
Abstract
India has the task of eliminating tuberculosis (TB) by 2025. This translates to curing about two million TB cases present today as well as reducing the TB infection rates rapidly (WHO Global Tuberculosis Report 2017). The ‘standard treatment regimen’ being administered today is a combination of drugs discovered and developed in the 1950s and 1960s. This regimen falls short of an ideal therapy in many ways including the requirement of prolonged treatment period of 6 months with unpleasant and toxic drug side effects (Yee et al. 2003). However, the characteristic of the TB patient population has changed considerably in the last few decades—coexistence of diabetes and HIV (human immunodeficiency virus) being the main drivers (Balganesh et al. 2008). In addition, India has a significant number of drug-resistant TB patients (Indian TB report 2018) who need novel drugs and regimens for faster and permanent cure. Thus there is an urgent unmet medical need for which TB drug discovery and development efforts globally and in India need to rise to this occasion.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


2.1 Approaches to TB Drug Discovery: The Past and the Present
2.1.1 A Historical Perspective of the TB Drug Regimen
India has the task of eliminating tuberculosis (TB) by 2025. This translates to curing about two million TB cases present today as well as reducing the TB infection rates rapidly (WHO Global Tuberculosis Report 2017). The ‘standard treatment regimen’ being administered today is a combination of drugs discovered and developed in the 1950s and 1960s. This regimen falls short of an ideal therapy in many ways including the requirement of prolonged treatment period of 6 months with unpleasant and toxic drug side effects (Yee et al. 2003). However, the characteristic of the TB patient population has changed considerably in the last few decades—coexistence of diabetes and HIV (human immunodeficiency virus) being the main drivers (Balganesh et al. 2008). In addition, India has a significant number of drug-resistant TB patients (Indian TB report 2018) who need novel drugs and regimens for faster and permanent cure. Thus there is an urgent unmet medical need for which TB drug discovery and development efforts globally and in India need to rise to this occasion.
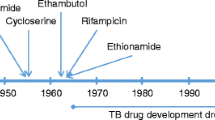
The unique biological aspects of the disease causing microbe, Mycobacterium tuberculosis (MTB) and its relevance to discovering new drugs were unravelled in the course of the development of the standard treatment of tuberculosis. The first drug used for the treatment of tuberculosis was streptomycin (Murray et al. 2015). In a limited clinical trial in 1946 it became obvious that treating TB patients with a single drug like streptomycin, though led to initial clinical improvement, rapidly resulted in the appearance of ‘streptomycin resistant’ microbe and the reappearance of the disease (Doll 1998; Yoshioka 1998). This gave an early indication of a requirement of combination therapy which led to the introduction of isoniazid (I), rifampicin (R), and pyrazinamide (Z) within a decade. A fourth drug, ethambutol (E), was added to the regimen in 1961 (D’Ambrosio et al. 2015; Mitchison and Davies 2012) which became ‘standard of care’ which is still in use as the first line of defence. The introduction of these drugs was a trial and error exercise, and led to the hypothesis that the drugs in the combination served different roles towards ‘curing’ the disease. The Standard Regimen, also referred to as short course chemotherapy (SCC), consisted of the above four drugs (Table 2.1), given over a period of 6 months divided in two phases; (a) all four drugs were given for first 2 months, called the intensive phase, followed by (b) two drugs, I and R for the following 4 months, called the continuation phase (Mitchison 1992).
TB cure is defined as the eradication of the microbe from all regions of the body, which leads to the prevention of ‘relapse’ of the disease and its symptoms even after 12 months of stopping the treatment—this is referred to as achieving ‘sterilisation’ (Mitchison 1992). As hypothesised by Mitchison, apart from preventing the appearance of drug-resistant mutants, drugs in the combination were active on different populations of the MTB bacilli in the patient. I and R acted on both intracellular and extracellular tubercule bacilli, while Z played a role in eliminating the ‘dormant’ bacilli, thus contributing to the prevention of ‘relapse’. The cure achieved with this ‘drug combination’ has set the tone and the bar for aiming better cure through the introduction of newer drugs. Although all the clinical trials that led to the standardisation of the therapy were carried out on patients with pulmonary TB, the same regimen also became the ‘standard of care’ for the treatment of extra-pulmonary TB.
The main challenge with the short course therapy is patient ‘compliance’: successful therapy requires continuing the treatment for 6 months. The main challenge is the ‘safety’ profile of the drugs that have significant unpleasant side effects (Simon et al. 1991) necessitating patient monitoring and counselling throughout the treatment period.
Before the advent of the ‘drug resistance’ against the standard regimen, new drug discovery efforts were largely directed towards finding novel anti-tuberculosis (anti-TB) molecules that can shorten duration of therapy. Understanding the physiology of the MTB bacilli in the different tissue niches had revealed the presence of the extracellular and the intracellular (within the alveolar macrophages) microbes which had differential sensitivity to the drugs in the regimen. Further studies on the physiology of the microbe also revealed that the hypoxic environment induces specific sets of genes which programmes ‘adapted mycobacteria’ with altered physiological properties, as well as a changed drug sensitivity pattern (Simon et al. 1991; Bagehi et al. 2003). Therefore, the working hypothesis developed was that new drugs must be active on different subpopulations of the microbe in the TB lesion, and this was necessary to achieve a faster cure.
The overall success rate of >90% achieved with the ‘standard regimen’ against drug sensitive TB infections (Rockwood et al. 2016) shifted the emphasis for new drugs from just the ‘potency’ perspective to the need on ‘shortening of therapy’ as the urgent unmet medical need, because shortening of treatment duration would increase patient compliance. Therefore the TPP (Therapeutic Product Profile) of a new anti-TB compound included the following critical properties:
-
Compatibility with the standard drug regimen
-
Compatibility with DOTS (Directly Observed Therapy, short course)
-
Affordable
-
Reduction in the treatment duration
Furthermore, it was also envisaged that the new drug will have to be ‘trialled’ as an add-on to the current regimen to investigate if the new combination was capable of reducing the treatment duration.
WHO declared TB a ‘global emergency’ in 1993 in response to the increasing prevalence of drug-resistant TB encountered globally (Klaudt 1994). Several independent factors contributed to the rapid dissemination of drug resistance in MTB clinical isolates, one of them being co-infection of HIV and TB. Treatment of patients infected with I and R resistant MTB immediately became a challenge; the loss of these two front-line drugs necessitated the use of less potent and toxic second-line drugs leading to not only more complex treatments but also a steep decline in the cure rates. Unfortunately, the drug discovery pipeline in 2000 was thin and the development cycle for introducing new drugs was, and is, complex—together they precipitated an unprecedented crisis, spurring reinforced drug discovery efforts worldwide.
2.2 TB Drug Discovery: Past and Present—A Perspective
Drug Discovery efforts leading to the advent of chemotherapy for treating TB was mainly driven by medicinal chemistry efforts. These efforts involved exploring the anti-TB activities of different scaffolds and their derivatives resulting in the discovery Para Amino Salicylate (Table 2.1). This was also the period where natural products obtained from various Streptomyces species and related genera were being investigated (Waksman et al. 1946). These efforts led to the identification of Streptomycin, which was the first chemotherapeutic agent that was tested on TB patients. The most potent and useful drug Rifampicin was discovered in 1966. With time, drug discovery paths have changed dramatically with the coming of the genome—sequencing era. The changes and the fresh challenges are discussed under the ‘modern drug discovery’ section.
