
Abstract
Crystal polymorphism has a significant effect on the luminescence of materials. Using 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP) as the key compound, crystal structure-dependent luminescence, also known as polymorph-dependent luminescence (PDL), is described. HPIP shows excited-state intramolecular proton transfer (ESIPT) luminescence, which is quite efficient in the solid state, yet very weak in solution; it is mainly ascribed to the suppression of efficient radiationless decay via the S0/S1–conical intersection when the molecular conformation is fixed coplanar. The two polymorphic forms of the crystals display blue-green and yellow luminescence and can be interconverted via heat transfer between the two states. Three-color PDL (yellow, orange, and red) is observed in 6-cyano HPIP. The relationship between crystal structure and luminescence color was rationally explained by TD-DFT calculations using dimer models. It is remarkable, that this dimer model is reasonably supported by the femtosecond time-resolved transient absorption spectroscopy. These results will contribute to the understanding of structure–property relationships in solid-state luminescence at the molecular level and would further the design of new polymorph-dependent luminescent materials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Luminescent crystal
- Polymorph-dependent luminescence
- Quantum chemical calculation
- ESIPT
- Fluorescence
- Femtosecond time-resolved transient absorption spectroscopy
1 Introduction
Organic solid-state luminescent materials have garnered considerable interest in various areas of science including OLEDs [1, 2], sensors [3], solid lasers [4], near-infrared light sources [5], and so on [6, 7]. In most cases, the electronic properties of materials are controlled via synthetic modification of luminescent molecules.
However, the luminescence of organic compounds is often quenched in the solid state, because the non-radiative deactivation pathway from the photoexcited state is enhanced through molecular interactions [8]. Common strategies to overcome this problem include the introduction of bulky groups [9,10,11] or design of non-planar structures [12,13,14] in order to avoid unfavorable molecular interactions. Therefore, these materials are primarily used as amorphous solids, and their luminescence is generally a reflection of that of a single molecule.
The luminescence of bulk solids is regulated not only by that of single molecules, but also by collective effects owing to the arrangement and interactions of small molecules. In other words, the luminescence of solid materials could be controlled by altering the molecular packing mode within the crystal structure, instead of by chemical modification.
Various examples of organic solid-state luminescent compounds have been developed in recent years. For example, aggregation-induced emission (AIE) [15, 16] is a phenomenon in which luminescence is enhanced rather than quenched by the aggregation of the molecules. In 2001, B. Z. Tang and co-workers reported [17] 1-methyl-1,2,3,4,5-pentaphenylsilole (Scheme 14.1a), which is non-luminescent in solution, exhibits remarkably enhanced emission (300 times) owing to the formation of nano-aggregates upon addition of a certain amount of a poor solvent. S. Y. Park and co-workers reported the enhanced luminescence of 1-cyano-trans-1,2-bis-(4′-methylbiphenyl)ethylene upon the formation of nanoparticles [18]. Tetraphenylethylene has also been a well-studied AIE-active core [19].

a Molecules displaying aggregation-induced emission (AIE). From left to right: silole, 1-cyano-trans-1,2-bis-(4′-methylbiphenyl)ethylene, and tetraphenylethylene. b Molecules displaying polymorph-dependent luminescence (PDL). From left to right: 2,2′:6′,2″-terpyridine, 3-(9-anthryl)pyrazole, and di(p-methoxyphenyl)dibenzofulvene
Most of AIE-active compounds are composed of two or more aromatic rings connected by a single bond or bonds. The aromatic rings can freely rotate in solution, and the excited state can be easily deactivated through thermal dissipation due to molecular rotation. In aggregates, where molecular rotation is suppressed, thermal deactivation is much less feasible and results in enhanced emission.
1.1 Polymorph-Dependent Luminescence (PDL)
Controlling the luminescence of solid materials by changing or altering the polymorphs, i.e., different molecular packing modes in the crystalline phase of a specific compound resulting from a different arrangement of the molecules within the solid state, is called polymorph-dependent luminescence (PDL) (Scheme 14.2).

Schematic representation of polymorph-dependent luminescence (PDL)
PDL has attracted considerable interest, because it can be used to modify the luminescence of materials and provides insight into the relationship between the molecular packing mode and bulk optical properties of said materials [20, 21]. Moreover, it may have applications in the development of new organic luminescent materials based on supramolecular chemistry. The most well-known is the stimuli–response emission, in which the emission properties are changed by external stimuli (heat, pressure, solvent vapor, and so on) without chemical modification.
The ability to tune the luminescence of materials by exploiting molecular packing has been studied extensively, and the number of reports on this subject has rapidly increased in recent years [22,23,24,25,26,27,28]. In these systems, alteration of inter-luminophore interactions [22,23,24,25] or packing-induced conformational changes of dye molecules [26,27,28] allows for the modulation of the luminescence.
One of the early examples of PDL-active molecules is 2,2′:6′,2″-terpyridine (tpy), which was reported in 2005 [29]; it is non-fluorescent in solution, but forms two polymorphic crystals, which show bright blue luminescence (quantum yield Φ ~ 0.2) and very weak luminescence (Φ < 0.01). Moreover, these two crystalline phases can be interconverted via heat-mode phase transfer, and the luminescence can be switched “on” or “off”. The other example is 3-(9-anthryl)pyrazole [30], which exhibits different colored PDL, owing to differences in the intermolecular hydrogen bonding between the pyrazolyl units. The PDL of di(p-methoxyphenyl)dibenzofulvene [31] is also a noteworthy example, where the twisted conformation of the two anisole planes is responsible for the PDL.
However, the development of such systems remains a challenge because of the lack of effective mechanisms that can modulate the molecular packing and lead to different luminescence properties.
Detailed studies on the relationship between the molecular packing mode and luminescence have also garnered significant interest. Systems with multiple-color PDL are desirable in order to study structure–property relationships; however, most reports to date describe two-color systems [32, 33]. Systems exhibiting three or more luminescence colors are rare [30, 34, 35]. One of the reasons for this is the difficulty in preparing multiple polymorphic phases and obtaining appropriate crystal data, because some phases often have amorphous or microcrystalline states.
2 Molecular Design
2.1 Designing Polymorph-Dependent Luminescent Compound
The ability to (i) form polymorphic phases and (ii) transduce conformational changes and molecular interactions into altered luminescence is required for a molecule to display PDL. It would be interesting if the polymorphic phases could be interconverted by external stimuli, because it may lead to luminescence-switchable materials.
Our strategy to realize the above points is to introduce adequate flexibility in the molecular conformation, especially around the aromatic luminescent core. As shown in Scheme 14.2, two π-electronic planes connected by a covalent single bond are “moderately” fixed with a weaker hydrogen bond, so that various conformations and packing modes could be tolerated during crystallization.
The second strategy is to take advantage of a characteristic luminescence that stands on the intramolecular hydrogen bond: in this case, N···H–O. It has been demonstrated that the phenolic proton transfers to the nitrogen atom promptly after photoexcitation, which is termed excited-state intramolecular proton transfer (ESIPT), and the generated excited species is deactivated upon emitting low-energy light (see next section for details). ESIPT luminescence, especially in our system, is supposed to be susceptible to the molecular conformation and the effects of surrounding molecules; therefore, different ESIPT luminescence might be expected in different polymorphic phases.
2.2 Mechanism of ESIPT Luminescence
A photoinduced ESIPT [36] is reported to be a remarkably fast process (rate constant k ≈ 1 × 1013 s−1) [37, 38]. In the ground state, the acidities (pKa) of phenolic–OH and hydrogen-bonded nitrogen are about 9 and 6, respectively; therefore, they may exist as an enol (N···H–O). Upon photoexcitation, the phenolic –OH becomes quite acidic (pKa ~ 2) and the proton transfer to the nitrogen (ESIPT) and subsequent tautomerization to the keto form are promoted (Scheme 14.3) [39]. Emission from the keto form is characterized by a large Stokes shift (≈10,000 cm−1), which enables long-wavelength fluorescence upon excitation with UV light. However, molecules which exhibit efficient ESIPT fluorescence are rather limited. The most studied compounds to date are 2-(2′-hydroxyphenyl)benzimidazole (HBI) [33, 40,41,42,43,44], 2-(2′-hydroxyphenyl) benzoxazole (HBO) [45,46,47,48], 2-(2′-hydroxyphenyl)benzothiazole (HBT) [49,50,51,52,53,54], salicylideneaniline [55,56,57], and their analogs.

