
Abstract
A simplistic view of food is that food molecules are assembled into hierarchical structures. As the two main components in foods, the self-assembling properties of food proteins and polysaccharides determine the nutritional value, texture, appearance, taste, odour, and shelf-life of foods. In this chapter efforts are first made to provide an overview of the concepts, mechanisms, and forces of self-assembly in a broad context, followed by the specific discussion of the self-assembly of food proteins and polysaccharides. The advancements of the self-assembled nanostructures with various morphologies and functionalities are summarized and discussed to provide a guideline for designing desired food structures and broadening the applications of food proteins and polysaccharides. These self-assemblies may also benefit the health of the consumer, when considering their journey in the body, i.e. the disassembly and reassembly processes. We hope that a better understanding of the self-assembly rules of food proteins and polysaccharides will spark food scientists to develop novel functional foods to meet future consumer demands.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Self-assembly is a ubiquitous process throughout nature and technology (Whitesides and Boncheva 2002; Whitesides and Grzybowski 2002; Mendes et al. 2013). Nature uses specific self-assembly of molecules to organize elaborate structures that possess unique biological functions (Luo et al. 2016). From ordered protein aggregates (e.g., viral capsids, collagen and actin filaments, flagella), topologically programmed nucleic acids (e.g., DNA duplexes, RNA triplexes), to complicated nucleosomes and ribosomes, these self-assembled structures could perform a number of functions, such as genome packaging, structural support, force generation, and information storage and transmission (Goodsell and Olson 2000; Saenger 2008; Luo et al. 2016). An in-depth understanding of molecular self-assembly is important not only to reveal the mechanisms of these beneficial biological processes, but also to provide valuable treatments for human diseases, such as the neurodegenerative diseases that are caused by abnormal protein self-assembly (Dobson 2003; Chiti and Dobson 2006). Self-assembly is also in the forefront of biotechnology and nanotechnology, as it provides an excellent tool to build a broad of complex architectures that cannot be easily achieved by other methods (Lee 2007).
Self-assembly is not a new concept in the food sector, and is omnipresent in both natural and processed foods. Typical examples include the formation of casein micelles in milk, oil-bodies in seeds and starch spherulites in plants, and the gelation of pectins in jelly, micelles in yogurt and soy proteins in tofu (Dickinson and Leser 2007; Ravichandran 2010; Sagalowicz et al. 2017). Two food components, protein and polysaccharide, are the main self-assembly units in the foodstuffs. The assembly of food proteins and polysaccharides has attracted much attention over the past two decades, mainly due to the excellent tech-functionalities of the resultant nanostructures, such as the assembled nanofibrils that form transparent hydrogels at low concentration and room temperature, aggregates that stabilize emulsions and foams, and nanoparticles that deliver drugs and nutrients (Veerman et al. 2003; Kroes-Nijboer et al. 2012; Yao et al. 2015; Hu et al. 2019). In addition, self-assembly is correlated to food texture, taste, and appearance. For instance, the beverage appearance is greatly marred by the self-assembly-induced precipitation. More importantly, as mentioned above, the hierarchical structure of biopolymers is directly linked to their unique biological functions. As such, the elaborate structures generated from the self-assembly of proteins and polysaccharides may bring specific nutritional values or functions to the consumer. All these examples point out that food scientists should have a comprehensive understanding of the self-assembly of proteins and polysaccharides. In this chapter, efforts are made to provide the concepts, mechanisms, and molecular forces for the self-assembly of food proteins and polysaccharides, to summarize and discuss the assembled nanostructures in the food sector, as well as to explain how the self-assembly affects food quality and functions.
2 Physical Aspects of Self-assembly
2.1 Self-assembly
Self-assembly is a special kind of aggregation whereby this process occurs toward the state of minimum free energy, mainly through non-covalent interactions, such as electrostatic interactions, van der Waals interactions, hydrogen bonding, hydrophobic interactions, metal coordination bonds, and steric and depletion forces (Lindoy and Atkinson 2000; Lee 2007; Ninham and Nostro 2010; Jiang et al. 2011; Padua and Nonthanum 2012; Billon and Borisov 2016; Sundararajan 2016; Wang et al. 2016; Sorrenti et al. 2017). Despite the entropy loss as the ordering of self-assembly building units, the self-assembly is energetically favourable because the entropic cost is greatly offset by the enthalpy gained from the non-covalent interactions (Rajagopal and Schneider 2004; Padua and Nonthanum 2012).
From the thermodynamic point of view, self-assembly is a process that minimizes Gibbs free energy. In general, it brings the entity closer to a thermodynamic equilibrium state (#1 in Fig. 9.1) (Whitesides and Boncheva 2002; Roy et al. 2016; Sorrenti et al. 2017). Many involved intermolecular interactions, as mentioned above, enable the system to explore different configurations (i.e. walk along the energy landscape), and to find the most stable one (Sorrenti et al. 2017). In some cases, the self-assembly process may cause metastable or kinetically trapped states (i.e. non-equilibrium states, #2 and #3 in Fig. 9.1) (Sorrenti et al. 2017). Due to the low energy barrier, the metastable structures will eventually evolve into the thermodynamic equilibrium state, even without any intervention. In contrast, the system will be kinetically trapped in state #3 because of the high energy barrier, if there is no intervention (Wang et al. 2016; Sorrenti et al. 2017). This means that the outcome of the self-assembly process, e.g. the morphology of assembled nanostructures, strongly depends on the experimental parameters and protocols (Sorrenti et al. 2017). In other words, the desired pathway can be rationally selected by appropriate preparation methodologies, resulting in the assemblies with targeted features starting from the same building blocks. This is crucial to develop materials with optimized functionalities (Sorrenti et al. 2017).

Schematic illustration of Gibbs free energy landscape of the different thermodynamic states in self-assembly process. Adapted with permission from Sorrenti et al. (2017). Copyright 2017 RSC
From the force point of view, self-assembly can be defined as a delicate balance of the attractive (driving force) and repulsive (opposition force) intermolecular forces (Whitesides and Boncheva 2002; Lee 2007; Kedracki 2015; Billon and Borisov 2016; Roy et al. 2016). The driving forces bring the self-assembly units together, while opposition forces are in balance with the driving forces (Lee 2007; Kedracki 2015; Billon and Borisov 2016). Besides, many biological and bio-mimetic systems show a unique directionality during self-assembly processes, as well as in many food systems, e.g. the formation of protein nanofibrils and nanotubes, and polysaccharides helices (Graveland-Bikker and de Kruif 2006; Lee 2007; Cao and Mezzenga 2019; Fittolani et al. 2019). The force responsible for these directional self-assembly processes is known as directional force or functional force (Lee 2007; Kedracki 2015; Billon and Borisov 2016). Hydrogen bond, coordination bond, electrostatic interaction, and π-π stacking are the most common directional forces (Lee 2007; Wang et al. 2016). These forces can be a part of a driving or opposition force, but sometimes act exclusively as directional force (Lee 2007).
Therefore, self-assembly is an equilibrium of three classes of forces: driving, opposition, and directional forces, as displayed in Table 9.1 (Lee 2007). The self-assembly process is quite random when only the first two classes of forces are in action. Most of the colloidal self-assembly processes belong to this category. When the third class of forces is involved with the first two classes of forces, the self-assembly processes are directional and often functional, leading to the formation of highly ordered or specific functional structures. In engineering, these three classes of forces can be greatly affected by a number of external parameters, such as pH, ionic strength and type, temperature, solvent type, mechanical treatments (pressure, shear, extension, sonication), or chemical treatments (Lee 2007). Therefore, self-assembly can be triggered, altered, or terminated by controlling these external parameters, thereby managing the desired assembly and the assembled nanostructures.
2.2 Forces in Self-assembly
In the self-assembly process, whether it occurs at an atomic-, molecular-, colloidal-, or macro-length scale, the non-covalent forces rather than the chemical forces play vital roles (Lee 2007). Even though these non-covalent forces are weak individually, a large number of such forces in the final can be significant. We first briefly illustrate these forces and then give an example of a combination of two forces—DLVO (Derjaguin–Landau–Verwey–Overbeek) theory.
