
Abstract
In this chapter, the direct visualization of crystal formation and growth probed by organic fluorescent molecules exhibiting mechanofluorochromism and displaying aggregation-induced emission (AIE) is discussed. The fluorescence observations of the evaporative crystallization can reveal a two-step nucleation model for nuclei formation. The fluorescence from the droplets showed dramatic changes depending on the molecular state, such as monomer, amorphous, and crystal polymorph. The quartz crystal microbalance (QCM) measurement also revealed the changes in the mechanical properties during the solvent evaporation. These methods provide a useful and convenient fluorescence tool for in situ crystal analysis, from which detailed experimental evidence and mechanistic insights into crystal formation and transformation can be obtained through direct fluorescence visualization with real-time, on-site, and nondestructive methods.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Evaporative crystallization
- Fluorescence color changes
- Mechanofluorochromism
- Aggregation-induced emission (AIE)
- Quartz crystal microbalance (QCM)
- Two-step nucleation model
- Liquid-like cluster
1 Introduction
Crystal formation from solution is essential in fundamental science as well as in the fabrication of pharmaceuticals, food, polymers, and organic solid materials; however, it remains poorly understood. In solution crystallization, the formation of crystal nuclei plays an important role in determining the crystal structure, size, and polymorph, controlling crystallization and crystal quality. In classical nucleation theory, molecules are added one-by-one to extend the crystal lattice and form an embryonic nucleus in a one-step process. The classical model for crystallization visualizes the formation of a metastable crystalline nucleus that reaches a critical size through density fluctuations and grows into a stable crystal. Some computational and experimental results, however, cannot be explained based only on classical nucleation theory [1]. Recently though, a two-step nucleation model involving a liquid-like cluster intermediate prior to nucleation has been developed to explain protein crystallization and has been shown to be of more general validity [2]. It is postulated that liquid-like clusters originate from disordered liquid or amorphous metastable clusters in homogeneous solutions [3]. There have been many reports supporting this two-step nucleation model, in which the intermediated phases play an important role in crystallization.
Nucleation is the initial step of crystallization. The nuclei could not be directly observed because the nuclei exist in the transition state. Understanding and controlling nuclei formation will provide a suitable process for crystallization because the organic molecular crystals generally are formed by weak intermolecular interaction such as van der Waals, π–π interaction, or hydrogen bonding [4]. However, direct observation of such processes under realistic conditions in the real time remains a challenge because of the lack of advanced techniques to discriminate the phase boundaries and capture the intermediate states.
In this chapter, studies of the direct visualization of crystal formation and growth, probed by organic fluorescent molecules by using fluorescence microscopy and spectroscopy, are introduced, with a particular focus on the fluorescence spectral change of a dibenzoylmethane boron difluoride complex exhibiting mechanofluorochromism and a cyanostilbene derivative displaying aggregation-induced emission (AIE).
2 Fluorescence Detection of Molecular Assembling
The fluorescence spectra of materials are sensitive to molecular environment and aggregation. In principle, fluorescence spectroscopy can be used to probe the progress of molecular assembly on the scale of just a few molecules or that of a bulk process. In this section, the studies of molecular assembling probed by fluorescence detection are described.
Yu et al. [5] monitored an amorphous-to-crystalline transformation through fluorescence color changes by the in situ microscopic observation of the crystallization of molecular microparticles. As a molecule, tetra-substituted ethene with novel morphology-dependent fluorescence was applied, which can distinguish the interface between the crystalline and amorphous phase by fluorescence color, providing a simple and practical method to probe the inner processes of a molecular microparticle. The fluorescence images of the crystallization due to contact between microparticles were categorized into three cases by monitoring the crystallization evolution of these defective microspheres. This method can clearly record the inhomogeneous crystallization of amorphous microparticles, whereby the perfect microparticles and those with defects demonstrate diverse destinies. The study presents a realistic picture of the microscopic kinetics of not only solid–solid transitions but also crystallizations that occur spontaneously in atmosphere or under external stimuli, such as mechanochromic behavior. Furthermore, this facile method may provide practical opportunities and utilizations for other molecules employing fluorescence microscopy and fluorescent materials.
Pansu et al. attempted fluorescence lifetime microscopy imaging (FLIM) of the nucleation and growth processes during fluorogenic precipitation in a microflow mapping, the schematic representation of which is as shown in Fig. 2.1 [6]. This is the first observation, enumeration, and mapping of the early stages of crystallization during antisolvent precipitation. As a molecule, (2Z, 2′Z)-2,2′-(1,4-phenylene)-bis-(3-(4-butoxyphenyl)acrylonitrile), DBDCS was chosen, which exhibits aggregation-induced emission enhancement (AIEE), namely, the molecules are non-fluorescent and the nuclei should appear as bright objects on a dark background. THF and water were used as good and poor solvents, respectively, for DBDCS precipitation.

Reproduced from Ref. [6] by permission of The Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS) and the RSC
Schematic illustration of FLIM imaging in a 3D hydrodynamic mixing setup.
The precipitation of a fluorescent dye in a microfluidic 3D hydrodynamic mixing setup was performed concomitant with the FLIM imaging. The FLIM images of the precipitation process are shown in Fig. 2.2. A short fluorescence lifetime of the DBDCS molecule and the long lifetime of its crystal, with nuclei of intermediate lifetime, are observed. We show that the precipitation is slowed down by the presence of a viscous skin at the interface between water and THF. From the analysis of the decays, we map the concentrations of the three species with over half a million pixels and show that nucleation and growth occur all along the device by the slow diffusion of water into the THF inner flow.