2.2.1 The Modern Day Drug Discovery Process
Target-based screening is driven by the availability of the genome sequences of multiple MTB strains including that of Mycobacterium bovis (BCG). The process usually starts with a ‘target identification and validation’ step. The availability of large databases of genome sequences from various microbes as well as the human counterpart has aided in choosing targets by detailed bioinformatic analysis (Vashist et al. 2012; Raman et al. 2008; Vaishali et al. 2019). This has helped in either identifying targets unique to MTB, or targets sufficiently different (by genome analysis or by their crystal structures) to allow the identification of selective compounds. A variety of MTB targets have been validated (shown to be essential for the viability of the microbe) using gene deletion techniques or by using chemical inhibitors, both in vitro and in vivo (Sassetti et al. 2003). Compounds identified through target-based screening, are labelled as ‘hits’ which may or may not have antimicrobial properties. GSK070 currently in phase 2 trials was initially discovered using the ‘target screening approach’ (Li et al. 2017).
Phenotypic screening involves screening compounds directly on the microbe, MTB or surrogate non-pathogenic mycobacteria like M. smegmatis (Msm) or BCG. This approach had yielded leads like TMC 207, which was progressed through the drug discovery process as bedaquiline and has been registered as Sirturo (Matteelli et al. 2010). Several other compounds such as Q203, BTZ043 (ClinicalTrials.gov, NCT02858973 and NCT02530710, 2018a, b) are in the late development phase (Phase 2). Compounds that have advanced through the drug discovery path were also identified through phenotypic screening on the MTB microbe and were progressed through different phases of drug discovery based on their inhibitory properties (MIC). These include compounds like delamanid and pretomanid. Delamanid and pretomanid (PA824) originated from a library of nitroimidazofuran derivatives that was part of a CIBA-GIEGY effort to discover anti-TB as well as antiparasitic drugs against entamoeba (Laurenzi et al. 2007).
It is interesting to note that the majority of compounds in the current clinical development phase have been identified through phenotypic screening and progressed through conventional medicinal chemistry efforts with limited contributions to their progression from the target perspective (Laurenzi et al. 2007; Singh and Mizrahi 2017). The limited success of target based lead discovery is not a reflection of the inability to identify inhibitors of ‘essential target’ function but is more of a reflection on the inability to convert the target inhibition into antitubercular activity. This limitation of translating enzyme inhibition to microbial inhibition has been instrumental to shift focus of anti-TB drug discovery efforts to starting points of compounds that have antimicrobial activity (MIC).
Schematic representation of the drug discovery process is shown below:
Drug discovery is a stepwise process: the different phases are shown in Fig. 2.1, wherein the first two steps focus on discovering molecules, while the last two steps involve fine-tuning the chemical structure to meet acceptable potency, safety and physicochemical properties resulting in a molecule that is ready for clinical evaluation (candidate drug). The criteria for progressing molecules in each of these steps are carefully designed keeping in mind the final characteristics of the drugs (TPP) to treat TB infections. While the essentials of the process in terms of the critical points to transition from one phase to the next remains almost unchanged, modern drug discovery has benefitted from advances in technology as well as the learning from past experiences. This has in turn increased the success rates in our ability to find novel molecules and have increased the chances of identifying a clinical candidate with a greater potential for success as effective therapeutics.

Schematic representation of the Drug Discovery Path
2.2.2 Hit Identification
In a nutshell, hit identification is finding starting chemical molecules which are usually weak to moderately potent against specific biochemical targets or against MTB cultured cells. In the early years of anti-infective drug discovery soil extracts were tested to identify active molecules (Clardy et al. 2009). The reason for this is that soils contain bacteria and other microbes that secrete their metabolites in order to protect themselves from the invading pathogen. Soil samples from all over the world including from the marine floor were collected and tested for antibacterial leads. This approach has yielded a large number of early antibiotics such as penicillin, vancomycin, streptomycin, rifampicin, etc. This effort continues to be used for discovering new antibiotics (von Bubnoff 2006; Durand et al. 2019).
Screening for ‘starting points’ in the modern drug discovery process now relies on ‘libraries of compounds’ that have been collected either through in-house research efforts or chemicals synthesized specifically against chosen targets, or compounds designed using starting points known to yield drugs, e.g. drug like molecules. Hit identification effort is further enhanced by the availability and deployment of the genome sequences of multiple MTB strains that has created opportunities to identify and use novel and specific biochemical pathways as targets. Availability of the genome information has also facilitated rapid identification of the molecular target/pathway for compounds that have inhibitory activity (MIC) on the cultured microbe (Mendes and Blundell 2017; Chim et al. 2011). Testing ‘libraries of compounds’ is not only applicable to biochemical screening but also to phenotypic screening (Manjunatha and Smith 2015). The phenotypic screening approach in the modern discovery context is used to find inhibitors of not only the ‘multiplying’ microbe but also against the MTB microbe in in vitro models corresponding to the different physiological states predicted in vivo.
Yet another hit identification technology that is impacting drug discovery in multiple ways is the significant increase in computational prowess, which has enabled ‘structure-based screening’ capability using appropriate software. This approach builds on the availability of the atomic structures/reliable models of the targets of interest and the ability to dock, and evaluate the binding energies of compounds that putatively bind to the desired pockets on the target. Compound structures that show high binding affinity in this approach are synthesized and are directly tested on the pathogen. Compounds found to be active are channelled into the outlined drug discovery path (Musa et al. 2009).
2.2.3 Lead Identification
The main aims of this phase of the programme can be summarized under the following broad headings and are applicable to compounds identified through either of the screening approaches:
-
Increase the robustness of the ‘hits’ through structural modifications: establish a structure–activity relationship with respect to the kill kinetics of the compound.
-
Understand the drug metabolism and pharmacokinetic properties of the compound—Establish a Mode of Action (MoA) of the molecule.
Lead identification has also been impacted by learning from the decades of drug discovery in the pharmaceutical industry. In 1997 Christopher Lipinsky, after analyzing various physical and structural characteristics of all the FDA-approved drugs, devised rules that aid in designing new drugs irrespective of the therapeutic areas (Walters 2012; Lipinski et al. 2001). These rules today are referred to as Lipinsky rules. While the original four TB drugs (I, R, Z, E) and most of the second-line TB drugs, like clofazimine, amikacin, cycloserine, ethionamide, etc., comply with these rules, bedaquiline, pretomanid, and delamanid, the recently discovered anti-TB drugs violate almost or all of these rules.
Current day drug discovery also lays significant emphasis on the quality of the ‘Lead’ molecules, especially regarding possible safety issues that may be embedded in the molecule. Thus, chemical functionalities like the ‘nitro’ group and reactive groups like Michael acceptors, acid chlorides, sulphonyl chlorides, etc., are avoided. In addition to potency, properties like solubility and metabolic stability are key parameters for a good lead (Lipinski et al. 2001). However, many of the anti-TB drugs in the current pipeline carry ‘nitro’ groups, e.g. pretomanid, delamanid, BTZ, where the nitro group is essential for its anti-TB activity. The recently introduced drug into our anti-TB armamentum, bedaquiline is highly lipophilic, is metabolically unstable (readily metabolized by CYP3A4) and is reported to prolong the QT interval (Koide et al. 2008). Moxifloxacin, a broad spectrum quinolone antibiotic which is also being used to treat MDR TB is known to prolong the QT interval (Fox and Menzies 2013; Van Heeswijk et al. 2014; Koide et al. 2008). Thus, it appears that the ideal lead molecule for anti-TB drug discovery remains elusive; however the potency of these compounds on MDR MTB strains and their acceptable safety margins are the properties driving the choice of these molecules as drugs.