2.2.1 Electrostatic Interaction
Electrostatic interaction appears universally for charged objects. In nonpolar media (e.g. vacuum, air, organic nonpolar liquids), the electrostatic interaction is governed by the Coulomb’s law (Lee 2007; Sundararajan 2016). The interaction potential U(x) between two charges of Q1 and Q2 is expressed as: \( U(x)=\frac{Q_1{Q}_2}{4\pi {\varepsilon}_0\varepsilon\ x}=\frac{z_1{z}_2{e}^2}{4\pi {\varepsilon}_0\varepsilon\ x} \), where z1 and z2 are the ionic valence of each charge, e is the elementary charge, ε0 is the dielectric permittivity of vacuum, ε is the relative dielectric permittivity, and x is the distance between two charges (Lee 2007). Due to U(x)~x−1, the electrostatic interactions in ion-free media can extend over long distance. In polar media (e.g. water, polar organic liquids), free ions in solutions are able to move to oppositely charged interfaces, resulting in the formation of a kind of molecular condenser, known as electrical double layer (Tadros 2013). The thickness of the double layer (i.e. screening length or Debye length) decreases with the increase in free ion concentration, written as \( {\kappa}^{-1}=\sqrt{\frac{\varepsilon_0\varepsilon {k}_{\mathrm{B}}T}{2\times {10}^3{N}_{\mathrm{A}}{e}^2I}} \), here I is the ionic strength (mol/L), kB is the Boltzmann constant, T is the absolute temperature, NA is the Avogadro number. The range of the electrostatic forces is then typically: κ−1 = 10 nm (at I = 1 mM), 3 nm (at 10 mM), 0.8 nm (at 150 mM), and 0.3 nm (at 1000 mM) at room temperature (Ninham and Nostro 2010). Different from the long-range electrostatic forces in nonpolar media, the electrostatic forces between objects in polar media become short-ranged, or even can be eliminated by increasing salt content due to the screening effect (Ninham and Nostro 2010; Tadros 2013). Therefore, the strength of electrostatic interactions is largely dependent on the solution ionic strength and pH. The flexibility of polymer chain and the charge distribution in polymer chain are also significant factors (Cao et al. 2016a).
2.2.2 Van der Waals Interaction
Van der Waals force is generated by dipole or induced-dipole interaction at the atomic and molecular levels, including three contributions: Keesom interaction (dipole–dipole), Debye interaction (dipole–induced dipole), and London interaction (instantaneous induced dipole–induced dipole) (Lindoy and Atkinson 2000; Parsegian 2005; Lee 2007; Ninham and Nostro 2010). All these interactions have a scaling of U(x)~x−6, thus the van der Waals force quickly vanishes at long distances between interacting atoms (Lee 2007; Ninham and Nostro 2010). The van der Waals force between two atoms is weak, but its total collective contribution to molecular interactions can be substantial (Lindoy and Atkinson 2000; Li and Alessandra 2018). For instance, for two identical interacting colloids with a radius of R, the van der Waals interaction potential is \( U(x)=-\frac{AR}{12x} \), here A is Hamaker constant (A ≈ 3kBT for proteins) (Hamaker 1937; Parsegian 2005; Lee 2007; Israelachvili 2011). Thus, the interaction between two protein monomers could be evident (U(x) = kBT) by considering R = 4 nm and x = 1 nm.
2.2.3 Hydrogen Bonding
Hydrogen bond is an attractive force between a hydrogen atom which is covalently bound to an electronegative atom (X-H, donor) and another electronegative atom bearing a lone pair of electrons (Y, acceptor), depicted as X-H···Y (Lindoy and Atkinson 2000; Lee 2007; Ninham and Nostro 2010; Sundararajan 2016). The common hydrogen bond donors include C-H, O-H, N-H, P-H, F-H, Cl-H, Br-H, I-H, while N, O, P, S, F, Cl, Br, I, alkenes, alkynes, aromatic π-clouds are the common acceptors (Lindoy and Atkinson 2000). Hydrogen bond is considered to be a quite strong and directional interaction (Lee 2007). It is generally stronger than the van der Waals interaction, but weaker than covalent and ionic bonds (Lee 2007). The directionality of hydrogen bond results from the hydrogen bond–capable molecules always interacting only through specific sites (Lee 2007). It is a key player in the assembly of protein and polysaccharide systems as almost all constituent units in protein and polysaccharide are capable of forming hydrogen bonding.
2.2.4 Hydrophobic Interaction
Hydrophobic interaction describes the energetic preference of nonpolar objects to interact with other nonpolar objects in the presence of aqueous solution (Motiejunas and Wade 2006). This short-range attractive interaction is mainly an entropic effect, but also affected by enthalpy contribution (Motiejunas and Wade 2006; Lee 2007). When a nonpolar molecule is present in the aqueous solution, a highly ordered hydrogen bond network around the nonpolar molecule is formed to minimize the disruption of this nonpolar object, i.e. the formation of a structured water “cage” (Schaeffer 2008). In the process of nonpolar molecular association, the nonpolar molecule system obviously loses its entropy, but the water system gains a significant increase of entropy that overcomes the entropy loss of nonpolar molecules (i.e. total ΔS is positive) (Lee 2007; Schaeffer 2008). Moreover, the enthalpy is increased as some of hydrogen bonds that form the “cage” are broken in the association process, but this effect is limited compared to the entropic effect (Lee 2007; Schaeffer 2008). Therefore, the Gibbs free energy change is negative (ΔG = ΔH − TΔS, ΔS = large positive value, ΔH = small positive value), implying that the hydrophobic effect is spontaneous. Similar to van der Waals forces, hydrophobic interactions are individually weak, but the total contribution to molecular interactions can be significant (Schaeffer 2008). The strength of hydrophobic interaction is associated with the solubility of the nonpolar molecules as well as the quality of the steric match between the molecules (Schaeffer 2008). Most proteins (possessing nonpolar amino acids) and some polysaccharide derivatives (such as MC and HPMC) show a significant hydrophobic character.
2.2.5 Steric Repulsion
Steric repulsion is a common force between colloidal particles when water-soluble polymers are tightly adsorbed or grafted onto the surface of colloidal objects (Fig. 9.2a) (Lee 2007). It mainly arises from the loss of configuration entropy when two polymer layers are overlapped (Cooper 2014). Steric repulsion is a long-range force and can reach up to ~10Rg (Rg is the gyration radius of the polymer chain) (Lee 2007). For effective steric repulsion, water-soluble polymers in the diffuse layer must satisfy the following three requirements: firstly, the polymers should be tightly anchored to the colloidal particles; secondly, part of the polymer chain should extend into the bulk solution; thirdly, there is no significant exposure of the colloidal surface (Cooper 2014).

Schematic illustration of steric repulsion and depletion attraction between two colloidal spheres. (a) Steric repulsion between polymer-adsorbed colloids. (b) Depletion attraction between two colloidal particles in the presence of non-adsorbing polymers or molecules
2.2.6 Depletion Attraction
Depletion force is the common force for the colloidal particles considering the presence of non-adsorbing polymers (Fig. 9.2b) (Lee 2007; Lekkerkerker and Tuinier 2011). A depletion region of polymers is generated when the colloidal particles are close enough to each other (smaller than the size of polymers, ∼2Rg), as the polymers are being squeezed out of this region (Lee 2007). The osmotic pressure force that is exerted by the polymers in the outside region (outside of depletion region) exceeds that in the inside region. This, therefore, induces a net attractive force between the colloidal particles (Lee 2007; Lekkerkerker and Tuinier 2011). The depletion force can determine the stability of colloids when there are no other significant attractive forces (Lee 2007). It can be strengthened by increasing polymer concentration and molecular weight (Lee 2007). Colloids with low curvature (e.g. nanorods) experience this attraction more strongly as the depletion attraction scales with the excluded volume of the colloids.
2.2.7 DLVO Theory: A Case of the Combination of van der Waals Attraction and Electrostatic Repulsion
The DLVO theory is a useful tool to describe the self-assembly process of charged colloids, which is the combination of two inter-colloidal forces: van der Waals force that acts as the attractive force and electric double-layer interaction as the repulsive force. Their total interaction potential U(x) can be written as (Adair et al. 2001; Mezzenga and Fischer 2013):
σ and R are the net surface charge density and radius of colloidal particles, respectively, x is the separated distance of a pair of colloids. The plots of this equation at different ionic strengths are shown in Fig. 9.3, by considering R = 5 nm, σ = 20 mC/m2, T = 298 K. The electrostatic force gives a positive term and varies with the solution ionic strength. In contrast, the van der Waals attraction gives a negative term and is independent from ionic strength (Fig. 9.3a) (Adair et al. 2001). The sum of these two forces at different ionic strengths is given in Fig. 9.3b. At low ionic strengths, particles have net repulsion at large and intermediate separations, and the approaching of colloids requires high kinetic energy due to the high energy barrier. Thus, in these cases, colloids are separately suspended in the solution. At high ionic strengths, the energy barrier is lowered and particles can overcome it more easily, leading to the aggregation or self-assembly (Adair et al. 2001).