Reproduced from Ref. [6] by permission of The Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS) and the RSC
FLIM images of the microprecipitation of DBDCS inside the microfluidic device; Qs/Qc = 10/0.5 (μL min−1); C = 0.21 × 10−3 mol L−1; inner flow diameter = 30 μm. The color of the pixel codes for the average lifetime.
A new method to synthesize and observe the precipitation of sub-micrometer particles was optimized to study and control the early stage of microprecipitation. The developed device is easy to construct and fully compatible with a wide range of solvents. We used fluorescence lifetime imaging microscopy to detect not only the oligomers of molecules that precede the formation of crystals but also the nucleation and growth kinetics simultaneously.
There has been demand for a real-time, on-site, nondestructive, fluorescence imaging technique to monitor the crystal formation and transformation processes of organic fluorescent molecules. Hu and Tang et al. reported the fluorescent visualization of crystal formation and transformation processes of organic luminogens with crystallization-induced emission characteristics [7]. In this work, (Z)-1-phenyl-2-(3-phenylquinoxalin-2(1H)-ylidene)ethanone (PPQE) with crystallization-induced emission properties was reported. Three polymorphs of PPQE with various emission behaviors were obtained with good reproducibility under controlled conditions (Fig. 2.3). With the crystallization-induced emission characteristics and polymorph-dependent luminescence of PPQE, a real-time, on-site, nondestructive fluorescence imaging technique to monitor crystal transformation processes and crystal formation from the amorphous state and dilute solution, respectively, was achieved. This study provides a useful and convenient fluorescence tool for in situ crystal analysis, from which detailed experimental evidence and mechanistic insights into crystal formation and transformation can be obtained through direct fluorescence visualization with real-time, on-site, and nondestructive capabilities. It is a powerful and convenient tool for crystal analysis, providing detailed and valuable information about the crystal formation and transformation processes.

Reproduced from Ref. [7] by permission of The Royal Society of Chemistry
Crystal formation and transformation processes. A real-time, on-site, nondestructive, fluorescence imaging technique has been reported to monitor the crystal formation and transformation processes of organic luminogens.
3 Evaporative Crystallization of Mechanofluorochromic Molecules
Dibenzoylmethanatoboron difluoride (BF2DBM) derivatives have excellent optical properties, such as two-photon absorption cross sections [8, 9], high fluorescence quantum yields in the solid state [10], multiple fluorescence colors [11,12,13,14,15], and reversible mechanofluorochromic properties [16, 17]. In particular, BF2DBM based on the 4-tert-butyl-4′-methoxydibenzoylmethane (avobenzone) boron difluoride complex (BF2AVB) exhibits different emission depending on the crystal phase (polymorph) [18]. BF2AVB also has excellent fatigue resistance by photoirradiation and a high fluorescence quantum yield, even in the solid state (~0.5), which is advantageous for photonics applications.
In this chapter, the fluorescence properties of BF2DBM in PMMA films and solution during evaporative crystallization were investigated to reveal the two-step nucleation model.
3.1 Fluorescence Visualization of Crystallization of Dibenzoylmethanatoboron Difluoride Complex
We have investigated the fluorescence properties of 4,4′-di-tert-butyldibenzoylmethanatoboron difluoride (BF2DBMb, Fig. 2.4a) in PMMA films and solution during evaporative crystallization to reveal the two-step nucleation model [19]. BF2DBMb has a mechanofluorochromic property, [11, 20] which originates from the different emission properties between the amorphous state and the crystal one [17]. The two-step nucleation model can be clarified by fluorescence detection, such that the detection of the amorphous state prior to crystallization by fluorescence color change can be expected. As described above, Yu et al. more recently reported the amorphous-to-crystalline transformation monitored by the fluorescence color change [5].

Reprinted by permission from Macmillan Publishers Ltd.: Ref. [19], copyright 2016
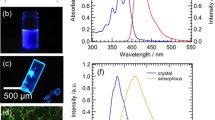
a Molecular structure of BF2DBMb. Fluorescence images of BF2DBMb in b 1,2-dichloromethane, c crystalline state, and d amorphous state under 365 nm UV irradiation, e absorption and fluorescence spectra of BF2DBMb, and f fluorescence spectra of crystal and amorphous states of BF2DBMb following excitation at 380 nm.
First, we confirmed the fluorescence properties of BF2DBMb in dilute solution, crystal and amorphous states, the fluorescence images of which are exhibited in Fig. 2.4b–d. The fluorescence exhibits purple, blue, and greenish-orange colors, for the dilute solution, crystal state, and amorphous state, respectively. The absorption and fluorescence spectra of BF2DBMb in 1,2-dichroloethane are shown in Fig. 2.4e. The absorption peaks were observed at 350, 370, and 390 nm and were in a mirror image of the fluorescence spectra with peaks at 413 and 430 nm and shouldered at 460 nm, which can be assigned to the vibrational modes of BF2DBMb monomer. The fluorescence showed peaks near 445 and 470 nm for the crystal, and near 550 nm for the amorphous state, as shown in Fig. 2.4f. The crystal and amorphous states were confirmed by XRD measurement. These findings indicate that molecular forms, such as monomer (isolated state) and aggregated state, can be distinguished by fluorescence color change.
Up to now, we have reported the evolution of the molecular assembling by fluorescence change in polymer matrix, which can provide information on the aggregation and segregation processes. To confirm the molecular assembling of BF2DBMb in polymer matrix (poly(methyl methacrylate), PMMA) by static trail, the concentration dependence of the fluorescence changes was as shown in Fig. 2.5. The close-up picture of the fluorescence image uniformly shows blue in 0.01 mol%. With increasing concentration, the fluorescence color changes to greenish-orange via light blue until 3.0 mol%. The needle with sky blue emission was confirmed for film of over 4.0 mol%, for which the segregation of BF2DBMb crystal in the polymer film most probably occurred. At the lower concentrations of 0.01 mol%, BF2DBMb fluorescence bands were observed near 430 nm, originating from the monomer as shown in Fig. 2.4e. The fluorescence peak was red-shifted until 0.5 mol%. At a concentration of 1.0 mol%, the fluorescence peak was located at 450 nm concomitant with a broad new fluorescence band appearing near 530 nm as a shoulder. Although the broad emission band near 530 nm increased with increasing concentration up to 3.0 mol%, it suddenly disappeared, and the fluorescence peak was shifted to the wavelength near 450 nm. The fluorescence spectrum is identical to that of the emission from the BF2DBMb crystal shown in Fig. 2.6f. These phenomena imply that the molecular assembling can be isolated or frozen by the polymer matrix, the dynamics of which revealing the formation of the crystal from the monomer state via the amorphous state of BF2DBMb. These findings are supported by the XRD results in polymer films. The fluorescence excitation spectra of BF2DBMb in PMMA films indicate the exciton splitting of the band, suggesting the formation of H- and J-aggregates with increasing concentration. Based on the band splitting, the average number of aggregated molecules was estimated to be 12 molecules.

Reprinted by permission from Macmillan Publishers Ltd.: Ref. [19], copyright 2016
Close-up fluorescence images of BF2DBMb in PMMA films with a concentration of a 0.01, b 0.05, c 0.1, d 0.5, e 1, f 2, g 3, and h 4 mol%, and i fluorescence spectra as a function of concentration obtained by excitation at 380 nm.