2.2.4 Lead Optimization (LO)
This is an important stage of drug discovery process. Reaching LO stage entails that a robust chemical class has been identified with cellular activity and robust SAR and SPR (structure property relationship) characterized (Rajarshi 2013). The availability of information on the molecular interaction between the inhibitor and its target is helpful in optimizing both the physical properties as well as in weeding out ‘problem chemical groups’ on the molecule. A TB structural genomics consortium of publicly available atomic structures of a large number of proteins has been useful in facilitating optimization of the binding potency of molecules to selected targets (Chim et al. 2011).
The major aim of the ‘LO’ efforts is to identify one or two molecules from the lead molecule cluster and demonstrate efficacy in an animal model of the disease with acceptable PK, PD and safety properties. TB drug discovery today not only requires testing of the LO compounds in various in vitro models representing different physiological states of the MTB microbe but also in in vivo models that represent these different physiological states. Compounds reaching this stage of discovery are also tested in different combinations with the standard regimen to ensure compatibility, overall efficacy as well as to rule out any antagonism between the compounds.
2.2.5 Challenges of Anti-Mycobacterial Chemistry and Progressing Leads into Clinical Candidates
The challenge to discovering new anti-TB drugs is to be able to design molecules that are active against the pathogen in its multiple metabolic states, or in niche environments (Dartois and Barry 2013).
Knowledge of the MTB microbe’s pathophysiology has established the presence of different physiological niches in human host where the microbes reside in different physiological states with altered response to drugs. Current drug discovery approaches use this information to develop several in vitro/ex vivo models to reproduce the MTB microbe target-gene functions in order to enhance the success in identifying and prioritizing high quality lead molecules. For example, all the compounds that have been introduced into the anti-TB regimen recently like bedaquiline, pretomanid, delamanid or those in late clinical development such as BTZ169 or sutezolid are not only active on the extracellular and the intracellular microbe, but are also active on the non-replicating microbe—the non-replicating property being an adaptive/induced response of the microbe to conditioned growth environment (Shehzad et al. 2013; Kaul et al. 2011).
The mouse model has been the work horse for ‘TB drug discovery’ and has been a robust reflection of the cidal activity of compounds (Chen et al. 2017; Devis et al. 2007). However, this model is not a true reflection of the immune responses that are seen in humans following an MTB infection, hence a disconnect between results obtained in the mouse model vis a vis in human trials. This is best exemplified by the lack of correlation seen in the ‘shortening of therapy’ studies in moxifloxacin containing regimens between the mouse and human (Lanoix et al. 2016). Several new modified models have currently been developed that are expected to be a closer approximation to that of the human host (Zhan et al. 2017; Gumbo et al. 2015).
Modern drug discovery uses PKPD (pharmacokinetic and pharmacodynamic) data modelling as the major tool to progress compounds rapidly through the preclinical stages. Based on the model, the dose and the optimal dosing intervals are calculated and confirmatory data is generated in appropriate models of infection (Vaddady et al. 2010; Naveen Kumar et al. 2014). This model is the basis for planning the dose and the frequency of dosing in humans both for safety studies as well as for efficacy demonstration. Unfortunately, success in predicting therapy outcomes in an MTB infection using this PKPD modelling has been elusive. This is probably due to our incomplete understanding of the processes that govern ‘cure’. The hallmark of an antibacterial compound is the correlation between the observed PK parameters and the efficacy in the animal model—this has been and continues to be a challenge for anti-TB drugs when moving from animal models to treating infected humans. While the PK parameters in the mouse model do indeed correlate with a dose-dependent reduction of microbial counts in the mouse lungs, these parameters have little value in predicting the extent of ‘sterilisation’ that can been achieved as well as the overall effect on the duration of therapy when translated to the human (Gengenbacher et al. 2017; Dartois 2014). One of the key factors that influence the ability of a drug to ‘sterilize’ the tubercle is the cellular architecture of the ‘lesion’ that plays a major role in modulating the entry of the drug into the lesion, this in turn induces/selects the multiple physiological states of the tubercule bacilli residing in the lesion; for e.g.: the presence of a low oxygen environment will result in the induction of the ‘anaerobic response’ in the microbe which results in the ‘non-replicating’ state (Lebardo et al. 2018).
Determination of the actual drug permeability into the infected tissues requires elaborate studies using radiolabelled drugs; additionally there is a dearth of standard data in different animal models and thus our inability to predict the real-life permeability in the human host. The classical examples are the drugs in the standard regimen of the first-line therapy. These are dosed more as a logistical convenience rather than their PK properties, e.g. isoniazid has a half-life of 1–3 h but is dosed once daily or twice weekly (Handbook of Anti-Tuberculosis agents 2008). However, knowing that these drugs have been effective, the effectiveness of these drugs is attributed to the slow multiplication time of the microbe and the Post Antibiotic Effect (PAE) of the compound (Chan et al. 2004). In contrast, the newer drugs, surtero, delamanid or pretomanid are dosed based on their PK parameters, where an MIC and/or exposure against the multiplying bacteria has been taken as the PK parameter to achieve cure (Esposito et al. 2015; Tiberia et al. 2018).
Last but not the least is the increased emphasis in understanding the ‘side effects’ as well as the ‘drug–drug interactions’ of newer TB drugs in standard of care regimen. The ability to predict possible ‘off target’ interactions based on the extensive databases available on the behaviour of different classes of compounds in human, has made putative drug–drug interactions predictable. This in turn has helped in designing drug combinations which would not only be efficacious but would also be safer.
The current compounds/drugs in various phases of Clinical trials are shown in Table 2.2. It is interesting to note that while protein synthesis inhibitors are drawn from the oxazolidinone class, the other major target of several compounds is DprE1, an enzyme involved in cell wall biosynthesis.
2.2.6 Newer Therapeutic Approaches
Modern anti-TB drug discovery includes two novel approaches which have yielded interesting results in appropriate animal models of TB infection. The first can be broadly labelled as ‘Adjunct therapy’, which as the name suggests would be drugs administered in combination with any Standard of Care anti-TB regimen and works synergistically with the former. The second approach is the identification of compounds that are registered as drugs for the treatment of diseases other than TB, but also have potent activity on MTB.
-
Adjunct therapy
This approach includes drugs that are not microbicidal on their own but modulate host pathways that combat MTB. The MTB bacilli survival strategy in the human host is known to include influencing/suppressing various inflammatory and cell-mediated immunogenic pathways that have the potential to eliminate the microbe. Compounds/drugs that can relieve this inhibition or augment activity of these anti-TB pathways can play a major role in combating the disease. Several compounds/drugs with such a potential have been identified using animal models (Rayasam and Balganesh 2015). In fact, it has been suggested that this may be a potent approach to reduce the duration of therapy.
-
Repurposed drugs as anti-TB therapy
Several drugs that have been approved for treating indications other than TB have shown potent anti-TB activity in both in vitro and in vivo models (Mishra et al. 2018). The main advantage with these leads is the ability to ‘fast track’ these compounds through the early development stages, provided their safety profile is compatible with the dose and dosing regimen for the anti-TB indication.
2.3 A Paradigm Shift in TB Drug Therapy: Finding New Combinations
The standard tuberculosis treatment regimen since its design, validation and acceptance as the ‘Standard of Care’ has been made up of I, R, Z and E. This combination continues to be the therapy for the treatment of infection caused by drug-sensitive MTB strains. The ‘standard regimen’ was arrived at systematically through testing multiple combinations, and either by adding or omitting new drugs in clinical trials (Aquinas 1982). This regimen has been effective and has been the first-line treatment of TB for nearly four decades since its introduction. Unfortunately, this was also the period [1963–2002] where no new drugs were discovered for the treatment of tuberculosis even though WHO had called ‘TB a global emergency’ in 1993 because of the emergence of multiple drug-resistant strains.