An example of DLVO interaction potential. (a) Electrostatic repulsion potential versus particle distance x at different ionic strengths, and van de Waals attraction potential versus particle distance x. (b) The sum interaction potential of electrostatic and van der Waals forces. R = 5 nm, σ = 20 mC/m2, T = 298 K are used for the calculation
2.3 Self-assembly of Food Proteins and Polysaccharides
2.3.1 Protein Self-assembly
Understanding the mechanisms and processes of protein self-assembly is essential to biologists and medical scientists, since it is related to many biological and physiological activities, as mentioned above. The understanding of food protein self-assembly is equally important to food scientists due to a broad range of food-related implications and applications. For instance, protein self-assembly can be harnessed for protein purification through phase separation or crystallization, or can be problematic during storage (Carpenter and Manning 2002; Flickinger 2013; McManus et al. 2016).
Most natural food proteins possess globular or fibrillar conformations (Mezzenga and Fischer 2013; Nicolai 2019). These protein structures result from a combination of the numerous interactions between amino acids, i.e. the self-assembly of polypeptide chain. Depending on the side group, the amino acids in food proteins can be divided into: nonpolar, polar, and ionic, which mainly contribute to hydrophobic interactions, hydrogen bonding, and electrostatic interactions, respectively. In globular conformations, the polypeptide chain is folded into compact spherical shape with most of the nonpolar amino acids buried in the interior, and the polar and ionic amino acids predominately located at the surface (Fig. 9.4a) (Mezzenga and Fischer 2013; Jones 2015; McManus et al. 2016; Cho and Jones 2019). The driving forces for this configuration include the hydrophobic effect as well as other forces, such as hydrogen bonding that contributes to the formation of protein secondary structure (α-helices and β-sheets) (Jones 2015). Indeed, hydrogen bonding is prevalent in proteins by considering all amino acids containing amine- and carbonyl-groups. Fibrous proteins often have specific amino acid sequences that favour twisting of the polypeptide. For example, gelatins are rich in Glycine-X-Y sequence that supports the twisting of gelatin chains with the formation of triple helixes; X and Y are mostly proline and hydroxyl-proline (Russell et al. 2007; Hafidz et al. 2011; Cao et al. 2015; Jones 2015).

Schematic illustration of food protein self-assembly and the resultant nanostructures. (a) The self-assembly of polypeptide chain into globular proteins. (b, c) The self-assembly of natural proteins into crystals (b) or amorphous aggregates (c). These processes are often reversible since the protein structure is remained. (d) Protein denaturation leads to the (partial) unfolding and/or hydrolysis of proteins. (e) The unfolded and hydrolyzed proteins can further assemble into amyloid fibrils or amorphous aggregates. These processes are often irreversible
Since globular proteins are generally viewed as colloids, a number of models for interpreting colloid self-assembly can be used to understand the self-assembly behaviour of globular proteins. The simplest model is the DLVO model, as discussed above, which could interpret many protein behaviours, such as the aggregation of proteins by adjusting pH or adding salts. However, DLVO cannot explain certain protein behaviour, such as protein crystallization at high salt contents (Piazza 1999; Mezzenga and Fischer 2013). Other studies indicate that the protein aggregation may originate from the presence of depletion forces (Mezzenga and Fischer 2013; McManus et al. 2016). It is worth noting that many models are employed to understand protein self-assembly with varying degree of success, yet no model captures all protein aggregation features (Mezzenga and Fischer 2013). This is possibly caused by the existence of many other forces (e.g. hydrophobic effect, hydrogen bonds, specifically ionic bindings) and effects (e.g., surface charge distributions, molecular recognition) that contribute to the complexity of protein self-assembly (Mezzenga and Fischer 2013). The self-assembly of globular proteins, in native state, can lead to the formation of crystals or amorphous aggregates (Fig. 9.4b and c), and this process is often reversible.
Protein self-assembly could also start from the denatured, unfolded, or hydrolyzed proteins, which is actually more frequently observed in the food systems (Fig. 9.4d). Loss of native structures leads to changes in the capacity of those proteins to interact with each other, and further determine their ability to form supramolecular assemblies (Jones 2015). For instance, the exposure of nonpolar amino acids by unfolding the globular proteins supports the formation of intermolecular forces (Jones 2015; Li et al. 2018). Knowledge of protein characteristics in the chosen environment is essential to the desired assembly, and there are mainly two routes for the protein self-assembly at the denaturation condition: fibrillization and random aggregation, leading to the formation of amyloid fibrils and amorphous aggregates (Fig. 9.4e and f). The most studied condition for triggering this type of self-assembly is heating proteins at various pHs and ionic strengths (van der Linden and Venema 2007; Nicolai and Durand 2013; Jones 2015; Schmitt et al. 2016; Cao and Mezzenga 2019). At pH in the neighbourhood of protein isoelectric point (IEP) or at high ionic strengths, the effective charge of the protein is remarkably suppressed so that amorphous aggregates (i.e. large fractal dimension) generate during thermal treatment, arising from the loss of opposition electrostatic repulsion (right side of Fig. 9.5). As the solution pH leaves from the protein IEP, the effective charge of the protein increases and aggregates become relatively less amorphous (middle of Fig. 9.5). Instead, protein aggregation produces fibrous structures when the pH is significantly far from IEP, since the highly effective charge on the protein surfaces makes them favourable for interactions only among discrete regions on the protein surface (left side of Fig. 9.5). A notable example can be found in β-lactoglobulin protein system. The morphology of protein aggregates remarkably depends on the solution pH: amyloid fibrils and fibrous strands formed at pH 2 and pH 7, respectively (far from β-lactoglobulin IEP); particulates formed at pH 5.2 (≈ IEP); microgels formed at pH 4.7 and 5.9 (near IEP) (Schmitt et al. 2016).

Schematic representation of the effect of pH and ionic strength on protein self-assembly during heat treatment. Adapted with permission from van der Linden and Venema (2007). Copyright 2007 Elsevier
2.3.2 Polysaccharide Self-assembly
Based on the charge nature, polysaccharides can be classified into cationic (chitosan), anionic (alginate, pectin, carrageenan, gellan gum, hyaluronic acid), and neutral (agarose, pullulan, dextran) (Kontogiorgos 2015; Stephen et al. 2016). Despite that most food polysaccharides only consist of 1–3 constituent units, the type of linkages, isomeric forms, esterification, the branching and periodicity of constituent units, and the wide range of molecular weight contribute to their great diversity in structure and property (Kontogiorgos 2015; Stephen et al. 2016). A notable example is that of cellulose and amylose. They have the same repeating unit (glucose), but the different linkage (β-D-(1→4) in cellulose and α-D-(1→4) in amylose), which leads to their extremely different digestion and assembly behaviours. Repulsive interactions in polysaccharide self-assembly often are the steric repulsion and sometimes electrostatic repulsion for charged ones. Attractive forces are the van der Waals interaction, hydrogen bonding, and sometimes the hydrophobic interaction and ionic binding for certain polysaccharides. Hydrogen bonding and ionic binding are the important directional forces in polysaccharide self-assembly.
Similar to food proteins, temperature, pH, and salt are the most common triggers for the self-assembly of food polysaccharides. Either increasing or lowering temperature can induce the self-assembly (Nishinari and Zhang 2004; Stephen et al. 2016). Agaroses, carrageenans, or gellan gums experience the transitions of coil-to-helix and helix-to-super helix in the cooling process, driven by the formation of hydrogen bonding and ionic binding. The helical structures in polysaccharides are still controversial, could be single, double, or triple helixes (Fig. 9.6b) (Schefer et al. 2014; Cao et al. 2016b; Stephen et al. 2016; Fittolani et al. 2019). In contrast, methylcellulose and hydroxypropyl methylcellulose are assembled in the heating process, mainly driven by the hydrophobic force. During heating, the solvated cage-like structures (formed through hydrogen bonds between water and cellulose derivatives) are destroyed and thereby hydrophobic regions are exposed, resulting in the formation of hydrophobic junction zones (Fig. 9.6c) (Li et al. 2001, 2002; Shen et al. 2016).