Molecular structure of intramolecular hydrogen-bonded benzazoles (HBI, HBO, and HBT), and enol-to-keto tautomerization via ESIPT by photoexcitation
ESIPT luminescence in the solid state [33, 49, 58,59,60,61] is a significant topic, and there have been some recent reports of basic applications in electroluminescent devices [49, 62,63,64,65,66], as well as white luminescent materials [64,65,66,67,68,69,70].
2.3 2-(2′-Hydroxyphenyl)Imidazo[1,2-a]Pyridine (HPIP)
2-(2′-Hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP, 1Enol; Scheme 14.4a) is an analog of HBI, i.e., isomeric heterocyclic system with varied heteroatom placement. The photoexcitation of HPIP similarly causes ESIPT, but contrary to the benzazoles mentioned above (HBI, HBO, and HBT), subsequent tautomerization to a typical keto form does not occur. Instead, HPIP proceeds to a zwitterionic IPT* emitting state (1IPT*), which further forms a twisted conformation in a fluid solution.

a Molecular structure of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP, 1), and its open-enol, hydrogen-bonded enol (1Enol), and intramolecular proton-transferred (IPT, 1IPT) forms. b Energy diagram of normal (left) and ESIPT (right) luminescence
As Douhal et al. reported [71, 72], HPIP exhibits ESIPT fluorescence (1IPT* → 1IPT) [37, 38, 73] in nonpolar organic solutions, albeit with a low quantum yield (Φ = 0.01–0.08). The fluorescence appears at a significantly lower energy (~600 nm) than that of HBI (~480 nm), probably because of the decreased energy level of 1IPT* and increased energy level of 1IPT owing to solvent rearrangement and/or conformational changes. This explanation is supported by the ESIPT fluorescence observed in rigid media. In a polymer matrix [74, 75] and frozen organic solution [61], where effects based on molecular motion are suppressed, the fluorescence is largely blue-shifted to ~520 nm.
In polar environments, where intramolecular hydrogen bonding is prevented, HPIP exists predominantly as the “open-enol” species and shows normal fluorescence (Scheme 14.4b).
3 Polymorph-Dependent Luminescence of HPIP
3.1 Overview
Two crystal polymorphs of HPIP (1) exhibit bright photoluminescence of different colors, blue-green and yellow, both of which are ESIPT luminescence. X-ray crystallographic analysis revealed different molecular conformations and packing in these two crystal polymorphs, and interconversion between the polymorphs can be realized via a heat-mode dry process.
Ab initio quantum chemical calculations were performed to investigate the enhanced ESIPT luminescence in the solid state.
3.2 Crystalline Polymorphism of HPIP
Slow and fast cooling of a heated aqueous ethanol solution of 1 gives polymorphic crystals 1-BG and 1-Y, respectively. In the infrared spectra of 1-BG and 1-Y, the O–H stretching band appears at around 3135 cm−1 in both crystals, indicating the intramolecular hydrogen bonding between the O–H and nitrogen (N1) in the imidazopyridine ring.
X-ray crystallographic analysis of 1-BG (Pbca, Z = 8, Fig. 14.1) and 1-Y (P21/c, Z = 8, Fig. 14.2) was performed at room temperature [76]. The asymmetric unit of 1-BG and 1-Y is composed of one and two molecules, respectively, and each molecule clearly participates in an intramolecular hydrogen bond (O–H···N1). The most notable conformational difference was the torsion angle θ (N1–C2–C1′–C2′) composed of the phenyl ring and the imidazopyridine ring. While the two aromatic rings are nearly coplanar (θ = –1.0° and 1.3°) in 1-Y, a larger torsion angle was found in 1-BG (θ = 5.8°). The distance between O and N1, representing the length of the intramolecular hydrogen bond, showed only a slight difference (<0.02 Å).

a ORTEP drawing of molecular structure of 1 in crystal 1-BG. b Molecular packing in 1-BG viewed from bc (b) and ab (c) planes

a ORTEP drawing of molecular structure of 1 in crystal 1-Y. b Molecular packing in 1-Y viewed from ac (b) and (101) (c) planes
In these crystals, two molecules are packed close enough to form a stacked pair in an antiparallel manner, and their inter-ring distances are the typical stacking distance of 3.4 Å. There is sufficient overlap between the stacked molecules in 1-BG and 1-Y. These molecular pairs are aligned in the edge-to-face mode in 1-BG (Fig. 14.1b, c), or slipped edge-to-edge mode in 1-Y (Fig. 14.2b, c).
3.3 Polymorph-Dependent Luminescence
The absorption spectra of 1-BG and 1-Y obtained by the diffuse reflectance method (Kubelka-Munk spectra) exhibited a maximum at around 340 nm (Fig. 14.3), which was close to that which is observed in a dilute THF solution. Upon excitation at 330 nm, 1-BG and 1-Y exhibited blue-green (496 nm) and yellow luminescence (529 nm), respectively. The luminescence quantum yields were considerably higher (Φ = 0.37–0.50) compared to those in solution. An amorphous solid prepared by the freeze-dry method showed yellow luminescence, similar to that of 1-Y (Table 14.1). The Stokes shift of the luminescence was large enough (9300–10,800 cm−1) to be well within the range of typical ESIPT fluorescence [39, 77]. Thus, ESIPT luminescence is dependent on the molecular packing mode in the solid state, probably because the emitting species has zwitterionic character, which is sensitive to the molecular conformation and field effects of surrounding molecules.

Modified from Mutai et al. [61] with permission from WIELY-VCH
Absorption and luminescence spectra of 1-BG (dashed line) and 1-Y (solid line).
On the other hand, the 2′-methoxy derivative MeOPIP (Scheme 14.5), which does not form intramolecular hydrogen bonds, showed only blue, normal fluorescence (382 nm) in the solid state.

Molecular structures of 1Enol, 1IPT, and MeOPIP
3.4 Thermal Properties and Luminescence Color Switching
Blue-green and yellow ESIPT luminescence is switchable by heat-mode control of the polymorphic states.
The thermal properties of both crystals will firstly be described. In the first differential scanning calorimetry (DSC) trace, 1-BG showed an endothermic peak at the melting point (141 °C) (Fig. 14.4, left). On the other hand, 1-Y and the amorphous solid displayed small peaks at around 131 °C, in addition to the melting peak at 141 °C; the former peaks were attributed to the phase transition of 1-BG. When powdered crystals of 1-Y or the amorphous solid were placed on a glass plate and heated at 135 °C for 1 min, the XRD pattern of the heated powder measured at the room temperature (Fig. 14.4d) was not the same as that of the starting crystal of 1-Y (Fig. 14.4c) or the amorphous solid, but was comparable to that of 1-BG (Fig. 14.4a). Accordingly, the luminescence turned from yellow to blue-green.

Modified from Mutai et al. [61] with permission from WIELY-VCH
Left: First-heating curves of DSC of 1-BG and 1-Y. Right: XRD profiles of 1-BG (a) and 1-Y (b) calculated from corresponding crystal structures, and the observed profiles of 1-Y at room temperature (c) and after heating at 135 °C, 1 min (d).
The resulting blue-green emitting powder was further heated above the melting point and kept at 150 °C for 1 min. Instant solidification of the heated material using ice cubes yielded a yellow-emitting solid, which showed only a vague XRD image, indicating that this solid was primarily amorphous.
Based on the above findings, we examined the repetitive-switching ability of the PDL of 1. [Yellow → blue-green] Powdered crystals of 1-Y were kept at 135 °C for 1 min, in order to convert them to the blue-green-emitting state. [Blue-green → yellow] Next, they were heated at 150 °C for 3 min and rapidly cooled, to generate the yellow-emitting state. Several color-switching cycles were successfully carried out, demonstrating that the color of the ESIPT luminescence of 1 could be interconverted via the heat-mode dry process.
3.5 Quantum Chemical Analysis
The luminescence property of 1 was further analyzed quantum chemically [78]. The ground-state geometries (S0) of 1 were optimized using density functional theory (DFT) with the 6-31G(d,p) basis set in conjunction with the B3LYP functional. For the optimized S0 geometries, the vertical excitation energies were evaluated with time-dependent (TD) DFT at the B3LYP/6-31+G(d,p) level of theory [79]. As a reference, the equation-of-motion with coupled-cluster singles and doubles (EOM-CCSD) method was performed to estimate the transition energies using the cc-pVDZ basis set [79]. The viability of the DFT calculations was verified by several higher-level methods. The quantitative vertical excitation energies including dynamical electron correlations were estimated by multi-state complete active-space second-order perturbation theory (MS-CASPT2) [80]. The singlet excited-state (S1) geometries and energy profile as a function of torsion angle (θ) were evaluated by the complete active-space self-consistent field method (CASSCF) using the atomic natural orbital small basis set (CASSCF(6,6)/ANO-S) [80]. The fully relaxed S1 potential energy surface (PES) was examined at the CASSCF(6,6)/ANO-S-MB (minimal base) level of theory.
3.6 S0 and S1 PES Scan Along the Central C-C Bond Torsion
In the ground state, the enol form (S0-enol) was energetically stable than the IPT form (S0-IPT) at all torsional angles (0–90°). The lowest-energy geometry in both states was the coplanar conformation, which could be due to the intramolecular hydrogen bond. The coplanar S1-IPT form was more stable (32.5 kJ mol−1) than the S1-enol form, indicating that ESIPT could undergo from the enol form. The observed decay times of the ESIPT luminescence were 0.52 and 5.26 ns in fluid and frozen THF solutions, respectively. Because ESIPT is an extremely fast process (<1 ps) [81,82,83], the conversion from S1-enol to S1-IPT proceeds instantaneously.
The effects of torsion angle on the energy level of the S1- and S0-IPT state were evaluated. As shown in Fig. 14.5, an increase in the torsion angle from 0 to 45° resulted in a relatively small change in the energy level of the S1-IPT state with a small local minimum at around 30°, which then decreased gradually as the torsion angle increased beyond 45°. On the other hand, a monotonic elevation in the S0-IPT energy level was observed as the torsion angle increased across the entire range examined (0 to 90°). The S0 → S1 energy gap of the IPT form became sufficiently small at 60°, and the conical intersection (CI) was observed at 90°. In the CI model [84, 85], it acts as a “funnel” for an efficient deactivation to the ground state within a single vibrational period. Therefore, the S0/S1-CI facilitated the fast radiationless decay process of the excited state, which explains the low quantum yield of the ESIPT fluorescence in solution.

