
Abstract
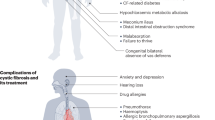
Cystic fibrosis could be a common life-bound autosomal recessive hereditary condition, with highest occurrence in Europe, North America, and Australia. The root of illness is mutation of a gene that encodes a chloride-conducting transmembrane channel known as the cystic fibrosis transmembrane conductance regulator (CFTR) that regulates anion transfer and mucociliary clearance within the airways. Operational failure of CFTR ends up in mucus withholding and chronic contagion, followed by local airway swelling that is harmful to the lungs. CFTR operational impairment principally affects epithelial cells, though there is proof of a function in immune cells. Cystic fibrosis influences numerous body systems, and morbidity and mortality are typically due to bronchiectasis, tiny airways obstacle, and progressive respiratory abnormality. Necessary comorbidities due to epithelial cell operational impairment occur within the pancreas (malassimilation), liver (biliary cirrhosis), sweat glands (heat shock), and vas deferens (sterility). The progress and delivery of medication that recover the clearance of mucus from the lungs and treat the ensuing infection, together with rectification of pancreatic insufficiency and malnutrition via multidisciplinary requisites, have resulted in noteworthy enhancements of life and clinical conclusion in patients with cystic fibrosis. Inventive and transformational treatments that aim on the fundamental defect in cystic fibrosis have currently been grown and are useful in lung surgery and dropping pulmonary exacerbations. Advance petite molecule and gene-based treatment are being developed to revive CFTR operation; these remedies pledge to transform illness and enhance the lives of individuals with cystic fibrosis disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
3.1.1 About the Disease
Cystic fibrosis (CF) respiratory organ malady is characterized by early organization and infection of the airways. Though structural changes within the CF airways will be ascertained at birth in each human and therefore the CF pig, modest inflammation is ascertained [1,2,3]. However, infection happens terribly apace and therefore the inflammatory response to pathogens is harsh [4]. Free and leap airway neutrophil elastase is detected terribly early in CF infants and predicts the event of bronchiectasis later in life [5]. No further respiratory organ malady is understood to induce such an untimely, persistent, and intense inflammatory method as seen within the CF airway. People with CF conjointly suffer from an extreme general inflammation characterized by inflated serum acute-phase reactants, high antibody titers to various exogenous and endogenous antigens, and an elevated occurrence of ileum inflammation with Crohn’s malady, instant hypersensitivity, and heightened Th2 responses [6, 7].
3.1.2 Epidemiology
The global occurrence and dominance of CF show important geographical inconsistency, as illustrated by the detection rates seen globally. Within the USA, the incidence of CF is reported to be 1 in each 3500 births [8]. Though a better incidence is noted in European nations at a rate of 1 in each of 2000–3000 births, in Africa and Asia, though CF is sternly beneath diagnosed, proof indicates that the prevalence of CF is low down to atypical [8, 9].
3.1.3 Causes
CF is caused by a severe functional scarcity of the cystic fibrosis transmembrane conductance regulator (CFTR) protein [10]. CFTR is mostly expressed within the apex membranes of epithelial cells that line the cylindrical structures of tissues that secrete fluids typically made in mucous secretion and different proteins. The airways are amid the tissues with the utmost expression of CFTR. The scarcity of functional CFTR causes scarce cAMP-dependent chloride and hydrogen carbonate secretion into airway secretions. Consequently, mucins are bound to the bronchial apex surfaces, and airway surface fluid pH scale is weakened [11].
3.2 Pathophysiology
The types of complications in patients with CF vary depending on the extent of mutation of CFTR. Also, some patients do not feel the pathological changes altogether as the system is typically affected with CF.
3.2.1 Biology of Disease
3.2.1.1 Respiratory System
Typically, critically sick patients who have CF feel acute respiratory failure owing to pneumonia or acute hemoptysis. The foremost common infecting organisms in patients with CF which have pneumonia include Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa. Noteworthy, patients with CF additionally tend to own nasal polyps that may trigger sinus infections. Thus, these patients may have longer and completely special antibiotic treatments than do patients who have pneumonia exclusively. It is hypothesized that the pH scale level within the cells of patients with cystic fibrosis differs from the extent in patients devoid of the disease. This distinction results in magnified numbers of asialoGM1 molecules that are receptors intended for bacterial respiratory organisms, and thereby ends up in magnified binding of P. aeruginosa and S. aureus. The decline within the quantity of CFTR to bind with the bacteria results in colonization of the airways [12]. In a very few patients with CF who have pneumonia, the pneumonia is because of Burkholderia cepacia (previously referred to as Pseudomonas cepacia), which is extremely immune to most antibiotics.