Reprinted by permission from Macmillan Publishers Ltd.: Ref. [19], copyright 2016
a Fluorescence images of a droplet of BF2DBMb in 1,2-dichloroethane during evaporation under 365 nm UV irradiation. The droplet diameter is approximately 5 mm. b Changes in fluorescence spectra of BF2DBMb in 1,2-dichloroethane during solvent evaporation.
Next, we attempted to measure the fluorescence change during evaporative crystallization from solution. The detection of the molecular assembling through the fluorescence changes can be expected, particularly liquid-like clusters, such as the amorphous aggregated state. Figure 2.6a shows the fluorescence images during the solvent evaporation from 3.1 × 10–2 mol dm−3 BF2DBMb in a 1,2-dichloroethane droplet, and the movie of which is shown in the supporting information [19]. In this trial, the time origin is set to after the complete focusing of the CCD camera, which describes the reason for the indicated time difference between the movie and the spectral change described below. At a time of 0 s, the fluorescence color is purple. The emission color changes to orange from the edge of droplet after 25 s. With the elapse of time, the emission color transiently becomes orange all over the droplet at approximately 32 s. The solvent evaporated, which resulted in a doughnut-shaped orange emission formed at approximately 34 s. The region of purple emission shrank from both inside and outside after 34 s. Finally, the entire droplet region turned to blue emission with small parts of the orange emission remaining. The evaporation of solvent from the inner region most probably originates from the analogous mechanism to gas bubbles, which has been reported for the molecular assembling during the evaporation of low vapor pressure solvent [21]. The orange emission is exhibited only in solution with non-equilibrium state. Actually, the concentration dependence of fluorescence spectra of BF2DBMb in solution does not exhibit orange emission, even under concentrated conditions. It is strongly suggested that the molecular form or assembly with orange emission can be like a supersaturated solution. To obtain the spectroscopic information for solvent evaporation, we observed the fluorescence spectra of BF2DBMb in 1,2-dichroloethane during evaporation as a function of time, which are shown in Fig. 2.6b. The fluorescence spectrum acquired immediately after applying the droplet exhibits peaks at 433 nm with shoulders at 415, 460, 550 nm, corresponding to the emission spectra of the monomer state. The peak near 550 nm corresponding to the amorphous state monotonically increased with time up to 91 s. The fluorescence peak at 433 nm decreased from 91 to 95 s. After 95 s, the peaks near 445 and 470 nm appeared concomitant with decreasing band intensity near 550 nm. The series of fluorescence spectral changes correspond to the fluorescence image change. Based on the information of the fluorescence properties of BF2DBMb as described above, we can explain the molecular assembling by solvent evaporative crystallization. The crystal of BF2DBMb formed from solution via the amorphous state. The dynamic fluorescence change is identical to observations when increasing the concentration in PMMA films during static trials. The Raman spectra of each species were identical to the fluorescence spectral change, which strongly supports the changes in molecular species in the ground state.
The fluorescence spectra were analyzed by nonlinear least squares fitting by six Gaussians. All of the observed spectra matched these values well. Thus, we plotted the relative abundances of monomer, crystal, and amorphous state as a function of time, which is shown in Fig. 2.7a. Afterward, the dropping the fraction of monomer species fell to approximately 0.9. The monomer fraction monotonically decreased, whereas the amorphous fraction increased up to 95 s. The amorphous fraction reached approximately 0.6 at 95 s; then, the fraction decreased considerably. The crystal fraction was not observed before 95 s. The fraction suddenly increased after 95 s, concomitant with the decrease of the amorphous state. These findings indicate that the crystal can be formed from monomer species via the amorphous state, which is presumed to show hierarchical change like a consecutive reaction, as schematically shown in Fig. 2.7b.

Reprinted by permission from Macmillan Publishers Ltd.: Ref. [19], copyright 2016
a Change in the relative abundance of monomer, amorphous, and crystal states based on time-resolved fluorescence spectral measurements. b Schematic representation of the molecular assembly based on the changes in fluorescence spectra.
Based on the observed phenomena by fluorescence change both depending on the dispersion concentration in PMMA films and during the solvent evaporative crystallization of BF2DBMb, we can conclude that the direct visualization of proposed a mechanism of the two-step nucleation model. The fluorescence color change from purple to blue via orange corresponds to the molecular formation change from monomer to crystal via amorphous state. The amorphous state is transiently formed prior to the crystal formation. The two-step nucleation model suggests that there is the liquid-like cluster as a crystal nucleus, which has been established based on the induction time of crystal formation [22], NMR spectroscopy [23, 24], electron microscopy [25], and non-photochemical laser-induced crystallization [26]. In the present case, the observed orange emission originating from amorphous species demonstrates the existence of the liquid-like cluster before crystallization, the state of which is only in the supersaturated region during the solvent evaporation, i.e., the non-equilibrium state. The time evolution of the relative abundance of the molecular form of BF2DBMb clearly reveals that the formation of the amorphous species acts as a precursor to crystal formation. We have verified that the fluorescence visualization during solvent evaporative crystallization agrees with the previously known two-step model for crystal formation [2, 27].
3.2 Changes in Optical and Mechanical Properties During Evaporative Crystallization
Recently, there have been many reports supporting the two-step nucleation model, which is relatively understood, with the intermediate phases playing an important role in crystallization [28, 29]. Tsarfati et al. mentioned the crystallization paths involving the three main stages—initial densification from the solvent-rich precursor, early ordering, and concurrent evolution of order and morphology [30]. This finding indicates that the liquid-like cluster state contains the solvent in the solution. It is unknown that the orange emission of BF2DBM originates from the amorphous phase as either highly dense solvent-rich state or solid aggregates. Based on the above background, we focus on the changes in optical and mechanical properties during evaporative crystallization in real time, particularly to clarify the orange emissive state as the intermediate phase [31].
To assess the optical and mechanical properties of the mutual state during evaporative crystallization in real time, we considered the droplet observation under the crossed Nicol condition [32]. The quartz crystal microbalance (QCM) is a tool that is used to perform real-time monitoring of the mass and the viscoelastic changes based on the change in the frequency (∆f) and resistance (∆R) from the adsorption onto the quartz substrate [33]. It is utilized in the evaluation of the deposited film thickness in the vacuum deposition.
We first observed the fluorescence images and polarized optical image, simultaneously, during the solvent evaporation. Figure 2.8 shows the photographs of the droplet between the polarizer under the crossed Nicol condition, with (upper side) and without (lower side) UV irradiation during the solvent evaporation. The polarized optical images with UV irradiation correspond to the fluorescence color changes of the droplet, which exhibits a purple color just after the dropping. However, there was no transparency in the image without UV irradiation, indicating that the droplet of BF2DBMb solution has no crystalline region. With time, the fluorescence color changed to orange from the peripheral of the droplet; this change was associated with the solvent evaporation from 20 to 40 s after the dropping, which can be assigned to the emission of the amorphous phase according to the previous report. After 60 s, birefringence was observed via the polarized optical image, and the texture in both the polarized optical images with and without UV irradiation gradually propagated throughout the droplet. The texture in the images implies the phase transformation of the amorphous state, followed by the formation of a crystalline state. Compared with both images at 85 s, the region with orange emission shows no birefringence, suggesting the optically isotropic phase. We have postulated that the amorphous phase as an intermediate can be identical to the liquid-like cluster state proposed in the two-step nucleation model of the crystallization process. These findings indicate that the photophysical and optical properties of the liquid-like cluster state have an isotropic aggregated state similar to an amorphous phase.