A fresh attempt at finding novel treatments that can reduce the duration of therapy was made by the introduction of the ‘quinolone antibiotic—oflaxacin’ in a trial conducted at TRC, Chennai in 2002 (Tuberculosis Research Centre Chennai 2002). This study showed that an oflaxacin containing regimen for 4 or 5 months was comparable in efficacy to the standard regimen of 6 months suggesting the possibility of a reduced treatment duration. In addition, the available limited data also showed that while the oflaxacin containing regimen administered in the 5 month therapy did have some efficacy against isoniazid resistant strains, the outcome with strains that were resistant to rifampicin and isoniazid simultaneously was poor.
With the appearance of resistance against the ‘standard regimen’, the approach for the development of a new drug through the clinical stages was to add the new drug into the existing regimen. This was dictated by the following pragmatic considerations:
-
Need for empirical treatment vs. ‘drug sensitivity’ based therapy: The isolation of the infecting TB microbe from the patient followed by drug sensitivity testing takes nearly 2 months (Dheda et al. 2013) thus necessitating empirical treatment before the drug sensitivity results could be obtained.
-
Need for a combination therapy vs. monotherapy: The need to treat TB with a combination of more than one drug is critical as monotherapy has been shown to rapidly induce drug resistance.
-
The assumption that because of the novel mode of action of the new drug, the new drug would be effective against all clinical strains of TB.
MDR TB is defined as MTB strains resistant to both I and R, and this resistance pattern makes the SCC ineffective because, the remaining two drugs in the regimen, Z is less effective against rapidly multiplying TB bacilli, whereas E has limited anti-mycobacterial activity. The second-line therapy for the treatment of such MDR strains consists of clofazimine, cycloserine, amikacin, ethionamide and pyrazinamide (Ramachandran 2019). Unfortunately, these drugs are mostly bacteriostatic, have severe side effects and require prolonged treatment for 24 months with a cure rate of only ~30–50% (Ramachandran and Swaminathan 2015). New drugs, bedaquiline and delamanid successfully completed the Phase 1 and 2 clinical developments in the year 2016 (Yang et al. 2019; Ferlazzo et al. 2018). The urgent need for an effective therapy for the treatment of MDR TB has prompted the reservation of these drugs for of MDR TB only.
For the first time in the history of TB drug development, bedaquiline and delamanid were trialled directly on patients infected with MDR strains in a cocktail of the background regimen. By 2007, thanks to the advent of the Line Probe Assay (Ruvandhi et al. 2017) and GeneXpert (Evans 2011) drug sensitivity testing protocols for detecting I and R resistance within 7 days were available. This enabled the segregation of patients into those infected with a DS and those infected with MDR strains. The ability to test new drugs in a cocktail of the background regimen on MDR patients and compare the efficacy with the background regimen opened up a completely new opportunity for developing new drugs for TB. This was a significant development because the currently available ‘second-line regimen’ is poorly effective requiring a 24 month therapy with limited success rates. The new drugs were trialled on MDR TB patients, as an ‘add-on’ to the 5–6 standard background drug cocktails to investigate efficacy (Lienhardt et al. 2010). The new treatment (cocktails of background regimen + delamanid or background regimen + bedaquiline) containing various cocktails were shown to be effective, and significantly better in comparison to the background therapy alone (Karekar and Marathe 2018; Olaru et al. 2017). However, even though a new regimen for the treatment of MDR TB became available, the side effects of the drugs in the background regimen continue to make the treatment problematic with concomitant non-compliance.
It was obvious by early 2000 that a completely new combination therapy was needed because of the rising number of MDR patients and the limitations of even the new cocktails made up of background therapy with the new drugs. Furthermore, shortening duration of therapy was an important milestone which was required to improve compliance. The anti TB-drug discovery pipeline in the year 2008–2009 included several novel and a few repurposed chemical entities. The novel compounds were pretomanid (Pre), bedaquiline (B), delamanid (D), which were in different stages of clinical development. In addition, the oxazolidinone class had three members, linezolid (LZ), sutezolid and AZD 5847, which were also in trials for the treatment of TB (Balasubramanian et al. 2014). Quinolones such as moxifloxacin (M), levofloxacin (L) and gatifloxacin (G) were yet another class being investigated as cocktails with standard care or second-line TB drugs (Johnson et al. 2006). During this unique ‘timeframe’, several novel compounds/drugs were available for development as anti-TB molecules providing opportunities to clinically evaluate new combinations. The TB Alliance pioneered this concept and tested a variety of cocktails against drug sensitive infections in an ‘Early Bactericidal Activity’ clinical study—a 14 day treatment trial (Joseph et al. 2011). This investigation demonstrated that the combination of Pre, M and Z to be clearly superior to the other combinations tested. Following this, GATB also launched a trial with Pre, M and Z on drug-resistant patients, in South Africa and Tanzania (Diacon et al. 2012).
It was envisaged that the new combinations should be able to treat DS as well as MDR infections, thus opening up the possibility of having a single simplified regimen to treat patients with DS, MDR and XDR infections. XDR is referred to MDR strains that are also resistant to the Quinolones and the injectables. Several cocktails, such as a combination of B, Pre, M, Z is being trialled on DS and MDR TB patients (Dawson et al. 2015), whereas a combination of B, Pre and L (linezolid) is being tried on MDR or XDR patients (ClinicalTrials.gov—NCT02333799, 2015). Other studies with cocktails containing B, P and L in combination with M or clofazimine aim to investigate shortening of therapy in MDR patients (Lebardo et al. 2018). A number of these trials are scheduled to be completed by 2020 paving the way for introducing new treatments. A novel combination of drugs with both improved efficacy, a shortened duration of therapy and suitable for both DS and MDR TB would be a major breakthrough.
Finally, testing cocktails of the newer compounds, which are under patent protection requires the consent from the concerned patent owners. The use of compounds in novel combinations would generally be covered by the respective patents and hence a discussion mechanism to obtain permission to conduct such trials needs to be streamlined.
2.4 Global and India Efforts on TB Drug Discovery
India has a number of global firsts as far as TB drug discovery is concerned. It was at the Tuberculosis Research Center (TRC), Chennai and currently named National Institute for Research in Tuberculosis (NIRT), an Institute under the Indian Council of Medical Research (ICMR) that a number of the early clinical studies to find a treatment for TB were carried out (Radhakrishna 2012). These studies led to recommendation of the current standard regimen of I, R, Z, and E. The very same institute was also responsible for bringing quinolone antibiotics into the anti-TB treatment to start shaping a new and effective second-line therapy. This establishes the fact that the clinical trial community in India is quite capable of bringing new molecules through the development path to the patient (Joseph et al. 2011).