As obstacle of the airway will amplify, it becomes harder for air to pass throughout exhalation. This condition results in enlargement of alveoli, wherever air lock happens and, over time, causes the barrel-shaped chest that is additionally general in patients with emphysema. Destruction of the pulmonary parenchyma results in magnified pulmonary blood pressure that, in turn, causes right-sided heart failure or core pulmonale.
Pulmonary performance testing may be a methodology that will be useful in establishing information that will assist in predicting deterioration in clinical standing in patients with CF [13]. One factor, forced expiratory volume in 1 s (FEV1), is usually used as an indicator of the worsening condition. The lower the FEV1, the effort of breathing increases; it is related with gas exchange [14]. The amplified work of breathing will embody any of the following: tachypnea, uneven breathing pattern, perspiration, spread nares, pursed lip breathing, intercostal muscle retractions, and use of retrofit muscles. Patients with lower FEV1 additionally tend to be in a very chronic state of acidosis and, similar to additional patients with chronic obstructive pulmonary disease, may have lower PaO2 levels to trigger the hypoxic ventilatory force, though they still want adequate oxygenation. Association or enlargement of the last joints of the toes and fingers, which have no definite cause, additionally happens in patients with CF.
Typically, patients with CF have declined levels of interleukin-10, a cytokine that has anti-inflammatory properties, particularly within the lungs [15]. The drop off levels disposes the patients to severe lung inflammation subsequent to infection. Occasionally, the pulmonary inflammation persists and becomes a chronic inflammatory situation. Chronic inflammation will cause hypertrophy of the bronchial arteries and ultimately, hemoptysis. This critical condition is more worsened by coagulopathies usually caused by malabsorption of vitamin K and perennial use of some antibiotic. The manifestation of hemoptysis varies from blood-tinged bodily sputum to enormous hemorrhaging. It is thought that 5–7% of patients with CF truly have enormous hemoptysis [16].
Patients with CF, who have elevated levels of thick and firm mucus secretion in their airways, are usually admitted to the intensive care unit on account of flow of air limitation. The foremost severe respiratory signs and symptoms are because of the creation of elevated levels of thickened mucus secretion that cause inflammation and swelling and therefore barred airways. The obstruction causes consolidation that results in pneumonia and respiratory failure. Often, patients have elevated withholding of mucus secretion within the right upper lobe, which is indicated by proof of hyperinflation on chest radiographs [17].
Approximately 10% of patients with CF too have infection caused by the fungus Aspergillus fumigatus, which might cause allergic bronchopulmonary aspergillosis and end up in a dramatic enhancement in secretions and an ultimate downward twist in the lung task [18]. Patients with sinobronchial allergic zymosis and/or allergic bronchopulmonary mycosis have terribly thick secretions and are immune to antibiotics. The patients with allergic bronchopulmonary do not show any sign and symptoms of Cystic fibrosis before sinobronchial allergic mycosis. In addition to/or else allergic bronchopulmonary mycosis develops, however once tested are found to own mutation(s) within the CFTR factor [18,19,20]. In several patients, CF is misdiagnosed as celiac disorder, asthma, or chronic bronchitis [21].
3.2.1.2 Hematopoietic System
Patients with CF own iron-deficit anemia and usually have anemia as a results of chronic hemoptysis in addition to or else colonization of resistant P. aeruginosa [22]. Blood loss usually comes from the hypertrophied and tortuous bronchial arteries as an outcome of chronic inflammation [16]. Pseudomonas aeruginosa, an antibiotic-resistant bacteria within the lungs addition to/or else superior airways of patients with CF, steals iron from the host for its self-growth. Additionally, sputum and bronchial airway cleaning fluid of patients with CF who have P. aeruginosa infection include a high iron content [23].
3.2.1.3 Gastrointestinal System
Some gastrointestinal issues in patients with CF are due to the lack of pancreas to produce digestive enzymes to the bowel. As a result of the quantity of pancreatic enzymes discharge decline, the pancreas oozes thick mucus that obstructs the pancreatic ducts and therefore the number of enzymes that may be secreted becomes yet lesser. This alteration causes malassimilation of proteins and persuades absorption of the fat-soluble vitamins A, D, E, and K. The pancreatic enzyme supplements that various patients with CF take could spoil iron assimilation [22]. It is suggested that patients with CF take supplements and vitamins individually.