From [31]. Reprinted with permission from Chemical Society of Japan
Photographs of the BF2DBMb in 1,2-DCE droplet between the polarizer arranged under the cross-Nicol condition together with and without UV irradiation during the solvent evaporation.
Next, we attempted QCM measurements during the evaporative crystallization of the droplet. Prior to discussing the results of the QCM measurements, to confirm that the Au electrode does not affect the fluorescence behavior in the evaporative crystallization of the droplet, we measured the fluorescence images and spectra during the solvent evaporation on Au-coated AT-cut quartz substrate, simultaneously. The behavior of the fluorescence spectral changes of BF2DBMb droplet onto Au electrode is almost the same as that in the previous report, indicating that Au electrode does not affect the spectral changes that occur during solvent evaporation, such as the surface plasmon resonance of Au thin film [34, 35].
We attempted the QCM measurements adapted for the evaporative crystallization of the BF2DBMb droplet, to assess the changes in the dynamic viscoelastic properties. First, to confirm the effects of just solvent evaporation, we measured ∆f and ∆R changes by the evaporation of 1,2-DCE as a function of time. Both values indicate the amount of change from before the dropping. Just after dropping, ∆f was −2 kHz and began to return to the initial value from approximately 50–156 s. Meanwhile, ∆R exhibited behavior similar to that of ∆f just after dropping and then monotonically recovered from 93 to 156 s. Changes in both values correspond to the solvent mass change ∆m based on the Sauerbrey equation described below; therefore, recovery to the initial values suggested that the solvent on the Au electrode fully evaporated with time.
Next, we performed the QCM measurements of BF2DBMb in the 1,2-DCE solution droplet on the Au-coated At-cut quartz electrode. Figure 2.9a shows the changes in ∆f and ∆R as a function of time after the dropping of the solution on the Au electrode. Just after dropping, ∆f showed a value of −2 kHz until 70 s, which is comparable to that in the 1,2-DCE solvent. From 70 to 86 s, ∆f temporarily decreased to −4.5 kHz and then reached −6.9 kHz at 95 s. This value was maintained constant between 95 and 107 s. Afterward, ∆f decreased again, ultimately exhibiting a value of −13.8 kHz. Just after dropping, ∆R exhibited a value of 0.37 kΩ until 85 s, which is also comparable to that in the 1,2-DCE solvent. From 85 to 93 s, ∆R temporarily increased to 1.2 kΩ and then steeply increased to 1.6 kΩ until 115 s. Afterward, ∆R is slightly decreased to 1.5 kΩ. As an overall trend, it is found that both values changed in two steps during the evaporative crystallization. It is possible to identify the three main stages concerning the fluorescence changes of BF2DBMb (purple to blue via orange emission).

