], while hnRNP C1/C2 interact with viral RNA and NS1 protein to facilitate virus reproduction [151,152,153,154]. hnRNP A2/B1 interact with NS1 of influenza virus, leading to the decrease of the viral NS1 RNA/protein levels as well as NS1 RNA nuclear export [155]. The negative regulator of splicing (NRS) element of gag gene in Rous sarcoma virus genome is required for 3′ long terminal repeat (LTR) polyadenylation. hnRNP H binds on NRS element and promotes polyadenylation.
The functions of RNA chaperones in DNA virus infections: To initiate viral DNA replication, hnRNP K interacts with DNA replication initiator protein UL84 to promote HCMV replication. hnRNPs regulate DNA virus gene expression at both transcriptional and post-transcriptional levels, either positively or negatively. At the transcription level, hnRNP D0B and hnRNP A/B bind the AAGTATGCA core element of HPV18 late promoter for suppressing the late genes, capsid proteins L1 and L2 [125, 156]. hnRNP K binds enhancer II (EN II) to promote HBV replication [157]. When APOBEC3B and hnRNP K form a complex, it alters the EN II binding activity and suppresses the S gene promoter activity [158]. At the posttranscriptional level, hnRNPs regulate the polyadenylation, splicing, and translation during DNA virus infections. In the process of HPV RNA splicing, hnRNP A1 directly binds with the splicing silencer element to inhibit the splicing of L1 mRNAs; hnRNP I also interferes with the splicing inhibitory elements and activates L1 gene expressions [159]. The genome of HPV consists of an early region and a late region followed by the proximal early (pAE) and the distal late (pAL) polyadenylation signals, respectively. The production of virions is controlled by the expression of L1 and L2 late genes in a differentiation-dependent way. hnRNP H binds the multiple GGG motifs at and promotes polyadenylation at an early polyadenylation signal, leading to inhibiting the L2 transcription. At the late infection stage, hnRNP H is captured by L1 to release the inhibitory effect on L2 [160, 161]. This process is called late gene autoregulation which enables rapid viral capsid protein production [161]. The SM polymerase (pol) mRNA of EBV early protein is cleaved and polyadenylated inefficiently. However, SM can harness hnRNP A1 and C for enhancing SM mRNA process under certain conditions [162]. In the translation process, hnRNP I and hnRNP K inhibit the L2 translation through binding on the 3′ end of L2 mRNA [163].
hnRNPs also exhibit some other functions during virus infections. During EBV infection, AUF1 can bind with the EBER1 noncoding RNA and compete with AUF1-interacting targets. In addition, AUF1/CBF2/EBNA2 forming a complex binds on the latency C promoter (Cp) [164]. Both EBNA2 and EBER1 are important for facilitating viral latency and cell transformation. hnRNPs are also able to regulate viral activities through modulating immune response. In the HSV-1 infection, viral DNA and hnRNP A/B form a complex, followed by homodimerization and demethylation. These events result in translocating the complex to the cytosol and activating natural immunity through type I interferon signaling.
4.8 Antiviral Drug Development and Challenge
Interest in targeting stress proteins in various diseases is increasing. First, targeting stress proteins would provide a wide spectrum of antiviral ability; this in turn increases the chances of curing related diseases. In cultured cells, for example, Hsp90 inhibitors display potent antiviral activity against many DNA and RNA viruses, including enteroviruses (EV-A71, coxsackievirus, poliovirus, rhinovirus, etc.), VSV, RSV, influenza viruses, CHIKV, HCV, HSV, HBV, EBV, HCMV, HTLV and SARS-CoV-2. GRp78 has been show a receptor of SARS-CoV-2. Target GRp78 would not only block viral entry, but aslo it must disrupt viral protein translation. It is also a wonderful strategy to target hnRNPs for antiviral. The depletion of hnRNP A1 inhibits viral reproductions of HPV, HCV, EV-A71, human T-cell lymphotropic virus 1 (HTLV-1), and HIV [165,166,167,168]; the depletion of hnRNP C combats viral infections of HCV, DENV, poliovirus, EBV, and HIV. The dysfunction of hnRNP I/PTB also impairs the reproduction of HCV, picornavirus, HPV, and HIV [169, 170]; the deprivation of hnRNP K significantly decreases the reproduction of influenza viruses, EV-A71, DENV, VSV, HCMV, HBV, HSV, HPV, HIV, and KSHV. Therefore, target of stress proteins is very attractive for drug development against viral infections that do not have effective therapies, especially newly emerging viral diseases such as COVID-19. Second, the affinity or dependence of stress proteins is commonly stronger under diseased situations than that in normal conditions. For example, mutant p53 relies much more on Hsp90 function than wild-type p53. Hsp90 inhibitor GM easily disturbs the association of mutant p53 with Hsp90 that causes the mutant p53 degradation only without affecting wild-type p53 [104]. Thirdly, little drug resistance would develop. No drug resistance will occur despite extensive passaging of the virus in the presence of Hsp90 inhibitors both in vitro cell culture and in vivo in animals.