The distal fraction of the bowel is usually expanded and crammed with fecal content in patients with CF. This alteration is manifested as vomiting, abdominal distention, anorexia, pain within the right lower quadrant of the abdomen, and cramping with a decline or no modification in intestinal movements [23]. Distal intestinal obstruction syndrome (DIOS) may be a result of defective oozing of salt and water from the intestinal epithelium, a state that causes dehydration of the intestinal material.
Some patients with cystic fibrosis even have gastroesophageal reflux disease (GERD) because of hypersecretion of gastric acid and hyposecretion of bicarbonate [23]. Postural emptying will worsen GERD, as will the negative pressures generated by strong coughing. GERD will likewise worsen bronchial reactivity.
3.2.1.4 Endocrine System
About 13% of all patients having CF own cystic fibrosis-associated diabetes that is most frequently diagnosed once the patients are 30 years old. Studies signify that the evaluation of glycated hemoglobin (hemoglobin A1C) does not seem to be a correct diagnostic assay for cystic fibrosis-associated diabetes; as a result, the turnover of red blood cells is more rapid in patients with cystic fibrosis than in patients with no cystic fibrosis [24]. The first downside in cystic fibrosis-associated diabetes is insulin deficit because of barrier of the pancreatic duct. Patients with cystic fibrosis-associated diabetes still need a high-energy diet that is contrary to the diet that others with diabetes mellitus should follow. Glucose metabolism is influenced by several reasons particular to CF, such as severe dehydration, administration of corticosteroids, malassimilation, repeated contamination, poor nutrition, amplified energy expenditure, slowed gastrointestinal travel time, and liver dysfunction [25].
3.2.1.5 Sweat Glands
On account of the declined levels of the protein CFTR, which assists to control salt in sweat, patients with CF will feel too much salt loss from extreme temperature or compared to tremendous work out. Several patients feel dehydration or high-temperature prostration manifested by sluggishness, weakness, and loss of hunger.
3.2.1.6 Reproductive System
Most men with CF are sterile; as a result, they do not have vas deferens or it is misshapen. Women have a tendency to be fertile; however, they usually need longer to become pregnant than do women devoid of CF. Mucus discharge plugs within the oviduct and thicker cervical mucus that lessen sperm movement are detected [23]. Puberty appears to be delayed for each men and women who have this problem.
3.2.2 Cell Type Involved
The lung is continuously exposed to both noxious and infectious agents, and a multi-tiered defense has evolved that is able to continuously cleanse airways without inciting a potentially harmful inflammatory response. The mucus clearance (MC) system appears to be paramount for airways defense and is the locus of defects that lead to genetic lung diseases such as CF and primary ciliary dyskinesia. Other important elements in this defense system include locally residing leukocytes (e.g., alveolar and airway macrophages), mucosal immunoglobulins, and secreted antimicrobial compounds (e.g., lysozyme and lactoferrin), all of which are available to neutralize microbes that escape the first line of defense, that is, mechanical MC. A normally functioning MC apparatus requires the coordinated activities of mucus secretion, salt and water transport, and ciliary beating. Mucus secretion creates a protective blanket that efficiently binds inhaled particles via its panoply of carbohydrate epitopes, where they become entrapped via turbulent flow. The mucus layer, which floats on top of a less viscous and physically distinct liquid layer, is propelled cephalad by a combination of coordinated cilia beating and airflow/cough. The underlying liquid layer, often referred to as the “sol” or “periciliary liquid” layer (PCL), is itself quite complex and specially structured to provide a low resistance environment for ciliary beating while allowing efficient mechanical coupling between the tips of cilia and the mucus layer.
3.2.3 Transcription Regulation
To understand the pathophysiology of CF, one of the approaches is to discover the CFTR expression pattern in different tissue surveys. CFTR is found to be expressed in the epithelial cells of a variety of tissues and organs, whose functions are significantly affected in CF patients: lung and trachea, pancreas, liver, intestines, and sweat glands. Low levels of CFTR transcripts can be found in kidney, uterus, ovary, thyroid, and even higher levels in salivary gland and bladder, but the epithelial cell function is not seriously compromised in tissues and organs of CF patients. It is possible that there is sufficient compensation of the missing function by other ion transporters. It is of interest to note that the low levels of CFTR expression in these tissues are driven off an alternative promoter.