Adapted with permission from Shigemitsu et al. [78]. Copyright 2019 American Chemical Society
S0 and S1 potential energy profiles for the enol and IPT forms at S1-optimized geometries (CASSCF(6,6)/ANO-S).
3.7 Enhanced Solid-State Luminescence of HPIP
HPIP 1 shows bright ESIPT luminescence not only in the crystalline form, but also in a dilute frozen solution and polymer matrices, whereas 1 shows weak luminescence in fluid solutions. The CASSCF study clearly demonstrated the presence of S0/S1-CI at θ = 90°, where efficient radiationless decay coupled with the twisting motion of an excited molecule could proceed (Fig. 14.5). This can reasonably explain the weak emission in solution. In rigid media (i.e., frozen solvents and polymer matrices), molecules are fixed in the coplanar enol conformation. In both crystals, when the torsion angle θ was below 6°, the excited species could not reach the S0/S1-CI. Therefore, the fixation of molecules in the coplanar conformation should be the origin of the observed emission enhancement. Suppression of the radiationless decay via the S0/S1-CI allows for emissive decay from the S1-IPT to the S0-IPT on a nanosecond timescale.
3.8 Polymorph-Dependent Luminescence Color of HPIP Crystals
From Fig. 14.5, it is apparent that the energy gap between the S1-IPT and S0-IPT states depends greatly on the torsion angle. Unless the energy gap becomes too small to induce efficient radiationless decay, or the torsion angle of the enol species is too large to block the ESIPT process, an increase in the torsion angle is expected to induce a redshift in the ESIPT luminescence. Notably, the potential energy of S1-IPT has a small local minimum at around 30°, and the energy gap does not decrease monotonically as a function of torsion angle.
As shown in Fig. 14.3, there is a slight difference in the torsion angles in the two crystals: molecules in 1-BG have a torsion angle of 5.8° and those of the two conformers, Y1 and Y2, in 1-Y are 1.3° and –1.0°. Because the differences in the emission energies and torsion angles in 1-BG and 1-Y crystals are small, higher-level ab initio calculations, MS-CASPT2(10,9)/ANO-L methods, were applied in order to examine the effect of torsional conformation on the emission energy. Although the results suggested a slightly lower energy for Y1 in 1-Y, there was no difference between 1-BG and Y2. Because the estimation is based only on the conformational difference in a single molecule, it may be necessary to include the effects of surrounding molecules, as well as the degree of conformational freedom in the crystal [86,87,88].
4 Conclusions
Owing to the ESIPT mechanism, which generates environment-sensitive zwitterionic excited species, HPIP 1 shows distinct PDL, i.e., blue-green (1-BG) and yellow (1-Y). Moreover, reproducible switching of the PDL was achieved by heat-mode control of the molecular packing, which was reasonably supported by power XRD analysis and DSC measurements. Ab initio calculations suggested that a free S1-IPT species could approach the S0/S1–CI coupled with the twisting motion of the central C–C bond, and return to the S0-enol state without emitting light.
The enhanced emission of HPIP in the solid state can be explained by the following factors: (1) blocking S1-IPT species (1IPT*) from the twisting motion that reach the CI and efficient radiationless decay, and (2) inhibition of excimer formation, because of the unmatched energy state between 1IPT* and surrounding 1Enol molecules.
5 Three-Color Polymorph-Dependent Luminescence
5.1 Overview
6-Cyano HPIP (2) (Fig. 14.6) forms three polymorphic crystals, which exhibit clearly different polymorph-dependent ESIPT luminescence (yellow, orange, and red) [89]. The effects of molecular packing on the ESIPT luminescence were studied by X-ray crystallographic analysis, quantum chemical calculations, and time-resolved transient absorption spectroscopy. TD-DFT calculations of the π-stacked dimer geometry extracted from each polymorphic crystal reasonably reproduced the luminescence energies. In addition, both the theoretical calculation and time-resolved measurement presented the similar result: interaction between the IPT* and neighbor enol species might be an important factor for PDL. The results give further insight into the relationship between luminescence and crystal structure and may contribute to the design of new PDL-active organic materials.

Molecular structure of 6-cyano HPIP 2 (a), and ORTEP drawings of 2 in 2-Y (b), 2-O (c), and 2-R (d)
5.2 X-Ray Crystallographic Analyses of 2-Y, 2-O, and 2-R
The prismatic yellow-luminescent crystal 2-Y and the platelet orange-luminescent crystal 2-O were prepared separately by controlling the recrystallization conditions. Needle-like red-luminescent crystals of 2-R were obtained manually from a mixture of polymorphic crystals.
In order to understand the role of molecular packing in PDL, X-ray crystallographic analyses of 2-Y, 2-O, and 2-R were performed [90]. All crystals were monoclinic and had the same space group (P21/n, Z = 4) and nearly identical cell densities. Figure 14.6b-d shows ORTEP drawings of a molecule in the polymorphic crystals. The distance between the oxygen and nitrogen (N1) atoms was 2.582–2.625 Å, indicating the formation of an intramolecular hydrogen bond (O–H···N1). The small torsion angle between the two aromatic rings (θ(N1–C2–C1′–C2′) = 1.4–2.6°) confirmed the coplanar conformation.
On the other hand, the molecular packing in 2-Y, 2-O, and 2-R was remarkably different, as illustrated in Figs. 14.7, 14.8, and 14.9, respectively. In 2-Y, molecules form parallel-stacked columns, in which molecules are slip-stacked along the long axis with an interplanar distance of 3.38 Å (Fig. 14.7a). The fused imidazolyl moiety overlaps with the phenyl moiety of the stacked molecule. The distance between an oxygen atom and the nearest nitrogen atom of a neighbor molecule is 3.64 Å, which ruled out the formation of an intermolecular hydrogen bond. As for the intercolumnar packing, every two columns aligned in an antiparallel fashion, to form a herringbone-like arrangement along the c-axis (Fig. 14.7b).

Mutai et al. [89]—Reproduced by permission of The Royal Society of Chemistry
Crystal structure of 2-Y. Top and side views of the columnar packing (a) and molecular packing viewed from bc plane (b).

Mutai et al. [89]—Reproduced by permission of The Royal Society of Chemistry
Crystal structure of 2-O. Top view of dimer (a) and molecular packing viewed from ab plane (b).

Mutai et al. [89]—Reproduced by permission of The Royal Society of Chemistry
Crystal structure of 2-R. Top and side views of the columnar packing (a) and molecular packing viewed from bc plane (b).
In 2-O, antiparallel π–π stacked dimeric units formed with an interplanar distance of 3.35 Å (Fig. 14.8a). The imidazolyl moieties overlapped, and the phenyl moiety overlaid the fused pyridyl group. Neighboring dimeric units packed in an edge-to-face type arrangement (Fig. 14.8b), which was similar to that observed in 1-BG (Fig. 14.1) [61].
Similar to 2-Y, parallel slip-stacked columnar packing was observed in 2-R (Fig. 14.9). The molecules were less slipped compared to 2-Y, and the same aromatic moieties overlapped (Fig. 14.9a). Formation of an intermolecular hydrogen bond could also be ruled out because of the large distance (3.53 Å) between oxygen atoms and the nearest nitrogen atom of surrounding molecules. As for the intercolumnar packing, the adjacent columns aligned in an antiparallel manner along the b- and c-axes (Figs. 14.9b).
In the three polymorphic crystals, the molecular packing was remarkably different, whereas the conformation of the molecules was nearly identical. Therefore, the intermolecular interactions that vary according to the different molecular packing modes may be a dominant factor in the PDL of 2.
5.3 Luminescence of 2-Y, 2-O, and 2-R
Figure 14.10 shows the luminescence spectra of the polymorphic crystals of 2-Y, 2-O, and 2-R upon excitation at 330 nm. The luminescence decays of the three polymorphic crystals were reasonably fitted with a mono- or bi-exponential curve (Table 14.2). The weighted average lifetimes (τM) of 2-Y, 2-O, and 2-R were 5.48, 5.51, and 2.26 ns, respectively, indicating singlet emission. The rate constants of the radiative (kem) and non-radiative (knr) deactivations were within the ordinary values for a π–π* transition. The low quantum yield of 2-R may be due to an efficient non-radiative deactivation pathway. Because the spectral data, Stokes’ shift values, and luminescence lifetimes were similar to those observed in a poly(methyl methacrylate) (PMMA) matrix [74], where 2 exists as an isolated molecule, the luminescence of these crystals was considered to be ESIPT luminescence from single molecules.

