
Abstract
Hepatitis B virus (HBV) is a DNA virus, belonging to the Hepadnaviridae family. It is a partially double-stranded DNA virus with a small viral genome (3.2 kb). Chronic HBV infection remains a global public health problem. If left untreated, chronic HBV infection can progress to end-stage liver disease, such as liver cirrhosis and hepatocellular carcinoma (HCC). In recent years, tremendous advances in the field of HBV basic and clinical research have been achieved, ranging from the HBV biological characteristics, immunopathogenesis, and animal models to the development of new therapeutic strategies and new drugs against HBV. In this overview, we begin with a brief history of HBV discovery and treatment milestones. We then briefly summarize the HBV research advances, which will be detailed in the following chapters.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Discovery of HBV
As early as the 1950s, clinicians observed the hepatitis that occurred after blood transfusion and proposed the concept of serum hepatitis [1]. In 1964, Blumberg and Alter collected blood samples from all over the world for the study of lipoprotein polymorphisms and serendipitously observed an unusual reaction between serum from a transfused hemophilic patient and an Australian aborigine, and the new antigen was designated as the “Australian antigen” [2, 3]. By 1967, accumulating evidences from Blumberg’s group and other research groups showed a strong correlation between the presence of the Australian antigen and serum hepatitis [4,5,6,7,8]. In 1970, David Dane used electron microscope to inspect the isolated virus particles from serum of patients with Australian antigen-associated hepatitis and demonstrated the famous “Dane particles” as the complete virus (42 nm in diameter) [9]. The outer surface protein of the Dane particle is the Australian antigen, which was later officially named by the World Health Organization (WHO) as the HBV surface antigen (HBsAg). In 1976, Blumberg was awarded the Nobel Prize in Physiology or Medicine for his scientific achievement in the discovery of HBV and his visionary concept in developing the first generation of plasma-derived HBsAg vaccine. In 1979, Galibert completed the whole genome sequence of HBV and demonstrated it as a partially double-stranded DNA virus with approximately 3200 base pairs of nucleotides in length [10], thus setting the stage for the second generation of HBV vaccine made from genetic engineering.
The development of HBV immunization began in 1971 by using HBV immunoglobulins to prevent HBV infection. In 1978, the efficacy of the first generation of HBV vaccine consisting of HBsAg particles made from the plasma of HBV carriers was successfully validated in clinical trials [11]. In 1979–1982, using modern molecular cloning methods, William Rutter cloned the HBsAg gene in Escherichia coli (E. coli) to express the HBsAg protein [12], thus opening the door for recombinant HBV vaccine [13]. Subsequently, the HBsAg gene was successfully transferred to yeast for the mass production of HBsAg as HBV vaccine [14]. In 1984, the yeast-expressed HBsAg vaccine prepared by Merck company was used to vaccinate chimpanzees, demonstrating that the vaccinated chimpanzees were totally protected from intravenous HBV challenge of human serum source [15]. Subsequently, the yeast-expressed HBsAg vaccine was approved by the US Food and Drug Administration (FDA) in 1986. In 1989, Dr. Roy Vagelos, on the behalf of Merck, provided all the technology needed to manufacture HBV vaccine to China with the largest burden of HBV infection. In 1991, HBV vaccine was incorporated into the neonatal immunization program by WHO, which significantly reduced the global HBV infection rate. Since 1992, HBV vaccination has been incorporated into the national routine immunization program by the Chinese government, which led to a markedly decline of HBV infection in China.
HBsAg is the most important marker for the diagnosis of HBV infection. Since the discovery of the “Australian antigen” by Blumberg, a variety of HBsAg qualitative detection methods have been developed, from the detection of HBsAg by immunoelectrophoresis in the 1960s to the enzyme-linked immunosorbent assay in the 1980s. Quantitative detection of HBsAg was introduced as early as in the 1990s, but fully automated and high-throughput quantitative assays have only been recently available. Serum HBsAg quantification not only serves as a useful test in clinical practice to define the specific immunological conditions of the single HBV carrier during the dynamic natural history of HBV infection but also a prediction marker of virological response to antiviral therapy and long-term prognosis [16, 17]. In addition to HBsAg, other commonly used HBV serum markers include anti-HBs, HBeAg and anti-HBe, and anti-HBc. HBV nucleic acid detection is mainly to detect HBV DNA level. HBV DNA testing has also undergone a process from qualitative to quantitative testing. The most widely used HBV DNA quantification method is the real-time polymerase chain reaction (PCR) method. Serum HBV DNA levels reflect the viral replication, and monitoring HBV DNA levels during treatment allows evaluation of antiviral therapy effect.
In the 1990s, interferon-α was approved by the US FDA for the treatment of HBV infection, marking the treatment of HBV infection into the era of antiviral therapy [18]. In the late 1990s, lamivudine became the first nucleos(t)ide analog (NA) approved for anti-HBV treatment, representing the treatment of HBV infection into the era of NAs [19]. At the beginning of the twenty-first century, the long-acting pegylated IFN (Peg-IFN) and the potent and low-resistant NAs entecavir (ETV) and tenofovir disoproxil fumarate (TDF) were approved for HBV treatment [18]. Long-term antiviral treatment with full suppression of serum HBV DNA to undetectable levels not only halts hepatic inflammation but also decreases the incidence of liver cirrhosis and HCC.
2 Epidemiology and Natural History of HBV Infection
HBV infection remains the most common chronic viral infection in the world. WHO estimates that globally, 2 billion people have been infected with HBV. In 2015, an estimated 257 million persons, or 3.5% of the world population, were living with chronic HBV infection [20]. About 887,000 people die each year from HBV-related liver disease with approximately half coming from China. In 2006, the epidemiology serosurvey of HBV in China revealed that the prevalence of HBsAg for population aged 1–59 years was 7.18% [21, 22]. Based on this calculation, there were about 93 million HBsAg carriers in China, of which approximately 20 million are chronic hepatitis B (CHB) patients [23]. The latest epidemiological data showed that the estimated national HBsAg prevalence in the general population was 6.1% in 2016 [24].