The majority of CFTR transcripts are driven from the key promoter, described in the previous section. However, although the canonical transcripts are found in cells with high CFTR expression, alternative transcription start sites are apparently used in cell lines with low expression levels. CFTR transcripts from even more distant transcription start sites between −868 and −794 can be found in CFPAC and T84 cell lines.
The immediate promoter region has also been characterized by consensus binding sites for several transcription factors: CTCF, AP-1, SP1, GRE, CRE, C/EBP, and Y-box proteins. DNase I hypersensitive site (DHS) mapping has been used to map various putative enhancer sequences within the CFTR intragenic regions. Presumably, multiple transcription factors can bind to chromatin at these sequences, opening the DNA and extending the physical interactions with the promoter, thereby affecting transcription. HNF1α binding sites, indicative of putative enhancer elements, can be found in multiple locations inside introns 10, 17a, and 20; it has been shown that RNAi-mediated inhibition of the HNF1α could lead to reduction of CFTR expression. Additional enhancer elements have been located in introns 1 and 11, and HNF1α and p300 are involved in the regulation of CFTR expression.
3.3 Current Treatment
The purpose of treatment mainly consists of the following:
3.3.1 Respiratory System
Stopping and regulating lung infections: Antibiotics are given. This primarily carries with it inhaled varieties of azithromycin, tobramycin, aztreonam and levofloxacin. Alternative antibiotics suggested are cipro, cephalexin, larotid, and doxycycline depending on the sensitivity patterns [26, 27].
Management of airway inflammation: NSAIDs, breathe in and systemic steroids and cromolyn [28].
Reducing viscoelasticity and eliminating thick, sticky mucus discharge from the lungs and expanding the airways: breathe in β-agonists with dampened oxygen; a 3–6% hypertonic saline solution and dornase alfa are suggested [29,30,31].
Additionally, workout and physiotherapy together with positive expiratory pressure (PEP) tool or an elevated occurrence chest wall swinging device (a percussion vest) is suggested [32].
3.3.2 Gastrointestinal Tract
Avoid or taking care of intestinal obstruction: oral rehydration and osmotic laxatives (unfinished obstruction) and hyperosmolar contrast enemas (total DIOS). An unbiased electrolyte bowel-cleaning solution or enema including diatrizoate meglumine and diatrizoate sodium, depending on vomiting condition [33]. To forestall repetition, regular administration of oral polythene glycol 3350 is also given for 6 months to 1 year.
Pancreatic insufficiency: pancreatic enzyme replacement therapy (PERT) containing multiple mixture of proteases, lipases, and amylases [34].
3.3.3 Nutrition and Electrolyte
Providing relevant nutrition and put off dehydration: A high-calorie-fat diet, add-on of vitamins ADEK, and minerals as well as fluoride and zinc are suggested. Furthermore, sodium chloride add-on is given customized to patient’s age and environmental conditions [35].
In the earlier period, Denufosol, an agonist of P2Y2 receptors was attempted in CF patients; however, it was eventually unsuccessful subsequent to early promising outcome.
3.3.4 Current and Future Medicinal Products
The present and upcoming therapeutic objects are primarily focused on exact structural and purposeful anomaly of CFTR protein.
3.3.4.1 CFTR Modulators
A new cluster of medicine, known as CFTR modulators, is offered that is ready to correct the essential defect in CF, that is, CFTR protein itself; however, the specific mechanism is not absolutely clarified.
3.3.4.2 Ivacaftor
Developed by vertex pharmaceuticals and permitted by FDA in 2012 for kids ≥6 years having rare mutation, G551D (class III), ivacaftor (Kalydeco) [36] was the primary doing-well medication to fix the malfunctioning protein and was tried to be terribly effective in two massive multicentric trials, STRIVE and ENVISION [37, 38]. Marked improvements in FEV1, weight and quality of life were ascertained. Currently, the FDA has enlarged its use in alternative mutations and additionally kids aged 2–5 years supported the results of KIWI trial [39]. In addition, a phase IV clinical trials study (GOAL) additionally reported development in FEV1 and FVC, BMI, quality of life and bated sweat chloride concentration in patients carrying a minimum of one G551D allele. Over 72% patients during this trial additionally carried F508del as second allele [40]. The G551D mutation causes the channel to act as sort of a secured gate, avoiding the transconductance of chloride and fluid. The site of channel is correct; however, the performance is impaired. Ivacaftor will increase the time of channel in an open state. However, the most limitation of this medical aid is that G551D mutation is there in just a pair of 2.3% patients [41]. It is not found to be effective within the commonest F508del (class II) mutation owing to reduced accessibility of protein. In addition, the high price of medical aid may additionally be a limiting reason (ICER: £335,000–£1,274,000/QALYs gained) [42].