Mutai et al. [89]—Reproduced by permission of The Royal Society of Chemistry
Luminescence spectra of 2-Y (solid line), 2-O (broken line), and 2-R (dotted line).
In contrast to the distinct luminescence colors, the excitation spectra were quite similar in shape and maximum wavelength, indicating that the light-absorption properties of 2 were not sensitive to molecular packing. Thus, differences in the molecular packing dominantly affected the electronic state of the IPT species and led to distinct ESIPT luminescence colors.
5.4 Heat-Mode Phase Transfer and Luminescence Color Switching
The thermal properties of the three polymorphs were examined by DSC. In the first-heating curve, 2-Y showed only one endothermic peak at 247 °C, which corresponded to the melting point (ΔH = 31 kJ mol−1). On the other hand, 2-O and 2-R displayed a small endothermic peak around 190 °C (ΔH ≈ 1 kJ mol−1) in addition to their melting peaks (247 °C). From optical microscopic observations, this thermal behavior was ascribed to a solid-solid phase transition, which was associated with a change in the luminescence color from orange (2-O) or red (2-R) to yellow.
When a microcrystalline powder of 2-O was heated at 200 °C for 3 min and then cooled to the room temperature, the powder XRD pattern of the resultant solid was obviously different from that obtained before heating and was comparable to that of 2-Y. It was therefore concluded that the small endothermic peak (190 °C) corresponded to the phase transition from 2-O to 2-Y. Although the XRD patterns of 2-R before and after heating could not be obtained because of a microcrystalline sample shortage, it is reasonable to assume that 2-R similarly undergoes a phase transfer to 2-Y at 190 °C.
5.5 Theoretical Study on Polymorph-Dependent Luminescence
5.5.1 Accuracy of TD-DFT Calculations
In order to obtain a deeper understanding of the polymorph-dependent ESIPT luminescence of 2, the ESIPT emission energies were evaluated using quantum chemical calculations. First, a series of computations, TD-DFT, CASSCF, and MS-CASPT2, were performed on the IPT form of the compound to validate the accuracies of the computed S1–S0 vertical transition energies. It was concluded that the TD-DFT level geometry optimization was sufficient to obtain an appropriate geometry of 2, and the Coulomb-attenuated functional CAM-B3LYP [91] was in quantitative agreement with the S1–S0 gap obtained from the MS-CASPT2 calculations, whereas the B3LYP functional substantially underestimated the S1–S0 gap.
5.5.2 Calculation of ESIPT Luminescence Energy
Two-layer ONIOM models [92] were applied to the molecular clusters extracted from 2-Y, 2-O, and 2-R in order to calculate the ESIPT state in different molecular aggregates. The effects of packing mode on ESIPT were taken into account through QM:MM calculations within the finite model. Each cluster model consisted of 17 molecules, in which one IPT species (2IPT) was surrounded with sixteen enol species (2Enol). The S1 state of the center 2IPT was geometrically optimized at the TD-DFT(B3LYP)/6-31G(d) level of theory with fixed geometries of the surrounding 2Enol molecules calculated at the semiempirical PM3 level.
The single point S1 → S0 emission energies were calculated at the TD(CAM-B3LYP)/6-31G(d). In the case of isolated monomer, optimized 2IPT without surrounding enols, the order of the computed emission energies (2-O < 2-R < 2-Y) did not coincide with the observed ESIPT luminescence (2-R < 2-O < 2-Y) (Table 14.3). On the other hand, when the π-stacked dimer geometries consisted of the optimized 2IPT and adjacent π-stacked 2Enol molecules (Fig. 14.11), the calculated emission energies were in accord with the observed ESIPT luminescence (Table 14.3). The calculations showed that the HOMO and LUMO existed on 2IPT, indicating that the dimer models were suitable to simulate the ESIPT-type S1 → S0 transition.

π-stacked dimers composed of 2IPT (highlighted) and 2Enol (grayscale) of 2-Y, 2-O, and 2-R
As a reference, the same TD-DFT calculation was also applied to the non-stacked dimer extracted from 2-O. The computed energy was similar to that of the isolated monomer, suggesting that the difference in the energy of the π-stacked dimer models could be attributed to the effects of the π-stacking modes.
The emission energies of the clusters were further calculated using ONIOM models and were found to be virtually identical to the corresponding isolated models (Table 14.3). These results suggested that the quantum chemical interaction between 2IPT and π-stacked 2Enol contributed to the emission energies of the polymorphic crystals of 2, while the effects of the surrounding enol molecules considered by the ONIOM models were limited.
5.6 Structure–Property Relationships in PDL
HPIP 1 has an intrinsic donor–acceptor nature due to the electron-rich and electron-deficient natures of phenyl and imidazopyridyl units, respectively (dipole moment of 1Enol: 5.40 D). Therefore, antiparallel stacking, which cancelled the dipole effect, was predominantly observed in the crystals of 1 [61]. On the other hand, parallel and antiparallel stacking was observed in the crystals of 2. DFT calculations showed that the dipole moment of 2Enol in the ground state was 1.66 D, which was much smaller than that of 1. The weaker donor–acceptor nature of 2 may be the reason that antiparallel stacking was not essential to cancel the dipole effect and may also be why slipped-parallel stacking was allowed.
As for the proton-transferred emitting species, the dipole moments of the excited (2IPT*) and ground states (2IPT) were calculated to be 4.99 and 3.70 D, respectively, and were oriented in a high-angled manner (ca. 150°) (Fig. 14.12). This could be the reason for the polymorph dependence of the luminescence color. Alterations in the molecular packing mode, namely changes in the environment polarity, would cause different trends in the energy levels of the two IPT states, resulting in a variation in the energy gap. The calculated energy levels (EHOMO and ELUMO) of the π-stacked dimers were consistent with the above discussion. While the EHOMO values increased in the order of 2-Y < 2-O < 2-R (ΔEHOMO = 0.36 eV), those of ELUMO increased in the order of 2-R ~ 2-Y < 2-O, with a much smaller difference (ΔELUMO = 0.12 eV).

Mutai et al. [89]—Reproduced by permission of The Royal Society of Chemistry
Dipole moment in excited and ground states of IPT species of 2.
5.7 Excited-State Dynamics in the Crystalline State
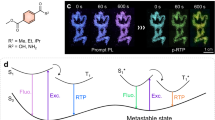
The excited-state dynamics of the PDL of 2 was further studied by means of a femtosecond pump-probe spectroscopic measurement [93]. By a femtosecond 400-nm pulse excitation of 2-Y, 2-O, and 2-R, two processes with the decay time of <1 ps, which corresponds to the ESIPT (2Enol* → 2IPT*), and a slower process (11–45 ps) were observed before the radiative deactivation (2–8 ns).
In the case of 2-O, the time-resolved profile of the transient absorbance was fitted with a triple-exponential function with lifetimes of 0.8 ± 0.3 ps (68%), 25 ± 5 ps (24%), and 5500 ps (8%). The third lifetime constant corresponding to the ESIPT emission was fixed to be 5500 ps. The shortest component (0.8 ps) was assigned to the ESIPT process (2Enol* → 2IPT*). In the crystalline solid, where molecules are densely packed, the twisting motion in the excited state is suppressed and the mutual interaction between excited-state and ground-state molecules should be easy. Therefore, we consider the 25 ps component to be the dynamics between the IPT* and enol species such as a heteroexcimer formation.
From the above discussion, the dynamics of the excited-state relaxation process of 2-O is shown schematically in Fig. 14.13. Upon excitation of 2-O by a fs 400 nm pulse, the 2Enol* generated and the ESIPT took place with a time constant of 0.8 ps. Then the heteroexcimer between 2IPT* and neighboring 2Enol species was formed with a time constant of 25 ps and deactivated with emission in a few nanoseconds.