The natural history of HBV infection depends on the interaction among the virus replication and evolution, the host immune response, and the environment factors. The age at which HBV is infected is the most important factor affecting chronicity. The risks of progression from acute to chronic HBV infection are approximately 95% if the infection occurs during the perinatal period, 20%–30% in children aged 1–5 years, and less than 5% in adults. The natural history of chronic HBV infection can be characterized into four phases: (1) immune-tolerant phase, (2) HBeAg-positive immune-active phase, (3) inactive CHB phase, and (4) HBeAg-negative immune reactivation phase [25, 26]. In the immune-tolerant phase, the HBV-infected subjects are characterized by high levels of HBV DNA, positive HBeAg, but normal ALT levels and the absence of significant inflammation or fibrosis in the liver. HBV-infected subjects in this phase can be defined as HBV carriers. In the HBeAg-positive immune-active phase, the HBV-infected patients are characterized by elevated ALT and HBV DNA levels together with liver injury by liver histology, and therefore, those patients are defined as HBeAg-positive CHB patients [27]. The transition from the immune-active phase to the immune-inactive phase is reflected by the seroconversion from HBeAg to antibody to HBeAg (anti-HBe), the normal ALT levels, and the low or undetectable HBV DNA levels. However, around 10–30% of those patients who have HBeAg seroconversion continue to have elevated ALT and high HBV DNA levels and, therefore, are defined as HBeAg-negative CHB patients [27]. Most of these patients have mutations in the pre-core or core promoter region. Patients with sustained liver injury and persistent HBV replication are prone to develop liver cirrhosis and HCC.
HBV infection not only leads to acute and chronic hepatitis but also is one of the most important etiological factors for liver cirrhosis and HCC. In the absence of antiviral treatment, the annual incidence of liver cirrhosis in CHB patients is 2–10% [28], the annual risk of progression from compensated to decompensated cirrhosis is 3–5%, and the 5-year survival rate in patients with decompensated cirrhosis is only 14%–35% [29]. The annual incidence of HCC in non-cirrhotic patients is about 0.5%–1.0% [29], whereas in cirrhotic patients, the annual incidence of HCC increases to 3–6% [28, 30, 31]. Liver cirrhosis has been classified by WHO as one of the top 10 causes of death in low-middle-income countries, and HCC as one of the top 10 causes of death in upper-middle-income countries [32].
3 Biological Characteristics and Immunopathogenesis of HBV
3.1 HBV Genome and Life Cycle
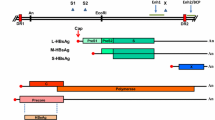
HBV particles are composed of the HBV genome, nucleocapsid, and the envelope proteins. The HBV genome is a partially double-stranded DNA with approximately 3200 base pairs. The longer-strand DNA is complementary to pregenomic RNA (pgRNA) and therefore is designated as minus (−) strand, whereas the shorter strand is designated as plus (+) strand. The (−) strand contains four overlapping open reading frames (ORFs) (PreC/C, P, PreS/S, and X). Under the joint regulation of the four promoters (the core promoter, the PreS1 promoter, the PreS2/S promoter, and the X promoter) and two enhancers (EnhI and EnhII), four distinct classes of HBV transcripts are transcribed: the 3.5 kb PreC/C mRNA, the 2.4 kb PreS1 mRNA, the 2.1 kb PreS2/S mRNA, and the 0.7 kb X mRNA. The PreC/C ORF is responsible for encoding HBeAg and HBcAg; the P ORF encodes the HBV DNA polymerase; the PreS/S ORF encodes the large (L), the middle (M), and the small (S) envelope proteins; and the X ORF is responsible for encoding the X protein (HBx).
The life cycle of HBV involves the viral entry into host cells; rcDNA’s entry into the nucleus to form cccDNA; the expressions of viral RNAs and proteins; viral capsid assembly; reverse transcription and rcDNA formation; and, finally, viral packaging, maturation, and budding. Mediated by the antigenic loop (AGL) present in the S domain of HBsAg, HBV is initially attached to heparan sulfate proteoglycan (HSPG) on the surface of hepatocyte membrane [33]. Subsequently, through the preS1 region of the L protein, HBV is tightly bound to the sodium-taurocholate cotransporting polypeptide (NTCP) on the surface of hepatocytes [34, 35]. After HBV entry into the hepatocytes, the nucleocapsid is released, and the rcDNA of HBV enters the nucleus to form cccDNA, which resides inside the hepatocyte as a microchromosome [36]. Current anti-HBV NAs have no direct effect on cccDNA, which explains why HBV infection is currently manageable but still incurable by current treatments. Under the action of host RNA polymerase II, cccDNA serves as the template for transcription of the abovementioned four HBV transcripts and the translation of seven viral proteins. Of note, in addition to encode the translation of HBcAg, HBeAg, and DNA polymerase, the 3.5 kb pgRNA also has an important function as a template for viral reverse transcription and replication. During viral replication, HBV DNA polymerase binds to the ε-stem-loop structure near the 5’ end of the pgRNA, forming a specific pgRNA-polymerase ribonucleoprotein (RNP) complex, which is encapsulated by the core antigen polypeptide dimer to form an immature nucleocapsid [37]. Under the catalysis of HBV DNA polymerase in the nucleocapsid, the 3.5 kb pregenomic RNA serves as the template for reverse transcription of the (−) strand DNA, which subsequently serves as the template for the synthesis of the (+) strand DNA, thus forming the progeny rcDNA. The recycling of the de novo synthesized rcDNA into the nucleus makes more cccDNA, maintaining the cccDNA reservoir. Double-stranded linear (dsl) DNA may be generated due to erroneous viral DNA replication [38]. Mature nucleocapsid and envelope protein aggregate in the endoplasmic reticulum to complete the packaging, maturation, and viral budding [39]. The discovery of HSPG and NTCP and their crucial roles in HBV entry into hepatocytes and the immature nucleocapsid containing the pgRNA and the dslDNA has significantly advanced our understanding of the HBV life cycle.