3.3.4.3 Lumacaftor
Another CFTR modulator, lumacaftor, has revealed favorable leads in F508del mutation. This is the main common mutation influencing more or less one-third of CF population in USA and nearly 70% in EU. This mutation affects the warmth steadiness because of misfolding of NBD1 field and limits the CFTR in ER for succeeding degradation. It did not succeed to localize to proper epithelial site and attain regular formation. Exaggerated transfer of protein to cell surface was ascertained in vitro by means of cultured individual bronchial epithelium [43]. Still, despite exaggerated transfer of protein to correct position, no rectification of the underlying functional impairment was ascertained. Besides, another in vitro study exposed disparity in negative results [44] that were more strengthened by a trial. No vital improvement was ascertained in FEV1, CFQR scores, and respiratory exacerbation rates [45].
3.3.4.4 Orkambi
The approval of CFTR modulators, Kalydeco™ (ivacaftor) and Orkambi™ (lumacaftor/ivacaftor), marks two mile stones in our pursuit of ‘a cure’ for people suffering from CF. Firstly, phase II trials were performed for each homozygous and heterozygous F508del patients >12 years old; however, solely homozygous patients confirmed clinically noteworthy results. Two massive phase III trials, TRAFFIC and TRANSPORT, were conducted with the mix medical aid (600 + 250 and 400 + 250 mg versus placebo) in patients ≥12 years with primary end as FEV1 improvement at 24 weeks. Patients finishing the study were progressed to 48 weeks PROGRESS trial. The isolated plus pooled outcomes showed a noteworthy improvement in parameters as well as FEV1, reduction of exacerbations, decline in hospitalizations, and rise in BMI and CFQR scores. The undesirable effects were comparable to placebo cluster except one case of death throughout the extension period [46, 47]. In addition, a phase I clinical trial study in homozygous kids ≤12 years showed promising results; however, more advanced phase studies are required [47]. However, in comparison to ivacaftor monotherapy in patients including G551D mutation during a separate study, there was considerably less improvement in pneumonic performed with combination medical aid [3]. Orkambi (lumacaftor + ivacaftor) is permitted recently for homozygous F508del patients ≥12 years. Orkambi acts by a two-step technique. Lumacaftor assists in moving the defective protein to its accurate site and ivacaftor rectifies and enhances its activity eventually escalating the conductance of ions and fluid.
3.3.5 Drawbacks of Current Treatment
Although over 2000 variants in the CFTR gene have been identified to date, F508del accounts for most CFTR alleles in patients with CF, there is still a little restriction that embody (a) non-considerable reply in F508del mutation heterozygotes by ivacaftor; (b) got to keep on further daily symptomatic cure; (c) contact with CYP3A inducers and inhibitors; (d) adverse effects together with elevated transaminases, cataract, oropharyngeal pain and URTI; (e) negligible profit in <12 years old; (f) require of upper prescribed amount up to 600 mg (during case of lumacaftor); and (g) common contact of lumacaftor and ivacaftor leading to improved metabolism of ivacaftor and require of a upper prescribed amount blend. Furthermore, on account of the multi domain formation and in order folding of CFTR, no solo “corrector drug” can repair all the misfolding in dissimilar domains, so a mixture of drugs is a necessity. Furthermore, from a clinical trial viewpoint, there are sample size problems, as precise criteria (major and minor endpoints) make choice more complicated before narrowed mutation precise inhabitants deserve exclusive adaptive trial designs [48].
3.3.6 Future Directions in Therapeutics
CF management does not solely need CFTR correction and modification; however, intensive symptomatic treatment targets inflammation, infection, bronchial hydration, and nutrition. Newer medicines targeting these problems are summed up below in short.