One of the major challenges for drug design against stress proteins is the off-target effects, which is due to structure similarities among different stress proteins. For instance, a drug targeting Hsp90α may affect Hsp90β and even other members of Hsp70s. Over 20 Hsp90 inhibitors for anticancer have been developed; however, clinical studies are often stopped because of strong toxicity and side effects. This challenge would become little importance for treating acute infections such as SARS-CoV infection. However, other challenges appear. Due to the importance of stress proteins in normal cells and virus infections variably dysfunction many biological processes (including stress protein responses) at certain time points, inhibitors targeting stress proteins must be used with great caution in clinical settings because it may cause unexpected side effects and even toxicity. The gone with wind infection manner (e.g., SARS-CoV) limits the opportunity of clinic settings, giving rise to additional challenges for stress protein-based antiviral drug development.
References
Molinari DNHAM. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2006;87:1377–408. https://doi.org/10.1152/physrev.00050.2006.-A.
Hendrick JP, F H. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem. 1993;62:349–84. https://doi.org/10.1146/annurev.bi.62.070193.002025.
Murshid A, Gong J, Calderwood S. The role of heat shock proteins in antigen cross presentation. Front Immunol. 2012;3:1–10. https://doi.org/10.3389/fimmu.2012.00063.
Asea A, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34.
Hoter A, El-Sabban ME, Naim HY. The HSP90 family: structure, regulation, function, and implications in health and disease. Int J Mol Sci. 2018;19:1–33. https://doi.org/10.3390/ijms19092560.
Wang X, Chen M, Zhou J, Zhang X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy. Int J Oncol. 2014;45:18–30. https://doi.org/10.3892/ijo.2014.2399.
Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14:630–42. https://doi.org/10.1038/nrm3658.
Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345–60. https://doi.org/10.1038/nrm.2017.20.
Stetler RA, et al. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol. 2010;92:184–211. https://doi.org/10.1016/j.pneurobio.2010.05.002.
Zhu X, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606.
Radons J. The human HSP70 family of chaperones: where do we stand? Cell. 2016;21:379–404. https://doi.org/10.1007/s12192-016-0676-6.
Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39:321–31. https://doi.org/10.1016/j.molcel.2010.07.012.
Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51. https://doi.org/10.1016/j.cell.2006.04.014.
Sharma SK, De Los Rios P, Christen P, Lustig A, Goloubinoff P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol. 2010;6:914–20. https://doi.org/10.1038/nchembio.455.
Mayer MP. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci. 2013;38:507–14. https://doi.org/10.1016/j.tibs.2013.08.001.
Kampinga HH, Craig EA. The Hsp70 chaperone machinery: J-proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579. https://doi.org/10.1038/NRM2941.
Garrido C, Paul C, Seigneuric R, Kampinga H. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44:1588–92.
van Montfort RL, et al. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–30.
MacRae TH. Structure and function of small heat shock/α-crystallin proteins: established concepts and emerging ideas. Life Sci. 2000;57:899–913.
Shashidharamurthy R, Koteiche HA, Dong J, Mchaourab HS. Mechanism of chaperone function in small heat shock proteins. J Biol Chem. 2005;280:5281–9. https://doi.org/10.1074/jbc.M407236200.
Hsu A-L. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–5. https://doi.org/10.1126/science.1083701.
Bruey J-M, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–52. https://doi.org/10.1038/35023595.
Guay J, et al. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110(Pt 3):357–68.
Parcellier A, et al. Small heat shock proteins HSP27 and αB-crystallin: cytoprotective and oncogenic functions. Antioxid Redox Signal. 2005;7:404–13.
Anckar J, Sistonen L. Regulation of HSF 1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–115. https://doi.org/10.1146/annurev-biochem-060809-095203.
Sakurai H, Enoki Y. Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 2010;277:4140–9. https://doi.org/10.1111/j.1742-4658.2010.07829.x.
McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–8. https://doi.org/10.1074/jbc.273.13.7523.
Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–63. https://doi.org/10.1002/jcp.10172.
Benham AM. The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal. 2012;16:781–9. https://doi.org/10.1089/ars.2011.4439.
Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell. 1999;10:2573–82.
Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–50. https://doi.org/10.1089/ars.2009.2466.
Bedard K, Szabo E, Michalak M, Opas M. Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int Rev Cytol. 2005;245:91–121. https://doi.org/10.1016/s0074-7696(05)45004-4.
Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–38. https://doi.org/10.1002/bies.20048.
Zuniga S, Sola I, Cruz JL, Enjuanes L. Role of RNA chaperones in virus replication. Virus Res. 2009;139:253–66. https://doi.org/10.1016/j.virusres.2008.06.015.
Ciocca RD, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell. 2005;10:86–113.
Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10:47–51. https://doi.org/10.1016/j.molmed.2003.12.005.
Calderwood SK, Gong J. Heat shock proteins promote cancer: it’s a protection racket. Trends Biochem Sci. 2016;41:311–23. https://doi.org/10.1016/j.tibs.2016.01.003.
Calderwood SK. Heat shock proteins and cancer: intracellular chaperones or extracellular signalling ligands? Philos Trans R Soc Lond Ser B Biol Sci. 2018;373:524. https://doi.org/10.1098/rstb.2016.0524.
Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron. 2019;102:294–320. https://doi.org/10.1016/j.neuron.2019.03.014.
Kampinga HH, Bergink S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016;15:748–59. https://doi.org/10.1016/s1474-4422(16)00099-5.
Munnink BBO, Hoek LVD. Viruses causing gastroenteritis: the known, the new and those beyond. Virus. 2016;8:42. https://doi.org/10.3390/V8020042.
Siebrasse EA, et al. Identification of MW polyomavirus, a novel polyomavirus in human stool. J Virol. 2012;86:10321. https://doi.org/10.1128/JVI.01210-12.
Jones MS, Lukashov VV, Ganac RD, Schnurr DP. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. J Clin Microbiol. 2007;45:2144. https://doi.org/10.1128/JCM.00174-07.
Kapoor A, et al. A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proc Natl Acad Sci U S A. 2008;105:20482–7. https://doi.org/10.1073/PNAS.0807979105.
Walter MJ, Wunderink GR. Severe respiratory viral infections: new evidence and changing paradigms. Infect Dis Clin N Am. 2017;31:455–74.
Manjarrez-Zavala ME, Rosete-Olvera DP, Gutiérrez-González LH, Ocadiz-Delgad R, Cabello-Gutiérrez C. Pathogenesis of viral respiratory infection. Resp Dis Infect. 2013; https://doi.org/10.5772/54287.
Messaoudi I, Basler CF. Immunological features underlying viral hemorrhagic fevers. Curr Opin Immunol. 2015;36:38. https://doi.org/10.1016/J.COI.2015.06.003.
Paessler S, Walker DH. Pathogenesis of the viral hemorrhagic fevers. Annu Rev Pathol Mech. 2013;8:411–40. https://doi.org/10.1146/annurev-pathol-020712-164041.
Karim S, et al. The role of viruses in neurodegenerative and neurobehavioral diseases. CNS Neurol Disord. 2014;13:1213–23. https://doi.org/10.2174/187152731307141015122638.
Karim S, et al. The role of viruses in neurodegenerative and neurobehavioral diseases. CNS Neurol Disord Drug Targets. 2014;13:1213.
Balin BJ, Hudson AP. Herpes viruses and Alzheimer’s disease: new evidence in the debate. Lancet Neurol. 2018;17:839–41. https://doi.org/10.1016/s1474-4422(18)30316-8.
Tsou Y-L, et al. Heat shock protein 90: role in enterovirus 71 entry and assembly and potential target for therapy. PLoS One. 2013;8:e77133. https://doi.org/10.1371/journal.pone.0077133.
Cabrera-Hernandez A, Thepparit C, Suksanpaisan L, Smith DR. Dengue virus entry into liver (HepG2) cells is independent of hsp90 and hsp70. J Med Virol. 2007;79:386–92. https://doi.org/10.1002/jmv.20786.
Okamoto T, et al. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–25. https://doi.org/10.1038/sj.emboj.7601367.
Momose F, et al. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem. 2002;277:45306–14. https://doi.org/10.1074/jbc.M206822200.
Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol. 2007;81:1339–49. https://doi.org/10.1128/JVI.01917-06.
Chase G, et al. Hsp90 inhibitors reduce influenza virus replication in cell culture. J Virol. 2008;377:431–9. https://doi.org/10.1016/j.virol.2008.04.040.
Zhou F, et al. Pim1 impacts enterovirus A71 replication and represents a potential target in antiviral therapy. Science. 2019;19:715. https://doi.org/10.1016/J.ISCI.2019.08.008.
Wan Q, Song D, Li H, He ML. Stress proteins: the biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct Target Ther. 2020;5(1):125.
Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci U S A. 2001;98:13931–5. https://doi.org/10.1073/pnas.241510898.
Geller R, Vignuzzi M, Andino R, Frydman J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007;21:195–205. https://doi.org/10.1101/gad.1505307.