3.2 HBV Transcription and Translation
Using the reporter gene system containing the HBV promoter, a series of cis-acting transcriptional regulatory sequence elements and trans-acting DNA-binding proteins have been discovered. The 3.5 kb pgRNA is primarily regulated by cis-acting regulatory sequence elements within the HBV genomic enhancer I (EnhI/Xp) and enhancer II (EnhII/Cp). HBV EnhI is located within the ORF P, between the ORF S and X, and overlaps with the X promoter, which enhances the transcription of C, SPI, SPII, and X promoters. The transcription factors reported bound to EnhI/Xp include C/EBP, P53, IRF, NF1, HNF3, HNF4, RXR, PPAR, COUPTF, RFX1, AP1, CREB, and ATF2 [40, 41]. EnhII is located to the upstream of Cp. The transcription factors bound to HBV EnhII/Cp include SP1, RFX1, C/EBP, FTF, HLF, E4BP4, HNF4, HNF3, RXR, PPAR, COUPTF1, and ARP1 [40, 41].
HNF4, RXR, PPAR, FXR, and LRH1 are liver-enriched transcription factors capable of supporting HBV replication in non-hepatoma cells [41, 42]. The identification of these liver-enriched transcription factors contributes significantly to the hepatocyte-specific tropism of HBV. In addition to the transcription factors, some transcriptional co-activators (including PGC1, CBP, SRC1, and PRMT1) and co-repressors (including SBP) are also involved in the regulation of HBV transcription and replication [43,44,45]. In addition, other host proteins including APOBEC3B, PRMT5, and PRKAA/AMPK have also been reported to participate in the HBV transcription and replication [46,47,48].
The viral protein HBx can also act as a trans-acting factor possibly through interacting with certain host proteins to enhance HBV transcription and replication. It has been reported that HBx not only enhances HBV DNA replication but also activates the transcription of 3.5 kb HBV RNA. The augmentation of HBV transcription is always observed in parallel to HBV replication, strongly suggesting that the stimulation of HBV replication is mainly due to the enhanced transcription by HBx [49, 50]. It has also been suggested that HBx enhances HBV replication through posttranscriptional regulation mechanisms, such as the regulation of intracellular calcium signaling and activation of downstream Pyk2/FAK kinase [51]. Recent studies have found that HBx can promote the degradation of SMC5/6 to enhance HBV replication [52, 53].
3.3 Immunopathogenesis of HBV
The immunopathogenesis of CHB is complex and has not yet been fully elucidated. Numerous studies have shown that HBV is a non-cytopathic hepatotropic virus. Persistent liver inflammation plays an important role in the progression of CHB to cirrhosis and HCC. The innate immune response plays a role in the early stages of HBV infection and induces subsequent adaptive immune responses [54]. HBV inhibits the innate immune response by interfering with Toll-like receptors and retinoic acid receptors through the viral components such as HBeAg and HBx [55,56,57,58,59]. Although the innate immune response of CHB patients is impaired, antiviral cytokines of the innate immune pathway can still inhibit HBV. This is supported by the ability of IFNα or TLR agonists to induce innate immunity to inhibit HBV replication [60]. At present, many novel anti-HBV drugs are being developed to inhibit HBV replication through the upregulation of innate immunity.
Many CHB patients are characterized with impaired frequency and function of myeloid dendritic cells (mDC) and plasmacytoid dendritic cells (pDC) in their peripheral blood. Immature mDC and pDC with decreased capacity to produce IFN-α are associated with tolerogenic T-cell responses and HBV persistence [61]. HBV-specific immune response plays a major role in HBV clearance. The virus-specific effector CD8+ T-cell response is central to HBV pathogenesis. Major histocompatibility complex (MHC) class I-restricted CD8+ cytotoxic T lymphocytes can induce hepatocyte apoptosis and secrete IFN-γ, thus inhibiting HBV replication through non-cytolytic mechanisms. In chronic HBV infection, apoptosis-prone HBV-specific CD8+ T cells, the reduced cytokine function and proliferative capacity, and the T-cell exhaustion contribute to HBV persistence [60].
4 Current Antiviral Drugs Against HBV and New Antiviral Drugs Under Development
4.1 Current Antiviral Drugs Against HBV
Current antiviral drugs for HBV include IFNs and NAs. The first milestone in CHB treatment is the utilization of IFN-α (IFN-α-2a, IFN-α-2b, and IFN-α-1b) produced by recombinant DNA technology. Meta-analysis has shown that conventional IFN-treated HBeAg-positive CHB patients have higher HBeAg seroconversion and HBsAg clearance, but lower cirrhosis and HCC incidence than the patients without IFN treatment [62, 63]. However, due to its limited efficacy, low sustained viral response, and frequent injections, the conventional IFN has been largely replaced with the long-acting Peg-IFN, which was approved by the US FDA for HBV treatment in 2002. International multicenter randomized controlled clinical trials showed that HBeAg-positive CHB patients treated with Peg-IFN-α-2a for 48 weeks resulted in HBeAg seroconversion rate of 32% at 24 weeks posttreatment follow-up [64] and the HBeAg seroconversion reached 41% at 48 weeks posttreatment follow-up [65]. Similar HBV DNA inhibition and HBeAg seroconversion can also be achieved in HBeAg-positive CHB patients treated with Peg-IFN-α-2b [66].