India has pioneered academic research in the field of mycobacteria, Prof T R Ramakrishnan and colleagues at the Indian Institute of Science, Bangalore extensively investigated the metabolism of MTB (Ramakrishnan et al. 1972). Their pioneering work on the mechanism of action of I on mycobacteria was also one of the first investigative biochemical studies of a drug action in MTB. Along these lines, over the years several important aspects of the MTB physiology, metabolism and their significance in the disease process have been discovered in several laboratories in India (Taneja et al. 2010; Anishetty et al. 2005). Detailed research on the biology of a variety of targets has resulted in the proposal of several targets for drug discovery effort (Nagaraja et al. 2017; Kurthkoti and Varshney 2012). A steady stream of Indian academic publications reveals design and discovery of several families of novel chemical moieties with inhibitory effects on M. bovis and M. semgmatis as well as studies on MTB (Singh et al. 2017; Puneet Chopra and Meena 2003). Some of this work has also been extended to identifying novel targets using repurposed compounds (Mishra et al. 2018). In addition, a significant progress has also been reported in the computational studies on metabolic pathways leading to identifying new targets (Chandra 2009; Balganesh and Furr 2007; Vashist et al. 2012). Indian scientists have been at the forefront in obtaining molecular structures of several putative target proteins of mycobacteria (Singh et al. 2018). The availability of the structural details of several proteins has also spurred computational studies involving the mapping of the binding of libraries of compounds on the protein structure to identify novel chemical starting points (Bhagavat et al. 2017; Syal et al. 2017). The progress made and the plethora of studies published by the Indian scientists suggests an ample background for TB drug discovery to capitalize on. However, very few of these studies have been progressed into the drug discovery path. This is perhaps a reflection of several India specific challenges including infrastructure and the limited drug development experience.
Tuberculosis drug discovery involves working with a highly infectious organism that requires advanced biosafety containment facilities and animal facilities which are also compliant with safety requirements (Singh 2013). The availability and access to such facilities in India is a limiting factor. This limitation restricts drug discovery in academic institutions to either carrying out studies with only the purified components of biochemical assays, or to use surrogate microbes like M. smegmatis. Unfortunately, this surrogate microbe cannot be used to progress anti-TB drug discovery through studies involving the ‘physiologically relevant’ models or in the animal models. Given this scenario it is only expected that the main contribution from the academic research is in the early parts of discovery.
Drug discovery requires an integrated team with expertise from different disciplines apart from those mentioned above and including pharmacology and clinical development. It is difficult to envisage such a setup of a multidisciplinary team in an academic environment. Advances made in the academic institutions will need to be capitalized upon by partnering with dedicated drug discovery teams either in industry or in consortia consisting of global players which include both academic and industry partners like the European Union Frame Work programmes (Lång et al. 2010). At the same time, it is imperative that the understanding of the basic biology of the microbe and the host continue in the academic institutions as these are the key enablers for drug discovery programmes.
Historically the first contribution of Pharma based in India to the anti-TB pipeline was by CIBA-GIEGY, India, where researchers identified CGI17341, a nitroimidazole, the development of which was halted because of ‘mutagenicity concerns’ (Mukherjee and Boshoff 2011). Examples of pharma in India involved in the discovery of anti-TB drugs are Lupin Limited and AstraZeneca R and D India (AZI); these companies have played important roles in the discovery of novel drugs for TB. Sudoterb, a pyrrole derivative discovered at the Lupin Labs in India was found to be effective in the mouse models of TB infection with potential for the reduction of therapy duration. Sudoterb was designed and synthesised at the Lupin Labs. Thus both the discovery and development was an in-house effort. Sudoterb was progressed through Phase 1 studies in 2004 (Ginsberg 2010) and Phase 2 in 2013 [LL3858, Sudoterb https://newdrugapprovals.org/2017/12/05/ll-3858-sudoterb/]. However, the outcomes of these trials have not been made public. AZI’s in-house research has contributed several molecules to clinical development. For example, AZD5847, an oxazolidinone moiety, was progressed to Phase 2 (Balasubramanian et al. 2014) while AZ 7371 an azaindole moiety, which was also discovered at AZI is being progressed through Phase 1 by the Global Alliance (Chatterji et al. 2014). AZI was also involved in the discovery of benzothiazinones, a novel class of anti-TB molecules (Makarov et al. 2009) whose derivative PBTZ-169 is currently in clinical trials (ClinicalTrials.gov—NCT03334734, 2017). Given the fact that the overall profit potential of anti-TB drugs globally is limited, it would be of interest to examine the reasons for the three companies, Ciba-Giegy India, Lupin Limited and AstraZeneca India, to be involved in research activities towards finding new drugs for TB treatment. During 1980s and 1990s the India based Ciba-Giegy unit was involved in the design and discovery of novel drugs for the treatment of anaerobic infections both bacterial and parasitic from which the anti-TB compound CGI17341 was identified. Lupin on the other hand continues to be a market leader in the sales of first-line anti-TB drugs and had an in-house commitment to find new drugs for the treatment of TB, which would also help enhance their sales portfolio. Sudoterb was one such compound, which was identified in this process. AstraZeneca India had a remit for finding new drugs for the treatment of neglected diseases and the compounds described were discovered in the projects for TB. Fast forward 2015, all these ‘drivers’ have changed and none of these three companies are currently focused on discovering new molecules for the treatment of TB.
The current list of clinical trials on TB being conducted in India since 2016 is shown in Table 2.3. For a country with a dire need to find new therapeutic options for treating TB, the number of trials are indeed limited. However, it is heartening to note that some of these are indeed exploring novel approaches—like the use of ‘adjunct therapy’, VPM 1002—a vaccine trial, as well trials exploring the use verapamil and metformin to augment existing therapy. While the paucity of TB compounds in clinical trials may be a concern, the fact remains that the ‘duration’ taken to trial a new drug through Phase 2 and 3 is prolonged, but the trialling procedure and duration have not changed over several decades. What we urgently need, in addition to new drugs, are biomarkers that are predictive of successful treatment—this could be a single most productive ‘game changer’ for goals of TB eradication in the near future.
2.5 Going Forward: TB Drug Discovery and India
With Lupin Labs and AZI no longer contributing to the discovery of novel anti-TB molecules it becomes even more imperative that alternative avenues are explored to continue populating the anti-TB drug discovery pipeline. Funding agencies in India, DBT, DST, CSIR and ICMR continue to invest in both the discovery and early development of potential anti-TB compounds. However given the understanding of the bottlenecks in making this happen, the major hurdle is in converting laboratory-based research into a directed product-based research. It is well established that the highest risk phase of the drug discovery path is in the translation of laboratory in vitro and in vivo animal data into effective and safe medicines in man which includes the regulatory toxicology studies, followed by the safety studies in man, and finally efficacy studies in patients. The Govt. of India under the auspices of its multiple funding agencies has been funding schemes that bring specific partners belonging to industry and academic to develop products. However, there have been limited takers from the Indian large pharma in this endeavour. The need to attract big pharma in India into this initiative is because of the ‘capabilities’ that reside ‘only’ in these units. Lupin developed Sudoterb demonstrating its in-house capacity to bring such molecules through the discovery and development chain. Knowing that the commercial market for a TB drug is limited, especially when half the sales are through government agencies (Arinaminpathy et al. 2016) and the drugs have to be given in pre-existing combinations, de-risking the progression of novel compounds through the ‘translation phase’ becomes imperative to attract pharma partners. This de-risking can be achieved through partnerships with academic institutions which have the necessary expertise or with industry partners who have brought drugs through the early clinical phases. Partnership with big pharma can be discussed once there is successful transition of compounds through the early development phase.
Indian research establishments need to find such a translation capacity through novel models. In this context it is worth examining global models that have addressed this topic:
2.5.1 European Union Framework 6 and 7 Projects
New Medicines for TB (NM4TB) and more medicines for TB (MM4TB)—were multi-partner consortia, which included both publicly and privately funded institutions which came together under this umbrella to take advantage of the discovery work being carried out in the academic laboratories and probe the translational aspects with help from the industry partners. NM4TB delivered a novel benzothiozinone into clinics in which AZI was a key player in this consortia. MM4TB too delivered a derivative of benzothiazinones, BTZ043 (ClinicalTrials.gov—NCT03590600, 2018c), which is currently in the late stage clinical development.