3.3.6.1 Inflammation
Andecaliximab, which is a protein to matrix metalloproteinase 9 (MMP9), is undergoing phase IIb and is expected to cut back inflammation and improve lung task. However, the baseline FEV1 needed for this drug is between 40% and 80% limiting its use in terribly severe CF [49]. An additional compound in phase 1 is POL6014 that is synthesized to dam neutrophil elastase operation, finally reducing the tissue damage and lung inflammation. LAU-7b, perhaps a fenretinide, is a component of retinoid compounds associated with vitamin A. Phase 2 study is thus far to start and it is expected to cut back the inflammatory response in CF lungs. CTX-4430 decreases the making of leukotriene B4, an inflammatory intermediary enhances in CF. It is currently undergoing a phase 2 trial [50]. Additional anti-inflammatory compounds within clinical progress pipeline are α-1 anti-trypsin, CTX-4430, enzyme substance AZD9668, JBT-101 (phase 2) for reducing inflammation.
3.3.6.2 Hydration and Mucus Secretion Clearance
AZD5634 is undergoing section 1b study. It is anticipated to dam the metal channel in CF airway, therefore rehydrating and tapering the mucus secretion within the lungs, creating it easier to clear. SPX-101 is one more compound designed to dam sodium channel operation within the lungs, presently undergoing phase 2 study. OrPro (ORP-100) may be a changed variety of thioredoxin, probably to lessen mucus thickness within the lungs and recover clearance from the CF airway. OligoG (Alginate Oligosaccharide) has revealed to decline mucus viscosity in CF airway. It is presently being tested in phase IIb in Europe and UK. It is often used either as a dry powder or fluid meant for nebulization [51].
Additional agents for rehydration of airway secretions comprise of bronchitol presently in phase 3 in USA and by now permitted in UK, Australia, and Russia (for patients >18 years); VX-371 (P1037) presently in phase 2 for obstructing sodium channel and delaying the length of hypertonic saline alone in subjects with cystic fibrosis [52]; GSK2225745 acting by calming ENaC during RNA intervention is ongoing to get in touch with the patients.
3.3.6.3 Nutrition
Liprotamase (Anthera AN-EPI 3332), perhaps pancreatic enzyme substitution for CF-associated pancreatic insufficiency, is undergoing phase 3 study [53].
AquADEKs-2 experience phase 2 may be a balanced blend of fat-soluble vitamins and numerous antioxidants as well as beta-carotene, mixed tocopherols, coenzyme Q10, mixed carotenoids, and minerals like zinc and selenium. Oral glutathione is being tried in phase 2 because this antioxidant is mainly for usual lung GIT operation.CF patients have delineated inferior glutathione levels and oral glutathione is probable to enhance growth and reduce gut inflammation [54]. Additional agents like protein burlulipase for pancreatic deficiency, lubiprostone for constipation and roscovitine for pulmonary contamination are presently being assessed at numerous centers.
3.4 Perspective
Gene engineering skills and novel molecular objective could also be explored further examination of this region led to the identification of the gene itself and the prediction of the amino acid sequence of the encoded protein, which was termed the CFTR (cystic fibrosis transmembrane conductance regulator). Assistance of current biology moves toward like DNA engineering, systems biology, metabolomics, ailment modeling, and intracellular protein kinetics could facilitate to unknot novel pathways and networks associated with cystic fibrosis and ultimately novel therapeutic targets. Also, the focus ought not to be reduced on new treatment techniques, new medicine for symptomatic progression and difficulty avoidance.