The second milestone in CHB treatment is the use of the lamivudine (LAM), which revolutionized the treatment of CHB. LAM exhibits good antiviral effects in HBeAg-positive and HBeAg-negative CHB patients, even in CHB patients with advanced liver diseases. However, resistance to LAM can be easily developed. It has been reported that LAM resistance can reach up to 80% after 5 years of treatment [67]. Following LAM, adefovir dipivoxil (ADV) was the second antiviral drug approved for anti-HBV therapy. However, like LAM, ADV has low genetic barrier and drug resistance can be easily developed and another drawback for ADV is its nephrotoxicity [18]. Telbivudine (LdT) is another nucleoside analog for antiviral treatment of CHB. With a proven safety profile, LdT is a pregnancy category B medication and has been applied to prevent mother-to-child-transmission (MTCT) in mothers with HBV infection [68,69,70]. However, similar with LAM and ADV, long-term LdT treatment leads to high rate of drug resistance (34% after 3-year TBV therapy) [71].
The third milestone in CHB treatment is the clinical use of the potent and low-resistant NAs: ETV and TDF. Both ETV and TDF can strongly inhibit HBV replication and have high genetic barrier to drug resistance. It has been shown that the 3-year cumulative ETV resistance rate is 1.7–3.3% [72]. Resistance to TDF was not detected in CHB patients after 6 years of TDF monotherapy [73]. Most CHB patients with long-term use of ETV or TDF can achieve histological improvement and even the reversal of liver fibrosis. Tenofovir alafenamide (TAF) has been recently approved for treatment of CHB in adults. Compared to TDF, TAF has a better safety profile (lower rates of bone and renal abnormalities) and similar antiviral efficacy. Therefore, current international guidelines recommend the use of Peg-IFN, ETV, TDF, and TAF as first-line therapeutic options for CHB, while the NAs with low genetic barriers (LAM, ADV, and LdT) are no longer recommended as the first-line antiviral agents in treatment-naïve CHB patients.
4.2 Optimization Treatment Strategies Based on Current Antiviral Drugs
Currently, most CHB patients treated with NAs or Peg-IFN monotherapy can achieve sustained viral suppression, whereas the difficulty in achieving HBsAg loss or the elimination of cccDNA remains the major obstacle for the cure of CHB. Theoretically, the combination of NAs and Peg-IFN may have a synergistic therapeutic effect to enable more CHB patients achieving HBsAg loss. Many clinical studies have been conducted to investigate the efficacies of different optimization strategies of NAs and Peg-IFN combination [64, 74,75,76,77,78,79,80,81,82,83].
One of the combination strategies is the simultaneous administration of NAs and Peg-IFN (the de novo combination). However, the initial de novo combination of LAM plus Peg-IFN and ADV plus Peg-IFN showed less-than-desirable results in treatment-naïve patients [64, 74, 75]. Recently, the de novo combination of TDF and Peg-IFN in treatment-naive CHB patients for 48 weeks led to increased rate of HBsAg loss at week 72 (9.1%) than those receiving Peg-IFN (2.8%) or TDF (0%) alone [78]. However, a recent randomized controlled, open-label study did not support the advantage of de novo combination of NA and Peg-IFN in CHB patients [77].
The other strategy to combine NA and Peg-IFN is the sequential combination, which means the “add-on” or “switch-to” strategy to CHB patients who are already on NA treatment. The “early add-on” strategy was investigated in the ARES study by comparing 24 weeks of ETV followed by 24 weeks of Peg-IFN add-on versus 48 weeks of ETV monotherapy for treatment-naïve HBeAg-positive CHB patients [79, 80]. The results showed no favorable effect of the combination strategy. The “late add-on” strategy was investigated in the PEGAN study enrolling only HBeAg-negative CHB patients with undetected HBV DNA by at least 1 year of NA treatment [81]. In the PEGAN study, patients were randomized to either continue NA or add on Peg-IFN treatment for 48 weeks. The results showed that HBsAg loss rates were significantly higher in the full-dose Peg-IFN add-on group than in the NA group [81].
The “early switch-to” strategy was investigated in the OSST study enrolling HBeAg-positive patients who had received 9 to 36 months of ETV therapy with HBeAg <100 PEIU/ml and HBV DNA ≤ 1000 copies/ml [82]. The enrolled patients in the OSST study were randomized to receive ETV or switch to Peg-IFN-α2a for 48 weeks. The “late switch-to” strategy was investigated in the New Switch study enrolling HBeAg-positive patients who achieved HBeAg loss and HBV DNA < 200 IU/mL with previous NA treatment (ADV, LAM or ETV). The patients were randomized to receive Peg-IFN for 48 or 96 weeks [83]. Both the OSST study and the New Switch study demonstrated a significant increase of HBsAg loss in the Peg-IFN switch group than the NA monotherapy group, and CHB patients with low baseline HBsAg level and on-treatment HBsAg response are more likely to benefit from the “switch-to” combination therapy [82, 83].
4.3 Advances in the Development of New Anti-HBV Drugs
The development of new anti-HBV drugs can be summarized into two categories: [1] new direct-acting antiviral drugs targeting the different steps of HBV life cycle, and [2] new indirect antiviral drugs modulating host immune response to inhibit or potentially eradicate HBV. The direct-acting antiviral drugs under development include HBV entry inhibitors, the therapeutic approaches targeting HBV cccDNA, RNA interference (RNAi)-based agents, capsid assembly inhibitors/modulators, new NAs targeting HBV polymerase, ribonuclease H (RNaseH) inhibitors, and HBsAg release inhibitors. The indirect antiviral drugs to suppress HBV via modulating the host innate or adaptive immunity include TLR-7 and TLR-8 agonists, retinoic acid-inducible gene 1 (RIG-1)/nucleotide-binding oligomerization domain protein 2 (NOD-2) agonists, programmed death receptor 1 (PD-1) inhibitors, and different kinds of therapeutic vaccines.