From [31]. Reprinted with permission from Chemical Society of Japan
Time evolution of a QCM results for ∆f and ∆R, b the mass change estimated by using the Sauerbrey equation, and c −∆f/∆R during the solvent evaporation of BF2DBMb in 1,2-DCE solution.
The change in f is related to the mass change (∆m) based on the Sauerbrey equation as shown in the following equation [36],
where f0, A, μq, and ρq are the resonant frequency, piezoelectrically active crystal area, density of quartz, and shear modulus of quartz for AT-cut crystal, respectively. In the present system, we used the following values: f0 = 8.947 MHz, A = 1.96 × 10−5 m2, μq = 2.95 × 1010 kg m−1 s−2, ρq = 2.65 × 103 kg m−3. Based on Eq. 2.1, we can estimate the ∆m of the BF2DBMb droplet during the evaporative crystallization, the time evolution of which is shown in Fig. 2.9b. Just after dropping, ∆m was 2 μg until 80 s, a value comparable with that for just 1,2-DCE solvent, which indicates that the Au electrode can recognize only the mass of the adsorbed solvent. From 80 to 86 s, ∆f temporarily decreased to 7.3 μg and then reached 15 μg at 95 s. The increase of ∆m is ascribed to the adsorption and precipitation of BF2DBMb onto the Au electrode. However, the estimated solute mass of the BF2DBMb solution was 34.65 μg, which is much larger than the estimated ∆m value. This causes the spreading out of the effective area of Au electrode and the viscoelastic property of the droplet as described the next paragraph. It is difficult to drop a smaller amount of solution within the electrode area, owing to the surface tension of the droplet.
The QCM results can be used to evaluate not only the ∆m but also the viscoelastic properties of the adsorbed materials. Kanazawa et al. reported that the ∆f with homogeneous viscous fluids is proportional to the square root of the viscosity coefficients [37]. Muramatsu et al. also represented the linear relation between ∆R and the square root of the viscosity coefficient [38]. However, in terms of ∆f for the viscoelastic medium, it is complicated to evaluate both the viscosity and elasticity separately, because ∆f depends both on the ∆m and viscosity. Based on the above findings, Kubono et al. semi-quantitatively formulated the relation assuming that the ∆f is the sum of the two contributions from the ideal frequency shift by adsorbed mass (∆fm) associated with the elasticity and that by viscous fluid (∆fv) with constants (a and b) as follows [39],
To estimate the contribution of the elasticity, the ratio −∆f/∆R can be suitable for the separation of viscoelastic properties from the ∆f value
where a′ and b′ are constants. If the viscous liquid adsorbs onto the electrode, a′ should be zero and −∆f/∆R is constant, which is independent of the viscosity coefficient. With an increase in elasticity, a′ increases, and −∆f/∆R becomes larger, which indicates that the two contributions (∆fm and ∆fv) to ∆f can be separated as the mass change and viscosity change. If ∆f decreased but −∆f/∆R remained constant, for example, this change would be attributed to the viscosity change, rather than the mass change.
The evaporation of 1,2-DCE led to a −∆f/∆R value of approximately 5.5, which indicates that the solvent adsorbed onto the electrode. Figure 2.9c shows changes of ∆f and ∆R as a function of time after the dropping of BF2DBMb in 1,2-DCE solution. The −∆f/∆R value was maintained at 5.5 from just after dropping until 70 s, which is comparable to that in 1,2-DCE solvent. From 70 to 86 s, −∆f/∆R temporarily increased to 9.2 at 84 s and then recovered to 5.5 until 112 s. Finally, the −∆f/∆R value increased to 9.2 from 114 to 134 s.
Crystallization occurs in the monomer molecular assemblies via the formation of the liquid-like cluster state with amorphous property, proposed by the two-step nucleation model, as described in the previous section. It is expected that the mechanical property will change from a viscous fluid to elastic media during the crystallization, that is, the −∆f/∆R value will increase after crystallization together with the fluorescence color change from orange to blue. In the early state after the dropping, the −∆f/∆R value indicates a viscous fluid owing to a large amount of solvent, which is considered to be due to the adhesion of the solvent to the substrate surface. This indicates that the molecule is dispersed in the solution and has a large amount of solvent attached to the substrate surface. The peak shape at 84 s of the −∆f/∆R value is considered to be due to the artifact caused by the adhesion of the aggregated amorphous state to the Au electrode, because ∆m also increased rapidly at the same time. The fluorescence color of the droplet at 84 s was orange, suggesting the formation of the amorphous phase. The −∆f/∆R value from 91 to 111 s was the same as that from 0 to 80 s, suggesting that the amorphous state has a similar viscosity as the solution. The sudden rise of −∆f/∆R at 111 s is ascribed to the transition to the elastic crystals.
We have reconsidered and proposed the schematic representation of the evaporative crystallization process combined with the two-step nucleation model and the present experimental results in terms of the viscoelastic properties, as shown in Fig. 2.10. Just after the dropping onto a substrate, the solute molecules exist in a monomeric state. With the elapse of time for the solvent evaporation, the monomer molecules aggregate with each other. The condensed monomer molecules form an amorphous state with an optically isotropic and viscous fluid; then, it adsorbs onto the substrate, which corresponds to a liquid-like cluster. The monomer molecules in the solvent are further adsorbed onto the amorphous state by solvent evaporation because the ∆m continually increases with time. It is also found that in the amorphous phase observed in the QCM measurement a solvent-containing state most probably exists during the evaporative crystallization stage based on the viscoelastic properties of the amorphous phase. It is suggested that the difference between the amorphous state and the liquid-like cluster state originates from the presence of the solvent in the aggregate.

From [31]. Reprinted with permission from Chemical Society of Japan
Schematic of the evaporative crystallization of BF2DBMb based on polarized optical images and viscoelastic measurements.
In previous reports, the relationship between the liquid-like cluster and amorphous state was only based on the fluorescence color. The results of the polarized optical images and QCM measurements indicate that the liquid-like cluster as an intermediate observed during the evaporative crystallization has high viscosity, despite the similar emission of the amorphous solid. The result of the two-step mass changes suggests that the liquid-like cluster has a finite size and then undergoes the phase transition to a crystal with desolvation during growth.
4 Molecular Assembling Probed by Aggregation-Induced Emission
Over the past decade, there have been many reports on aggregation-induced (or enhanced) emission (AIE or AIEE) in organic molecules [40], even though the fluorescence quantum yield is low in solution, as reported by Hong et al. [41]. AIEE enables selective detection of the assembly dynamics for aggregates and crystals. Here, for the first time, we apply AIEE to the dynamics of crystal formation. Specifically, we use a cyanostilbene derivative [1-cyano-trans-1,2-bis-(4′-methylbiphenyl)-ethylene (CN-MBE; Fig. 2.11)] for AIEE, as reported by An et al. [42]. Using a fluorescence microscope, we characterized the spectral and intensity changes of CN-MBE emission in solution during solvent evaporation [43, 44].

Molecular structure of CN-MBE
The absorption and fluorescence spectra of CN-MBE in solution and in nanoparticles were reported previously [42]. To suppress molecular rotation and aggregation for the observation monomer fluorescence, spectra of cast films of CN-MBE in PMMA were measured as a function of CN-MBE concentration (a polymer matrix in dilute conditions). Figure 2.12 plots normalized fluorescence spectra from 330-nm excitation of these films cast from DCE solutions. At concentrations <0.1 mol%, peak emission was observed at 440 nm, with a shoulder at 420–430 nm. At increased concentrations, the fluorescence peak shifts to 470 nm and narrows. Aggregate species were observed with the fluorescence microscope at concentrations >1.0 mol%. Thus, the spectral shift and narrowing of the fluorescence originated from CN-MBE J-aggregates, according to the previous report [42]. To identify the emissive species at lower concentrations, excitation spectra of the CN-MBE/PMMA films were acquired, indicating a broad excitation spectrum at 355 nm for concentrations <0.1 mol% is most likely a combination of the planar and twisted conformers that coexist in the films as a monomer.

Reproduced from Ref. [43] by permission of The Royal Society of Chemistry
Fluorescence spectra of CN-MBE in PMMA films as a function of concentration (excitation at 330 nm).
To confirm AIEE in the increased fluorescence intensity during crystal formation, we acquired fluorescence microscope images during solvent evaporation from 5.0 × 10–3 mol dm−3 CN-MBE in a DCE droplet (Fig. 2.13). During the initial 75 s, no emission was observed from the droplet. A violet-blue feature first appeared after 90 s. More features appeared over time, aggregated around the initial feature and accompanying the increased fluorescence intensity, until the completion of the evaporation at 100 s. These images indicated that CN-MBE emission in the solution phase was very weak. The polarization microscopy images were obtained with crossed Nicol polarizers, indicating the crystal formation. These observations suggest that AIEE allows us to follow crystal formation by changes in the fluorescence intensity. We can thus characterize the dynamics of organic crystal formation by the fluorescence intensity changes.