References
Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, Rector MV, Suter MJ, Kao S, McLennan G, Tearney GJ, Zabner J, McCray PB Jr, Welsh MJ (2010) Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med 182(10):1251–1261
Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA IV, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr, Zabner J, Welsh MJ (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2(29):29ra31
Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordoñez CL, Campbell PW, Ashlock MA, Ramsey BW (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363(21):1991–2003
Muhlebach MS, Stewart PW, Leigh MW, Noah TL (1999) Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am J Respir Crit Care Med 160(1):186–191
Sly PD (2013) Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 368(21):1963–1970
Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, Dehlink E, Nachbaur E, Horak F, Jaksch P, Döring G, Crameri R, Jung A, Rochat MK, Hörmann M, Spittler A, Klepetko W, Akdis CA, Szépfalusi Z, Frischer T, Eiwegger T (2013) A Th17-and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 187(6):621–629
Lloyd-Still JD (1994) Crohn’s disease and cystic fibrosis. Dig Dis Sci 39(4):880–885
Kumar S, Tana A, Shankar A (2014) Cystic fibrosis-what are the prospects for a cure? Eur J Intern Med 25(9):803–807
WHO (2018) Genes human disease-cystic fibrosis. WHO, Geneva, Switzerland
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922):1066–1073
Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J (2012) Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487(7405):109
Vonberg RP, Gastmeier P (2005) Isolation of infectious cystic fibrosis patients: results of a systematic review. Infect Cont Hosp Epidemiol 26(4):401–409
Liou TG, Adler FR, Fitzsimmons SC, Cahill BC, Hibbs JR, Marshall BC (2001) Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol 153(4):345–352
Hart N, Polkey MI, Clément A, Boulé M, Moxham J, Lofaso F, Fauroux B (2002) Changes in pulmonary mechanics with increasing disease severity in children and young adults with cystic fibrosis. Am J Respir Crit Care Med 166(1):61–66
Saadane A, Soltys J, Berger M (2005) Role of IL-10 deficiency in excessive nuclear factor-κB activation and lung inflammation in cystic fibrosis transmembrane conductance regulator knockout mice. J Allergy Clin Immunol 115(2):405–411
Antonelli M, Midulla F, Tancredi G, Salvatori FM, Bonci E, Cimino G, Flaishman I (2002) Bronchial artery embolization for the management of nonmassive hemoptysis in cystic fibrosis. Chest 121(3):796–801
Orenstein DM, Winnie GB, Altman H (2002) Cystic fibrosis: a 2002 update. J Pediatr 140(2):156–164
Marchand E et al (2001) Frequency of cystic fibrosis transmembrane conductance regulator gene mutations and 5T allele in patients with allergic bronchopulmonary aspergillosis. Chest 119(3):762–767
Venarske DL, de Shazo RD (2002) Sinobronchial allergic mycosis: the SAM syndrome. Chest 121(5):1670–1676
Elphick HE, Southern KW (2016) Antifungal therapies for allergic bronchopulmonary aspergillosis in people with cystic fibrosis. Cochrane Database Syst Rev (11):CD002204
Mange EJ, Mange AP (1999) Basic human genetics. Sinauer Associates, Sunderland, MA
Stites SW, Plautz MW, Bailey K, O’Brien-Ladner AR, Wesselius LJ (1999) Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am J Respir Crit Care Med 160(3):796–801
Taussig L (1999) Pediatric respiratory medicine. Mosby, St Louis, MO
Brunzell C, Schwarzenberg SJ (2002) Cystic fibrosis-related diabetes and abnormal glucose tolerance: overview and medical nutrition therapy. Diabet Spect 15(2):124–127
Moran A, Hardin D, Rodman D, Allen HF, Beall RJ, Borowitz D, Brunzell C, Campbell PW III, Chesrown SE, Duchow C, Fink RJ, Fitzsimmons SC, Hamilton N, Hirsch I, Howenstine MS, Klein DJ, Madhun Z, Pencharz PB, Quittner AL, Robbins MK, Schindler T, Schissel K, Schwarzenberg SJ, Stallings VA, Zipf WB (1999) Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract 45(1):61–73
Moss RB (2002) Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest 121(1):55–63
Konstan MW, Flume PA, Kappler M, Chiron R, Higgins M, Brockhaus F, Zhang J, Angyalosi G, He E, Geller DE (2011) Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: the EAGER trial. J Cyst Fibros 10(1):54–61
Flume PA, O’Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ Jr, Willey-Courand DB, Bujan J, Finder J, Lester M, Quittell L, Rosenblatt R, Vender RL, Hazle L, Sabadosa K, Marshall B, Cystic Fibrosis Foundation, Pulmonary Therapies Committee (2007) Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 176(10):957–969
Salvatore D, D’Andria M (2002) Effects of salmeterol on arterial oxyhemoglobin saturations in patients with cystic fibrosis. Pediatr Pulmonol 34(1):11–15
Robinson M, Regnis JA, Bailey DL, King M, Bautovich GJ, Bye PT (1996) Effect of hypertonic saline, amiloride, and cough on mucociliary clearance in patients with cystic fibrosis. Am J Respir Crit Care Med 153(5):1503–1509