Typical polysaccharide structures at molecular levels. (a) Left: “egg-box” model of alginate or pectin in the presence of divalent ions, e.g. Ca2+; Right: coordination of Ca2+ in a cavity created by a pair of guluronate sequences. The open circles represent Ca2+ ions and the black dots represent the oxygen atoms possibly involved in the coordination with Ca2+. Adapted with permission from Fang et al. (2007). Copyright 2007 ACS. (b) Single, double, or triple helical structures in polysaccharides, such as carrageenan (single helix), agarose (double helix), curdlan (triple helix). (c) Hydrophobic association in cellulose derivatives, (e.g. methylcellulose, hydroxypropyl methylcellulose). At relatively high temperatures, the solvated cage-like structures (formed through hydrogen bonds between water and cellulose derivative chains) are destroyed and thereby hydrophobic regions are exposed, causing the formation of hydrophobic junction zones. Adapted with permission from Li et al. (2001). Copyright 2001 ACS
For ionic polysaccharides, pH and salt not only affect the electrostatic force but also have other important effects. The effect of pH is correlated to the dissociation constant (pKa). The charge magnitude of ionic polysaccharides depends on the solution pH relative to the pKa. For instance, alginate (pKa ≈ 3.8) is slightly negative (or near neutral) at pH 2 and strongly negative at pH 7, by referring \( \mathrm{pH}-{\mathrm{p}K}_{\mathrm{a}}={\log}_{10}\Big(\left[{\mathrm{CO}}_2^{-}\right]/\left[\mathrm{C}{\mathrm{O}}_2\mathrm{H}\right] \)). The protonation of COO− at pH 2 not only weakens the electrostatic repulsion but also enhances the hydrogen bonding due to COOH with high hydrogen bond forming ability, resulting in the self-assembly (or gelation) of alginates at low pH (Draget 2009; Draget et al. 2016). The mechanism for the assembly of chitosan (pKa ≈ 6.3) through increasing pH is considered to be similar, i.e. transition of NH3+ to NH2 (Yi et al. 2005; Pillai et al. 2009; Zargar et al. 2015; Shen et al. 2016). Salt plays a vital role in the electrostatic force via altering the Debye screening length, as discussed above. More importantly, some specifically ionic bindings make polysaccharides with a complex self-assembly behaviour. For example, multivalent cations, e.g. Ca2+, specifically bind to alginate or pectin, causing the formation of an ordered “egg-box” structure (Fig. 9.6a) (Fang et al. 2007). It should be noted that, different from protein systems, the assembly induced by temperature, pH, or salt is often reversible for polysaccharide systems.
3 Self-assembled Nanostructures
Nanostructured materials are the forefront of many fields due to their unique and outstanding properties, and self-assembly is broadly considered as a promising approach to produce these nanostructures. In principle, the nanostructures generated from food proteins and polysaccharides could further enrich the versatility of nanostructured materials in terms of category and function due to their nutritional value, biodegradability, biocompatibility, safety, etc. Here we summarize the food protein and polysaccharide nanostructures with different morphologies, and their formation conditions and potential applications.
3.1 Protein Self-assembled Nanostructures
Under certain conditions, proteins can self-assemble into a variety of structures with different sizes and morphologies, including crystals, nanofilaments, nanotubes, and amorphous aggregates. In this part, we will first introduce a naturally assembled protein nanostructure and then discuss the nanostructures produced by the processing.
3.1.1 Natural Self-assembled Nanostructure—Casein Micelles
Milks contain large quantities of protein-based nanostructures, known as casein micelles. These colloidal particles, typically have an average diameter of ~200 nm, are naturally assembled from the phosphoproteins—caseins (αs1-casein, αs2-casein, β-casein, κ-casein) and calcium phosphate, driven by the forces of hydrogen bonding, ionic bridging, hydrophobic interaction, electrostatic interaction, and van der Waals attraction (Dalgleish 2011; de Kruif et al. 2012; Jones 2015). Within casein micelles, the balance of the hydrophobic and hydrophilic amino acids not only allows formation of this micelle nanostructure, but also helps in retaining the individual character of the micelles (i.e. adequately stable as a suspension) (Kontogiorgos 2015). Although the composition and forces in casein micelles are well understood, their structure, especially the interior structure, remains a matter of debate (Dalgleish 2011; Mezzenga and Fischer 2013). Various models have been proposed, but it is generally agreed that calcium phosphate is responsible for forming salt bridges between phosphoseryl residues on the β- and α-caseins, and κ-casein is predominantly distributed on the micelle surface and contributes to stabilizing micelles (Mezzenga and Fischer 2013; Cho and Jones 2019). Although it is not possible to duplicate the exact assembly of the casein micelles, casein proteins have been demonstrated to assemble micelles-like structures by reincorporating calcium phosphate and citrate ions at milk-relevant contents (Jones 2015). Besides, many methods are available to disassemble and reassemble natural casein micelles, which are useful to create novel nanostructures for controlled delivery purposes (Jones 2015; Cho and Jones 2019).
3.1.2 Amorphous Aggregates
Amorphous protein aggregates could be generated from the self-assembly of native proteins (i.e. in a mild condition without the protein denaturation process). This type of aggregates is often reversible due to the lack of significant changes in protein structures and the absence of strong forces. For instance, the clusters formed in high concentration lysozyme protein solution are reversible; the clusters are disassembled by lowering protein concentration (Lu et al. 2008). In contrast, most amorphous aggregates in the food sector are produced by protein unfolding and then assembly, which are generally irreversible because of the significant changes in protein structures and the significant aggregation interactions between proteins. The formation of stable suspensions of amorphous aggregates requires an intermediate surface charge and low protein concentration, otherwise leading to the formation of precipitates or bulk gels (Nicolai and Durand 2013; Schmitt et al. 2016). In most cases, it is not straightforward to form homogeneous nanoaggregates by simply heating globular protein solution. Particularly, Schmitt et al. (Schmitt et al. 2009) found that stable suspensions of roughly spherical protein nanoparticles (with a hydrodynamic radius of ~200 nm) can be formed by heating β-lactoglobulin without added salt in two narrow pH ranges (around pH 4.6 and around pH 5.8).
3.1.3 Nanofilaments
The formation of filamentous nanostructures needs more specified and stringent conditions than amorphous aggregates. Typically, two common filamentous structures could be produced from food proteins: strand-like objects formed when heating proteins at neutral pH and low salt content; amyloid fibrils formed when heating proteins at low pH and low salt content. These filamentous structures are the promising materials owing to their unique properties. For instance, the high aspect ratio allows them to form gels or significantly increase solution viscosity at very low protein concentration (Veerman et al. 2003; Kroes-Nijboer et al. 2012).
In the formation of strand-like objects, the intermolecular interactions at certain sites (e.g. disulphide bonds) bring protein monomers together and lead to directional growth, where electrostatic repulsion is in balance with these forces to prevent structure collapse (van der Linden and Venema 2007; Nicolai and Durand 2013; Nicolai et al. 2011). The strands are structurally less ordered than amyloid fibrils due to the fact that the protein chains are confined (low unfolding and hydrolysis extent) (Nicolai and Durand 2013). The resultant strands at neutral pH often have diameters less than 10 nm and lengths between tens and hundreds of nanometres (Nicolai and Durand 2013).