Kumar P, Gaur P, Kumari R, Lal SK. Influenza A virus neuraminidase protein interacts with Hsp90, to stabilize itself and enhance cell survival. J Cell Biochem. 2019;120:6449–58. https://doi.org/10.1002/jcb.27935.
Zhong M, et al. Heat-shock protein 90 promotes nuclear transport of herpes simplex virus 1 capsid protein by interacting with acetylated tubulin. PLoS One. 2014;9:e99425. https://doi.org/10.1371/journal.pone.0099425.
Burch AD, Weller SK. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol. 2005;79:10740–9. https://doi.org/10.1128/JVI.79.16.10740-10749.2005.
Li Y-H, Tao P-Z, Liu Y-Z, Jiang J-D. Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother. 2004;48:867–72. https://doi.org/10.1128/aac.48.3.867-872.2004.
Sun X, Kenney SC. Hsp90 inhibitors: a potential treatment for latent EBV infection? Cell Cycle. 2010;9:1665–6. https://doi.org/10.4161/cc.9.9.11594.
Kawashima D, et al. Nuclear transport of Epstein-Barr virus DNA polymerase is dependent on the BMRF1 polymerase processivity factor and molecular chaperone Hsp90. J Virol. 2013;87:6482–91. https://doi.org/10.1128/JVI.03428-12.
Kotsiopriftis M, Tanner JE, Alfieri C. Heat shock protein 90 expression in Epstein-Barr virus-infected B cells promotes gammadelta T-cell proliferation in vitro. J Virol. 2005;79:7255–61. https://doi.org/10.1128/JVI.79.11.7255-7261.2005.
Sciammas R, Bluestone JA. TCRgammadelta cells and viruses. Microbes Infect. 1999;1:203–12.
Taguwa S, et al. Zika virus dependence on host hsp70 provides a protective strategy against infection and disease. Cell Rep. 2019;26:906–20. https://doi.org/10.1016/j.celrep.2018.12.095.
Pujhari S, et al. Heat shock protein 70 (Hsp70) mediates Zika virus entry, replication, and egress from host cells. Emerg Microbes Infect. 2019;8:8–16. https://doi.org/10.1080/22221751.2018.1557988.
Triantafilou K, Fradelizi D, Wilson K, Triantafilou M. GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J Virol. 2002;76:633–43. https://doi.org/10.1128/jvi.76.2.633-643.2002.
Manzoor R, et al. Heat shock protein 70 modulates influenza A virus polymerase activity. J Biol Chem. 2014;289:7599–614. https://doi.org/10.1074/jbc.M113.507798.
Dong Q, et al. Hsc70 regulates the IRES activity and serves as an antiviral target of enterovirus A71 infection. Antivir Res. 2018;150:39–46. https://doi.org/10.1016/j.antiviral.2017.11.020.
Hovanessian AG, Galabru J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur J Biochem. 1987;167:467–73. https://doi.org/10.1111/j.1432-1033.1987.tb13360.x.
Gale J, Michael J, Korth MJ, Katze MG. Repression of the PKR protein kinase by the hepatitis C virus NS5A protein: a potential mechanism of interferon resistance. Clin Diagn Virol. 1998;10:157–62. https://doi.org/10.1016/S0928-0197(98)00034-8.
Lee TG, Tang N, Thompson S, Miller J, Katze MG. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol Cell Biol. 1994;14:2331–42. https://doi.org/10.1128/mcb.14.4.2331.
Lee TG, Tomita J, Hovanessian AG, Katze MG. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc Natl Acad Sci U S A. 1990;87:6208–12. https://doi.org/10.1073/pnas.87.16.6208.
Lee TG, Tomita J, Hovanessian AG, Katze MG. Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J Biol Chem. 1992;267:14238–43.
Melville MW, Hansen WJ, Freeman BC, Welch WJ, Katze MG. The molecular chaperone hsp40 regulates the activity of P58IPK, the cellular inhibitor of PKR. Proc Natl Acad Sci U S A. 1997;94:97–102. https://doi.org/10.1073/pnas.94.1.97.
Melville MW, et al. The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J Biol Chem. 1999;274:3797–803. https://doi.org/10.1074/jbc.274.6.3797.
Leone G, et al. C-terminal trimerization, but not N-terminal trimerization, of the reovirus cell attachment protein Is a posttranslational and Hsp70/ATP-dependent process. J Biol Chem. 1996;271:8466–71. https://doi.org/10.1074/jbc.271.14.8466.
Macejak DG, Sarnow P. Association of heat shock protein 70 with enterovirus capsid precursor P1 in infected human cells. J Virol. 1992;66:1520–7.