Reproduced from Ref. [43] by permission of The Royal Society of Chemistry
Fluorescence microscope images as a function of time for 5.0 × 10–3 mol dm−3 CN-MBE in a DCE droplet during solvent evaporation.
To investigate the molecular dynamics of crystal formation, fluorescence spectra were acquired during solvent evaporation for droplets of 5.0 × 10–3 mol dm−3 CN-MBE in DCE (Fig. 2.14a). As above, the fluorescence intensity was weak just after the drop was formed. With time, the fluorescence intensity gradually increased by a factor of 100; normalized spectra are shown in Fig. 2.14b. After 97 s, the peak emission was at 480 nm, with a shoulder at 440 nm. Other peaks that originate from the monomer structure are observed at 443, 474, and 505 nm. The spectra became narrower with time, especially over 410–450 nm, as the solvent completely evaporated, confirmed by microbalance. After 112 s, the spectra are identical to those in Fig. 2.12, suggesting that solvent evaporation results in the formation of molecular assemblies owing to increasing concentrations of CN-MBE.

Reproduced from Ref. [43] by permission of The Royal Society of Chemistry
Fluorescence spectral changes (a) and normalized spectra (b) of 5.0 × 10–3 mol dm−3 CN-MBE in a DCE droplet during solvent evaporation as a function of time. c The observed spectra after 97 s and simulated spectra from summation of the relative abundance of the planar conformational monomer and the J-aggregates by least-square fitting.
The fluorescence spectra were analyzed as a function of concentration as shown in Fig. 2.14c. We assumed that the spectra are from two species: a planar conformational monomer and J-aggregates. The spectrum of J-aggregates was from CN-MBE crystals. The spectrum of the planar conformational monomer was calculated from the difference between observed spectra at different time intervals. The observed spectra at each time were reproduced by nonlinear least squares fitting of the summation of emission from the planer monomer and J-aggregates.
Figure 2.15 shows a plot of the time evolution of the total fluorescence intensity of CN-MBE and the relative abundance of J-aggregates in a DCE droplet during solvent evaporation. The total fluorescence intensity significantly and monotonically increased after 98 s and became constant after 110 s. The relative abundance of the J-aggregates increased after 97 s and became constant (0.97) after 102 s. Therefore, the rate of J-aggregate formation is faster than the increase in total fluorescence intensity, indicating a time lag for the increase in fluorescence intensity for J-aggregates. The time at which a constant fraction of J-aggregates is reached (102 s) appears to coincide with the half maximum of the total fluorescence intensity (dashed vertical line as shown in Fig. 2.15). As mentioned above, the total fluorescence intensity is probably attributable to the amount of CN-MBE crystals, which implies that fluorescence spectral and intensity changes reflect crystal nuclei formation and crystal growth. This phenomenon was observed not only in the solvent evaporation process, but also for the water fraction dependence of the nanoparticle formation for CN-MBE, which supports our findings.

Reproduced from Ref. [43] by permission of The Royal Society of Chemistry
Change in fluorescence intensity (red circles) and relative abundance of J-aggregate (blue circles) of CN-MBE as a function of time. The solid line is a curve fit based on the sigmoidal function (see main text).
On the basis of classical nucleation theory, the time evolution of CN-MBE fluorescence spectra during solvent evaporation can be used to model droplet growth [45, 46]. Initially, the concentration in solution was relatively low; thus, an equilibrium state was established between the monomer and J-aggregates. The J-aggregates begin from the smallest cluster of two monomers formed via intermolecular interactions. This small cluster is unstable because of the unfavorable surface free energy and dissociates before crystal formation. As the concentration increases by solvent evaporation, there are aggregates (concentration fluctuations) in supersaturated solutions. As apparent in Fig. 2.15, the abundance of J-aggregates during solvent evaporation indicates the formation of subcritical clusters. In general, the nucleus formation rate J is given by the Arrhenius reaction rate equation: J = Aexp(−∆G/kBT), where kB is Boltzmann’s constant, A is the pre-exponential factor, and ∆G is the Gibbs energy of cluster formation. Because of the energy barrier, critical nuclei formation is a competition between growth and dissolution. Therefore, the time lag between the J-aggregate abundance, and the total fluorescence intensity indicates that the growth from J-aggregates to crystal nuclei is the rate-determining step of nucleation. Whether nuclei formation occurs depends on whether there is classical nucleation theory or not, although clearly only in the present results.
Amyloid fibril formation has been probed with thioflavin T (ThT) fluorescent dye [47], which is essentially nonfluorescent in solution [48]. An interaction with, or binding to, the amyloid fibril results in fluorescence enhancement; thus, ThT is a powerful tool for studying the kinetics of fibril formation, which is analogous to the organic crystal formation. We can use the fibrillation kinetics to model the initial stage of CN-MBE crystal formation. A sigmoidal function can be used to obtain an estimate of the time required for aggregation to level off [49],
where I is fluorescence intensity, Ibase and Imax are, respectively, the fluorescence intensity before and after the change, k is the apparent rate constant for the growth, and thalf is the time to half of maximal fluorescence. The time evolution of the fluorescence intensity and the relative abundance of J-aggregates were well reproduced by Eq. 2.1. The larger values of k and thalf for growth of the J-aggregates relative to those for the fluorescence intensity strongly support the notion that J-aggregates act as precursors for crystal nuclei. The solvent dependence for the time evolution depends on the kinetic values associated with the boiling point and polarity of solvents, although it did not depend on the sigmoidal behavior. It is suggested that the molecular assembly kinetics mainly govern the increase in the local concentration of the droplets. The initial stages of crystal nuclei and crystal formation are schematically depicted in Fig. 2.16.