Amyloid fibrils are characteristic with a cross-β structure in which continuous hydrogen-bonded β-sheets run along the fibrils (McManus et al. 2016; Chiti and Dobson 2017; Eisenberg and Sawaya 2017). These nanofibrils formed from different proteins are similar: unbranched filamentous structures with a few nanometres in diameter and up to several micrometres in length (McManus et al. 2016; Chiti and Dobson 2017; Eisenberg and Sawaya 2017). Heating proteins at low pH and low ionic strength is often used to prepare food protein amyloid fibrils. In this procedure, the protein monomers are first hydrolyzed and unfolded, and then assembled into amyloid fibrils. Hydrophobic interactions, hydrogen bonding, and sometimes π-π stacking are the dominant attractive forces for holding the protein nanofibril structures. In contrast, electrostatic repulsion is the main opposition force to prevent the structure collapse. Indeed, the final fibril morphology is largely affected by the solution ionic strength, since it modulates the strength of electrostatic interactions. Long semiflexible fibrils are generated at low ionic strength, whereas short wormlike fibrils prevail at higher ionic strength (Loveday et al. 2010, 2017; Cao and Mezzenga 2019). This arises from the fact that the strong electrostatic repulsion at low ionic strength leads to the attachment of building blocks to the growing fibrils in a well-ordered arrangement, whereas at higher ionic strength the growth of fibrils is more haphazard and chaotic (Loveday et al. 2017; Cao and Mezzenga 2019). Other factors, such as pH, temperature, protein concentration, stirring speed, co-solvent, and some chemicals could also greatly affect the protein fibrillization process and the final nanofibril morphology (Cao and Mezzenga 2019).
3.1.4 Nanotubes
Nanotube is one of the most promising materials from the last century. Carbon nanotubes could be used to build probes and sensors, to store energy and hydrogen gas, and to serve as field emission displays and radiation sources, etc. (Baughman et al. 2002; de Volder et al. 2013). Peptide nanotubes are also of immense interest due to their diverse bio-functionalities which lead to numerous potential applications in nanotechnology as well as in biomedicine (Scanlon and Aggeli 2008; Hamley 2014). Indeed, it has been proved that the nanotube structure provides superior drug loading and uptake, and improved release profiles of therapeutics, compared to the spherical counterparts (Geng et al. 2007; Tiwari et al. 2017). Nanotubes, generated from food proteins, could even have new possibilities in food, pharmaceutical, and cosmetic fields, due to their nutritional value, biodegradability, and biocompatibility. Unfortunately, food protein-derived nanotubes are relatively less studied and less of concern. To the best of author’s knowledge, food protein nanotubes have so far only been reported for α-lactalbumin, lysozyme, and albumin (Graveland-Bikker and de Kruif 2006; Lara et al. 2013; Tiwari et al. 2017).
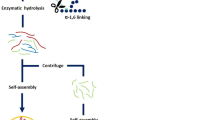
The formation of α-lactalbumin nanotubes includes two steps: first, the proteins are partially hydrolyzed by enzymes, second, the hydrolyzed proteins are self-assembled into nanotubes in the presence of suitable multivalent ions (Fig. 9.7) (Graveland-Bikker and de Kruif 2006). These assembled nanotubes typically have a diameter of ~20 nm and few micrometres in length. The prerequisites to form these nanotube structures are at an intermediate protein concentration and in the presence of appropriate cations. Various di- and tri-valent cations could trigger this self-assembly, including Ca2+, Mn2+, Zn2+, Cu2+, and Al3+, except Ba2+ and Mg2+.

The formation of α-lactalbumin nanotubes. (a) Schematic illustration of the self-assembly of partially hydrolyzed α-lactalbumin into nanotubes in the presence of Ca2+. (b) TEM image of negatively stained nanotubes. The dark line in the middle corresponds to the hollow of the nanotube. Reproduced with permission from Graveland-Bikker and de Kruif (2006). Copyright 2006 Elsevier
Lysozyme nanotubes are generated by heating proteins at pH 2 and 90 °C for 30 h (Lara et al. 2013). Under this condition, lysozyme proteins are first hydrolyzed and then assembled into amyloid fibrils with multi-stranded helical ribbon morphology. In the final stage, the helical ribbons progressively closed into nanotubes. Hence, these lysozyme nanotubes can also be recognized as a state of amyloid fibrils. The nanotube diameter is dominated by the initial helical ribbons width and the folding angle, which ranges from ~40 to 150 nm. It should be noted that many protein amyloid fibrils possess multi-stranded helical ribbon morphology (Lara et al. 2011), and thereby could be the source to produce nanotube structures.
Albumin nanotubes are formed by heating proteins (80–85 °C) in the presence of glutathione and paclitaxel, which respectively function as the accelerator of protein unfolding and the nucleation core of self-assembly (Tiwari et al. 2017). By exposing buried nonpolar residues, glutathione greatly boosts the interaction of albumin and hydrophobic paclitaxel. Afterwards, the crystallization of the paclitaxels that are located in the core contributes to the growth of nanotubes. In this case, nanotubes have a diameter of 70–120 nm and length of up to few micrometres.
3.2 Polysaccharide Self-assembled Nanostructures
The most common function of food polysaccharides is food structuring, which requires the creation of structures up to millimetres or centimetres with specific mechanical properties, that is, the formation of three-dimensional macrostructures (such as bulk gels) (Kontogiorgos 2015; Stephen et al. 2016; Foegeding et al. 2017). Therefore, in food structuring applications, the assembly of polysaccharides involves several length scales, ranging from atomic, molecular, microscopic to macroscopic scales (Kontogiorgos 2015). Current functions related to polysaccharides are not only limited to food structuring, but also include nanoplatforms for targeted delivery and biomedical imaging (Saravanakumar et al. 2012; Debele et al. 2016; Swierczewska et al. 2016). For example, due to the specific cellular recognition of some polysaccharide colloidal nanoparticles, the drug, gene, or nutrient delivery systems derived from these nanoparticles show superior performances (Saravanakumar et al. 2012; Salatin and Yari Khosroushahi 2017). In these applications, individual nanoparticles should be retained and their aggregation must be avoided (Kontogiorgos 2015). Some assembling approaches, e.g., gelation triggered by salt, pH, temperature alteration, electrostatic complexation of opposite charged polysaccharides, have been employed to prepare polysaccharide nanoparticles, but often in a mechanical intervention and/or a low polymer concentration to prevent the bulk gelation. Controlling polysaccharide self-assembly in nanometre length scale is not as easy as that in protein system, because dispersions of polysaccharides in aqueous solutions exhibit very low interfacial tension (Kontogiorgos 2015). Therefore, most of self-assembly-derived polysaccharide nanoparticles are formed by the aid of other methods or chemical modification.
A common approach to produce polysaccharide nanoparticles with controllable size or shape is to first establish the liquid droplets and then self-assemble polysaccharides in these confined droplets (Burey et al. 2008; Shewan and Stokes 2013; Joye and McClements 2014). Extrusion and emulsification always are used to produce these droplets, and the size and shape of nanoparticles are controllable by altering the applied experimental conditions. It should be noted that a “switching” effect can be built into these polysaccharide nanostructures that respond to stimulation in vitro or in vivo, due to the reversible feature of polysaccharide assembly process (Myrick et al. 2014). Another prominent approach to produce polysaccharide nanoparticles, especially when designing delivery nanoplatforms, is through the assembly of hydrophobic segment-grafted hydrophilic polysaccharides, i.e. the assembly of amphiphilic copolymers, as discussed below (Myrick et al. 2014; Debele et al. 2016; Swierczewska et al. 2016). Such copolymer assembled nanostructures is known as promising drug carriers, and even could lower drug toxicity because the hydrophilic polysaccharide parts are often less absorbed by normal tissues but can accumulate in cancerous tissues through the enhanced permeability and retention effect (EPR effect) (Myrick et al. 2014; Keservani and Sharma 2018).
Besides the above-mentioned methods, the assembled nanostructures can also be separated from many natural materials since they are already existent in nature but are highly structured. For example, cellulose nanofibrils and cellulose nanocrystals can be dissociated from fibre cell walls by mechanical, chemical, enzymatic treatment, or a combination thereof (Fig. 9.8) (Xu et al. 2013; Salas et al. 2014; Patel 2018). The abundance of OH groups in cellulose chains facilitates the formation of hydrogen bonding, resulting in the assembly into highly ordered structures (i.e. crystalline regions) that alternate with disordered structures (i.e. amorphous regions) (Salas et al. 2014; Patel 2018). Cellulose nanocrystals are usually produced through ultrasonic acid hydrolysis, in which most of the amorphous regions are degraded and the crystalline parts are remained (Salas et al. 2014; Patel 2018). The yielded cellulose nanocrystals often possess a diameter of 10–50 nm and a length of several hundred nm (Habibi et al. 2010; Xu et al. 2013). In contrast, cellulose nanofibrils contain both amorphous and crystalline regions and have a larger aspect ratio than cellulose nanocrystals (Salas et al. 2014; Patel 2018). These cellulose nanostructures have gained great attention in the scientific community, including in food science, due to a wide spectrum of unique properties such as high aspect ratio, excellent mechanical strength and inherent abundance. They can be used to stabilize emulsions and foams, to prepare hydrogels and aerogels, and to fabricate food-grade packing materials, etc. (Salas et al. 2014; Coffey et al. 2016; Ullah et al. 2016; Patel 2018).