4.3.1 Direct Antiviral Drugs Against the Life Cycle of HBV
HBV entry inhibitor Myrcludex-B targeting the NTCP receptor is currently under phase II clinical trial. It has been shown that Myrcludex-B not only inhibits HBV DNA replication but also reduces cccDNA formation [84, 85]. Genome-editing technologies including transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9) system, which can be designed to target HBV cccDNA sequences, represent highly promising therapeutic tools to achieve the ultimate goal of curing CHB [86,87,88,89,90,91]. However, specific and efficient delivery of the gene editing sequences to HBV-infected hepatocytes and the risk of the intrinsic off-target effects of the genome-editing technologies are big challenges that need to be met in the future. The next-generation RNAi agents that target HBV transcripts to reduce viral antigen, HBV DNA, and cccDNA levels are also being developed. For example, the RNAi-based agent ARC-520 is currently in phase II clinical trial [92]. The nucleocapsid assembly inhibitors including NVR 3–778 [93,94,95], JNJ-6379 [96], GLS4 [97], and ABI-H0731 [98] are currently under different phases of clinical trials. Several new NAs currently under different phases of clinical trials include Besifovir (LB80380/BSV, a new acyclic nucleotide analog) [90, 99,100,101] and CMX157 (phase II) [102, 103]. The RNaseH inhibitors are also promising candidates for developing new anti-HBV drugs [104, 105]. The HBsAg release inhibitors include REP2139 or its analog REP2165 which have progressed to phase II clinical trials. The results showed that the combination of REP2139-Mg or REP2165-Mg (250 mg iv qW) with TDF and Peg-IFN led to HBsAg loss or seroconversion in a high proportion of CHB patients (http://replicor.com/, as of August 2019).
4.3.2 Indirect Antiviral Drugs that Modulate Host Immune Response to Control CHB
Indirect antiviral drugs that exhibit anti-HBV effect may function through modulating the host innate or adaptive immune response. The rationale behind the development of indirect antiviral drugs to modulate the host innate immunity is that the HBV-infected hepatocytes have impaired innate immune response; thus reactivating the host innate immune response may lead to the control of HBV infection. For example, Toll-like receptor 7 (TLR-7) and TLR-8 agonists are currently under clinical trials [106, 107]. In chronic HBV infection, T-cell immune tolerance and the T-cell exhaustion contribute to HBV persistence. Sustained high expression of the programmed cell death-1 (PD-1) in T cells plays an important role in T-cell exhaustion; thus, blocking the PD-1 pathway using the anti-PD-1 antibody (nivolumab) could be a major immunotherapeutic strategy to treat HBV infection [108, 109]. In addition, the therapeutic vaccines to treat HBV infection are also being developed [110,111,112,113,114].
In summary, since the discovery of HBV by Blumberg and Alter in the 1960s, tremendous advances in the field of HBV basic research, prevention, and clinical control of HBV infection have been achieved. A deeper understanding of the HBV life cycle and HBV immunopathogenesis, together with the development of cell culture models and animal models for HBV study, will further drive the development and testing of new therapeutic agents against HBV infection. Future treatment options for HBV cure may be a combination of multiple antiviral drugs, either the combination of different direct antiviral drugs targeting the various steps of HBV life cycle or the combination of direct antiviral drugs with host immune modulators. With the optimization treatment strategies based on current antiviral drugs and the newly developed antiviral agents, the ultimate cure of HBV infection will be achieved in the foreseeable future.
References
Maccallum FO (1946) Homologous serum hepatitis. Proc R Soc Med 39(10):655–657
Blumberg BS (1964) Polymorphisms of the serum proteins and the development of Iso-precipitins in transfused patients. Bull N Y Acad Med 40:377–386
Blumberg BS, Alter HJ, Visnich S (1965) A “New” antigen in leukemia sera. JAMA 191:541–546
Blumberg BS, Gerstley BJ, Hungerford DA, London WT, Sutnick AI (1967) A serum antigen (Australia antigen) in Down’s syndrome, leukemia, and hepatitis. Ann Intern Med 66(5):924–931
Bayer ME, Blumberg BS, Werner B (1968) Particles associated with Australia antigen in the sera of patients with leukaemia, down’s syndrome and hepatitis. Nature 218(5146):1057–1059
Prince AM, Fuji H, Gershon RK (1964) Immunohistochemical studies on the etiology of anicteric hepatitis in Korea. Am J Hyg 79:365–381
Prince AM (1968) An antigen detected in the blood during the incubation period of serum hepatitis. Proc Natl Acad Sci U S A 60(3):814–821
Prince AM (1968) Relation of Australia and SH antigens. Lancet 2(7565):462–463
Dane DS, Cameron CH, Briggs M (1970) Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1(7649):695–698
Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P (1979) Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281(5733):646–650
Szmuness W, Stevens CE, Harley EJ, Zang EA, Oleszko WR, William DC et al (1980) Hepatitis B vaccine: demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N Engl J Med 303(15):833–841
Valenzuela P, Gray P, Quiroga M, Zaldivar J, Goodman HM, Rutter WJ (1979) Nucleotide sequence of the gene coding for the major protein of hepatitis B virus surface antigen. Nature 280(5725):815–819
Edman JC, Hallewell RA, Valenzuela P, Goodman HM, Rutter WJ (1981) Synthesis of hepatitis B surface and core antigens in E. coli. Nature 291(5815):503–506
Valenzuela P, Medina A, Rutter WJ, Ammerer G, Hall BD (1982) Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature 298(5872):347–350
McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR (1984) Human hepatitis B vaccine from recombinant yeast. Nature 307(5947):178–180