Khachatoorian R, et al. The NS5A-binding heat shock proteins HSC70 and HSP70 play distinct roles in the hepatitis C viral life cycle. J Virol. 2014;454-455:118–27. https://doi.org/10.1016/j.virol.2014.02.016.
Khachatoorian R, et al. Structural characterization of the HSP70 interaction domain of the hepatitis C viral protein NS5A. J Virol. 2015;475:46–55. https://doi.org/10.1016/j.virol.2014.10.011.
Chroboczek J, Gout E, Favier AL, Galinier R. Novel partner proteins of adenovirus penton. Curr Top Microbiol Immunol. 2003;272:37–55.
Gout E, Gutkowska M, Takayama S, Reed JC, Chroboczek J. Co-chaperone BAG3 and adenovirus penton base protein partnership. J Cell Biochem. 2010;111:699–708. https://doi.org/10.1002/jcb.22756.
Pater A, Kumar KU, Pater MM, Devireddy LR. BAG-1, a novel Bcl-2-interacting protein, activates expression of human JC virus. J Gen Virol. 2000;81:351–7. https://doi.org/10.1099/0022-1317-81-2-351.
Takahashi N, et al. BAG-1M, an isoform of Bcl-2-interacting protein BAG-1, enhances gene expression driven by CMV promoter. Biochem Biophys Res Commun. 2001;286:807–14. https://doi.org/10.1006/bbrc.2001.5473.
Sullivan CS, Cantalupo P, Pipas JM. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol Cell Biol. 2000;20:6233–43. https://doi.org/10.1128/mcb.20.17.6233-6243.2000.
Sullivan CS, Gilbert SP, Pipas JM. ATP-dependent simian virus 40 T-antigen-Hsc70 complex formation. J Virol. 2001;75:1601–10. https://doi.org/10.1128/JVI.75.4.1601-1610.2001.
White E, Spector D, Welch W. Differential distribution of the adenovirus E1A proteins and colocalization of E1A with the 70-kilodalton cellular heat shock protein in infected cells. J Virol. 1988;62:4153–66.
Young P, Anderton E, Paschos K, White R, Allday MJ. Epstein-Barr virus nuclear antigen (EBNA) 3A induces the expression of and interacts with a subset of chaperones and co-chaperones. J Gen Virol. 2008;89:866–77. https://doi.org/10.1099/vir.0.83414-0.
Fazakerley JK. Neurovirology and developmental neurobiology. Adv Virus Res. 2001;56:73–124.
Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95–119.
Kuzyk MA, et al. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol Cell Probes. 2009;8:1860. https://doi.org/10.1074/mcp.M800540-MCP200.
Bromberg Z, Weiss Y. The role of the membrane-initiated heat shock response in cancer. Front Mol Biosci. 2016;3:12. https://doi.org/10.3389/fmolb.2016.00012.
Martine P, et al. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019;10:256. https://doi.org/10.1038/s41419-019-1491-7.
Zhang R, et al. Ts-Hsp70 induces protective immunity against Trichinella spiralis infection in mouse by activating dendritic cells through TLR2 and TLR4. PLoS Negl Trop Dis. 2018;12:e0006502. https://doi.org/10.1371/journal.pntd.0006502.
Lehner T, et al. Functional domains of HSP70 stimulate generation of cytokines and chemokines, maturation of dendritic cells and adjuvanticity. Biochem Soc Trans. 2004;32:629–32. https://doi.org/10.1042/BST0320629.
Fang H, et al. Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J Biol Chem. 2011;286:30393–400. https://doi.org/10.1074/jbc.M111.266528.
Al-Huseini LM, et al. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem. 2014;289:16442–51. https://doi.org/10.1074/jbc.M113.532069.
Pathak SK, et al. Activated apoptotic cells induce dendritic cell maturation via engagement of Toll-like receptor 4 (TLR4), dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC-SIGN), and beta2 integrins. J Biol Chem. 2012;287:13731–42. https://doi.org/10.1074/jbc.M111.336545.
Chen Y, et al. IL-1β induction of MUC5AC gene expression is mediated by CREB and NF-κB and repressed by dexamethasone. Lung Cell Mol Phys. 2014;306:L797–807. https://doi.org/10.1152/ajplung.00347.2013.
Wong CP, Bray TM, Ho E. Induction of proinflammatory response in prostate cancer epithelial cells by activated macrophages. Cancer Lett. 2009;276:38–46. https://doi.org/10.1016/j.canlet.2008.10.025.
Jäättelä M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261–71.
Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–6. https://doi.org/10.1038/nsmb993.
Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemist. 2008;47:7001–11.
Dan X, et al. Hsp27 responds to and facilitates enterovirus A71 replication by enhancing viral internal ribosome entry site-mediated translation. J Virol. 2019;93:e02322–18. https://doi.org/10.1128/JVI.02322-18.