Schematic illustration of cellulose nanofibrils and nanocrystals produced from fibre cell walls by mechanical and chemical treatments, respectively. Reproduced with permission from Salas et al. (2014). Copyright 2014 Elsevier
3.3 Protein-co-polysaccharide Self-assembled Nanostructures
Surfactants (e.g. mono- or diglycerides) have the unique property of self-assembling into a broad range of nanostructures, from micelles and vesicles to membranes and cubic phases, as these molecules possess discrete hydrophobic and hydrophilic moieties (Smart et al. 2008; Jones 2015). Despite that proteins and polysaccharides could self-assemble into several nanostructures as discussed above, these structures are not as diverse and controllable as the specific structures assembled from surface-active agents. This is due to the lack of a significant anisotropic distribution of hydrophobic and hydrophilic moieties in food polymers (Jones 2015). One method to increase this anisotropy is to attach a second component, forming copolymers. A number of chemical techniques could be used to generate these copolymers, but many of them are unfavourable for food formulations (Jones 2015).
Maillard reaction, as one of most common food chemical reactions, is widely used in the food sector to improve food tastes and appearances. It is also an ideal approach to produce food-grade copolymers, typically protein-co-polysaccharide. During Maillard reaction, a reducing end of a polysaccharide and a free amine of a protein are conjugated, with the formation of covalently bonded protein-co-polysaccharide (Kato 2002; Oliver et al. 2006; Jones 2015; de Oliveira et al. 2016). In this type of copolymers, the protein part is often the relatively hydrophobic component that forms the internal phase of micelle-like structures (Fig. 9.9) (Smart et al. 2008; Jones 2015). These food-grade copolymers are the ideal platforms to encapsulate and deliver bioactive compounds. For instance, casein-co-maltodextrin assembled colloids have very high stability, and could reduce the oxidization of encapsulated vitamin D (Markman and Livney 2012; Jones 2015).

Different geometries could be produced from protein-co-polysaccharide in selective conditions. Adapted with permission from Smart et al. (2008). Copyright 2008 Elsevier
4 Tech-functionalities
Proteins and polysaccharides are widely used in the food sector as thickeners, gelling agents, emulsifiers, foam stabilizers, fat replacers, and so on (Phillips and Williams 2009). Self-assembly could happen at different length scales, produce diverse structures, continuum at different time scales (Whitesides and Grzybowski 2002). The appropriate control of self-assembly can produce novel foods and enable new applications. For example, the self-assembly-induced phase separation could produce diverse structures and thus lead to different texture properties. Moreover, the assembled nanostructures generally possess better tech-functionalities than the individual proteins and polysaccharides, owing to their specific morphologies and structural alterations (e.g. high aspect ratio and heat resistance for protein nanofibrils). Many examples are mentioned above, such as forming cold-set gels, stabilizing emulsions and foams, and delivering drugs and nutrients. It is worth to note that some polysaccharides are capable of binding with a particular group of receptors at the cell surfaces, thereby can be engineered to prepare desired platforms to enhance the bioavailability of the loaded biomolecules, including food nutrients and bioactive compounds (Schmitt et al. 2016; Swierczewska et al. 2016; Salatin and Yari Khosroushahi 2017).
5 Disassembly and Reassembly in the Gastrointestinal Tract
As discussed above, the self-assembly is extremely vital to both natural and processed foods as it determines food appearance, texture, shelf-life, etc. However, the final functions of foods mainly depend on their biological fates in the gastrointestinal tract. As is well known, the macromolecular assembly plays a key role in biological phenomena; analogously, food polymer assembly process in the body certainly affect food functions. Assembly-related processes, such as self-assembly, disassembly, and reassembly, are present in the human digestion and absorption processes. During eating, foods are first broken down in the mouth and then entered into stomach and intestine. Afterwards, foods are degraded into small molecules that are absorbed by intestinal walls and eventually enter into the bloodstream. The optimal control of the disassembly of food protein and polysaccharide nanostructures can bring not only the basic nutritional value but also other additional functions, e.g., by delivering incorporated bioactive compounds to the targeted sites (Mcclements et al. 2009; Joye and McClements 2014; McClements 2014; Yao et al. 2015). In contrast, inappropriate disassembly could cause some safety issues. For example, both high-speed (leading to high local concentration) and wrong-site release (such as in the colon altering the gut microbiota) of vitamins will harm the consumer health (McClements and Xiao 2017). Moreover, due to the condition change in the gastrointestinal tract, e.g. pH, the assembled nanostructures could further self-assemble into advanced nanostructures that alter the biological fates of food proteins and polysaccharides by controlling their digestion and absorption abilities. Besides, some degraded molecules can reassemble into nanostructures in the gastrointestinal tract, such as the amyloid fibrils formation in the stimulated gastric condition (Bateman et al. 2011). Understanding the self-assembly, disassembly, and reassembly of food protein and polysaccharide nanostructures during digestion and absorption processes is vital to maximize food nutritional value and improve food quality and even safety.
6 Summary, Challenges, and Future Scope
Self-assembly is commonly seen in both natural and processed foods, which is of vital importance to control food quality, functions, and even safety. It can be employed to innovate functional foods, whereas uncontrollable self-assembly may lower food quality and even cause safety issues. Toward the state of minimum free energy and the equilibrium of three classes of forces—driving, opposition, and directional forces, is the basic principle of the self-assembly of proteins and polysaccharides. The specific forces and mechanisms in certain food systems are not well understood, which arises from the complexity of food systems, and requires a deeper investigation in the future by learning from synthetic polymer systems. A variety of protein and polysaccharides nanostructures with unique properties and functionalities, such as micelles, nanofibrils, and nanotubes, could be produced by the self-assembly approach. Yet, some nanostructures are only reported to be generated from a few food sources under very specific experimental conditions. Future research needs to understand the generic feature of these nanostructures, and thereby to extend their sources as well as categories. It is worthy of noticing that many nanostructures, such as amyloid fibrils and amorphous aggregates, are recently considered to be a generic feature of proteins. The self-assembly behaviour of copolymers has been significantly investigated in polymer science but is far from thorough understanding in food science, which calls for more efforts in the future. Milliard reaction is not the only chemical reaction in the field of food chemistry. In addition, an increasing number of food-grade cross-linkers were found in recent years; this may also open the doors to produce protein-co-polysaccharides. In order to successfully exploit self-assembly in practical applications and to ensure efficient scale-up, a high level of control is also required.
Although some progress has been made in understanding how proteins and polysaccharides assemble into nanostructures and how external factors determine nanostructures properties, studies on how self-, dis-, and re-assembly in gastrointestinal tract control the specific functions of foods are still in the infant stage. They are crucial for human health as these assembly processes can modulate food-body interactions and the biological fate of food components. Know-how on them could provide the guidance to produce foods and generate new functions in food products to benefit the consumer. In summary, future research on the assembly of food polymers and the resultant nanostructures is extremely indispensable.