2.5.2 TB Accelerator Programme
Yet another initiative—Bill and Melinda Gates Foundation, which is at the forefront of tackling the diseases of the developing and underdeveloped countries, brings together both academic and industry players as partners to accelerate the identification and development of novel compounds for the treatment of TB. Several academic institutions and pharmaceuticals are members of this effort.
India has its own example of such a successful collaborative effort. The development of RotaVac, a low cost, rotavirus vaccine through a collaborative ‘public–private partnership’ effort between the Department of Biotechnology and Bharat Biotech is indeed a major milestone achievement for India’s drug/vaccine research community (Glass et al. 2005). One of the significant contributors to this successful partnership was the detailed preclinical and early clinical development that has been carried out before the final Phase 3 trial. This preclinical work had been supported by various funding agencies and expertise both in India and the USA (Press Information Bureau, Government of India 2013). The question is, is such a model workable for developing anti-TB drugs? The involvement of industry in a less ‘commercially attractive’ area like tuberculosis will be generally driven by a ‘risk analysis’ on the chances of success. The RotaVac team was able to mitigate most of the risk involved with a network of global partners. This model is similar to the EU Framework or the TB Drug Accelerator models.
Two models India can experiment with: The first is exemplified by the University of Dundee Drug Discovery Unit who have been successful in developing new drugs for treating malaria (Norcross et al. 2016). The Dundee unit is made up of joint faculty from the University of Dundee with extensive domain expertise are also part of the Drug Discovery Unit. They undertake Drug Discovery programmes in a project mode and progress molecules through the late preclinical stage to clinic readiness. India has institutes like CSIR-CDRI (Council for Scientific and Industrial Research-Central Drug Research Institute), which can be mandated to take leads identified across different academic institutions in India and progress the molecules to be clinic ready. This would require a commitment of the faculty of the institute to form a project team which would be responsible for planning and implementing every aspect projects from conception to clinical ready stage. The second model would be for funding agencies to form ‘facilitation groups’ which are made of experienced drug hunters who are mandated to evaluate and progress a portfolio of starting points. The starting points would be from various academic institutes, and the facilitation group would generate data on the putative leads through CRO’s (Contract Research Organisations) as needed. A variation on this model has been the Open Source Drug Discovery (OSDD) approach for finding new therapies for Neglected Diseases (Rayasam and Balganesh 2015; Ummanni et al. 2014). OSDD built a network of projects starting from lead discovery to clinical trial that was facilitated with expert panels and funding. The fundamental tenet of OSDD was that all data generated should be in the public domain. As of today, OSDD is a part of the India TB Research and Development Consortium (ITRDC) initiative (Koshy 2016) which in turn is being run under the auspices of ICMR (ICMR press release 2017), and is mandated to bring new therapies faster into India, as well as fast track projects from the Indian research community that can yield additional candidate drugs. It will be of interest to follow up the workings of this consortium in the coming years.
The regulatory framework in India has been active and has responded to the unmet needs. However innovative paths need to be found/improvised to enable new drugs to be tested in a regulatory acceptable manner (Vaidyanathan 2019). Several new drug combinations are being trialled globally. It will be helpful if this testing is accelerated in India which would enable faster drug approvals resulting in benefits to our patients in a timely manner. Given that the big pharma interests in developing new drug for TB in India is limited because of the narrow commercial potential of anti-TB compounds, it will finally rest on the appropriate agencies to take the responsibility of finding ways to not only trial the new compounds in the Indian scenario but also of bringing new drugs to TB patients in India.
Finally it has to be understood that there are two faces to Drug Discovery and Development—one dealing with relieving human suffering and the other, the commercial value of the investment. Drug companies are conscious of the Return on Investment (RoI) that can be generated from successful drug development. Antibiotics generally have limited commercial attractiveness as compared to therapies for metabolic diseases because of several reasons, including:
-
the treatment course is short
-
clinical trials are challenging
-
new antibiotics will always be used as last resort
These challenges have led to many pharmaceutical companies exiting the ‘anti-infective’ therapy area. Taking cognizance of this, Governments in the US and the EU have launched several initiatives to incentivize research in the anti-infective area (Simpkin et al. 2017).
Another important factor to note is that the TB market is also impacted by the socio-economic status of the patient population and that the treatments are a combination therapy. This has led to the US FDA to formulate ways to incentivize research into finding new anti-TB molecules, as well as a ‘voucher’ system, for example, to help compensate for low RoI to pharma companies (Beith et al. 2009). India too needs to find ways to encourage investment into anti-TB drug development to incentivise large pharma companies to participate in this effort.
There is an overall need for integrated thinking involving strengthening discovery research, building consortia which include India’s big pharma, finding ways to trial newer anti-TB drugs in India and finally incentivizing investment in TB research with schemes that help the commercialization. Such a discussion would involve several government and regulatory authorities while concerted and sustained discussion on the various issues would help overcome this bottleneck—it is high time we make this happen in India.
2.6 Closing the Loop
India has the maximum number of TB patients in the world and also has a high percentage of MDR patients. TB therapy in India has to adapt to tackle several aspects that contribute to the prevalence—awareness, access to treatments and counselling, diagnostics, drug sensitivity testing, prolonged treatment duration, affordability and a host of several related factors like nutrition, stigma, and long-term patient support to help patient compliance, each of which has a bearing on the ability to treat a TB patient successfully. Newer TB drugs have to be robust enough to be effective under some of the conditions mentioned above, for e.g. active against DS and MDR TB, and at the same time be cost effective. Diagnostic tests including drug sensitivity tests will play a major role in our ability to not only treat the patient and contain the disease, but will also be critical to ensure ‘long-term effectiveness’ of the drugs. Patient centric counselling and subsidised medical support is also essential to increase compliance. It must also be accepted that there are logistical challenges when dealing with the large numbers of TB cases, newer TB drugs need to be free from aggravating these pre-existing encumbrances.
Clearly there are several strings that need to be knitted to achieve progress. The disease poses an immense threat to the nation and has direct bearing on the health economics of the nation. The regimens in use fall short of being user friendly in terms of the need for reduced treatment duration and their side effects, yet the aim is to achieve a TB free India by 2025. Therefore, it is imperative that Indian scientists from both the academic and industry along with the appropriate executive machinery as well as a political commitment need to get together to achieve this task.