Sun M, et al. Down-regulating heat shock protein 27 is involved in porcine epidemic diarrhea virus escaping from host antiviral mechanism. Vet Microbiol. 2017;205:6–13. https://doi.org/10.1016/j.vetmic.2017.04.031.
Tong S-W, et al. HSPB1 is an intracellular antiviral factor against hepatitis B virus. J Cell Biochem. 2013;114:162–73. https://doi.org/10.1002/jcb.24313.
Preville X, et al. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res. 1999;247:61–78. https://doi.org/10.1006/excr.1998.4347.
Wan SW, et al. Endothelial cell surface expression of protein disulfide isomerase activates beta1 and beta3 integrins and facilitates dengue virus infection. J Cell Biochem. 2012;113:1681–91. https://doi.org/10.1002/jcb.24037.
Diwaker D, Mishra KP, Ganju L, Singh SB. Protein disulfide isomerase mediates dengue virus entry in association with lipid rafts. Viral Immunol. 2015;28:153–60. https://doi.org/10.1089/vim.2014.0095.
Calderon MN, Guerrero CA, Acosta O, Lopez S, Arias CF. Inhibiting rotavirus infection by membrane-impermeant thiol/disulfide exchange blockers and antibodies against protein disulfide isomerase. Intervirology. 2012;55:451–64. https://doi.org/10.1159/000335262.
Gallina A, et al. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J Biol Chem. 2002;277:50579–88. https://doi.org/10.1074/jbc.M204547200.
Wang M, et al. Oblongifolin M, an active compound isolated from a Chinese medical herb Garcinia oblongifolia, potently inhibits enterovirus 71 reproduction through downregulation of ERp57. Oncotarget. 2015;7:8797–808.
Dobmeyer TS, et al. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte loss in HIV infection. Free Radic Biol Med. 1997;22:775–85. https://doi.org/10.1016/s0891-5849(96)00403-0.
Knobil K, Choi AM, Weigand GW, Jacoby DB. Role of oxidants in influenza virus-induced gene expression. Am J Phys. 1998;274:L134–42. https://doi.org/10.1152/ajplung.1998.274.1.L134.
Acar A, et al. Investigation of oxidative stress and antioxidant defense in patients with hepatitis B virus infection and the effect of interferon-alpha plus lamivudine combination therapy on oxidative stress. Mikrobiyol Bul. 2009;43:411–23.
Korenaga M, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–8. https://doi.org/10.1074/jbc.M506412200.
Ano Y, et al. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett. 2010;469:39–43. https://doi.org/10.1016/j.neulet.2009.11.040.
Olagnier D, et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014;10:e1004566. https://doi.org/10.1371/journal.ppat.1004566.
Liao SL, Raung SL, Chen CJ. Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci Lett. 2002;324:133–6. https://doi.org/10.1016/s0304-3940(02)00236-7.
De Marco F, et al. Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One. 2012;7:e34366. https://doi.org/10.1371/journal.pone.0034366.
Tatu U, Hammond C, Helenius A. Folding and oligomerization of influenza hemagglutinin in the ER and the intermediate compartment. EMBO J. 1995;14:1340–8.
Zai J, et al. N-glycosylation of the premembrane protein of Japanese encephalitis virus is critical for folding of the envelope protein and assembly of virus-like particles. Acta Virol. 2013;57:27–33.
Dubuisson J, Rice CM. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J Virol. 1996;70:778–86.
Shabman RS, Gulcicek EE, Stone KL, Basler CF. The Ebola virus VP24 protein prevents hnRNP C1/C2 binding to karyopherin α1 and partially alters its nuclear import. J Infect Dis. 2011;204:S904–10. https://doi.org/10.1093/infdis/jir323.
Pettit Kneller EL, Connor JH, Lyles DS. hnRNPs Relocalize to the cytoplasm following infection with vesicular stomatitis virus. J Virol. 2009;83:770–80. https://doi.org/10.1128/JVI.01279-08.
Shi ST, Lai MM. Viral and cellular proteins involved in coronavirus replication. Curr Top Microbiol Immunol. 2005;287:95–131. https://doi.org/10.1007/3-540-26765-4_4.
Clerte C, Hall KB. Characterization of multimeric complexes formed by the human PTB1 protein on RNA. RNA. 2006;12:457–75. https://doi.org/10.1261/rna.2178406.
Chung RT, Kaplan LM. Heterogeneous nuclear ribonucleoprotein I (hnRNP-I/PTB) selectively binds the conserved 3′ terminus of Hepatitis C viral RNA. Biochem Biophys Res Commun. 1999;254:351–62. https://doi.org/10.1006/bbrc.1998.9949.