References
Adair J, Suvaci E, Sindel J (2001) Surface and colloid chemistry. In: Cahn R, Flemings M, Ilschner B et al (eds) Encyclopedia of materials: science and technology, 2nd edn. Elsevier Inc., pp 1–10
Bateman L, Ye A, Singh H (2011) Re-formation of fibrils from hydrolysates of β-lactoglobulin fibrils during in vitro gastric digestion. J Agric Food Chem 59:9605–9611
Baughman RH, Zakhidov AA, De Heer WA (2002) Carbon nanotubes - the route toward applications. Science 297:787–792
Billon L, Borisov O (2016) Macromolecular self-assembly. Wiley
Burey P, Bhandari BR, Howes T, Gidley MJ (2008) Hydrocolloid gel particles: formation, characterization, and application. Crit Rev Food Sci Nutr 48:361–377
Cao Y, Mezzenga R (2019) Food protein amyloid fibrils: origin, structure, formation, characterization, applications and health implications. Adv Colloid Interface Sci 269:334–356
Cao Y, Wang L, Zhang K et al (2015) Mapping the complex phase behaviors of aqueous mixtures of κ-carrageenan and type B gelatin. J Phys Chem B 119:9982–9992
Cao Y, Fang Y, Nishinari K, Phillips GO (2016a) Effects of conformational ordering on protein/polyelectrolyte electrostatic complexation: ionic binding and chain stiffening. Sci Rep 6:23739
Cao Y, Li S, Fang Y et al (2016b) Conformational transition of polyelectrolyte as influenced by electrostatic complexation with protein. Biomacromolecules 17:3949–3956
Carpenter JF, Manning MC (2002) Rational design of stable lyophilized protein formulations: theory and practice. Springer
Chiti F, Dobson CM (2006) Protein misfolding, functional Amyloid, and human disease. Annu Rev Biochem 75:333–366
Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem 86:27–68
Cho Y-H, Jones OG (2019) Assembled protein nanoparticles in food or nutrition applications. In: Toldrá F (ed) Advances in food and nutrition research. Academic Press, pp 47–84
Coffey DG, Bell DA, Henderson A (2016) Cellulose and cellulose derivatives. In: Stephen AM, Phillips GO, Williams PA (eds) Food polysaccharides and their applications, 2nd edn. CRC Press, pp 147–180
Cooper S (2014) Polymer biomaterials in solution, as interfaces and as solids. CRC Press
Dalgleish DG (2011) On the structural models of bovine casein micelles - review and possible improvements. Soft Matter 7:2265–2272
de Kruif CG, Huppertz T, Urban VS, Petukhov AV (2012) Casein micelles and their internal structure. Adv Colloid Interface Sci 171:36–52
de Oliveira FC, dos R Coimbra JS, de Oliveira EB et al (2016) Food protein-polysaccharide conjugates obtained via the Maillard reaction: a review. Crit Rev Food Sci Nutr 56:1108–1125
de Volder MFL, Tawfick SH, Baughman RH, Hart AJ (2013) Carbon nanotubes: present and future commercial applications. Science 339:535–539
Debele TA, Mekuria SL, Tsai H-C (2016) Polysaccharide based nanogels in the drug delivery system: application as the carrier of pharmaceutical agents. Mater Sci Eng C 68:964–981
Dickinson E, Leser ME (2007) Food colloids: self-assembly and material science. Royal Society of Chemistry
Dobson CM (2003) Protein folding and misfolding. Nature 426:884–890
Draget KI (2009) Alginates. In: Phillips GO, Williams PA (eds) Handbook of hydrocolloids, 2nd edn. Woodhead Publishing, pp 807–828
Draget KI, Moe ST, Skjak-Bræk G, Smidsrød O (2016) Alginates. In: Stephen AM, Phillips GO, Williams PA (eds) Food polysaccharides and their applications, 2nd edn. CRC Press, pp 289–334
Eisenberg DS, Sawaya MR (2017) Structural studies of amyloid proteins at the molecular level. Annu Rev Biochem 86:69–95
Fang Y, Al-Assaf S, Phillips GO et al (2007) Multiple steps and critical behaviors of the binding of calcium to alginate. J Phys Chem B 111:2456–2462
Fittolani G, Seeberger PH, Delbianco M (2019) Helical polysaccharides. Pept Sci:e24124
Flickinger MC (2013) Downstream industrial biotechnology: recovery and purification. Wiley
Foegeding EA, Stieger M, van de Velde F (2017) Moving from molecules, to structure, to texture perception. Food Hydrocoll 68:31–42
Geng Y, Dalhaimer P, Cai S et al (2007) Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol 2:249–255
Goodsell DS, Olson AJ (2000) Structural symmetry and protein function. Annu Rev Biophys Biomol Struct 29:105–153
Graveland-Bikker JF, de Kruif CG (2006) Unique milk protein based nanotubes: food and nanotechnology meet. Trends Food Sci Technol 17:196–203
Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 110:3479–3500
Hafidz RNRM, Yaakob CM, Amin I, Noorfaizan A (2011) Chemical and functional properties of bovine and porcine skin gelatin. Int Food Res J 18:813–817
Hamaker HC (1937) The London-van der Waals attraction between spherical particles. Physica 4:1058–1072
Hamley IW (2014) Peptide nanotubes. Angew Chemie - Int Ed 53:6866–6881
Hu J, Yang J, Xu Y, Zhang K, Nishinari K, Phillips GO, Fang Y (2019) Comparative study on foaming and emulsifying properties of different beta-lactoglobulin aggregates. Food Funct
Israelachvili JN (2011) Van der Waals forces between particles and surfaces. In: Intermolecular and surface forces. Academic Press, pp 253–289
Jiang XC, Zeng QH, Chen CY, Yu AB (2011) Self-assembly of particles: some thoughts and comments. J Mater Chem 21:16797–16805
Jones OG (2015) Protein nanostructures. In: Marangoni AG, Pink DA (eds) Edible nanostructures: a bottom-up approach. Royal Society of Chemistry, pp 69–113
Joye IJ, McClements DJ (2014) Biopolymer-based nanoparticles and microparticles: fabrication, characterization, and application. Curr Opin Colloid Interface Sci 19:417–427
Kato A (2002) Industrial applications of Maillard-type protein-polysaccharide conjugates. Food Sci Technol Res 8:193–199
Kedracki D (2015) DNA-copolymers structure formation: beyond self-assembly. University of Geneva
Keservani RK, Sharma AK (2018) Nanodispersions for drug delivery. CRC Press
Kontogiorgos V (2015) Polysaccharide nanostructures. In: Marangoni AG, Pink DA (eds) Edible nanostructures: a bottom-up approach. Royal Society of Chemistry, pp 41–68
Kroes-Nijboer A, Venema P, van der Linden E (2012) Fibrillar structures in food. Food Funct 3:221–227
Lara C, Adamcik J, Jordens S, Mezzenga R (2011) General self-assembly mechanism converting hydrolyzed globular proteins into giant multistranded amyloid ribbons. Biomacromolecules 12:1868–1875
Lara C, Handschin S, Mezzenga R (2013) Towards lysozyme nanotube and 3D hybrid self-assembly. Nanoscale 5:7197–7201
Lee YS (2007) Self-assembly and nanotechnology: a force balance approach. Wiley
Lekkerkerker HNW, Tuinier R (2011) Colloids and the depletion interaction. Springer
Li T, Alessandra M (2018) Pharmaceutical crystals: science and engineering. Wiley
Li L, Thangamathesvaran PM, Yue CY et al (2001) Gel network structure of methylcellulose in water. Langmuir 17:8062–8068
Li L, Shan H, Yue CY et al (2002) Thermally induced association and dissociation of methylcellulose in aqueous solutions. Langmuir 18:7291–7298
Li C, Qin R, Liu R et al (2018) Functional amyloid materials at surfaces/interfaces. Biomater Sci 6:462–472
Lindoy LF, Atkinson I (2000) Self-assembly in supramolecular systems. Royal Society of Chemistry
Loveday SM, Wang XL, Rao MA et al (2010) Tuning the properties of β-lactoglobulin nanofibrils with pH, NaCl and CaCl2. Int Dairy J 20:571–579
Loveday SM, Anema SG, Singh H (2017) β-Lactoglobulin nanofibrils: the long and the short of it. Int Dairy J 67:35–45
Lu PJ, Zaccarelli E, Ciulla F et al (2008) Gelation of particles with short-range attraction. Nature 453:499–503
Luo Q, Hou C, Bai Y et al (2016) Protein assembly: versatile approaches to construct highly ordered nanostructures. Chem Rev 116:13571–13632
Markman G, Livney YD (2012) Maillard-conjugate based core-shell co-assemblies for nanoencapsulation of hydrophobic nutraceuticals in clear beverages. Food Funct 3:262–270
McClements DJ (2014) Nanoparticle- and microparticle-based delivery systems: encapsulation, protection and release of active compounds. CRC Press
McClements DJ, Xiao H (2017) Is nano safe in foods? Establishing the factors impacting the gastrointestinal fate and toxicity of organic and inorganic food-grade nanoparticles. NPJ Sci Food 1:6