Reproduced from Ref. [43] by permission of The Royal Society of Chemistry
Schematic of CN-MBE crystal formation based on changes in fluorescence spectra.
5 Summary
The present method allows crystal formation to be observed using a conventional optical detection system under ambient conditions, making it attractive to study the control of polymorphism of organic emissive materials with multiple emissive states or colors depending on their phase, such as mechanofluorochromic materials.
The study has clearly confirmed that the two-step nucleation model is based on the fluorescence color change. The intermediate state, such as the liquid-like cluster, is an important indicator of polymorphic expression, the origin of which will be key to understanding. The present method has low requirements for experimental equipment because the crystal formation can be observed using a conventional optical detection system under the ambient atmosphere, especially the control of polymorphism for organic emissive materials with multiple emissive states or color depending on the phase such as mechanofluorochromic molecules. The method represents a direct visualization of Ostwald’s rule of stages during the phase change of organic molecular solids, including the inhomogeneous phenomena.
References
Wolde, P.R., Frenkel, D.: Enhancement of protein crystal nucleation by critical density fluctuations. Science 277, 1975–1978 (1997)
Erdemir, D., Lee, A.Y., Myerson, A.S.: Nucleation of crystals from solution: classical and two-step models. Acc. Chem. Res. 42, 621–629 (2009)
Vekilov, P.G.: The two-step mechanism of nucleation of crystals in solution. Nanoscale 2, 2346–2357 (2010a)
Schwoerer, M., Wolf, H.C.: Organic Molecular Solids. Wiley-VCH, Weinheim, Germany (2007)
Ye, X., Liu, Y., Lv, Y., Liu, G., Zheng, X., Han, Q., Jackson, K.A., Tao, X.: In situ microscopic observation of the crystallization process of molecular microparticles by fluorescence switching. Angew. Chem. Int. Ed. 54, 7976–7980 (2015)
Tran, V.L., Genot, V., Audibert, J.F., Prokazov, Y., Turbin, E., Zuschratter, W., Kim, H.J., Jung, J., Park, S.Y., Pansu, R.B.: Nucleation and growth during a fluorogenic precipitation in a micro-flow mapped by fluorescence lifetime microscopy. New J. Chem. 40, 4601–4605 (2016)
Zheng, C., Zang, Q., Nie, H., Huang, W., Zhao, Z., Qin, A., Hu, R., Tang, B.Z.: Fluorescence visualization of crystal formation and transformation processes of organic luminogens with crystallization-induced emission characteristics. Mater. Chem. Front. 2, 180–188 (2018)
Halik, M., Wenseleers, W., Grasso, C., Stellacci, F., Zojer, E., Barlow, S., Bredas, J.L., Perry, J.W., Marder, S.R.: Bis(dioxaborine) compounds with large two-photon cross sections, and their use in the photodeposition of silver. Chem. Commun. 1490–1491 (2003)
Cogne-Laage, E., Allemand, J.F., Ruel, O., Baudin, J.B., Croquette, V., Blanchard-Desce, M., Jullien, L.: Diaroyl(methanato)boron difluoride compounds as medium-sensitive two-photon fluorescent probes. Chem. Eur. J. 10, 1445–1455 (2004)
Ono, K., Yoshikawa, K., Tsuji, Y., Yamaguchi, H., Uozumi, R., Tomura, M., Taga, K., Saito, K.: Synthesis and photoluminescence properties of BF2 complexes with 1,3-diketone ligands. Tetrahedron 63, 9354–9358 (2007)
Zhang, G.Q., Lu, J.W., Sabat, M., Fraser, C.L.: Polymorphism and reversible mechanochromic luminescence for solid-state difluoroboron avobenzone. J. Am. Chem. Soc. 132, 2160–2162 (2010)
Zhang, G.Q., Singer, J.P., Kooi, S.E., Evans, R.E., Thomas, E.L., Fraser, C.L.: Reversible solid-state mechanochromic fluorescence from a boron lipid dye. J. Mater. Chem. 21, 8295–8299 (2011)
Samonina-Kosicka, J., DeRosa, C.A., Morris, W.A., Fan, Z.Y., Fraser, C.L.: Dual-emissive difluoroboron naphthyl-phenyl beta-diketonate polylactide materials: effects of heavy atom placement and polymer molecular weight. Macromolecules 47, 3736–3746 (2014)
Sakai, A., Tanaka, M., Ohta, E., Yoshimoto, Y., Mizuno, K., Ikeda, H.: White light emission from a single component system: remarkable concentration effects on the fluorescence of 1,3-diaroylmethanatoboron difluoride. Tetrahedron Lett. 53, 4138–4141 (2012)
Sakai, A., Ohta, E., Yoshimoto, Y., Tanaka, M., Matsui, Y., Mizuno, K., Ikeda, H.: Novel fluorescence domain “excited multimer” formed upon photoexcitation of continuously-stacked diaroylmethanatoboron difluoride molecules with fused π-orbital in crystals. Chem. Eur. J. 21, 18128–18137 (2015)
Sagawa, T., Ito, F., Sakai, A., Ogata, Y., Tanaka, K., Ikeda, H.: Substituent-dependent backward reaction in mechanofluorochromism of dibenzoylmethanatoboron difluoride derivatives. Photochem. Photobiol. Sci. 15, 420–430 (2016)
Ito, F., Sagawa, T.: Quantitative evaluation of thermodynamic parameters for thermal back-reaction after mechanically induced fluorescence change. RSC Adv. 3, 19785–19788 (2013)
Zhang, G., Chen, J., Payne, S.J., Kooi, S.E., Demas, J.N., Fraser, C.L.: Multi-emissive difluoroboron dibenzoylmethane polylactide exhibiting intense fluorescence and oxygen-sensitive room-temperature phosphorescence. J. Am. Chem. Soc. 129, 8942–8943 (2007)
Ito, F., Suzuki, Y., Fujimori, J., Sagawa, T., Hara, M., Seki, T., Yasukuni, R., de la Chapelle, M.L.: Direct visualization of the two-step nucleation model by fluorescence color changes during evaporative crystallization from solution. Sci. Rep. 6, 22918 (2016)
Galer, P., Korosec, R.C., Vidmar, M., Sket, B.: Crystal structures and emission properties of the BF2 Complex 1-Phenyl-3-(3,5-dimethoxyphenyl)-propane-1,3-dione: multiple chromisms, aggregation- or crystallization-induced emission, and the self-assembly effect. J. Am. Chem. Soc. 136, 7383–7394 (2014)