Adapted with permission from Ishibashi et al. [93]. Copyright 2019 American Chemical Society
Schematic diagram of ESIPT and subsequent relaxation processes of crystalline solid of 2.
In the case of 2-Y and 2-R, the time-resolved profiles show similar behaviors as 2-O, while the time constants obtained by fitting with the triple-exponential function were different. The component due to ESIPT process is in the order 2-Y (0.3 ps) < 2-R (0.5 ps) < 2-O (0.8 ps). The X-ray crystallographic analysis (Fig. 14.6) showed the distance between O and N1 as 2.582 Å (2-Y), 2.622 Å (2-R), and 2.625 Å (2-O), suggesting that the difference in the rate of ESIPT process might be related to the length of the intramolecular hydrogen bond (O–H···N1).
The rate of the heteroexcimer formation is in the order 2-R (11 ps) < 2-O (25 ps) < 2-Y (45 ps). The excimer formation process might be affected by two factors: the π–π interplanar distance and the longitudinal slippage of the dimeric unit. Because the interplanar π–π distances of the three polymorphs are nearly identical, the rate of the excimer formation would mainly depend on the longitudinal slippage (the overlap) of the π–π stacked dimeric unit as well as the small dihedral angle between the two aromatic rings (Fig. 14.6). The π–π overlap of dimer unit of 2-Y was the smallest, resulting in the slowest excimer formation rate (45 ps). On the other hand, both 2-O and 2-R show larger π–π overlap, while the dihedral angle of 2-O of the ground-state enol form was larger (2.6°) compared to that of 2-R (1.4°). The less planarity of the molecule might result in the slower excimer formation time of 2-O compared to 2-R. From the above results, we can conclude that the difference in the molecular packings is somewhat capable of “controlling” the excimer formation.
6 Conclusion
In conclusion, the three-color, polymorph-dependent luminescence of 6-cyano HPIP (2) (2-Y (yellow), 2-O (orange), and 2-R (red)) was investigated. X-ray crystallographic analysis indicated a remarkable difference in the molecular packing, which should be a dominant factor in the polymorph-dependent ESIPT luminescence. Theoretical studies indicated that intermolecular interactions between π-stacked IPT and enol molecules may contribute to the emission energy of crystal polymorphs. The different directions of the dipole moments in the ESIPT and IPT states may be the reason for the sensitive polymorph-dependent luminescence color, as the energy level of the two states would shift differently with variations in the environment, resulting in changes in the energy gap. It is also remarkable that the excited-state dynamics of the three polymorphs elucidated by the femtosecond transient absorption and fluorescence lifetime measurements presented the consistent model with the quantum chemical simulations. Namely, between the rapid ESIPT (<1 ps) and the nanosecond emissive decay processes, the heteroeximer formation process, whose rate is obviously dependent on the molecular packing mode (11–45 ps), is found. The result would open the way for detailed experimental analysis of the mechanism of PDL.
7 Other HPIP Derivatives
To date, various derivatives of HPIP have been synthesized, and the fluorescence of some derivatives has been reported [74]. Similar to 1 and 2, the derivatives exhibited bright ESIPT fluorescence in a PMMA matrix (Φ = 0.1–0.6), although they showed weak emission (Φ ≈ 0.01) in a THF solution. The introduction of electron-donating and electron-withdrawing groups onto the imidazopyridine moiety (R1) caused blue and redshifts in the ESIPT fluorescence, respectively. On the contrary, substitution at the phenyl ring (R2) shifted the ESIPT fluorescence in the opposite direction, i.e., electron-donating and electron-withdrawing groups caused red and blue shifts, respectively (Scheme 14.6).