Mcclements DJ, Decker EA, Park Y, Weiss J (2009) Structural design principles for delivery of bioactive components in nutraceuticals and functional foods. Crit Rev Food Sci Nutr
McManus JJ, Charbonneau P, Zaccarelli E, Asherie N (2016) The physics of protein self-assembly. Curr Opin Colloid Interface Sci 22:73–79
Mendes AC, Baran ET, Reis RL, Azevedo HS (2013) Self-assembly in nature: using the principles of nature to create complex nanobiomaterials. Wiley Interdiscip Rev Nanomed Nanobiotechnol 5:582–612
Mezzenga R, Fischer P (2013) The self-assembly, aggregation and phase transitions of food protein systems in one, two and three dimensions. Reports Prog Phys 76:046601
Motiejunas D, Wade RC (2006) Structural, energetic, and dynamic aspects of ligand-receptor interactions. In: Comprehensive medicinal chemistry II. Elsevier Inc., pp 193–212
Myrick JM, Vendra VK, Krishnan S (2014) Self-assembled polysaccharide nanostructures for controlled-release applications. Nanotechnol Rev 3:319–346
Nicolai T (2019) Gelation of food protein-protein mixtures. Adv Colloid Interface Sci 270:147–164
Nicolai T, Durand D (2013) Controlled food protein aggregation for new functionality. Curr Opin Colloid Interface Sci 18:249–256
Nicolai T, Britten M, Schmitt C (2011) β-Lactoglobulin and WPI aggregates: formation, structure and applications. Food Hydrocoll 25:1945–1962
Ninham BW, Nostro PL (2010) Molecular forces and self assembly: in colloid, nano sciences and biology. Cambridge University Press
Nishinari K, Zhang H (2004) Recent advances in the understanding of heat set gelling polysaccharides. Trends Food Sci Technol 15:305–312
Oliver CM, Melton LD, Stanley RA (2006) Creating proteins with novel functionality via the Maillard reaction: a review. Crit Rev Food Sci Nutr 46:337–350
Padua GW, Nonthanum P (2012) Material components for nanostructures. In: Padua GW, Wang Q (eds) Nanotechnology research methods for food and bioproducts. Wiley-Blackwell, pp 5–17
Parsegian VA (2005) Van der Waals Forces: a handbook for biologists, chemists, engineers, and physicists. Cambridge University Press
Patel AR (2018) Functional and engineered colloids from edible materials for emerging applications in designing the food of the future. Adv Funct Mater 1806809:1–34
Phillips GO, Williams PA (2009) Handbook of hydrocolloids, 2nd edn. Woodhead Publishing
Piazza R (1999) Interactions in protein solutions near crystallisation: a colloid physics approach. J Cryst Growth 196:415–423
Pillai CKS, Paul W, Sharma CP (2009) Chitin and chitosan polymers: chemistry, solubility and fiber formation. Prog Polym Sci 34:641–678
Rajagopal K, Schneider JP (2004) Self-assembling peptides and proteins for nanotechnological applications. Curr Opin Struct Biol 14:480–486
Ravichandran R (2010) Nanotechnology applications in food and food processing: innovative green approaches, opportunities and uncertainties for global market. Int J Green Nanotechnol Phys Chem 1:72–96
Roy A, Shrivastava SL, Mandal SM (2016) Self-assembled carbohydrate nanostructures: synthesis strategies to functional application in food. In: Novel approaches of nanotechnology in food. Academic Press, pp 133–164
Russell JD, Dolphin JM, Koppang MD (2007) Selective analysis of secondary amino acids in gelatin using pulsed electrochemical detection. Anal Chem 79:6615–6621
Saenger W (2008) Principles of nucleic acid structure. Springer Science & Business Media
Sagalowicz L, Michel M, Blank I et al (2017) Self-assembly in food — a concept for structure formation inspired by nature. Curr Opin Colloid Interface Sci 28:87–95
Salas C, Nypelö T, Rodriguez-Abreu C et al (2014) Nanocellulose properties and applications in colloids and interfaces. Curr Opin Colloid Interface Sci 19:383–396
Salatin S, Yari Khosroushahi A (2017) Overviews on the cellular uptake mechanism of polysaccharide colloidal nanoparticles. J Cell Mol Med 21:1668–1686
Saravanakumar G, Jo DG, Park JH (2012) Polysaccharide-based nanoparticles: a versatile platform for drug delivery and biomedical imaging. Curr Med Chem 19:3212–3229
Scanlon S, Aggeli A (2008) Self-assembling peptide nanotubes. Nano Today 3:22–30
Schaeffer L (2008) The role of functional groups in drug-receptor interactions. In: Wermuth CG (ed) The practice of medicinal chemistry, 3rd edn. Elsevier/Academic Press, pp 464–480
Schefer L, Adamcik J, Mezzenga R (2014) Unravelling secondary structure changes on individual anionic polysaccharide chains by atomic force microscopy. Angew Chemie - Int Ed 53:5376–5379
Schmitt C, Bovay C, Vuilliomenet AM et al (2009) Multiscale characterization of individualized β-lactoglobulin microgels formed upon heat treatment under narrow pH range conditions. Langmuir 25:7899–7909
Schmitt C, Bovay C, Kolodziejczyk E et al (2016) Functional dairy protein-based aggregates for designing food formulations. In: Williams PA, Phillips GO (eds) Gums and stabilisers for the food industry 18 - hydrocolloid functionality for affordable and sustainable global food solutions. Royal Society of Chemistry, pp 321–330
Shen X, Shamshina JL, Berton P et al (2016) Hydrogels based on cellulose and chitin: fabrication, properties, and applications. Green Chem 18:53–75
Shewan HM, Stokes JR (2013) Review of techniques to manufacture micro-hydrogel particles for the food industry and their applications. J Food Eng 119:781–792
Smart T, Lomas H, Massignani M et al (2008) Block copolymer nanostructures. Nano Today 3:38–46
Sorrenti A, Leira-Iglesias J, Markvoort AJ et al (2017) Non-equilibrium supramolecular polymerization. Chem Soc Rev 46:5476–5490
Stephen AM, Phillips GO, Williams PA (2016) Food polysaccharides and their applications., 2nd edn. CRC Press
Sundararajan PR (2016) Physical aspects of polymer self-assembly. Wiley
Swierczewska M, Han HS, Kim K et al (2016) Polysaccharide-based nanoparticles for theranostic nanomedicine. Adv Drug Deliv Rev 99:70–84
Tadros T (2013) Encyclopedia of colloid and interface science. Springer
Tiwari AP, Joshi MK, Maharjan B et al (2017) Formation of lipophilic drug-loaded human serum albumin nanofibers with the aid of glutathione. Chem Eng J 313:753–758
Ullah H, Santos HA, Khan T (2016) Applications of bacterial cellulose in food, cosmetics and drug delivery. Cellulose 23:2291–2314
van der Linden E, Venema P (2007) Self-assembly and aggregation of proteins. Curr Opin Colloid Interface Sci 12:158–165
Veerman C, Baptist H, Sagis LMC, van der Linden E (2003) A new multistep Ca2+-induced cold gelation process for β-lactoglobulin. J Agric Food Chem 51:3880–3885
Wang J, Liu K, Xing R, Yan X (2016) Peptide self-assembly: thermodynamics and kinetics. Chem Soc Rev 45:5589–5604
Whitesides GM, Boncheva M (2002) Beyond molecules: self-assembly of mesoscopic and macroscopic components. Proc Natl Acad Sci U S A 99:4769–4774
Whitesides GM, Grzybowski B (2002) Self-assembly at all scales. Science 295:2418–2421
Xu X, Liu F, Jiang L et al (2013) Cellulose nanocrystals vs. Cellulose nanofibrils: a comparative study on their microstructures and effects as polymer reinforcing agents. ACS Appl Mater Interfaces 5:2999–3009
Yao M, McClements DJ, Xiao H (2015) Improving oral bioavailability of nutraceuticals by engineered nanoparticle-based delivery systems. Curr Opin Food Sci 2:14–19
Yi H, Wu LQ, Bentley WE et al (2005) Biofabrication with chitosan. Biomacromolecules 6:2881–2894
Zargar V, Asghari M, Dashti A (2015) A review on chitin and chitosan polymers: structure, chemistry, solubility, derivatives, and applications. ChemBioEng Rev 2:204–226
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zeng, H. (2021). Self-assembling Properties. In: Fang, Y., Zhang, H., Nishinari, K. (eds) Food Hydrocolloids. Springer, Singapore. https://doi.org/10.1007/978-981-16-0320-4_9
Download citation
DOI: https://doi.org/10.1007/978-981-16-0320-4_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0319-8
Online ISBN: 978-981-16-0320-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)