References
Anishetty S, Pulimi M, Pennathur G (2005) Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput Biol Chem 29:368–378
Anon (2008) Handbook of anti-tuberculosis agents. Tuberculosis 88:85–170
Aquinas M (1982) Short course chemotherapy for tuberculosis. Drugs 2:118–132
Arinaminpathy N et al (2016) The number of privately treated tuberculosis cases in India-an estimation from drug sales data. Lancet Infect Dis 16(11):1255–1260
Bagehi G, Jaya M, Tyagi S (2003) Hypoxia- responsive expression of Mycobacterium tuberculosis Rv3134c and devR promoters. Microbiology 149:2303–2305
Balasubramanian V et al (2014) Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob Agents Chemother 58(1):495–502
Balganesh TS, Furr BJA (2007) Molecular approaches to target discovery - evaluating targets for antituberculosis drug discovery programmes. Infect Disord Drug Targets 7:120–126
Balganesh TS, Alzari PM, Cole ST (2008) Rising standards for tuberculosis drug development. Trends Pharmacol Sci 29(11):576–581
Beith A, Eichler R, Weil D (2009) Case study. In: Worldwide - incentives for tuberculosis diagnosis and treatment. Centre for Global Development, Washington, DC
Bhagavat R et al (2017) A genome-wide structure-based survey of nucleotide binding proteins in M. tuberculosis. Nat Sci Rep 7:12489
von Bubnoff A (2006) Seeking new antibiotics in the nature’s backyard. Cell 127:867–869
Central TB Division (2018) India TB report 2018. Annual status report. Central TB Division, New Delhi
Chan C-Y et al (2004) In vitro post-antibiotic effects of rifapentine, isoniazid, and moxifloxacin against mycobacterium tuberculosis. Antimicrob Agents Chemother 48:340–343
Chandra N (2009) Computational systems approach for drug target discovery. Expert Opin Drug Discovery 4(12):1221–1236
Chatterji M et al (2014) 1, 4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob Agents Chemother 58(9):5325–5331
Chen X et al (2017) Delamanid kills dormant mycobacteria in vitro and in a guinea pig model of tuberculosis. Antimicrob Agents Chemother 61(6):e02402–e02416
Chim N et al (2011) The TB Structural Genomics Consortium: a decade of progress. Tuberculosis (Edinb) 91(2):155–172
Clardy J, Fischbach MA, Currie CR (2009) The natural history of antibiotics. Curr Biol 19(11):R437–R441
ClinicalTrials.gov (2015) A phase 3 study assessing the safety and efficacy of bedaquiline plus PA-824 plus linezolid in subjects with drug resistant pulmonary tuberculosis. NCT02333799
ClinicalTrials.gov (2017) Multicenter, open, randomized study with active control (isoniazid) to evaluate the early antibacterial activity, safety and pharmacokinetics of the drug PBTZ169 (capsules 80 mg) when used in patients with first-diagnosed tuberculosis of the respiratory system with bacterial excretion and saved bacterial susceptibility to isoniazid and rifampicin. https://clinicaltrials.gov/ct2/show/NCT03334734
ClinicalTrials.gov (2018a) A dose-escalation study to evaluate safety, tolerability and pharmacokinetics of multiple doses of Q203 in normal healthy male and female volunteers. NCT02858973
ClinicalTrials.gov (2018b) A single ascending dose study of BTZ043. NCT02530710
ClinicalTrials.gov (2018c) A single ascending dose study of BTZ043. https://www.clinicaltrials.gov/ct2/show/NCT03590600
Crofton J (1959) Chemotherapy of pulmonary tuberculosis. Br Med J 1:1610–1614
D’Ambrosio L et al (2015) New anti-tuberculosis drugs and regimens: 2015 update. ERJ Open Res 1:00010-2015
Dartois V (2014) The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol 12(3):159–167
Dartois V, Barry CE (2013) A medicinal chemists’ guide to lead optimization for tuberculosis. Bioorg Med Chem Lett 23(17):4741–4750
Dawson R et al (2015) Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of anti-tuberculosis treatment: a phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 385(9979):1738–1747
Devis GR et al (2007) Evaluation of new antituberculosis drugs in mouse models. Antimicrob Agents Chemother 51:403–404
Dheda K, Ruhwold M, Theron G, Peter J, Yam WC (2013) Point of care diagnosis of tuberculosis: past, present and future. Respirology 18(2):217–232
Diacon AH et al (2012) 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 380(9846):986–993
Doll R (1998) Controlled trials - the 1948 watershed. BMJ 317:1217–1220
Doster B et al (1973) Ethambutol in the initial treatment of pulmonary tuberculosis. Am Rev Respir Dis 107:177–190
Durand GA, Raoult D, Dubourg G (2019) Antibiotic discover: history, methods and perspectives. Int J Antimicrob Agents 53:371–352
Esposito S, Bianchini S, Blasi F (2015) Bedaquiline and delamanid in tuberculosis. Expert Opin Pharmacother 16(15):2319
Evans CA (2011) GeneXpert—a game-changer for tuberculosis control. PLoS Med 8(7):e1001064
Ferlazzo G et al (2018) Early safety and efficacy of the combination of bedaquiline and delamanid for the treatment of patients with drug-resistant tuberculosis in Armenia, India, and South Africa: a retrospective cohort study. Lancet Infect Dis 18(5):536–544
Fox GJ, Menzies D (2013) A review of the evidence for using bedaquiline to treat multi drug resistant tuberculosis. Infect Dis Ther 2:123–144
Gengenbacher M et al (2017) NOS2-deficient mice with hypoxic necrotizing lung lesions predict outcome of tuberculosis chemotherapy in humans. Sci Rep 7:8853
Ginsberg A (2010) Drugs in development for tuberculosis. Drugs 70:2201–2214
Glass RI et al (2005) Development of candidate Rotavirus Vaccines derived from neonatal strain in India. J Infect Dis 192:530–535
Gumbo T et al (2015) Nonclinical models for anti-tuberculosis drug development: a landscape analysis. J Infect Dis 211(Suppl 3):S83–S95
ICMR (2017) Press release
Johnson JL et al (2006) Early and extended early bactericidal activity of levofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis 10(6):605–612
Joseph P et al (2011) Outcome of standardized treatment for patients with MDR-TB from Tamil Nadu, India. Indian J Med Res 133(5):529–534
Karekar SR, Marathe PA (2018) Current status of delamanid in the management of MDR tuberculosis. J Assoc Physicians India 66:72
Kaul A et al (2011) The challenge of new drug discovery for tuberculosis. Nature 469:483–490
Klaudt K (1994) TB-a global emergency. WHO, Geneva
Koide T et al (2008) Severe QT interval prolongation associated with moxifloxacin- a case study. Cases J 1:409
Koshy J (2016) TB drug project gets a lease of life. The Hindu
Kumar N et al (2014) Pharmacokinetics and dose response of anti-TB drugs in rat infection model of tuberculosis. Tuberculosis 94:282
Kurthkoti K, Varshney U (2012) Distinct mechanisms of DNA repair in mycobacteria and their implications in attenuation of the pathogen growth. Mech Ageing Dev 133:138–146
Lång H, Quaglio GL, Olesen OF (2010) Tuberculosis research in the European Union: past achievements and future challenges. Tuberculosis 90:1–6
Lanoix JP, Chaisson RE, Nuermberger EL (2016) Shortening tuberculosis treatment with flouroquinolones- lost in translation. CID 62:484–490
Laurenzi M, Ginsberg A, Spigelman M (2007) Challenges associated with current and future TB treatment. Infect Disord Drug Targets 7:105–119
Lebardo MDJ, Helena BIM, Clifton BE (2018) The present state of the tuberculosis drug development pipe-line. Curr Opin Pharmacol 42:81–94
Lehmann J (1946) para amino salicylic acid in the treatment of tuberculosis. Lancet 1:15–16
Li X et al (2017) Discovery of a potent and specific M. tuberculosis leucyl-t-RNA synthetase inhibitor- (S) 3- (aminoomethyl)-4-chloro-7-(2-hydoxyethoxy) benzo(c) (1,2) oxoborol-1(3H)-ol (GSK656). J Med Chem 60:8011–8026