Molecular structure of HPIP substituted at imidazopyridine (R1) and phenyl (R2) moieties
Furthermore, some derivatives display polymorph-dependent ESIPT luminescence: for example, 6-bromo HPIP (λem = 539 and 575 nm), 5′-methyl HPIP (λem = 521 and 536 nm), and 6-chloro-5’-methyl HPIP (λem = 541 and 576 nm). On the other hand, polymorphism in aryl-substituted HPIPs has not been observed.
The luminescence of HPIP may be controlled by chemical modifications and supramolecular methods (polymorphism). Thus, HPIP could be a good model to evaluate the effects of substituents and polymorphism on solid-state luminescence.
8 Conclusions and Future Outlook
Crystal engineering toward the formation of polymorphic phases of luminophores is a powerful strategy for controlling the luminescence of molecular solids, and it may lead to new photofunctional materials.
Owing to ESIPT, HPIP 1 exhibits environment-sensitive zwitterionic excited species, which leads to PDL, i.e., blue-green (1-BG) and yellow (1-Y). Furthermore, 6-cyano HPIP 2 emits three-color PDL, i.e., yellow (2-Y), orange (2-O), and red (2-R). Switching luminescence color via heat-mode phase transfer indicates that ESIPT is a useful mechanism for converting information regarding molecular packing into a significant change in luminescence. ESIPT luminescence of HPIP can also be controlled by common synthetic modifications, and the effects of substituents on luminescence can be reasonably explained by the calculated electronic configurations and energy levels of S0-IPT and S1-IPT states [36].
Theoretical studies of HPIP derivatives have presented valuable information on the electronic state of molecular crystals that may affect PDL. Femtosecond time-resolved measurement is also promising technique that will offer much clear and convincing experimental data for discussing the excited dynamics in crystals.
Thus, the remarkable polymorph dependence of the luminescence color demonstrates that ESIPT is a promising mechanism for packing-directed control of luminescence and will improve the understanding of the structure–property relationships observed in PDL. It should be possible to design a library of compounds based on the combination of conventional synthetic strategies and supramolecular chemistry (i.e., polymorph dependence).
Currently, the number of compounds that show PDL is still limited. Therefore, the development of a series of luminescent core-based PDL-active compounds should contribute to the understanding of crystal structure–luminescence relationships and may lead to the design of polymorphic materials displaying specific and desirable luminescence properties. These findings should also offer a novel design concept toward tunable organic luminescent solids and thus provide a roadmap for new functional materials.
References
Sasabe, H., Kido, J.: Multifunctional materials in high-performance OLEDs: challenges for solid-state lighting. Chem. Mater. 23, 621–630 (2011)
Kamtekar, K.T., Monkman, A.P., Bryce, M.R.: Recent advances in white organic light-emitting materials and devices (WOLEDS). Adv. Mater. 22, 572–582 (2010)
Yanai, N., Kitayama, K., Hijikata, Y., Sato, H., Matsuda, R., Kubota, Y., Takata, M., Mizuno, M., Uemura, T., Kitagawa, S.: Gas detection by structural variations of fluorescent guest molecules in a flexible porous coordination polymer. Nat. Mater. 10, 787–793 (2011)
Samuel, I.D.W., Turnbull, G.A.: Organic semiconductor lasers. Chem. Rev. 107, 1272–1295 (2007)
Qian, G., Wang, Z.Y.: Near-infrared organic compounds and emerging applications. Chem. -Asian J. 5, 1006–1029 (2010)
Mishra, A., Bäuerle, P.: Small molecule organic semiconductors on the move: promises for future solar energy technology. Angew. Chem. Int. Ed. 51, 2020–2067 (2012)
Zhao, Y.S., Fu, H., Peng, A., Ma, Y., Liao, Q., Yao, J.: Construction and optoelectronic properties of organic one-dimensional nanostructures. Acc. Chem. Res. 43, 409–418 (2010)
Birks, J.B.: Photophysics of Aromatic Molecules. Wiley-Interscience, London (1970)
Moorthy, J.N., Natarajan, P., Venkatakrishnan, P., Huang, D.-F., Chow, T.J.: Steric Inhibition of π-stacking: 1,3,6,8-tetraarylpyrenes as efficient blue emitters in organic light emitting Diodes (OLEDs). Org. Lett. 9, 5215–5218 (2007)
Qin, T., Zhou, G., Scheiber, H., Bauer, R.E., Baumgarten, M., Anson, C.E., List, E.J.W., Müllen, K.: Polytriphenylene dendrimers: a unique design for blue-light-emitting materials. Angew. Chem. Int. Ed. 47, 8292–8296 (2008)
Li, B., Li, J., Fu, Y., Bo, Z.: Porphyrins with four monodisperse oligofluorene arms as efficient red light-emitting materials. J. Am. Chem. Soc. 126, 3430–3431 (2004)
Li, M., Niu, Y., Zhu, X., Peng, Q., Lu, H.-Y., Xia, A., Chen, C.-F.: Tetrahydro[5]helicene-based imide dyes with intense fluorescence in both solution and solid state. Chem. Commun. 50, 2993–2995 (2014)
Shimizu, M., Kaki, R., Takeda, Y., Hiyama, T., Nagai, N., Yamagishi, H., Furutani, H.: 1,4-bis(diarylamino)-2,5-bis(4-cyanophenylethenyl)benzenes: Fluorophores exhibiting efficient red and near-infrared emissions in solid state. Angew. Chem. Int. Ed. 51, 4095–4099 (2012)
Shimizu, M., Takeda, Y., Higashi, M., Hiyama, T.: 1,4-Bis(alkenyl)-2,5-dipiperidinobenzenes: minimal fluorophores exhibiting highly efficient emission in the solid state. Angew. Chemie Int. Ed. 48, 3653–3656 (2009)
Qin, A., Tang, B.Z. (eds.): Aggregation-Induced Emission: Fundamentals. Wiley, Chichester, UK (2014)
Mei, J., Hong, Y., Lam, J.W.Y., Qin, A., Tang, Y., Tang, B.Z.: Aggregation-induced emission: the whole is more brilliant than the parts. Adv. Mater. 26, 5429–5479 (2014)
Luo, J., Xie, Z., Lam, J.W.Y., Cheng, L., Tang, B.Z., Chen, H., Qiu, C., Kwok, H.S., Zhan, X., Liu, Y., Zhu, D.: Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 381, 1740–1741 (2001)
An, B.K., Kwon, S.K., Jung, S.D., Park, S.Y.: Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 124, 14410–14415 (2002)
Tong, H., Hong, Y., Dong, Y., Häussler, M., Lam, J.W.Y., Li, Z., Guo, Z., Guo, Z., Tang, B.Z.: Fluorescent “light-up” bioprobes based on tetraphenylethylene derivatives with aggregation-induced emission characteristics. Chem. Commun. 3705–3707 (2006)
Varughese, S.: Non-covalent routes to tune the optical properties of molecular materials. J. Mater. Chem. C 2, 3499–3516 (2014)
Yan, D., Evans, D.G.: Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids. Mater. Horiz. 1, 46–57 (2014)
Yoon, S.J., Chung, J.W., Gierschner, J., Kim, K.S., Choi, M.G., Kim, D., Park, S.Y.: Multistimuli two-color luminescence switching via different slip-stacking of highly fluorescent molecular sheets. J. Am. Chem. Soc. 132, 13675–13683 (2010)
Hinoue, T., Shigenoi, Y., Sugino, M., Mizobe, Y., Hisaki, I., Miyata, M., Tohnai, N.: Regulation of π-stacked anthracene arrangement for fluorescence modulation of organic solid from monomer to excited oligomer emission. Chem. Eur. J. 18, 4634–4643 (2012)
Yamaguchi, S., Yoshikawa, I., Mutai, T., Araki, K.: Solid-state luminescence of tetraphenylpyrene derivatives: mechano/vapochromic luminescence of 1,3,6,8-tetra(4′-carboxyphenyl)pyrene. J. Mater. Chem. 22, 20065 (2012)
Avobenzone, D., Zhang, G., Lu, J., Sabat, M., Fraser, C.L.: Polymorphism and reversible mechanochromic luminescence for solid-state. J. Am. Chem. Soc. 132, 2160–2162 (2010)
Shida, T., Mutai, T., Araki, K.: Sterically induced polymorphism: ON–OFF control of excited-state intramolecular proton transfer (ESIPT) luminescence of 1-methyl-2-(2′-hydroxyphenyl)benzimidazole. CrystEngComm 15, 10179–10182 (2013)
Luo, X., Li, J., Li, C., Heng, L., Dong, Y.Q., Liu, Z., Bo, Z., Tang, B.Z.: Reversible switching of the emission of diphenyldibenzofulvenes by thermal and mechanical stimuli. Adv. Mater. 23, 3261–3265 (2011)
Li, Y.-X., Zhou, H.-B., Miao, J.-L., Sun, G.-X., Li, G.-B., Nie, Y., Chen, C.-L., Chen, Z., Tao, X.-T.: Conformation twisting induced orientational disorder, polymorphism and solid-state emission properties of 1-(9-anthryl)-2-(1-naphthyl)ethylene. CrystEngComm 14, 8286–8291 (2012)
Mutai, T., Satou, H., Araki, K.: Reproducible on-off switching of solid-state luminescence by controlling molecular packing through heat-mode interconversion. Nat. Mater. 4, 685–687 (2005)
Zhang, H.Y., Zhang, Z.L., Ye, K.Q., Zhang, J.Y., Wang, Y.: Organic crystals with tunable emission colors based on a single organic molecule and different molecular packing structures. Adv. Mater. 18, 2369–2372 (2006)
Gu, X., Yao, J., Zhang, G., Yan, Y., Zhang, C., Peng, Q., Liao, Q., Wu, Y., Xu, Z., Zhao, Y., Fu, H., Zhang, D.: Polymorphism-dependent emission for Di(p-methoxylphenyl)dibenzofulvene and analogues: optical waveguide/amplified spontaneous emission behaviors. Adv. Funct. Mater. 22, 4862–4872 (2012)