Ertel KJ, Brunner JE, Semler BL. Mechanistic consequences of hnRNP C binding to both RNA termini of poliovirus negative-strand RNA intermediates. J Virol. 2010;84:4229–42. https://doi.org/10.1128/JVI.02198-09.
Brunner JE, Ertel KJ, Rozovics JM, Semler BL. Delayed kinetics of poliovirus RNA synthesis in a human cell line with reduced levels of hnRNP C proteins. J Virol. 2010;400:240–7. https://doi.org/10.1016/J.VIROL.2010.01.031.
Perwitasari O, et al. MicroRNA-555 has potent antiviral properties against poliovirus. J Gen Virol. 2016;97:659–68. https://doi.org/10.1099/jgv.0.000372.
Tsai P-L, et al. Cellular RNA binding proteins NS1-BP and hnRNP K regulate influenza A virus RNA splicing. PLoS Pathog. 2013;9:e1003460. https://doi.org/10.1371/journal.ppat.1003460.
Wolff T, O’Neill RE, Palese P. NS1-binding protein (NS1-BP): a novel human protein that interacts with the influenza A virus nonstructural NS1 protein is relocalized in the nuclei of infected cells. J Virol. 1998;72:7170–80.
Thompson MG, et al. Co-regulatory activity of hnRNP K and NS1-BP in influenza and human mRNA splicing. Nat Commun. 2018;9:2407. https://doi.org/10.1038/s41467-018-04779-4.
Paek KY, Kim CS, Park SM, Kim JH, Jang SK. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol. 2008;82:12082–93. https://doi.org/10.1128/JVI.01405-08.
Bokinsky G, et al. Two distinct binding modes of a protein cofactor with its target RNA. J Mol Biol. 2006;361:771–84. https://doi.org/10.1016/j.jmb.2006.06.048.
Williams MC, et al. Mechanism for nucleic acid chaperone activity of HIV-1 nucleocapsid protein revealed by single molecule stretching. PNAS. 2001;98:6121–6. https://doi.org/10.1073/pnas.101033198.
Lin J-Y, et al. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5' untranslated region and participates in virus replication. J Gen Virol. 2008;89:2540–9. https://doi.org/10.1099/vir.0.2008/003673-0.
Anderson EC, Hunt SL, Jackson RJ. Internal initiation of translation from the human rhinovirus-2 internal ribosome entry site requires the binding of Unr to two distinct sites on the 5' untranslated region. J Gen Virol. 2007;88:3043–52. https://doi.org/10.1099/vir.0.82463-0.
Kim CS, Seol SK, Song O-K, Park JH, Jang SK. An RNA-binding protein, hnRNP A1, and a scaffold protein, septin 6, facilitate hepatitis C virus replication. J Virol. 2007;81:3852–65. https://doi.org/10.1128/JVI.01311-06.
Lin J-Y, et al. hnRNP A1 interacts with the 5' untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J Virol. 2009;83:6106–14. https://doi.org/10.1128/JVI.02476-08.
Leong SY, Ong BKT, Chu JJH. The role of Misshapen NCK-related kinase (MINK), a novel Ste20 family kinase, in the IRES-mediated protein translation of human enterovirus 71. PLoS Pathog. 2015;11:e1004686. https://doi.org/10.1371/journal.ppat.1004686.
Jagdeo JM, et al. N-terminomics TAILS identifies host cell substrates of poliovirus and Coxsackievirus B3 3C proteinases that modulate virus infection. J Virol. 2018;92:e02211–7. https://doi.org/10.1128/JVI.02211-17.
Chang C-J, et al. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) interacts with dengue virus core protein. DNA Cell Biol. 2001;20:569–77. https://doi.org/10.1089/104454901317094981.
Miau L-H, Chang C-J, Shen B-J, Tsai W-H, Lee S-C. Identification of heterogeneous nuclear ribonucleoprotein K (hnRNP K) as a repressor of C/EBP-mediated gene activation. J Biol Chem. 1998;273:17789–97.
Dechtawewat T, et al. Role of human heterogeneous nuclear ribonucleoprotein C1/C2 in dengue virus replication. J Virol. 2015;12:14. https://doi.org/10.1186/s12985-014-0219-7.
Kanlaya R, Pattanakitsakul S-N, Sinchaikul S, Chen S-T, Thongboonkerd V. Vimentin interacts with heterogeneous nuclear ribonucleoproteins and dengue nonstructural protein 1 and is important for viral replication and release. Mol BioSyst. 2010;6:795. https://doi.org/10.1039/b923864f.
Noisakran S, et al. Identification of human hnRNP C1/C2 as a dengue virus NS1-interacting protein. Biochem Biophys Res Commun. 2008;372:67–72. https://doi.org/10.1016/J.BBRC.2008.04.165.