Brunetto MR (2010) A new role for an old marker, HBsAg. J Hepatol 52(4):475–477
Sonneveld MJ, Zoutendijk R, Janssen HL (2011) Hepatitis B surface antigen monitoring and management of chronic hepatitis B. J Viral Hepat 18(7):449–457
Trepo C (2014) A brief history of hepatitis milestones. Liver Int 34(Suppl 1):29–37
Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z et al (1999) Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med 341(17):1256–1263
WHO (2017) Global hepatitis report, 2017. [updated April 2017; cited 2019 June 11]
Liang X, Bi S, Yang W, Wang L, Cui G, Cui F et al (2009) Epidemiological serosurvey of hepatitis B in China--declining HBV prevalence due to hepatitis B vaccination. Vaccine 27(47):6550–6557
Liang X, Bi S, Yang W, Wang L, Cui G, Cui F et al (2009) Evaluation of the impact of hepatitis B vaccination among children born during 1992–2005 in China. J Infect Dis 200(1):39–47
Lu FM, Zhuang H (2009) Management of hepatitis B in China. Chin Med J 122(1):3–4
Polaris Observatory C (2018) Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol 3(6):383–403
Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH et al (2016) AASLD guidelines for treatment of chronic hepatitis B. Hepatology (Baltimore, MD) 63(1):261–283
Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM et al (2018) Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology (Baltimore, MD) 67(4):1560–1599
European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL 2017 (2017) Clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol 67(2):370–398
Hsu YS, Chien RN, Yeh CT, Sheen IS, Chiou HY, Chu CM et al (2002) Long-term outcome after spontaneous HBeAg seroconversion in patients with chronic hepatitis B. Hepatology (Baltimore, MD) 35(6):1522–1527
Fattovich G, Bortolotti F, Donato F (2008) Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol 48(2):335–352
Chu CM, Liaw YF (2006) Hepatitis B virus-related cirrhosis: natural history and treatment. Semin Liver Dis 26(2):142–152
Chen YC, Chu CM, Yeh CT, Liaw YF (2007) Natural course following the onset of cirrhosis in patients with chronic hepatitis B: a long-term follow-up study. Hepatol Int 1(1):267–273
WHO (2018) The top 10 causes of death. [updated May 24 2018; cited 2019 June 11]
Sureau C, Salisse J (2013) A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology (Baltimore, MD) 57(3):985–994
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z et al (2012) Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. elife 1:e00049
Hayes CN, Zhang Y, Makokha GN, Hasan MZ, Omokoko MD, Chayama K (2016) Early events in hepatitis B virus infection: from the cell surface to the nucleus. J Gastroenterol Hepatol 31(2):302–309
Nassal M (2015) HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64(12):1972–1984
Jones SA, Hu J (2013) Protein-primed terminal transferase activity of hepatitis B virus polymerase. J Virol 87(5):2563–2576
Nassal M (2008) Hepatitis B viruses: reverse transcription a different way. Virus Res 134(1–2):235–249
Hu J, Liu K (2017) Complete and incomplete hepatitis B virus particles: formation, function, and application. Viruses 9(3):56
Tang H, Banks KE, Anderson AL, McLachlan A (2001) Hepatitis B virus transcription and replication. Drug News Perspect 14(6):325–334
Tang H, McLachlan A (2001) Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci U S A 98(4):1841–1846
Reese V, Ondracek C, Rushing C, Li L, Oropeza CE, McLachlan A (2011) Multiple nuclear receptors may regulate hepatitis B virus biosynthesis during development. Int J Biochem Cell Biol 43(2):230–237
Shalaby RE, Iram S, Cakal B, Oropeza CE, McLachlan A (2017) PGC1 alpha transcriptional adaptor function governs hepatitis B virus replication by controlling HBcAg/p21 protein-mediated capsid formation. J Virol 91(20)
Shalaby RE, Iram S, Oropeza CE, McLachlan A (2019) Peroxisome proliferator-activated receptor gamma coactivator family members competitively regulate hepatitis b virus biosynthesis. Virology 526:214–221
Reese VC, Moore DD, McLachlan A (2012) Limited effects of bile acids and small heterodimer partner on hepatitis B virus biosynthesis in vivo. J Virol 86(5):2760–2768
Chen Y, Hu J, Cai X, Huang Y, Zhou X, Tu Z et al (2018) APOBEC3B edits HBV DNA and inhibits HBV replication during reverse transcription. Antivir Res 149:16–25
Xie N, Yuan K, Zhou L, Wang K, Chen HN, Lei Y et al (2016) PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy 12(9):1507–1520
Zhang W, Chen J, Wu M, Zhang X, Zhang M, Yue L et al (2017) PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology (Baltimore, MD) 66(2):398–415
Gong DY, Chen EQ, Huang FJ, Leng XH, Cheng X, Tang H (2013) Role and functional domain of hepatitis B virus X protein in regulating HBV transcription and replication in vitro and in vivo. Viruses 5(5):1261–1271
Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F et al (2005) The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol 79(9):5548–5556
Bouchard MJ, Wang L, Schneider RJ (2006) Activation of focal adhesion kinase by hepatitis B virus HBx protein: multiple functions in viral replication. J Virol 80(9):4406–4414
Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK et al (2016) Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531(7594):386–389
Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X et al (2016) Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16(11):2846–2854
Guidotti LG, Chisari FV (2001) Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol 19:65–91
Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R et al (2009) Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology (Baltimore, MD) 49(4):1132–1140
Chen J, Wu M, Zhang X, Zhang W, Zhang Z, Chen L et al (2013) Hepatitis B virus polymerase impairs interferon-alpha-induced STA T activation through inhibition of importin-alpha5 and protein kinase C-delta. Hepatology (Baltimore, MD) 57(2):470–482
Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z et al (2010) The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol (Baltimore, MD: 1950) 185(2):1158–1168
Jiang J, Tang H (2010) Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1(12):1106–1117
Han Q, Zhang C, Zhang J, Tian Z (2011) Reversal of hepatitis B virus-induced immune tolerance by an immunostimulatory 3p-HBx-siRNAs in a retinoic acid inducible gene I-dependent manner. Hepatology (Baltimore, MD) 54(4):1179–1189
Bertoletti A, Ferrari C (2012) Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut 61(12):1754–1764
Isogawa M, Tanaka Y (2015) Immunobiology of hepatitis B virus infection. Hepatol Res 45(2):179–189
Lin SM, Yu ML, Lee CM, Chien RN, Sheen IS, Chu CM et al (2007) Interferon therapy in HBeAg positive chronic hepatitis reduces progression to cirrhosis and hepatocellular carcinoma. J Hepatol 46(1):45–52
Wong DK, Cheung AM, O’Rourke K, Naylor CD, Detsky AS, Heathcote J (1993) Effect of alpha-interferon treatment in patients with hepatitis B e antigen-positive chronic hepatitis B. A meta-analysis. Ann Intern Med 119(4):312–323
Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G et al (2005) Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med 352(26):2682–2695
Piratvisuth T, Lau G, Chao YC, Jin R, Chutaputti A, Zhang QB et al (2008) Sustained response to peginterferon alfa-2a (40 kD) with or without lamivudine in Asian patients with HBeAg-positive and HBeAg-negative chronic hepatitis B. Hepatol Int 2(1):102–110
Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y et al (2005) Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 365(9454):123–129
Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C et al (2003) Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 36(6):687–696
Han GR, Cao MK, Zhao W, Jiang HX, Wang CM, Bai SF et al (2011) A prospective and open-label study for the efficacy and safety of telbivudine in pregnancy for the prevention of perinatal transmission of hepatitis B virus infection. J Hepatol 55(6):1215–1221
Han GR, Jiang HX, Yue X, Ding Y, Wang CM, Wang GJ et al (2015) Efficacy and safety of telbivudine treatment: an open-label, prospective study in pregnant women for the prevention of perinatal transmission of hepatitis B virus infection. J Viral Hepat 22(9):754–762
Marcellin P, Chang TT, Lim SG, Sievert W, Tong M, Arterburn S et al (2008) Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology (Baltimore, MD) 48(3):750–758
Seto WK, Lai CL, Fung J, Wong DK, Yuen JC, Hung IF et al (2011) Significance of HBV DNA levels at 12 weeks of telbivudine treatment and the 3 years treatment outcome. J Hepatol 55(3):522–528
Yokosuka O, Takaguchi K, Fujioka S, Shindo M, Chayama K, Kobashi H et al (2010) Long-term use of entecavir in nucleoside-naive Japanese patients with chronic hepatitis B infection. J Hepatol 52(6):791–799
Kitrinos KM, Corsa A, Liu Y, Flaherty J, Snow-Lampart A, Marcellin P et al (2014) No detectable resistance to tenofovir disoproxil fumarate after 6 years of therapy in patients with chronic hepatitis B. Hepatology (Baltimore, MD) 59(2):434–442
Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R et al (2004) Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 351(12):1206–1217
Stelma F, van der Ree MH, Jansen L, Peters MW, Janssen HLA, Zaaijer HL et al (2017) HBsAg loss after peginterferon-nucleotide combination treatment in chronic hepatitis B patients: 5 years of follow-up. J Viral Hepat 24(12):1107–1113
Marcellin P, Ahn SH, Ma X, Caruntu FA, Tak WY, Elkashab M et al (2016) Combination of tenofovir disoproxil fumarate and peginterferon alpha-2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology 150(1):134–144. e10
de Niet A, Jansen L, Stelma F, Willemse SB, Kuiken SD, Weijer S et al (2017) Peg-interferon plus nucleotide analogue treatment versus no treatment in patients with chronic hepatitis B with a low viral load: a randomised controlled, open-label trial. Lancet Gastroenterol Hepatol 2(8):576–584
Marcellin P, Ahn SH, Chuang WL, Hui AJ, Tabak F, Mehta R et al (2016) Predictors of response to tenofovir disoproxil fumarate plus peginterferon alfa-2a combination therapy for chronic hepatitis B. Aliment Pharmacol Ther 44(9):957–966
Brouwer WP, Xie Q, Sonneveld MJ, Zhang N, Zhang Q, Tabak F et al (2015) Adding pegylated interferon to entecavir for hepatitis B e antigen-positive chronic hepatitis B: a multicenter randomized trial (ARES study). Hepatology (Baltimore, MD) 61(5):1512–1522
van Campenhout MJH, Brouwer WP, Xie Q, Guo S, Chi H, Qi X et al (2019) Long-term follow-up of patients treated with entecavir and peginterferon add-on therapy for HBeAg-positive chronic hepatitis B infection: ARES long-term follow-up. J Viral Hepat 26(1):109–117
Bourliere M, Rabiega P, Ganne-Carrie N, Serfaty L, Marcellin P, Barthe Y et al (2017) Effect on HBs antigen clearance of addition of pegylated interferon alfa-2a to nucleos(t)ide analogue therapy versus nucleos(t)ide analogue therapy alone in patients with HBe antigen-negative chronic hepatitis B and sustained undetectable plasma hepatitis B virus DNA: a randomised, controlled, open-label trial. Lancet Gastroenterol Hepatol 2(3):177–188
Ning Q, Han M, Sun Y, Jiang J, Tan D, Hou J et al (2014) Switching from entecavir to PegIFN alfa-2a in patients with HBeAg-positive chronic hepatitis B: a randomised open-label trial (OSST trial). J Hepatol 61(4):777–784
Hu P, Shang J, Zhang W, Gong G, Li Y, Chen X et al (2018) HBsAg loss with peg-interferon Alfa-2a in hepatitis B patients with partial response to Nucleos(t)ide analog: new switch study. J Clin Transl Hepatol 6(1):25–34
Blank A, Markert C, Hohmann N, Carls A, Mikus G, Lehr T et al (2016) First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J Hepatol 65(3):483–489
Blank A, Eidam A, Haag M, Hohmann N, Burhenne J, Schwab M et al (2018) The NTCP-inhibitor Myrcludex B: effects on bile acid disposition and Tenofovir pharmacokinetics. Clin Pharmacol Ther 103(2):341–348