See for example, (a) Krishna, G.R., Kiran, M.S.R.N., Fraser, C.L., Ramamurty, U., Reddy, C.M.: The relationship of solid-state plasticity to mechanochromic luminescence in difluoroboron avobenzone polymorphs. Adv. Funct. Mater. 23, 1422–1430 (2013); (b) Mikhlina, Y.A., Bolotin, B.M., Kuz’mina, L.G.: Relationship between the structure and luminescent properties for two crystal modifications of N-butyl-2-cyano-3-[4-(dimethylamino)phenyl]-2-propenamide. Crystallogr. Rep. 58, 687–691 (2013); (c) Abe, Y., Karasawa, S., Koga, N.: Crystal structures and emitting properties of trifluoromethylaminoquinoline derivatives: thermal single-crystal-to-single-crystal transformation of polymorphic crystals that emit different colors. Chem. -Eur. J. 18, 15038–15048 (2012); (d) Abe, Y., Karasawa, S., Koga, N.: Crystal structures and emitting properties of trifluoromethylaminoquinoline derivatives: thermal single-crystal-to-single-crystal transformation of polymorphic crystals that emit different colors. Chem. Eur. J. 18, 15038–15048 (2012); (e) Kohmoto, S., Tsuyuki, R., Masu, H., Azumaya, I., Kishikawa, K.: Polymorphism-dependent fluorescence of 9,10-bis(pentafluorobenzoyloxy)anthracene. Tetrahedron Lett. 49, 39–43 (2008)
Konoshima, H., Nagao, S., Kiyota, I., Amimoto, K., Yamamoto, N., Sekine, M., Nakata, M., Furukawa, K., Sekiya, H.: Excited-state intramolecular proton transfer and charge transfer in 2-(2′-hydroxyphenyl)benzimidazole crystals studied by polymorphs-selected electronic spectroscopy. Phys. Chem. Chem. Phys. 14, 16448–16457 (2012)
Dong, Y., Xu, B., Zhang, J., Tan, X., Wang, L., Chen, J., Lv, H., Wen, S., Li, B., Ye, L., Zou, B., Tian, W.: Piezochromic luminescence based on the molecular aggregation of 9,10-Bis((E)-2-(pyrid-2-yl)vinyl)anthracene. Angew. Chem. Int. Ed. 51, 10782–10785 (2012)
Luo, X., Zhao, W., Shi, J., Li, C., Liu, Z., Bo, Z., Dong, Y.Q., Tang, B.Z.: Reversible switching emissions of tetraphenylethene derivatives among multiple colors with solvent vapor, mechanical, and thermal stimuli. J. Phys. Chem. C 116, 21967–21972 (2012)
Douhal, A., Lahmani, F., Zewail, A.H.: Proton-transfer reaction dynamics. Chem. Phys. 207, 477–498 (1996)
Barbatti, M., Aquino, A.J.A., Lischka, H., Schriever, C., Lochbrunner, S., Riedle, E.: Ultrafast internal conversion pathway and mechanism in 2-(2′-hydroxyphenyl)benzothiazole: a case study for excited-state intramolecular proton transfer systems. Phys. Chem. Chem. Phys. 11, 1406–1415 (2009)
Lochbrunner, S., Schultz, T., Schmitt, M., Shaffer, J.P., Zgierski, M.Z., Stolow, A.: Dynamics of excited-state proton transfer systems via time-resolved photoelectron spectroscopy. J. Chem. Phys. 114, 2519–2522 (2001)
Ormson, S.M., Brown, R.G.: Excited state intramolecular proton transfer part 1: ESIPT to nitrogen. Prog. React. Kinet. 19, 45–91 (1994)
Furukawa, K., Yamamoto, N., Nakabayashi, T., Ohta, N., Amimoto, K., Sekiya, H.: Changes in the electric dipole moments and molecular polarizabilities of enol and keto forms of 2-(2’-hydroxyphenyl)benzimidazole along the proton transfer reaction path in a PMMA film. Chem. Phys. Lett. 539–540, 45–49 (2012)
Tsai, H.H.G., Sun, H.L.S., Tan, C.J.: TD-DFT study of the excited-state potential energy surfaces of 2-(2’-hydroxyphenyl)benzimidazole and its amino derivatives. J. Phys. Chem. A 114, 4065–4079 (2010)
Henary, M.M., Wu, Y.G., Cody, J., Sumalekshmy, S., Li, J., Mandal, S., Fahrni, C.J.: Excited-state intramolecular proton transfer in 2-(2’-arylsulfonamidophenyl)benzimidazole derivatives: The effect of donor and acceptor substituents. J. Org. Chem. 72, 4784–4797 (2007)
Ouyang, J., Ouyang, C., Fujii, Y., Nakano, Y., Shoda, T., Nagano, T.: Synthesis and fluorescent properties of 2-(1H-benzimidazol-2-yl)-phenol derivatives. J. Heterocycl. Chem. 41, 359–365 (2004)
Das, K., Sarkar, N., Majumdar, D., Bhattacharyya, K.: Excited-state intramolecular proton transfer and rotamerism of 2-(2′-hydroxyphenyl) benzimidazole. Chem. Phys. Lett. 198, 443–448 (1992)
Seo, J., Kim, S., Park, S., Park, S.Y.: Tailoring the excited-state intramolecular proton transfer (ESIPT) fluorescence of 2-(2′-Hydroxyphenyl)benzoxazole derivatives. Bull. Korean Chem. Soc. 26, 1706–1710 (2005)
Chen, W., Twum, E.B., Li, L., Wright, B.D., Rinaldi, P.L., Pang, Y.: Rotational energy barrier of 2-(2′,6′-Dihydroxyphenyl)benzoxazole: a case study by NMR. J. Org. Chem. 77, 285–290 (2012)
Ohshima, A., Momotake, A., Nagahata, R., Arai, T.: Enhancement of the large stokes-shifted fluorescence emission from the 2-(2′-hydroxyphenyl)benzoxazole core in a dendrimer. J. Phys. Chem. A 109, 9731–9736 (2005)
Wang, H., Zhang, H., Abou-Zied, O.K., Yu, C., Romesberg, F.E., Glasbeek, M.: Femtosecond fluorescence upconversion studies of excited-state proton-transfer dynamics in 2-(2′-hydroxyphenyl)benzoxazole (HBO) in liquid solution and DNA. Chem. Phys. Lett. 367, 599–608 (2003)
Ma, J., Zhao, J., Yang, P., Huang, D., Zhang, C., Li, Q.: New excited state intramolecular proton transfer (ESIPT) dyes based on naphthalimide and observation of long-lived triplet excited states. Chem. Commun. 48, 9720–9722 (2012)
Yao, D., Zhao, S., Guo, J., Zhang, Z., Zhang, H., Liu, Y., Wang, Y.: Hydroxyphenyl-benzothiazole based full color organic emitting materials generated by facile molecular modification. J. Mater. Chem. 21, 3568–3570 (2011)
Mohammed, O.F., Luber, S., Batista, V.S., Nibbering, E.T.J.: Ultrafast branching of reaction pathways in 2-(2′-Hydroxyphenyl)benzothiazole in polar acetonitrile solution. J. Phys. Chem. A 115, 7550–7558 (2011)
Rini, M., Dreyer, J., Nibbering, E.T.J., Elsaesser, T.: Ultrafast vibrational relaxation processes induced by intramolecular excited state hydrogen transfer. Chem. Phys. Lett. 374, 13–19 (2003)
Frey, F., Laermer, T.: Elsaesser, Femtosecond studies of excited-state proton and deuterium transfer in benzothiazole compounds. J. Phys. Chem. 95, 10391–10395 (1991)
Anthony, K., Brown, R.G., Hepworth, J.D., Hodgson, K.W., May, B., West, M.A.: Solid-state fluorescent photophysics of some 2-substituted benzothiazoles. J. Chem. Soc. Perkin Trans. 2, 2111–2117 (1984)
Amimoto, K., Kawato, T.: Photochromism of organic compounds in the crystal state. J. Photochem. Photobiol. C Photochem. Rev. 6, 207–226 (2005)
Mitra, S., Tamai, N.: Dynamics of photochromism in salicylideneaniline: a femtosecond spectroscopic study. Phys. Chem. Chem. Phys. 5, 4647–4652 (2003)
Harada, J., Fujiwara, T., Ogawa, K.: Crucial role of fluorescence in the solid-state thermochromism of salicylideneanilines. J. Am. Chem. Soc. 129, 16216–16221 (2007)
Kwon, J.E., Park, S.Y.: Advanced organic optoelectronic materials: harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 23, 3615–3642 (2011)
Seo, J., Kim, S., Lee, Y.S., Kwon, O.H., Park, K.H., Choi, S.Y., Chung, Y.K., Jang, D.J., Park, S.Y.: Enhanced solid-state fluorescence in the oxadiazole-based excited-state intramolecular proton-transfer (ESIPT) material: synthesis, optical property, and crystal structure. J. Photochem. Photobiol. A Chem. 191, 51–58 (2007)
Chuang, W.-T., Hsieh, C.-C., Lai, C.-H., Lai, C.-H., Shih, C.-W., Chen, K.-Y., Hung, W.-Y., Hsu, Y.-H., Chou, P.-T.: Excited-state intramolecular proton transfer molecules bearing o-hydroxy analogues of green fluorescent protein chromophore. J. Org. Chem. 76, 8189–8202 (2011)
Mutai, T., Tomoda, H., Ohkawa, T., Yabe, Y., Araki, K.: Switching of polymorph-dependent ESIPT luminescence of an imidazo[1,2-a]pyridine derivative. Angew. Chem. Int. Ed. 47, 9522–9524 (2008)
Park, S., Park, J., Seo, S.H., Kim, S.Y.: Park, tetraphenylimidazole-based excited-state intramolecular proton-transfer molecules for highly efficient blue electroluminescence. Adv. Funct. Mater. 18, 726–731 (2008)
Hu, Y., Zhang, Y., Liang, F., Wang, L., Ma, D., Jing, X.: Blue organic light-emitting diodes based on an oxadiazole-containing organic molecule exhibiting excited state intramolecular proton transfer. Synth. Met. 137, 1123–1124 (2003)
Park, S., Ji, E.K., Se, H.K., Seo, J., Chung, K., Park, S.Y., Jang, D.J., Medina, B.M., Gierschner, J., Soo, Y.P.: A white-light-emitting molecule: frustrated energy transfer between constituent emitting centers. J. Am. Chem. Soc. 131, 14043–14049 (2009)
Kim, S., Seo, J., Jung, H.K., Kim, J.-J., Park, S.Y.: White luminescence from polymer thin films containing excited-state intramolecular proton-transfer dyes. Adv. Mater. 17, 2077–2082 (2005)
Chang, S.M., Tzeng, Y.J., Wu, S.Y., Li, K.Y., Hsueh, K.L.: Emission of white light from 2-(2′-hydroxyphenyl) benzothiazole in polymer electroluminescent devices. Thin Solid Films 477, 38–41 (2005)
Shono, H., Ohkawa, T., Tomoda, H., Mutai, T., Araki, K.: Fabrication of colorless organic materials exhibiting white luminescence using normal and excited-state intramolecular proton transfer processes. ACS Appl. Mater. Interfaces. 3, 654–657 (2011)