Quan JM, Tiddens HA, Sy JP, McKenzie SG, Montgomery MD, Robinson PJ, Wohl ME, Konstan MW, Pulmozyme Early Intervention Trial Study Group (2001) A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. J Pediatr 139(6):813–820
McIlwaine MP, Alarie N, Davidson GF, Lands LC, Ratjen F, Milner R, Owen B, Agnew JL (2013) Long-term multicentre randomised controlled study of high frequency chest wall oscillation versus positive expiratory pressure mask in cystic fibrosis. Thorax 68(8):746–751
Colombo C, Ellemunter H, Houwen R, Munck A, Taylor C, Wilschanski M, ECFS (2011) Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros 10:S24–S28
Stern RC, Eisenberg JD, Wagener JS, Ahrens R, Rock M, do Pico G, Orenstein DM (2000) A comparison of the efficacy and tolerance of pancrelipase and placebo in the treatment of steatorrhea in cystic fibrosis patients with clinical exocrine pancreatic insufficiency. Am J Gastroenterol 95(8):1932
Borowitz D, Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, Spear SL, Michel SH, Parad RB, White TB, Farrell PM, Marshall BC, Accurso FJ (2009) Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr 155(6):S73–S93
Vertex Pharmaceuticals Inc (2012) Kalydeco™ (ivacaftor). Product Information. Cambridge
Ramsey BW (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365(18):1663–1672
Davies JC (2013) Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 187(11):1219–1225
Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, Southern KW, Robertson S, Green Y, Cooke J, Rosenfeld M, KIWI Study Group (2016) Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 4(2):107–115
Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, Sagel SD, Khan U, Mayer-Hamblett N, Van Dalfsen JM, Joseloff E, Ramsey BW, GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network (2014) Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med 190(2):175–184
(2016) http://www.who.int/genomics/publications/en/HGN_WB_04.02_report.pdf
Whiting P, Burgers L, Westwood M, Ryder S, Hoogendoorn M, Armstrong N, Allen A, Severens H, Kleijnen J (2014) Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess 18(18):1–106
Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci 108(46):18843–18848
Kopeikin Z, Yuksek Z, Yang HY, Bompadre SG (2014) Combined effects of VX-770 and VX-809 on several functional abnormalities of F508del-CFTR channels. J Cyst Fibros 13(5):508–514
Clancy J, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, De Boeck K, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordoñez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW (2012) Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67(1):12–18
Ramsey B, Elborn S (2014) Effect of lumacaftor in combination with ivacaftor in patients with cystic fibrosis who are homozygous for F508del-CFTR: pooled results from the phase 3 TRAFFIC and TRANSPORT studies. In: The 28th Annual North American Conference of the Cystic Fibrosis Foundation, Atlanta, GA
Wainwright CE (2015) Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 373(3):220–231
Rafeeq MM, Murad HAS (2017) Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med 15(1):84
Gilead Sciences (2018) A phase 2b, dose-ranging study of the effect of GS-5745 on FEV1 in adult subjects with cystic fibrosis
Steven Rowe M, Stuart Elborn MD (2019) A phase 2, multicenter, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy, safety, and tolerability of CTX-4430 administered orally once-daily for 48 weeks in adult patients with cystic fibrosis
Tacjana Pressler PM (2017) A double-blind, randomized, placebo-controlled cross over study of inhaled alginate oligosaccharide (OligoG) administered for 28 days in subjects with cystic fibrosis
Vertex Pharmaceuticals Inc (2017) A phase 2a, randomized, double-blind, placebo-controlled, incomplete block, crossover study to evaluate the safety and efficacy of VX-371 in subjects aged 12 years or older with cystic fibrosis, homozygous for the F508del-CFTR mutation, and being treated with Orkambi
Anthera Pharmaceuticals (2018) A phase 3, open-label study evaluating the efficacy and safety of liprotamase in subjects with cystic fibrosis-related exocrine pancreatic insufficiency
Sarah J, Schwarzenberg M, Sarah J Schwarzenberg MD (2018) A multi center placebo controlled double blind randomized study evaluating the role of oral glutathione on growth parameters in children with cystic fibrosis
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Verma, P., Vishwakarma, V.K., Paswan, S.K., Rao, C.V., Srivastava, S. (2020). Cystic Fibrosis: Biology and Therapeutics. In: Rayees, S., Din, I., Singh, G., Malik, F. (eds) Chronic Lung Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-15-3734-9_3
Download citation
DOI: https://doi.org/10.1007/978-981-15-3734-9_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-3733-2
Online ISBN: 978-981-15-3734-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)