Dinh PX, Das A, Franco R, Pattnaik AK. Heterogeneous nuclear ribonucleoprotein K supports vesicular stomatitis virus replication by regulating cell survival and cellular gene expression. J Virol. 2013;87:10059–69. https://doi.org/10.1128/JVI.01257-13.
Wang Y, Zhou J, Du Y. hnRNP A2/B1 interacts with influenza A viral protein NS1 and inhibits virus replication potentially through suppressing NS1 RNA/protein levels and NS1 mRNA nuclear export. J Virol. 2014;449:53–61. https://doi.org/10.1016/j.virol.2013.11.009.
Wang X, et al. Viral DNA replication orientation and hnRNPs regulate transcription of the human papillomavirus 18 late promoter. mBio. 2017;8:e00713–7. https://doi.org/10.1128/mBio.00713-17.
Ng LFP, et al. Host heterogeneous ribonucleoprotein K (hnRNP K) as a potential target to suppress Hepatitis B virus replication. PLoS Med. 2005;2:e163. https://doi.org/10.1371/journal.pmed.0020163.
Zhang W, et al. Cytidine deaminase APOBEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell Microbiol. 2007;10:112–21. https://doi.org/10.1111/j.1462-5822.2007.01020.x.
Somberg M, Zhao X, Fröhlich M, Evander M, Schwartz S. Polypyrimidine tract binding protein induces human papillomavirus type 16 late gene expression by interfering with splicing inhibitory elements at the major late 5' splice site, SD3632. J Virol. 2008;82:3665–78. https://doi.org/10.1128/JVI.02140-07.
Oberg D, Fay J, Lambkin H, Schwartz S. A downstream polyadenylation element in human papillomavirus type 16 L2 encodes multiple GGG motifs and interacts with hnRNP H. J Virol. 2005;79:9254–69. https://doi.org/10.1128/JVI.79.14.9254-9269.2005.
Zheng Z-Z, et al. Specific interaction between hnRNP H and HPV16 L1 proteins: Implications for late gene auto-regulation enabling rapid viral capsid protein production. Biochem Biophys Res Commun. 2013;430:1047–53. https://doi.org/10.1016/J.BBRC.2012.12.042.
Catherine Silver S, Yoshizaki T, Pagano JS. The Epstein-Barr virus (EBV) SM protein enhances pre-mRNA processing of the EBV DNA polymerase transcript. J Virol. 1998;72:8485–92.
Collier B, Goobar-Larsson L, Sokolowski M, Schwartz S. Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogenous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J Biol Chem. 1998;273:22648–56. https://doi.org/10.1074/jbc.273.35.22648.
Fuentes-Pananá EM, Peng R, Brewer G, Tan J, Ling PD. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J Virol. 2000;74:8166–75. https://doi.org/10.1128/jvi.74.17.8166-8175.2000.
Monette A, Ajamian L, López-Lastra M, Mouland AJ. Human immunodeficiency virus type 1 (HIV-1) induces the cytoplasmic retention of heterogeneous nuclear ribonucleoprotein A1 by disrupting nuclear import: implications for HIV-1 gene expression. J Biol Chem. 2009;284:31350–62. https://doi.org/10.1074/jbc.M109.048736.
Asai K, Platt C, Cochrane A. Control of HIV-1 env RNA splicing and transport: investigating the role of hnRNP A1 in exon splicing silencer (ESS3a) function. Virology. 2003;314:229–42. https://doi.org/10.1016/S0042-6822(03)00400-8.
Tange TO, Damgaard CK, Guth S, Valcárcel J, Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001;20:5748–58. https://doi.org/10.1093/emboj/20.20.5748.
Damgaard CK, Tange T s, Kjems J. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA. 2002;8:1401–15.
Gordon H, et al. Depletion of hnRNP A2/B1 overrides the nuclear retention of the HIV-1 genomic RNA. RNA Biol. 2013;10:1714–25. https://doi.org/10.4161/rna.26542.
Suh D, et al. Mapping of determinants required for the function of the HIV-1 env nuclear retention sequence. J Virol. 2003;310:85–99. https://doi.org/10.1016/S0042-6822(03)00073-4.
Acknowledgment
This chapter was modified from the paper published by our group in Signal Transduction and Target Therapy (Wan et al., 5(1): 125, 2020). Related contents are reused with permission.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
He, ML., Wan, Q., Song, D., He, B. (2021). Stress Proteins: Biological Functions, Human Diseases, and Virus Infections. In: Huang, C., Zhang, Y. (eds) Oxidative Stress. Springer, Singapore. https://doi.org/10.1007/978-981-16-0522-2_4
Download citation
DOI: https://doi.org/10.1007/978-981-16-0522-2_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0521-5
Online ISBN: 978-981-16-0522-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)