Tang, K.-C., Chang, M.-J., Lin, T.-Y., Pan, H.-A., Fang, T.-C., Chen, K.-Y., Hung, W.-Y., Hsu, Y.-H., Chou, P.-T.: Fine tuning the energetics of excited-state intramolecular proton transfer (ESIPT): white light generation in a single ESIPT system. J. Am. Chem. Soc. 133, 17738–17745 (2011)
Sun, W., Li, S., Hu, R., Qian, Y., Wang, S., Yang, G.: Understanding solvent effects on luminescent properties of a triple fluorescent ESIPT compound and application for white light emission. J. Phys. Chem. A 113, 5888–5895 (2009)
Chen, K.-Y., Hsieh, C.-C., Cheng, Y.-M., Lai, C.-H., Chou, P.-T.: Extensive spectral tuning of the proton transfer emission from 550 to 675 nm via a rational derivatization of 10-hydroxybenzo[h]quinoline. Chem. Commun. 4395–4397 (2006)
Douhal, A., Amat-Guerri, F., Acuna, A.U.: Photoinduced intramolecular proton transfer and charge redistribution in imidazopyridines. J. Phys. Chem. 99, 76–80 (1995)
Douhal, A.: The involvement of rotational processes in the intramolecular proton-transfer cycle. Ber. Bunsenges. Phys. Chem. 102, 448–451 (1998)
(a) Stasyuk, A.J., Banasiewicz, M., Cyrański, M.K., Gryko, D.T.: Imidazo[1,2-a]pyridines susceptible to excited state intramolecular proton transfer: one-pot synthesis via an Ortoleva-King reaction. J. Org. Chem. 77, 5552–5558 (2012)
Mutai, T., Sawatani, H., Shida, T., Shono, H., Araki, K.: Tuning of excited-state intramolecular proton transfer (ESIPT) fluorescence of imidazo[1,2-a]pyridine in rigid matrices by substitution effect. J. Org. Chem. 78, 2482–2489 (2013)
Douhal, A., Amat-Guerri, F., Acuña, A.U.: Probing nanocavities with proton-transfer fluorescence. Angew. Chemie Int. Ed. Eng. 36, 1514–1516 (1997)
(a) Crystallographic data for 1-BG: C13H10N2O, FW = 210.23, Orthorhombic, Pbca, a = 7.6560(2), b = 12.1630(3), c = 22.2380(5) Å, V = 2070.80(9) Å3, Z = 8, dcalcd = 1.349, T = 298(2) K, 18651 measured and 2030 independent reflections, R1 = 0.0548, wR2 = 0.1297, GOF = 1.104. CCDC No.: 698144 (b) crystallographic data for 1-Y: C13H10N2O, FW = 210.23, Monoclinic, P21/c, a = 18.2400(4), b = 5.77300(10), c = 20.3010(5) Å, b = 108.1650(10)°, V = 2031.15(8) Å3, Z = 8, dcalcd = 1.375, T = 298(2) K, 18482 measured and 3733 independent reflections, R1 = 0.0624, wR2 = 0.1732, GOF = 1.116. CCDC No.: 698145
Khan, A.U., Kasha, M.: Mechanism of four-level laser action in solution excimer and excited-state proton-transfer cases. Proc. Natl. Acad. Sci. U. S. A. 80, 1767–1770 (1983)
Shigemitsu, Y., Mutai, T., Houjou, H., Araki, K.: Excited-state intramolecular proton transfer (ESIPT) emission of hydroxyphenylimidazopyridine: computational study on enhanced and polymorph-dependent luminescence in the solid state. J. Phys. Chem. A 116, 12041–12048 (2012)
Guallar, V., Moreno, M., Lluch, J.M., Amat-Guerri, F., Douhal, A.: H-atom transfer and rotational processes in the ground and first singlet excited electronic states of 2-(2′-Hydroxyphenyl)oxazole derivatives: experimental and theoretical studies. J. Phys. Chem. 100, 19789–19794 (1996)
Organero, J.A., Moreno, M., Santos, L., Lluch, J.M., Douhal, A.: Photoinduced proton transfer and rotational motion of 1-Hydroxy-2-acetonaphthone in the S1 state: a theoretical insight into its photophysics. J. Phys. Chem. A 104, 8424–8431 (2000)
Fournier, T., Pommeret, S., Mialocq, J.-C., Deflandre, A.: Excited-state intramolecular proton transfer (ESIPT) and energy relaxation processes in hydroxyphenylbenzotriazole derivatives: a femtosecond laser study. In: De Schryver, F.C., De Feyter, S., Schweitzer, G. (eds.) Femtochemistry, pp. 323–333. Wiley-VCH, Weinheim, Germany (2001)
de Klerk, J.S., Bader, A.N., Ariese, F., Gooijer, C.: High-resolution fluorescence studies on excited-state intra- and intermolecular proton transfer. In: Geddes, C.D. (ed.) Reviews in Fluorescence, pp. 271–298. Springer, New York (2009)
Mitra, S., Tamai, N.: A combined experimental and theoretical study on the photochromism of aromatic anils. Chem. Phys. 246, 463–475 (1999)
(a) Gentili, P.L., Ortica, F., Romani, A., Favaro, G.: Effects of proximity on the relaxation dynamics of flindersine and 6(5H)-Phenanthridinone. J. Phys. Chem. A 111, 193–200 (2007); (b) Domcke, W., Yarkony, D.R., Köppel, H.: Conical Intersections: Electronic Structure, Dynamics & Spectroscopy, Advanced Series in Physical Chemistry. World Scientific, Singapore (2004); (c) Gustavsson, T., Improta, R., Markovitsi, D.: DNA/RNA: building blocks of life under UV irradiation. J. Phys. Chem. Lett. 1, 2025–2030 (2010)
(a) Merchán, M., Serrano-Andrés, L.: Ultrafast internal conversion of excited cytosine via the lowest ππ* electronic singlet state. J. Am. Chem. Soc. 125, 8108–8109 (2003); (b) Ismail, N., Blancafort, L., Olivucci, M., Kohler, B., Robb, M.A.: Ultrafast decay of electronically excited singlet cytosine via a π,π* to n0,π* state switch. J. Am. Chem. Soc. 124, 6818–6819 (2002); (c) Blancafort, L., Cohen, B., Hare, P.M., Kohler, B., Robb, M.A.: Singlet excited-state dynamics of 5-Fluorocytosine and cytosine: an experimental and computational study. J. Phys. Chem. A 109, 4431–4436 (2005); (d) Zgierski, M.Z., Fujiwara, T., Kofron, W.G., Lim, E.C.: Highly effective quenching of the ultrafast radiationless decay of photoexcited pyrimidine bases by covalent modification: photophysics of 5,6-trimethylenecytosine and 5,6-trimethyleneuracil. Phys. Chem. Chem. Phys. 9, 3206–3209 (2007); (e) Paterson, M.J., Robb, M.A., Blancafort, L., DeBellis, A.D.: Theoretical study of benzotriazole uv photostability: ultrafast deactivation through coupled proton and electron transfer triggered by a charge-transfer state. J. Am. Chem. Soc. 126, 2912–2922 (2004)
Fukunaga, H., Fedorov, D.G., Chiba, M., Nii, K., Kitaura, K.: Theoretical analysis of the intermolecular interaction effects on the excitation energy of organic pigments: solid state quinacridone. J. Phys. Chem. A 112, 10887–10894 (2008)
Filhol, J.-S., Deschamps, J., Dutremez, S.G., Boury, B., Barisien, T., Legrand, L., Schott, M.: Polymorphs and colors of polydiacetylenes: a first principles study. J. Am. Chem. Soc. 131, 6976–6988 (2009)
Wu, Q., Peng, Q., Niu, Y., Gao, X., Shuai, Z.: Theoretical insights into the aggregation-induced emission by hydrogen bonding: A QM/MM study. J. Phys. Chem. A 116, 3881–3888 (2012)
Mutai, T., Shono, H., Shigemitsu, Y., Araki, K.: Three-color polymorph-dependent luminescence: crystallographic analysis and theoretical study on excited-state intramolecular proton transfer (ESIPT) luminescence of cyano-substituted imidazo[1,2-a]pyridine. CrystEngComm 16, 3890–3895 (2014)
Crystallographic data for 2-Y: C14H9N3O, FW = 235.24, Monoclinic, P21/n, a = 6.052(6), b = 4.919(5), c = 36.98(3) Å, β = 94.155(4)°, V = 1098.0(17) Å3, Z = 4, dcalcd = 1.423, T = 113(2) K, 8137 measured and 2402 independent reflections, R1 = 0.0754, wR2 = 0.2201, GOF = 0.938. CCDC No.: 965203. Crystallographic data for 2-O: C14H9N3O, FW = 235.24, Monoclinic, P21/n, a = 13.654(6), b = 7.343(3), c = 14.200(6) Å, β = 130.054(2)°, V = 1089.8(8) Å3, Z = 4, dcalcd = 1.434, T = 113(2) K, 8291 measured and 2487 independent reflections, R1 = 0.0334, wR2 = 0.0963, GOF = 1.097. CCDC No.: 965204. Crystallographic data for 2-R: C14H9N3O, FW = 235.24, Monoclinic, P21/n, a = 3.8196(10), b = 11.356(3), c = 25.246(7) Å, β = 93.1961(11)°, V = 1093.3(5) Å3, Z = 4, dcalcd = 1.429, T = 113(2) K, 8368 measured and 2479 independent reflections, R1 = 0.0591, wR2 = 0.1328, GOF = 0.929. CCDC No.: 965205
Yanai, T., Tew, D.P., Handy, N.C.: A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004)
Dapprich, S., Komáromi, I., Byun, K.S., Morokuma, K., Frisch, M.J.: A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 461–462, 1–21 (1999)
Ishibashi, Y., Murakami, M., Araki, K., Mutai, T., Asahi, T.: Excited-state intramolecular proton-transfer process of crystalline 6-Cyano-2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine, as revealed by femtosecond pump-probe microspectroscopy. J. Phys. Chem. C 123, 11224–11232 (2019)
Acknowledgements
We thank Dr. Yasuhiro Shigemitsu at the Industrial Technology Center of Nagasaki (Japan) for the high level of quantum chemical calculations, and Prof. Tsuyoshi Asahi and Dr. Yukihide Ishibashi for the femtosecond pump-probe spectroscopic measurement. We also thank Emeritus Prof. Dr. Koji Araki at the Institute of Industrial Science, University of Tokyo.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mutai, T. (2020). Luminescent Crystal–Control of Excited-State Intramolecular Proton Transfer (ESIPT) Luminescence Through Polymorphism. In: Sakamoto, M., Uekusa, H. (eds) Advances in Organic Crystal Chemistry. Springer, Singapore. https://doi.org/10.1007/978-981-15-5085-0_14
Download citation
DOI: https://doi.org/10.1007/978-981-15-5085-0_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5084-3
Online ISBN: 978-981-15-5085-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)