Schenning, A., Benneker, F.B.G., Geurts, H.P.M., Liu, X.Y., Nolte, R.J.M.: Porphyrin wheels. J. Am. Chem. Soc. 118, 8549–8552 (1996)
Knezic, D., Zaccaro, J., Myerson, A.S.: Nucleation induction time in levitated droplets. J. Phys. Chem. B 108, 10672–10677 (2004)
Hughes, C.E., Harris, K.D.M.: A technique for in situ monitoring of crystallization from solution by solid-state C-13 CPMAS NMR spectroscopy. J. Phys. Chem. A 112, 6808–6810 (2008)
Hughes, C.E., Williams, P.A., Keast, V.L., Charalampopoulos, V.G., Edwards-Gau, G.R., Harris, K.D.M.: New in situ solid-state NMR techniques for probing the evolution of crystallization processes: pre-nucleation, nucleation and growth. Faraday Discuss. 179, 115–140 (2015)
Harano, K., Homma, T., Niimi, Y., Koshino, M., Suenaga, K., Leibler, L., Nakamura, E.: Heterogeneous nucleation of organic crystals mediated by single-molecule templates. Nat. Mater. 11, 877–881 (2012)
Garetz, B.A., Matic, J., Myerson, A.S.: Polarization switching of crystal structure in the nonphotochemical light-induced nucleation of supersaturated aqueous glycine solutions. Phys. Rev. Lett. 89, 175501 (2002)
Vekilov, P.G.: Nucleation. Cryst. Growth Des. 10, 5007–5019 (2010b)
Guo, C., Wang, J., Li, J., Wang, Z., Tang, S.: Kinetic Pathways and mechanisms of two-step nucleation in crystallization. J. Phys. Chem. Lett. 7, 5008–5014 (2016)
Zhang, T.H., Liu, X.Y.: How does a transient amorphous precursor template crystallization. J. Am. Chem. Soc. 129, 13520–13526 (2007)
Tsarfati, Y., Rosenne, S., Weissman, H., Shimon, L.J.W., Gur, D., Palmer, B.A., Rybtchinski, B.: Crystallization of organic molecules: nonclassical mechanism revealed by direct imaging. ACS Cent. Sci. 4, 1031–1036 (2018)
Ito, F., Saigusa, M., Kanayama, N.: Chem. Lett in press (2019)
Lu, F.N., Jang, K., Osica, I., Hagiwara, K., Yoshizawa, M., Ishii, M., Chino, Y., Ohta, K., Ludwichowska, K., Kurzydlowski, K.J., Ishihara, S., Nakanishi, T.: Supercooling of functional alkyl-p molecular liquids. Chem. Sci. 9, 6774–6778 (2018)
Ariga, K., Endo, K., Aoyama, Y., Okahata, Y.: QCM analyses on adsorption of gaseous guests to cast films of porphyrin-resorcinol derivatives. Colloids Surf. A 169, 177–186 (2000)
Lakowicz, J.R.: Radiative decay engineering: biophysical and biomedical applications. Anal. Biochem. 298, 1–24 (2001)
Ito, F., Ohta, R., Yokota, Y., Ueno, K., Misawa, H., Nagamura, T.: Near-infrared fluorescence enhancement by regularly arranged gold nanoblocks. Chem. Lett. 39, 1218–1219 (2010)
Sauerbrey, G.: Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Zeitschrift für Physik 155, 206–222 (1959)
Kanazawa, K.K., Gordon, J.G.: Frequency of a quartz microbalance in contact with liquid. Anal. Chem. 57, 1770–1771 (1985)
Muramatsu, H., Tamiya, E., Karube, I.: Computation of equivalent circuit parameters of quartz crystals in contact with liquids and study of liquid properties. Anal. Chem. 60, 2142–2146 (1988)
Kubono, A., Akiyama, R.: Viscoelastic analysis in the formation of organic thin films. Mol. Cryst. Liq. Cryst. 445, 213–222 (2006)
Tang, B.Z., Qin, A.: Aggregation-Induced Emission: Fundamentals. Wiley, United Kingdom (2013)
Hong, Y.N., Lam, J.W.Y., Tang, B.Z.: Aggregation-induced emission. Chem. Soc. Rev. 40, 5361–5388 (2011)
An, B.K., Kwon, S.K., Jung, S.D., Park, S.Y.: Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 124, 14410–14415 (2002)
Ito, F., Fujimori, J.I.: Fluorescence visualization of the molecular assembly processes during solvent evaporation via aggregation-induced emission in a cyanostilbene derivative. CrystEngComm 16, 9779–9782 (2014)
Ito, F., Fujimori, J., Oka, N., Sliwa, M., Ruckebusch, C., Ito, S., Miyasaka, H.: AIE phenomena of a cyanostilbene derivative as a probe of molecular assembly processes. Faraday Discuss. 196, 231–243 (2017)
Davey, R.J., Schroeder, S.L.M., ter Horst, J.H.: Nucleation of organic crystalsa molecular perspective. Angew. Chem. Int. Ed. 52, 2166–2179 (2013)
Okutsu, T.: Photochemically-induced crystallization of protein. J. Photochem. Photobiol. C 8, 143–155 (2007)
Buxbaum, J.N., Linke, R.P.: A molecular history of the amyloidoses. J. Mol. Biol. 421, 142–159 (2012)
Stsiapura, V.I., Maskevich, A.A., Kuzmitsky, V.A., Uversky, V.N., Kuznetsova, I.M., Turoverov, K.K.: Thioflavin T as a molecular rotor: fluorescent properties of Thioflavin T in solvents with different viscosity. J. Phys. Chem. B 112, 15893–15902 (2008)
Nielsen, L., Khurana, R., Coats, A., Frokjaer, S., Brange, J., Vyas, S., Uversky, V.N., Fink, A.L.: Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 (2001)
Acknowledgements
The authors thank Mr. Jun-ichi Fujimori, Ms. Yukino Suzuki, Ms. Mai Saigusa, and Prof. Naoki Kanayama (Shinshu University); Dr. Mitsuo Hara and Prof. Takahiro Seki (Nagoya University); and Dr. Ryohei Yasukuni, and Prof. Marc Lamy de la Chapelle (Paris 13 University) for their support for the experiment and kind discussion. We also thank Dr. Kennosuke Itoh (Shinshu University) for the NMR measurements. This work was partly supported by the Nanotechnology Platform Program, by JSPS KAKENHI Grants Numbers JP21750021, JP15H01081, JP17H05253, JP19H02686, by the Iketani Science and Technology Foundation, and by the JGC-S Scholarship Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ito, F. (2020). Direct Visualization of Crystal Formation and Growth Probed by the Organic Fluorescent Molecules. In: Sakamoto, M., Uekusa, H. (eds) Advances in Organic Crystal Chemistry. Springer, Singapore. https://doi.org/10.1007/978-981-15-5085-0_2
Download citation
DOI: https://doi.org/10.1007/978-981-15-5085-0_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5084-3
Online ISBN: 978-981-15-5085-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)