Lienhardt C, Vernon A, Raviglione MC (2010) New drugs and new regimens for the treatment of tuberculosis - review of the drug development pipeline and implications for national programmes. Curr Opin Pulm Med 16:186–193
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Makarov V et al (2009) Benzothiazinones Kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324(5928):801–804
Manjunatha UH, Smith PW (2015) Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg Med Chem 23:5087–5097
Matteelli A, Carvalho ACC, Dooley KE, Kritsk A (2010) TMC207- the first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol 5(6):849–858
Mendes V, Blundell TL (2017) Targeting tuberculosis using structure-guided fragment-based drug design. Drug Discov Today 22(3):546–554
Mishra A et al (2018) An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: implication to a novel combination therapy. EMBO Mol Med 10:e8038
Mitchison DA (1992) The Garrod lecture - understanding the chemotherapy of tuberculosis—current problems. J Antimicrob Chemother 29:477–493
Mitchison D, Davies G (2012) The chemotherapy of tuberculosis: past, present and future. Int J Tuberc Lung Dis 16:724–732
Mukherjee T, Boshoff H (2011) Nitroimidazoles for the treatment of TB: past, present and future. Future Med Chem 3(11):1427–1454
Murray JF, Schraufnagel DE, Hopewell PC (2015) Treatment of tuberculosis - a historical perspective. Ann Am Thorac Soc 12(12):1749–1759
Musa TL, Ioerger TR, Sacchettini JC (2009) The Tuberculosis Structural Genomics Consortium: a structural genomics approachto drug discovery. Adv Prot Chem Struct Biol 77:43–76
Nagaraja V, Godbole AA, Henderson SR, Maxwell A (2017) DNA topoisomerase 1 and DNA gyrase as targets for Drug Discovery. Drug Discov Today 22:510–518
Norcross NR et al (2016) Tri-substituted pyrimidines as efficacious and fast-acting antimalarials. J Med Chem 59(13):6101–6120
Olaru ID, Heyckendorf J, Andres S, Kalsdorf B, Lange C (2017) Bedaquiline-based treatment regimen for multidrug-resistant tuberculosis. Eur Respir J 49:1700742
Press Information Bureau, Government of India (2013) DBT announces phase III clinical trial results of rotavirus vaccine developed in India say vaccine demonstrates strong efficacy
Puneet Chopra LS, Meena YS (2003) New drug targets for Mycobacterium tuberculosis. Indian J Med Res 117:1–9
Radhakrishna S (2012) Contributions of The Tuberculosis Research Centre, Chennai in the field of epidemiology of tuberculosis. Indian J Tuberc 59:68–77
Rajarshi G (2013) Exploring structure activity relationships. Methods Mol Biol 993:81–84
Ramachandran G (2019) Pharmacokinetics of second-line anti-tuberculosis drugs. Indian J Pediatr 86:714
Ramachandran G, Swaminathan S (2015) Safety and tolerability of second line anti-tuberculosis medications. Drug Saf 38:253–269
Ramakrishnan T, Suryanarayana Murthy P, Gopinathan KP (1972) Intermediary metabolism of mycobacteria. Bacteriol Rev 36:65–108
Raman K, Yeturu K, Chandra N (2008) targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol 2:109
Rayasam GV, Balganesh TS (2015) Exploring the potential of adjunct therapy in tuberculosis. TIPS 36:506–513
Rockwood N, du Bruyn E, Morris T, Wilkinson RJ (2016) Assessment of treatment response in tuberculosis. Expert Rev Respir Med 10(6):643–654
Ruvandhi R et al (2017) Accuracy of line probe assays for the diagnosis of pulmonary and multidrug- resistant tuberculosis: a systematic review and meta-analysis. Eur Respir J 49:1601075
Sassetti CM, Boyd DH, Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84
Schatz A et al (1944) Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc Soc Exp Biol Med 55(1):66–69
Shehzad A, Rehman G, Ul-Islam M, Khattak WA, Lee YS (2013) Challenges in the development of drugs for the treatment of tuberculosis. Braz J Infect Dis 17(1):74–81
Simon GK, Lye MS, Ahmad N (1991) Side effects of short course tuberculosis chemotherapy. Med J Malaysia 46(1):88
Simpkin VL, Renwick MJ, Kelly R, Mossialos E (2017) Incentivising innovation in antibiotic drug discovery and development - progress, challenges and next steps. J Antib 70:1087–1096
Singh S (2013) Biosafety precautions in tuberculosis laboratory. Indian J Tuberc 60:135–137
Singh V, Mizrahi V (2017) Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov Today 22(3):503–509
Singh P et al (2017) S-enantiomer of the anti-tubrcular compound S006-830 complements activity of front line TB drugs and targets biogenesis of Mycobacterium tuberculosis cell wall. AECS Omega 2:8453–8465
Singh A, Vijayan M, Varsheny U (2018) Distinct properties of a hypoxia specific paralog of single stranded DNA (SSB) protein in mycobacteria. Tubreculosis 108:16–25
Syal K et al (2017) Synthetic analogue of (p) ppGpp analogue is an inhibitor of stringent response in mycobacteria. Antimicrob Agents Chemother 61(6):e00443–e00417
Taneja NK, Dhingra S, Mittal A, Naresh M, Tyagi JS (2010) Mycobacterial transcriptional adaptation, growth and dormancy phenotype is triggered by Vitamin C. PLoS One 5(5):e10860
Tiberia S et al (2018) New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 24(2):86–98
Tuberculosis Research Centre Chennai (2002) A randomised clinical trial for treatment of smear positive pulmonary tuberculosis with regimens using Ofloxacin in the intensive phase. Indian J Tuberc 49:27
Ummanni R, Ramchandran S, Kumar A, Rayasam GV (2014) Innovation at OSDD – unconventional vs conventional approaches. Sci Report 51:20
Vaddady PK, Lee RE, Meibohm B (2010) In vitro pharmacokinetic, pharmacodynamic models in anti-infective drug development - focus on TB. Future Med Chem 2(8):1355–1369
Vaidyanathan G (2019) India’s clinical-trial rules to speed up drug approvals. Nature
Vaishali P et al (2019) Mycobacterial genomics and structural bioinformatics: opportunities and challenges in drug discovery. Emerg Microb Infect 8:109–118
Van Heeswijk RP, Dannemann B, Hoetelmans RP (2014) Bedaquilne - a review of human pharmacokinetics and drug-drug interactions. JAC 69:2310–2318
Vashist R et al (2012) Crowd sourcing a new paradigm for Interactome driven drug target identification in Mycobacterium tuberculosis. PLoS One 7(7):e39808
Waksman SA, Reilly HC, Johnstone DB (1946) Isolation of streptomycin-producing strains of Streptomyces griseus. J Bacteriol 52(3):393–397
Walters WP (2012) Going further than Lipinski’s rule in drug design. Expert Opin Drug Discovery 7(2):99–107
WHO (2017) WHO global tuberculosis report. WHO, Geneva
Yang L, Sun F, Zhang W (2019) Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis; Promising but challenging. Drug Dev Res 80:98–105
Yeager RL et al (1952) Pyrazinamide (Aldinamide) in the treatment of pulmonary tuberculosis. Am Rev Tuberc Pulmo Dis 65(5):523–546
Yee D et al (2003) Incidence of serious side effects from first-line anti-tuberculosis drugs among patients treated for active tuberculosis. Am J Crit Care Med 167:1492
Yoshioka A (1998) Use of randomisation in the Medical Research Council’s clinical trial of streptomycin in pulmonary tuberculosis in the 1940s. BMJ 317:1220–1223
Yuan T, Simpson SN (2018) Hit generation in TB drug discovery: from genome to granuloma. Chem Rev 118:1887–1916
Zhan L, Tang J, Sun M, Qin C (2017) Animal models for tuberculosis in translational and precision medicine. Front Microbiol 8:717
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Balganesh, T.S., Bhat, J.J., Ugarkar, B. (2021). Modern Drug Discovery and Development for TB: The India Narrative. In: Dikshit, M. (eds) Drug Discovery and Drug Development. Springer, Singapore. https://doi.org/10.1007/978-981-15-8002-4_2
Download citation
DOI: https://doi.org/10.1007/978-981-15-8002-4_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-8001-7
Online ISBN: 978-981-15-8002-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)