Gaj T, Gersbach CA, Barbas CF 3rd. (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405
Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4):347–355
Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR (2012) Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol 86(17):8920–8936
Dong C, Qu L, Wang H, Wei L, Dong Y, Xiong S (2015) Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir Res 118:110–117
Kennedy EM, Bassit LC, Mueller H, Kornepati AVR, Bogerd HP, Nie T et al (2015) Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476:196–205
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14(1):49–55
Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D et al (2017) RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Trans Med 9(409)
Lam AM, Espiritu C, Vogel R, Ren S, Lau V, Kelly M et al (2019) Preclinical characterization of NVR 3-778, a first-in-class capsid assembly modulator against hepatitis B virus. Antimicrob Agents Chemother 63(1)
Klumpp K, Shimada T, Allweiss L, Volz T, Lutgehetmann M, Hartman G et al (2018) Efficacy of NVR 3-778, alone and in combination with pegylated interferon, vs entecavir in uPA/SCID mice with humanized livers and HBV infection. Gastroenterology 154(3):652–662. e8
Yuen MF, Gane EJ, Kim DJ, Weilert F, Yuen Chan HL, Lalezari J et al (2019) Antiviral activity, safety, and pharmacokinetics of capsid assembly modulator NVR 3-778 in patients with chronic HBV infection. Gastroenterology
Zoulim F, Yogaratnam J, Vandenbossche JJ, Moscalu I, Streinu-Cercel A, Lenz O et al (2019) Safety, pharmacokinetics and antiviral activity of a novel hepatitis B virus capsid assembly modulator, JNJ-56136379, in Patients with Chronic Hepatitis B. APASL. Adv Ther 36(9):2450–2462
Zhang HZX, Chen H et al (2018) Safety, Pharmacokinetics and anti-viral efficacy of novel core protein allosteric modifier GLS4 in patients with chronic hepatitis B: interim results from a 48 weeks phase 2a Study. AASLD 2018 Abstract LB-13
Ma XL Lalezari J et al (2019) Interim safety and efficacy results of the ABI-H0731 phase 2a program exploring the combination of ABI-H0731 with Nuc therapy in treatment-naive and treatment-suppressed chronic hepatitis B patients. EASL 2019 Abs LBO-06. Hepatology 70(1):e130
Lampertico P (2014) Oral antiviral therapy for hepatitis B: the case of besifovir, a new kid on the block with a long way to go. Gut 63(6):869–870
Lai CL, Ahn SH, Lee KS, Um SH, Cho M, Yoon SK et al (2014) Phase IIb multicentred randomised trial of besifovir (LB80380) versus entecavir in Asian patients with chronic hepatitis B. Gut 63(6):996–1004
Ahn SH, Kim W, Jung YK, Yang JM, Jang JY, Kweon YO et al (2019) Efficacy and safety of besifovir dipivoxil maleate compared with tenofovir disoproxil fumarate in treatment of chronic hepatitis B virus infection. Clin Gastroenterol Hepatol 17(9):1850–1859.e4
Feng S, Gao L, Han X, Hu T, Hu Y, Liu H et al (2018) Discovery of small molecule therapeutics for treatment of chronic HBV infection. ACS Infect Dis 4(3):257–277
Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, De Clercq E et al (2007) Evaluation of hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)propyl]- adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob Agents Chemother 51(10):3505–3509
Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F et al (2014) Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antivir Res 108:48–55
Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK et al (2017) Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antivir Res 143:205–217
Grant E, Joshi A, Ayithan N, Daffis S, Woo J, Lam T et al (2018) Pharmacodynamic response to oral administration of the selective toll-like receptor 8 agonist GS-9688 in healthy volunteers. Hepatology (Baltimore, MD) 68:270a–a
Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM et al (2013) GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 144(7).:1508–1517, 17.e1–17.e10
Wenjin Z, Chuanhui P, Yunle W, Lateef SA, Shusen Z (2012) Longitudinal fluctuations in PD1 and PD-L1 expression in association with changes in anti-viral immune response in chronic hepatitis B. BMC Gastroenterol 12:109
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C et al (2017) Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389(10088):2492–2502
Boni C, Janssen HLA, Rossi M, Yoon SK, Vecchi A, Barili V et al (2019) Combined GS-4774 and tenofovir therapy can improve HBV-specific T-cell responses in patients with chronic hepatitis. Gastroenterology 157(1):227–241.e7
Lok AS, Pan CQ, Han SH, Trinh HN, Fessel WJ, Rodell T et al (2016) Randomized phase II study of GS-4774 as a therapeutic vaccine in virally suppressed patients with chronic hepatitis B. J Hepatol 65(3):509–516
Martin P, Dubois C, Jacquier E, Dion S, Mancini-Bourgine M, Godon O et al (2015) TG1050, an immunotherapeutic to treat chronic hepatitis B, induces robust T cells and exerts an antiviral effect in HBV-persistent mice. Gut 64(12):1961–1971
Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT et al (2015) Present and future therapies of hepatitis B: from discovery to cure. Hepatology (Baltimore, MD) 62(6):1893–1908
Brahmania M, Feld J, Arif A, Janssen HL (2016) New therapeutic agents for chronic hepatitis B. Lancet Infect Dis 16(2):e10–e21
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (No.81772193).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Li, H. et al. (2020). Hepatitis B Virus Infection: Overview. In: Tang, H. (eds) Hepatitis B Virus Infection. Advances in Experimental Medicine and Biology, vol 1179. Springer, Singapore. https://doi.org/10.1007/978-981-13-9151-4_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-9151-4_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9150-7
Online ISBN: 978-981-13-9151-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)